

Abstract
Mutations in isocitrate dehydrogenases 1 and 2 (IDH) occur in the majority of WHO grade II and III gliomas. IDH1/2 active site mutations confer a neomorphic enzyme activity producing the oncometabolite D-2-hydroxyglutarate (D-2HG), which generates the glioma CpG island methylation phenotype (G-CIMP). While IDH1/2 mutations and G-CIMP are commonly retained during tumor recurrence, recent work has uncovered losses of the IDH1 mutation in a subset of secondary glioblastomas. Co-occurrence of the loss of the mutant allele with extensive methylation changes suggests a possible link between the two phenomena. Here, we utilize patient-derived IDH1R132H/WT glioma cell lines and CRISPR-Cas9 mediated gene knockout to model the genetic loss of IDH1R132H, and characterize the effects of this deletion on DNA methylation. After D-2HG production has been abolished by deletions within the IDH1 alleles, these models show persistent DNA hypermethylation at 7 CpG sites previously used to define G-CIMP-positivity in patient tumor samples. Despite these defining G-CIMP sites showing persistent hypermethylation, we observed a genome-wide pattern of DNA demethylation, enriched for CpG sites located within open sea regions of the genome as well as in CpG-island shores of transcription start sites, after loss of D-2HG production. These results suggest that inhibition of D-2HG from genetic deletion of IDH alleles is not sufficient to reverse hypermethylation of all G-CIMP defining CpG sites, but does result in more demethylation globally and may contribute to the formation of a G-CIMP-low like phenotype.
Keywords: IDH1, G-CIMP, D-2-hydroxyglutarate, glioma, DNA methylation
Introduction
Heterozygous missense mutations in isocitrate dehydrogenases 1 and 2 (IDHs) are among the most common genetic lesions in WHO grades II and III gliomas, and are responsible for the pathological accumulation of D-2-hydroxyglutarate (D-2HG) in these tumors [1–3]. D-2HG modulates the activity of alpha-ketoglutarate (α-KG) dependent enzymes due to structural similarity to α-KG, and by this mechanism causes pro-tumorigenic changes in chromatin structure and gene expression [4–6]. One of these IDH1 mutation-linked epigenetic changes is the glioma CpG island methylator phenotype (G-CIMP), which is a pattern of predominantly gene-promoter based CpG island hypermethylation found in WHO grade II, III, and IV glioma wherein IDH mutations commonly occur [7]. In previous reports, CpG promoter methylation in 7 different genes, most tightly linked to the proneural G-CIMP-positive status, were utilized to classify glioblastoma tumors as G-CIMP positive [7].
Although the G-CIMP pattern of methylation is usually stable at tumor recurrence, there are specific decreases in DNA methylation within G-CIMP positive tumors with important prognostic implications [8]. Specifically, among the G-CIMP positive astrocytic gliomas a pattern of hypomethylation termed G-CIMP-low has been identified in both low and high grade tumors [8]. This pattern shows enrichment for hypomethylation in CpG open seas [8], which are defined as regions more than 4 kilobases away from any annotated CpG island [9]. For the G-CIMP-low phenotype, patient tumors were classified using 131 CpG probes which were hypomethylated in a subset of IDH mutant-non-codel tumors that had been previously classified as G-CIMP positive [8]. Furthermore, G-CIMP-low status correlated with worse survival when compared to G-CIMP-high tumors [8, 10]. Understanding the genetic and environmental factors that contribute to the rise of the G-CIMP-low phenotype is therefore an important area of research for understanding glioma tumor progression and for prognostic purposes.
Recently, attempts to characterize progression of IDH1 mutant gliomas have identified copy number alterations in the IDH1 gene in secondary glioblastomas [11]. These copy number changes include losses of the wildtype allele of IDH1 [12], deletions of the mutant allele of IDH1 [11, 13], as well as amplifications of either allele [11]. Losses of the mutant IDH1 allele have also co-occurred with acquisition of a G-CIMP-low like DNA methylation phenotype [11], which has led to the hypothesis that loss of mutant IDH1-dependent D-2HG production is one path to a G-CIMP-low DNA methylation pattern [11]. Previous literature also suggests that oncogenic IDH1 mutations are not strictly required for continual proliferation of IDH1 mutant glioma cells in vitro [14, 15] and in in vivo orthotopic xenografts [11, 16]. We took advantage of this finding to engineer isogenic cell line models from patient-derived glioma cell lines with endogenous heterozygous IDH1R132H/WT, and generated knockouts of either the R132H or wildtype IDH1 allele. We and others have previously shown that loss of either mutant or wildtype IDH1 is sufficient to suppress D-2HG production [11, 17]. We utilized these genetically-engineered patient-derived models to ask whether the DNA methylation landscape in IDH1 mutant glioma relies on continual IDH1 mutation-dependent D-2HG production.
MATERIALS AND METHODS
Cell Culture
Cell Samples
Cell line IMA (also known as TB096 and TB09-0096-2), and cell line 08-0537 were isolated at the Preston Robert Tisch Brain Tumor Center at Duke University from a male Grade III anaplastic astrocytoma and secondary glioblastoma respectively and as previously described [17]. Adherent line IMA was cultured in a media containing a 1:1 combination of [(Stem Cell Technologies human complete Neurocult™ media supplemented with 20ng/mL EGF, 10ng/mL FGF, and 2ug/mL heparin) : (DMEM (Invitrogen cat#11965-082)] + 10% Invitrogen FBS16000-044 lot 168075 as previously described [17]. Cell line 08-0537 was grown in 1X human complete Neurocult™ media with 20ng/mL EGF, 10ng/mL FGF, and 2ug/mL heparin on laminin coated cell culture dishes for adherent culture. Cell media was refreshed every 4-6 days and cells were split approximately every 10 days or at 80% confluence. Cells were grown at 37°C and 5% CO2.
Cell Growth Assays
IMA cells were plated at 1000 cells per well in 6 well plates for long term growth assays or 2000 cells per well in 96 well plates for short term growth assays. For long term growth assays, plates were fixed in methanol and stained with 0.2% crystal violet solution before imaging plates on a Chemi-doc imager (Bio-Rad). Areas were determined in ImageJ using the Colony Area plugin. For short term growth assays cells were plated in 96-well black walled plates (Greiner), cultured for 6 days, and then processed following the manufacturer’s recommended protocol for the Cyquant assay (Invitrogen).
Cell Line authentication and mycoplasma testing
Genomic DNA was extracted from IMA, 08-0537, and derivative subclones for cell line authentication by STR profiling assay against matched patient blood genomic DNA. Mycoplasma testing was performed by the Duke Cell Culture facility using the MycoAlert PLUS test (Lonza) and all cells tested negative. Patient derived cell line IMA was used between passages 10-20 and Cas9-edited derived clones were utilized between passages 10-20 post-cloning. Cell Line 08-0537 was utilized between passages 20-32 and clones were utilized between passages 10-20 post-cloning.
Transfection of Patient Derived Glioma Cell Lines
Patient derived glioma cell lines with endogenous IDH1 mutations were transiently transfected in one of two ways. IMA and 08-0537 clones from experiment 1 were created using the Lonza 4D-Nucleofector X Unit using 20uL cuvette strips, program CA-137, and buffer P3 (Lonza). IMA clones from experiment 2 were produced using the Neon electroporator, 10uL R buffer, with program 1150V, 2 pulses, 30s pulse width (Invitrogen). All transfections were performed using 200,000 cells and 500ng of plasmid. Cells were allowed to recover following electroporation for 4 days by plating in full growth media in 24 well plates (1 electroporation per well and 500uL of media) before selecting for the transfected clones through flow cytometry for GFP positive cells at the Duke Flow Cytometry core facility. After sorting, cells were allowed to recover for 1 week before plating single clones using limiting dilution cloning. For cell line 08-0537, to overcome low plating efficiency, cells were initially cloned through first plating clones at 1000 cells/dish on a 10cm laminin coated plate wherein individual foci were eventually selected using Corning cloning cylinders, and promising polyclonal colonies were then passaged into limiting dilution plates to isolate true single clones.
Initial Screening and Allele Specific PCR of Single Clones
Initial screening of clones was performed using PCR primers that do not discriminate between mutant and wildtype IDH1 alleles to identify clones carrying insertions or deletions in either allele (Supplementary Table 4). After selection of promising clones, allele-specific PCR of single clones was performed to amplify either the wildtype or mutant IDH1 allele, and finally the resulting product was subjected to Sanger sequencing as reported previously [18].
Plasmid Constructs
pSpCas9(BB)-2A-GFP (PX458) was a gift from Feng Zhang (Addgene plasmid # 48138) [19], and guides were cloned into the vectors using FastDigest BbsI (Fermentas) following Feng Zhang’s Addgene supplementary cloning protocol and using synthesized oligonucleotides (IDT). The pCR-Blunt kit (Thermo-Scientific) was used according to the manufacturer’s cloning protocol.
Whole Exome-Sequencing
Cell line DNA and normal DNA isolated from patient blood was provided to Personal Genome Diagnostics Inc. for next-generation sequencing of captured coding regions of the genome as previously described [20].
Methylation Array Processing and Analysis
Genomic DNA was isolated from cells between passages 10 and 20 post-limiting dilution cloning using a Qiagen All Prep RNA/DNA kit following manufacturer’s recommended instructions. gDNA was then processed through the Zymo genomic DNA Clean & Concentrator kit and eluted into diH2O. Samples were further processed by the Duke Molecular Genomics Shared Resource using Zymo EZ DNA methylation kit and Illumina’s Infinium MethylationEPIC BeadChip following manufacturer’s recommended protocol. Arrays were captured using the Illumina iScan system and pre-processed in R version 3.5.2 using the ChAMP package (version 2.12.4) or minfi package (version 1.28.3) [21–23]. Briefly, ChAMP processing removes probes with detection p-values >.01, or <3 beads in 5% of samples, as well as SNP-related probes, multi-hit probes, and non-CpG probes. After ChAMP processing, 720551 probes passed filtering and were used for downstream analysis. Beta-mixture quantile dilation normalization method was used to correct for probe-type design bias [24]. To assess stability of findings in the face of different choices of normalization method we also processed the data through minfi and GenomeStudio using Functional normalization and Illumina’s proprietary normalization methods and results were consistent with ChAMP processed data. Differentially methylated probes were called using limma [25–27]. Copy number calls were performed by Conumee R package version 1.9.0 [28].
Western blotting
Total cell lysate in 1X RIPA buffer was quantitated by bicinchoninic acid assay and 20ug of total protein was resolved using 4-12% Bis-Tris-buffered SDS-PAGE gels (Invitrogen). Gels were soaked in transfer buffer + 20% methanol and transferred to PVDF membranes using a mini Trans-Blot transfer cell (Bio-Rad) following manufacturer’s protocol. After transfer, PVDF membranes were washed briefly in TBST and then blocked for 1 hour in TBST Protein-Free blocking buffer (Pierce catalog #37571). After blocking, primary antibodies were diluted either 1:500, for anti-IDH1R132H, or otherwise 1:1000 in blocking buffer and incubated overnight at 4°C. Membranes were washed and incubated with horseradish peroxidase (HRP)-conjugated secondary antibody 1:2000 for one hour in TBST. Detection was performed via chemiluminescence on a Bio-Rad ChemiDoc MP system. Antibodies used included total IDH1 antibody (D2H1, Cell Signaling #8137), COX IV antibody (3E11, Cell Signaling #4850), and IDH1R132H (DIA-H09, Dianova).
Analysis of D-2HG
Analysis of D-2HG in media and tissue was performed as previously described by the Duke Pharmacokinetics/Pharmacodynamics Laboratory [29]. Normalization to total protein for cell pellets was performed using bicinchoninic acid assay (Thermo-Fischer).
Statistical Analysis
Comparisons between two groups were evaluated using two-tailed Student’s t-test when only two groups were present. For categorical analysis of CpG genomic context a Chi square goodness of fit test was run against the frequency of CpG sites on the EPIC methylation chip to identify contexts enriched over CpG probe rate on the chip. Results are depicted as arithmetic mean +/− SD unless otherwise indicated in the figure legend. Experimenters were not systematically blinded to group assignment or outcome assessment.
Results
Generating isogenic IDH1R132H patient-derived cell lines
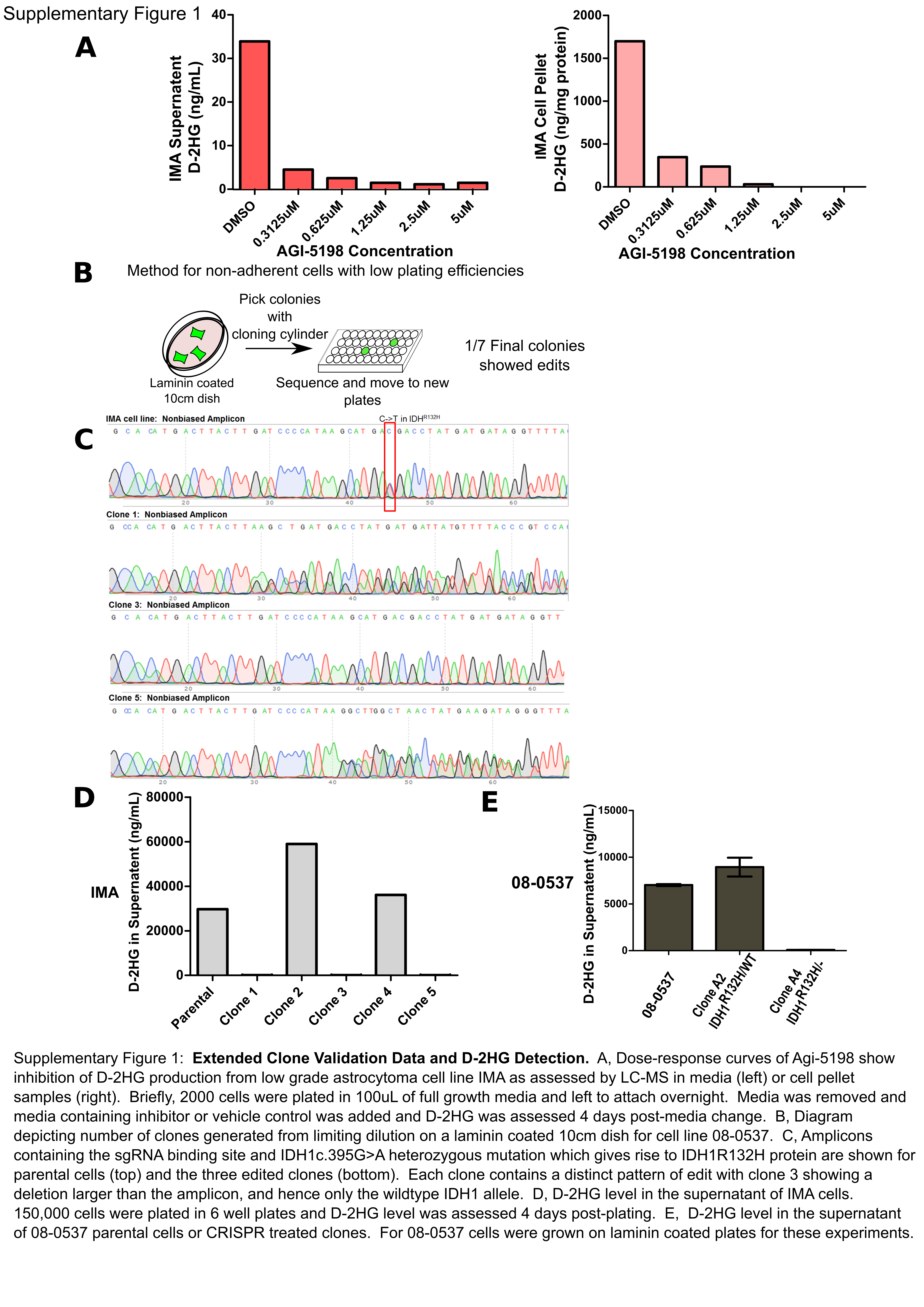
IDH1 mutations are thought to be an early initiating event in gliomagenesis [30], and mutant IDH1 activity has been shown to mediate transformation of normal human astrocytes [15]. However, previous studies using inhibitors of mutant IDH1 have demonstrated that the mutation can become non-essential for viability and continual proliferation in vitro [14] and in vivo [15, 16]. Using a mutant IDH1 astrocytoma cell line derived from a WHO Grade III anaplastic astrocytoma (IMA), which also has astrocytoma pathognomonic mutations in TP53 and ATRX (Supplementary Table 1), we assessed the effects of mutant IDH1 inhibitor AGI-5198 on proliferation and D-2HG production. As in previous reports, D-2HG production was suppressed by 90-99% when cells were treated with 1.25 μM of the selective IDH1R132H inhibitor AGI-5198 (Supplementary Figure 1A) [16, 31]. Despite this profound suppression of D-2HG, there was no difference in short term or long-term growth of this line, even upon treatment with 10 μM of AGI-5198 (Figure 1A and Figure 1B). In addition to the IMA cell line we have previously reported that the IDH1-mutant glioblastoma line 08-0537 does not show in vitro differences in proliferation upon treatment with AGI-5198, and has, similar to IMA, co-occurring mutations in TP53 and ATRX (Supplementary Table 1) [29].
Figure 1: IDH1R132H is not required by some glioma lines in vitro for proliferation.

A, Cell line IMA showed no difference in 6 day proliferation by Cyquant assay when treated with 5 μM Agi-5198 compared to DMSO vehicle treated cells used as control. B, Longer time course treatment of IMA with Agi-5198 does not inhibit growth from single cells into colonies, plate area as assessed in ImageJ Colony Area Plugin p=.23; Student’s two-tailed t-test C, Diagram showing electroporation protocol for transient expression of plasmid based transgenes in patient-derived glioma lines with endogenous IDH1R132H/WT genotype. D, IMA single cell clones with edits in the IDH1R132H allele producing putative non-sense transcripts do not have IDH1R132H protein that is detectable by Western blot; however, they do retain a band in total IDH1 protein. COX IV as loading control. E, Functionally, cells without detectable expression of IDH1R132H protein and with putative heterozygous knockouts of the IDH1R132H allele by Sanger sequencing do not produce pathological amounts of D-2HG as assessed by LC-MS in the cell pellets. F, Western blotting of 08-0537 clones isolated in this study for total IDH1, IDH1R132H, or COXIV loading control. IDH1R132H protein is still detectable in edited clone A4 suggesting wildtype allele specific editing. G, D-2HG levels as assessed by LC-MS in 08-0537 parental cells in the cell culture pellet. Values for cell culture pellets are normalized to total protein content of the cell pellet sample using bicinchoninic acid assay. For all experiments, N=3. Images are representative results. n.s. denotes non-significant p-value according to Student two-tailed t test when comparing the two conditions indicated by brackets.
We next used CRISPR/Cas9-mediated gene editing to generate knockouts of the IDH1 allele to create isogenic models of IDH1 loss. Because these lines are not amenable to chemical-based transfection methods, we utilized electroporation and transient expression of a plasmid encoding IDH1 targeting sgRNA and Cas9-2A-GFP (Figure 1C and Supplemental Figure 1B). To improve the likelihood of generating single cloned and edited cells we utilized flow cytometry to select for Cas9 activity using GFP expression as a surrogate marker, and strictly gated cells for GFP positivity before limiting dilution cloning. Screening IMA cells across two experimental attempts using two sgRNAs against IDH1 produced approximately 200 clones for further characterization, which ultimately yielded three distinct clones with three distinct edits in the mutant allele (Supplementary Figure 1C). Western blotting cell protein lysates using a specific anti-human IDH1R132H antibody or anti-total human IDH1 antibody showed IMA clones with nonsense mutations in the IDH1R132H allele did not have detectable IDH1R132H protein (Figure 1D and Supplementary Figure 2A). Furthermore, in IMA, D-2HG production levels were reduced by 100-1000 fold in clones with frameshift mutations in the IDH1R132H allele (Figure 1E and Supplementary Figure 1D). Likewise, cloning of 08-0537 cells generated seven sub-clones which contained at least one clone with a nonsense mutation in the wildtype IDH1 allele (Supplementary Figure 2B). 08-0537 clones with nonsense mutations in the wildtype IDH1 allele retained a band at the expected weight for mutant IDH1 in a Western blot specific for IDH1R132H but no longer produced elevated amounts of D-2HG (Figure 1F and 1G and Supplementary Figure 1E). This observation corroborates previous reports that heterozygosity for the IDH1 mutation is necessary for efficient production of D-2HG [17].
Loss of D-2HG is not sufficient to completely reverse the G-CIMP phenotype in patient-derived glioma cell lines
As D-2HG production is sufficient to induce a genome-wide pattern of CpG island hypermethylation and is necessary for the maintenance of this hypermethylation in previously described astrocyte models [32], we investigated whether our genetic models of IDH1 loss, of either WT or mutant allele, would show any widespread changes in CpG hypermethylation. We profiled CpG methylation using Illumina’s EPIC array for parental, IDH1 knockout, or unedited IMA clones and performed unsupervised hierarchical clustering of samples. We observed that clones with loss of D-2HG production clustered together while parental and non-edited clones clustered together, suggesting that loss of IDH1R132H may be sufficient to induce genome-wide methylation changes, and is consistent with the previous report in human astrocytes [32] (Figure 2A, and Supplementary Figure 3A). Similarly, in cell line 08-0537 the parental cells and non-edited clones cluster together, while the wildtype loss clone shows greater distance (Figure 2B). Next, we grouped the clones into edited and unedited subsets, and assessed the difference in beta value between clones for each CpG site individually using the DMP algorithm in the ChAMP processing pipeline. In our control condition we detected 6417 CpG sites showing differential methylation between parental and unedited clones, but between parental and edited clones we detected many more significantly differentially methylated probes, 16777CpGs in total. (Supplementary Table 2). We further refined this list by imposing a condition of delta Beta values ≤ −0.2 in all four D-2HG deficient clones across both IMA and 08-0537, and without recurrent delta Beta values ≤ −0.2 in any unedited clone. We identified a list of 971 probes most tightly associated with loss of mutant IDH1-mediated D-2HG production across both lines (Supplementary Table 3).
Figure 2: IDH1 edited clones retain G-CIMP methylation patterns at several G-CIMP defining sites but undergo greater genome-wide loss of methylation than matched IDH1R132H/WT clones.
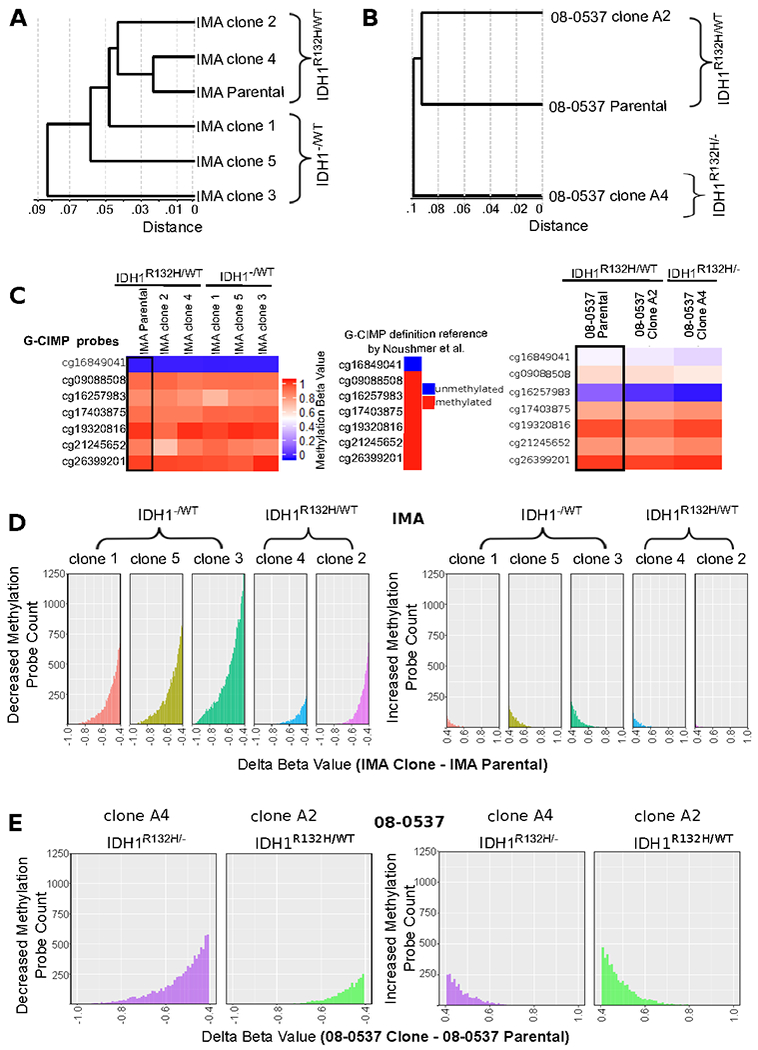
A, Unsupervised hierarchical clustering of IMA clones based on correlation of methylation beta values clusters IMA clones with IDH1R132H/WT genotype with parental IDH1R132H/WT IMA cells. Distance is given in 1-r (pearson correlation distance). B, Similarly, unsupervised hierarchical clustering of 08-0537 clones using the same methodology shows unedited clones cluster with parental cells. C, Observation that G-CIMP sites most indicative of G-CIMP retain G-CIMP linked alterations in methylation pattern in all clones irrespective of IDH1R132H allele status. Note that while 8 sites are typically used for G-CIMP identification on the 450K array only 7 of these sites are present in the methylation EPIC array used in this work. Therefore, conservatively 6/7 sites must display the G-CIMP pattern to enable a call of G-CIMP positive. Site cg16849041 is hypomethylated in G-CIMP and hypomethylated in all IMA samples analyzed. The other 6 sites are hypermethylated in G-CIMP and show hypermethylation (β≥.7) in all IMA clones analyzed. The 08-0537 cells may not fit the definition of G-CIMP positivity based on the 7 site definition; however, sites that are hypermethylated retain hypermethylation in all clones. D, Histogram plots of genome-wide beta value differences (Parental IMA cells – averaged values from either IDH1R132H/WT clones or IDH1−/WT clones) shows that methylation loss is the predominant change in methylation identified in the genome-wide pattern of clones; and, the IDH1−/WT cells show greater loss of genome-wide methylation compared to IDH1R132H/WT clones. Each clone was passaged at least 10 population doublings post-single cell cloning. E, The 08-0537 clone with loss of heterozygosity at the IDH1 locus also shows more pronounced CpG demethylation patterns than its matched unedited clone.
To determine what the observed DNA methylation differences mean in the context of known DNA methylation patterns conferred by the IDH1 mutation, we specifically focused on previously reported G-CIMP associated loci that are used for classification of G-CIMP status [7]. In cell line IMA we did not detect appreciable variation between clones in the methylation of the 7 CpG-loci previously identified as being most tightly linked to the G-CIMP phenotype (Figure 2C) [7]. Specifically, in IMA cells 6/6 hypermethylated sites used to classify tumors as G-CIMP retained hypermethylation following IDH1R132H loss (β>0.7) and 1/1 sites hypomethylated in G-CIMP retained hypomethylation (β<0.2). (Figure 2C) [7]. In comparison, the hypermutated line 08-0537 showed discrepancy in one probe used to classify G-CIMP hypermethylation and one probe for G-CIMP linked hypomethylation, yet knockout of wildtype IDH1, and consequent loss of D-2HG production, did not result in significant demethylation of those G-CIMP associated loci that were hypermethylated in the parental cells (Figure 2C). When we attempted to cluster samples using Euclidean distance and complete linkage using only the 7 G-CIMP loci under consideration, there was no grouping of clones based on IDH1 editing status (Supplementary Figure 3B). In looking at other CpG sites of potential clinical interest, such as MGMT-SPT27 [33], we observed methylation was retained, suggesting loss of IDH1R132H is not sufficient to completely remove the hypermethylation phenotype in human astrocytoma cell lines (Supplementary Figure 3C).
To observe differences in CpG methylation changes that might be driving the clustering of the edited clones, we binned extreme methylation changes (Beta value change of clone – parental <−0.4). We noted that in both IMA and 08-0537 cell lines the loss of the IDH1R132H/WT heterozygous state was associated with a greater number of probes showing dramatic demethylation (delta Beta <−0.4) than in unedited control cells (Figure 2D and 2E and Supplementary Figure 3B).
Loss of D-2HG production is associated with reductions in DNA methylation at specific G-CIMP-low associated sites
Having seen differences in levels of demethylation between edited and unedited clones, we hypothesized that the demethylation could indicate the acquisition of a G-CIMP-low methylation state. We extracted the 131 probes identified as being hypomethylated in G-CIMP-low tumors as previously reported [8] and performed unsupervised hierarchical clustering using Euclidean distance of the samples (Figure 3A). We did not observe an exact recapitulation of the 131 site hypomethylation previously identified in patient tumors as defining the G-CIMP-low state, and many probes retained hypermethylation (Beta>0.7). However, we did observe that clones with CRISPR-mediated IDH1 alterations seemed to have overall lower levels of methylation in the 131 sites when we compared edited sub-clones to parental cells or to unedited sub-clones of the same cell line (Figure 3B).
Figure 3: Demethylation is observed in a subset of the 131 CpG probes originally annotated as hypomethylated in G-CIMP-low.
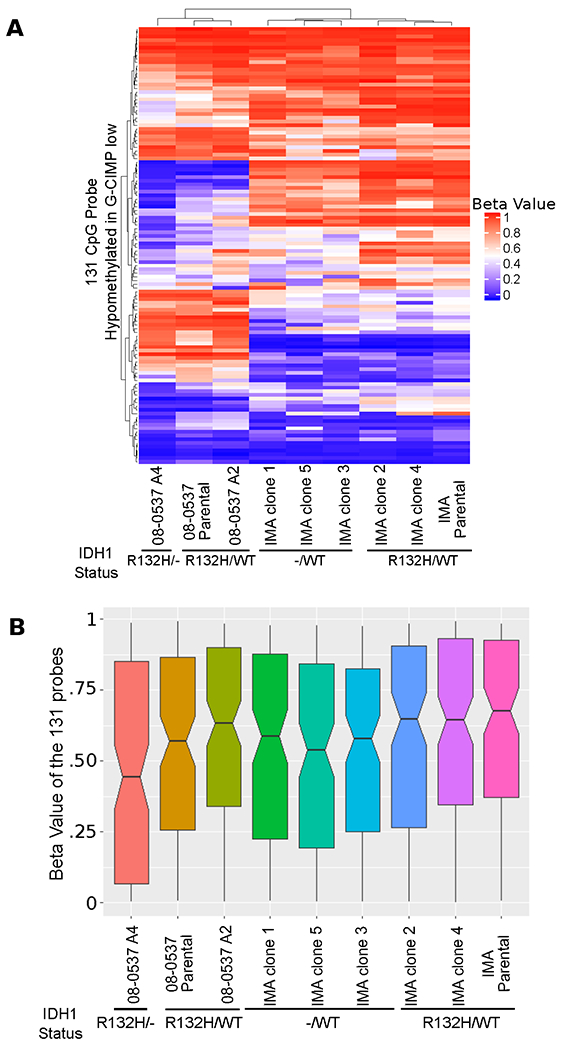
A, Unsupervised hierarchical clustering based on Euclidean distance of 131 probes hypomethylated in G-CIMP-low shows that, within cell lines, cells with intact heterozygous IDH1R132H/WT mutations tend to cluster together, and this clustering is likely driven by demethylation of a subset of probes in clones with altered IDH1. B, Notched boxplots showing median and confidence interval for methylation level of the 131 CpG probes. Clones with altered IDH1 show the largest decreases in median methylation level compared to their matched unedited controls.
Previous TCGA results characterizing the G-CIMP-low state also report a relative enrichment for demethylation within CpG open sea regions, and a relative absence of CpG demethylation in CpG islands [8]. While removing sites that showed methylation changes in unedited clones as a control, we explored the genomic context of all CpG sites showing conserved demethylation (delta Beta ≤−0.2) in all edited clones. In both IMA as well as 08-0537 there was a distinct overrepresentation of demethylated CpG sites at intergenic open seas, 5’-UTR open seas, and CpG shore sites 1500 base pairs from transcription start sites (Figure 4A and Figure 4B). We observed the relative absence of CpG demethylation in CpG islands, especially within the CpG regions 200bp from transcription start sites, as well as CpG sites located within gene bodies compared to the background frequency of these sites on the EPIC array. Previous reports, first describing a G-CIMP-low like state in astrocytoma, found similar patterns of demethylation when comparing global methylation patterns of identified G-CIMP-low cases with G-CIMP-high cases [8]. Specifically, it was shown that G-CIMP-low cases showed enrichment for demethylation in CpG open sea regions, and underrepresentation of demethylation in CpG islands when comparing differential probes of G-CIMP-low cases with all probes profiled [8]. The genomic context of CpG probes undergoing demethylation in our model of IDH1 loss is therefore similar to the genomic context of CpG probes showing hypomethylation in the G-CIMP-low pattern in patient tumors.
Figure 4: Genomic context of differentially methylated probes shows enrichment for open sea regions of the genome and CpG Island shores near transcription start sites.
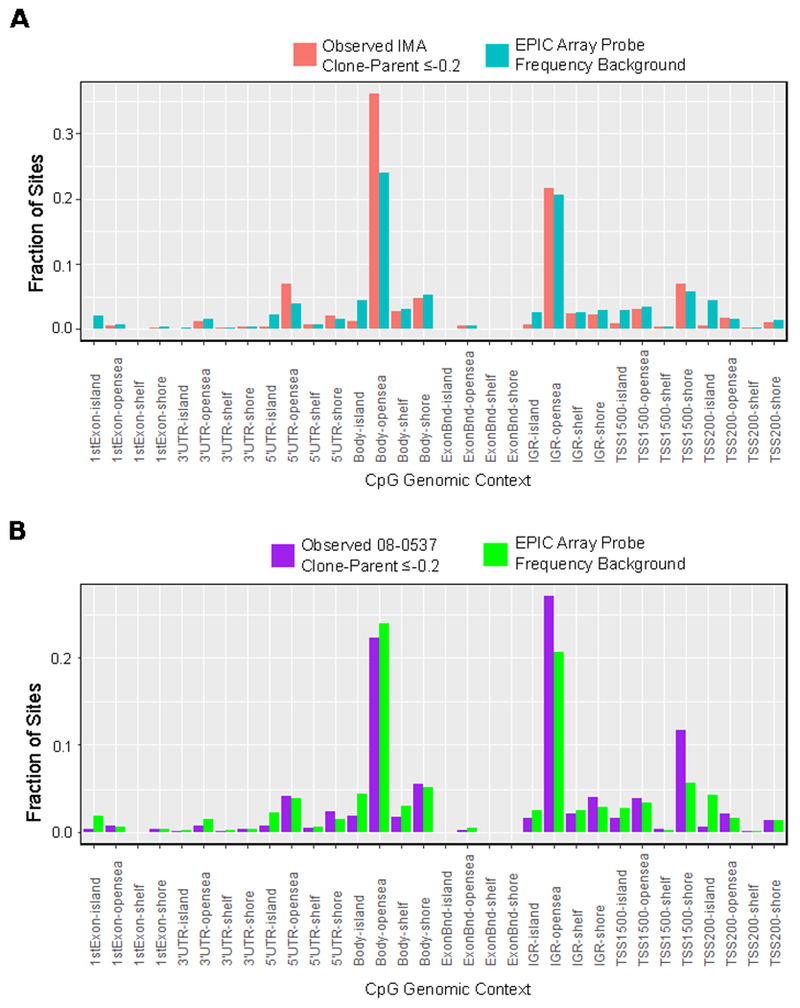
A, Fraction of CpG sites identified as differentially methylated with change in Beta value ≤−0.2 concurrent in all three IDH1−/WT clones of IMA and no corresponding change in Beta value ≤−0.2 in either unedited clone. Plotted in blue is the frequency of CpG sites within each genomic context on the EPIC array. Chi-squared goodness of fit p-value for difference from expected background frequency < 2.2x10^−16 B, Similar enrichment plot for 08-0537 using change in Beta value ≤−0.2 in the clone with loss of heterozygous IDH1R132H/WT status and removing probes showing a delta Beta value ≤−0.2 change in matched unedited control clone. Chi-squared goodness of fit p-value for difference from expected background frequency < 2.2x10^−16.
Discussion
Utilizing patient-derived grade III and grade IV glioma cell lines from IDH1 mutant tumors, we have generated isogenic glioma cell lines that differ in the mutation status of IDH1. We show that knockout of either wildtype or R132H encoding IDH1 allele was sufficient to eliminate elevated D-2HG production. We further characterize the DNA methylome of these isogenic pairs of cells to reveal that although some of the G-CIMP loci retain hypermethylation on loss of D-2HG, there is a genome-wide pattern of measurable demethylation especially in open sea regions and CpG island shores. While the G-CIMP-low phenotype does not strictly require loss of an IDH1 allele in patient tumors, there are identified subsets of tumors with copy number alterations at the IDH1 locus that display a G-CIMP-low phenotype [11]. Therefore, our results support the possibility that loss of an IDH1 allele and D-2HG production may be a driving factor of methylation changes in this subset of recurrent tumors [11].
In our IDH1-mutant glioma cell lines, we found that knockout of either IDH1WT (in 08-0537) or IDH1R132H (IMA) was sufficient to induce a trend of demethylation. This is consistent with loss of D-2HG mediated inhibition of alpha-ketoglutarate-dependent epigenetic enzymes, such as the ten-eleven translocation genes (TETs). Others have previously shown that in IDH1R132H/WT gliomas, loss of heterozygosity at Chr2q, which contains the IDH1 locus, leads to loss of methylation at a variety of sites outside of CpG islands [11]. Our results are consistent with this observation, indicating that loss of D-2HG production in IDH1 mutant cells can reprogram the DNA methylome. Additionally, our results are complementary to previously published models used in investigations that characterized methylation changes after inhibitor treatment or spontaneous loss of IDH1 alleles [11]. While the natural loss of IDH1 tends to co-occur with multiple gene deletions on Chr2q, our cell models have no conserved significant differences in Chr2q copy number between clones at the locus surrounding IDH1, which allows us to parse effects of IDH1R132H loss from passenger effects of co-deleted genes [11] (Supplementary Figure 4). Likewise, small molecule inhibitors may have their own off-target effects, and might be difficult to apply consistently over the long periods of in vitro culture that have been required to see D-2HG suppression-mediated demethylation effects [16, 31, 32]. For these reasons the use of genetically-engineered patient-derived IDH1 mutant cell lines provide a novel and complementary approach for studying the roles of IDH1 in maintenance of IDH1 mutation linked epigenetic changes over long time courses.
While we cannot rule out the possibility that the genome wide patterns of demethylation observed were pre-existing alterations in a subset of the parental cell population, our results do suggest that loss of D-2HG production is causally related to genome wide changes in methylation. These methylation changes are largely limited to decreased hypermethylation in sub-sets of the G-CIMP-low loci in open sea CpG contexts, and we did not observe significant reversions of DNA hypermethylation in most contexts within gene promoters. Nonetheless 19/1308 CpG loci used previously [8] to cluster IDH1 mutant gliomas from IDH1 wildtype gliomas did show changes in methylation (absolute change in Beta > 0.2) in the IMA edited clones as detected by ChAMP calling of differentially methylated probes. This is in comparison to 6/1308 CpG probes being detected as DMPs in the unedited IMA clones (Supplementary Table 2). The retained hypermethylation in promoters may be a combination of methylation and 5-hydroxymethylation due to the inability of the detection modality to discriminate between the two marks; however, the observed complete removal of methylation in the G-CIMP-low loci is not affected by this limitation.
Recent studies have suggested that low levels of demethylation in IDH1-mutant G-CIMP positive gliomas may occur via passive demethylation [34]. This interpretation is based on the observation that demethylated loci in G-CIMP-low gliomas correlate closely with late replicating genomic regions of neural stem cells, and these late replicating regions may have insufficient methylation maintenance activity by DNMT1, occurring due to increasing genome replication rate [34]. Importantly, our results show that suppression of D-2HG production is likely a modifier of DNA methylation in IDH1 mutant tumors; however, further experiments will be required to understand whether the demethylation we observed in the IDH1 edited clones is due to changes in the fidelity of DNA maintenance methylation and consequently passive demethylation or alternatively to changes in active demethylation.
In future work it will be important to determine if these losses of DNA methylation in G-CIMP-low loci affect the activity of enhancer elements located in intergenic open sea regions, and what effects these activity changes have on the tumorigenicity of IDH1 mutant glioma cells. Although previous pre-clinical reports suggest that these methylation reversions are not required to see therapeutic effect of small molecule IDH1 inhibitor treatment [31], there could be heterogeneity in the dependence of different IDH1 mutant gliomas to the various epigenetic changes induced by the IDH1 mutation. We speculate that this heterogeneity in dependence and stability of the promoter associated DNA methylation could contribute to the failure to achieve therapeutic benefits following inhibition of mutant IDH1 in some orthotopic xenograft models [16, 35].
In conclusion, this study provides new ways of modeling IDH1 mutant glioma in a genetically relevant background that complements existing methods. We show that these models exhibit hypermethylation at the most conserved G-CIMP loci which persists after loss of IDH1 mutation linked D-2HG production. Furthermore, we also show loss of neomorphic IDH1 mutation results in broad genome-wide changes in CpG methylation in the G-CIMP-low like genomic context of open seas, similar to patient tumors and astrocyte overexpression systems [11, 12, 32]. Future work that utilizes these genetically relevant models will help to elucidate how IDH1 mutations continue to contribute to the malignant phenotype of IDH1 mutant glioma.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Implications:
These findings show that loss of the IDH1 mutation in malignant glioma cells leads to a pattern of DNA methylation alterations, and shows plausibility of IDH1 mutation loss being causally related to the gain of a G-CIMP-low like phenotype.
Acknowledgements
We thank Paula K. Greer for technical assistance and lab management, and Carlee Hemphill for performing Identifiler profiling of cell lines. These studies were supported by a grant from the National Cancer Institute of the National Institutes of Health (Award Number R01CA140316) to H. Yan. Flow cytometry and cell sorting was performed in collaboration with the Duke Cancer Institute Flow Cytometry core facility. Illumina DNA Methylation Arrays were processed by the Duke Molecular Physiology Institute Molecular Genomics Core. We thank the Duke PK/PD core for LC-MS quantification of D-2HG in cell samples. The results shown here are in whole or part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. Casey Moure was supported by an NRSA Fellowship F31CA203399. Bill Diplas was supported by NRSA Fellowship F30CA206423.
Grant Support: H.Y. received NIH R01 grant R01CA140316. BHD received NIH F30CA206423. CJM received NIH F31CA203399.
Footnotes
Conflict of Interest Statement: H.Y. is a founder and chief scientific officer of Genetron Health and receives royalties from Personal Genome Diagnostics (PGDX). B.H.D., R.Y., Y.H., and H.Y. receive royalties from Genetron Health for technology discussed in this manuscript.
Accession Codes
Methylation array data in this study are MIAMI compliant and have been submitted to the MIAME compliant NCBI Gene Expression Omnibus (GEO) database, accession number GSE128032. Raw sequencing data will be available to qualified investigators upon request.
References
- 1.Yan H, et al. , IDH1 and IDH2 mutations in gliomas. N Engl J Med, 2009. 360(8): p. 765–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Network TCGAR, Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. New England Journal of Medicine, 2015. 372(26): p. 2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dang L, et al. , Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature, 2009. 462(7274): p. 739–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koivunen P, et al. , Transformation by the R Enantiomer of 2-Hydroxyglutarate Linked to EglN Activation. Nature, 2012. 483(7390): p. 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flavahan WA, et al. , Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature, 2016. 529(7584): p. 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waitkus MS, Diplas BH, and Yan H, Isocitrate dehydrogenase mutations in gliomas. Neuro Oncol, 2016. 18(1): p. 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noushmehr H, et al. , Identification of a CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell, 2010. 17(5): p. 510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ceccarelli M, et al. , Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell, 2016. 164(3): p. 550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sandoval J, et al. , Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics, 2011. 6(6): p. 692–702. [DOI] [PubMed] [Google Scholar]
- 10.de Souza CF, et al. , A Distinct DNA Methylation Shift in a Subset of Glioma CpG Island Methylator Phenotypes during Tumor Recurrence. Cell Rep, 2018. 23(2): p. 637–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mazor T, et al. , Clonal expansion and epigenetic reprogramming following deletion or amplification of mutant IDH1. Proceedings of the National Academy of Sciences, 2017. 114(40): p. 10743–10748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang L, et al. , Exome sequencing identifies somatic gain-of-function PPM1D mutations in brainstem gliomas. Nat Genet, 2014. 46(7): p. 726–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Favero F, et al. , Glioblastoma adaptation traced through decline of an IDH1 clonal driver and macro-evolution of a double-minute chromosome. Annals of Oncology, 2015. 26(5): p. 880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luchman HA, et al. , Spontaneous loss of heterozygosity leading to homozygous R132H in a patient-derived IDH1 mutant cell line. Neuro-Oncology, 2013. 15(8): p. 979–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johannessen T-CA, et al. , Rapid Conversion of Mutant IDH1 from Driver to Passenger in a Model of Human Gliomagenesis. Molecular Cancer Research, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tateishi K, et al. , Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell, 2015. 28(6): p. 773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin G, et al. , Disruption of wild-type IDH1 suppresses D-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res, 2013. 73(2): p. 496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diplas BH, et al. , Sensitive and rapid detection of TERT promoter and IDH mutations in diffuse gliomas. Neuro Oncol, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ran FA, et al. , Genome engineering using the CRISPR-Cas9 system. Nat Protoc, 2013. 8(11): p. 2281–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diplas BH, et al. , The genomic landscape of TERT promoter wildtype-IDH wildtype glioblastoma. Nature Communications, 2018. 9(1): p. 2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris TJ, et al. , ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics, 2014. 30(3): p. 428–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aryee MJ, et al. , Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics, 2014. 30(10): p. 1363–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou W, Laird PW, and Shen H, Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res, 2017. 45(4): p. e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teschendorff AE, et al. , A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics, 2013. 29(2): p. 189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smyth GK, Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol, 2004. 3: p. Article3. [DOI] [PubMed] [Google Scholar]
- 26.Wettenhall JM and Smyth GK, limmaGUI: a graphical user interface for linear modeling of microarray data. Bioinformatics, 2004. 20(18): p. 3705–6. [DOI] [PubMed] [Google Scholar]
- 27.Peters TJ, et al. , De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin, 2015. 8: p. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hovestadt V ZM, conumee: Enhanced copy-number variation analysis using Illumina DNA methylation arrays. 2017. p. R package. [Google Scholar]
- 29.Waitkus MS, et al. , Adaptive Evolution of the GDH2 Allosteric Domain Promotes Gliomagenesis by Resolving IDH1(R132H)-Induced Metabolic Liabilities. Cancer Res, 2018. 78(1): p. 36–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lai A, et al. , Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J Clin Oncol, 2011. 29(34): p. 4482–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rohle D, et al. , An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science, 2013. 340(6132): p. 626–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turcan S, et al. , Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nature Genetics, 2018. 50(1): p. 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Capper D, et al. , DNA methylation-based classification of central nervous system tumours. Nature, 2018. 555: p. 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nomura M, et al. , DNA demethylation is associated with malignant progression of lower-grade gliomas. Scientific reports, 2019. 9(1): p. 1903–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kopinja J, et al. , A Brain Penetrant Mutant IDH1 Inhibitor Provides In Vivo Survival Benefit. Scientific Reports, 2017. 7(1): p. 13853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.