

Abstract
Programmed death receptor-1/ligand 1 (PD-1/L1) antibodies can induce durable remissions in malignancies. However, response rates are only ~10–20% in unselected patients versus ~50% in microsatellite instability-high (MSI-high) tumors, probably related to high tumor mutational burden (TMB). Pembrolizumab is approved for MSI-high or deficient mismatch repair tumors. However, outside of colorectal and endometrial carcinoma, only a small subset of tumors are MSI-high, making this treatment option unavailable to most patients. It is not known if MSIstable tumors with high TMB respond to PD-1/PD-L1 blockade. Next generation sequencing (NGS) was performed on 60 patients (14 different histologies) treated with checkpoint blockade using the FoundationOne assay to determine the TMB and MSI status. TMB was dichotomized into two groups; low-to-intermediate (0–19 mutations/mb) vs. high (≥20 mutations/mb). . Benefit rate (stable disease for ≥6 months and partial or complete response) was determined:2,179 of 148,803 samples (1.5%) were MSI-high; 9,762 (6.6%), TMB-high (7,972, MS-stable/TMB-high). The majority (82.1%) of MSI-H tumors were TMB-high; however, only 18.3% of TMB-high tumors were MSI-H. Median progression-free survival for MS-stable/TMB-high versus MS-stable/TMB-Low/TMB-Intermediate tumors was 26.8 vs. 4.3 months (P = 0.0173). Thus, our data demonstrate that MS-stable/TMB-high tumors are more common than MSI-high cancers and may benefit from immunotherapy.
Introduction:
Programmed death receptor-1/ligand 1 (PD-1/L1) antibodies can induce durable remissions in solid and hematologic malignancies. However, only 10–20% of unselected patients respond to PD-1/L1 blockade. There is an unmet need for novel biomarkers that will identify patients more likely to respond to PD-1/PD-L1 inhibition. The utility of PD-L1 expression as a biomarker has been studied extensively, and in general response rates to PD-1/PD-L1 blockade are 0–17% for PD-L1 negative tumors and 36–100% for PD-L1 positive tumors (1). However, standardization of PD-L1 as a useful biomarker has been difficult as many detection methods are currently used (immunohistochemistry, flow cytometry, mRNA expression)(2). The most responsive cancers to PD-1/PD-L1 blockade have been melanoma and NSCLC, both of which have high tumor mutational burden (TMB) (3). Retrospectively and prospectively TMB can be an effective tissue agnostic biomarker in predicting responses to PD-1/PD-L1 blockade(4–6).
PD-1/PD-L1 blockade is also highly effective in microsatellite instability-high (MSI-high)/mismatch repair-deficient (dMMR) tumors (7,8). The sensitivity of MSI-high tumors to PD-1 blockade may be related to high TMB since TMB predicts checkpoint blockade response in many cancer types (5,9). Pembrolizumab is approved by the Food and Drug Administration (FDA) for MSI-high or dMMR solid tumors (www.fda.gov), representing the first tissue-agnostic approved as a cancer therapeutic (7,8). However, outside of colorectal and endometrial carcinoma, only a small subset of tumors are MSI-high(10), making this treatment option unavailable to most patients. Because a higher percentage of tumors are TMB-high than MSI-high(11), we sought to determine if MS-stable/TMB-high tumors (both tested on the same tissue sample) respond to checkpoint blockade.
Methods:
This study was performed in accordance with UCSD Institutional Review Board guidelines for data analysis () and for any investigational treatments for which patients provided consent. Approval for the Foundation Medicine dataset was obtained from the Western Institutional Review Board (Protocol number 20152817). Hybrid capture-based next generation sequencing (NGS) was performed on all samples using the FoundationOne assay (182, 236, 315, 327, or 405 genes, depending on the time period; http://www.foundationmedicine.com/). The average sequencing depth of coverage was greater than 250x, with >100x at >99% of exons(12). The pathologic diagnosis of each case was confirmed by review of hematoxylin and eosin stained slides and all samples that advanced to DNA extraction contained a minimum of 20% tumor cells. Sequencing was performed by Foundation Medicine on tumor samples between October 1, 2012 to April 1, 2018. TMB was calculated by interrogating up to 1.2 mb of the genome. The number of somatic mutations were enumerated and extrapolated to the whole exome using a validated algorithm(11). Alterations known to be oncogenic drivers were excluded. TMB was dichotomized into two groups; low-to-intermediate (0–19 mutations/mb) vs. high (≥20 mutations/mb). MSI status (stable vs. high) was determined using 114 intronic homopolymer repeat loci with adequate coverage on the NGS panel. These sequences were analyzed for length variability and compiled into an overall score using principal components analysis(13).
Our inclusion criteria for UCSD patients were that they were consented as required for the PREDICT study (), were seen and treated at UCSD at any time after October 2012, were adults (at least 18 years of age) and had cancer that was tested for microsatellite status and for TMB (by Foundation Medicine) and they were treated with checkpoint blockade with at least one evaluable follow up. For the Foundation Medicine dataset ( N = 148,803 tumor samples), all samples analyzed by Foundation Medicine were included.
Sixty patients (14 different histologies) treated with checkpoint blockade were evaluable. Benefit rate (stable disease for ≥6 months and partial or complete response) was determined (RECIST criteria). Authors reviewed clinical documentation and radiographic images for evidence of progression. Median progression-free survival (PFS) and overall survival (OS) were calculated from the start of checkpoint blockade, and data was censored at the last visit for patients still progression free or alive, respectively, for PFS and OS. PFS and OS were calculated by the method of Kaplan and Meier (P values by log-rank test). Patients were censored at date of last follow up for PFS and OS, if they had not progressed or died, respectively. The Fisher exact test was used to assess categorical variables. P=values ≤.0.05 were considered significant.
Results:
TMB and MSI status were analyzed on 148,803 tumor samples (Foundation Medicine (FM) dataset); 2,179 (1.5%) of 148,803 samples were MSI-high whereas 9,762 (6.6%) were TMB-high. The majority (82.1%) of MSI-high tumors were TMB-high; however, only 18.3% of TMB-high tumors were MSI-H. Therefore, of 148,803 patients, 2,179 were MSI-high whereas 7,972 patients were MS-stable but TMB-high (Fig. 1). Cutaneous malignancies had the highest TMB whereas endometrial, colorectal cancer, and small intestine cancer had the highest percent of MSI-H samples (Fig 1A).
Figure 1: The relationship between MSI status and TMB across diverse malignancies.
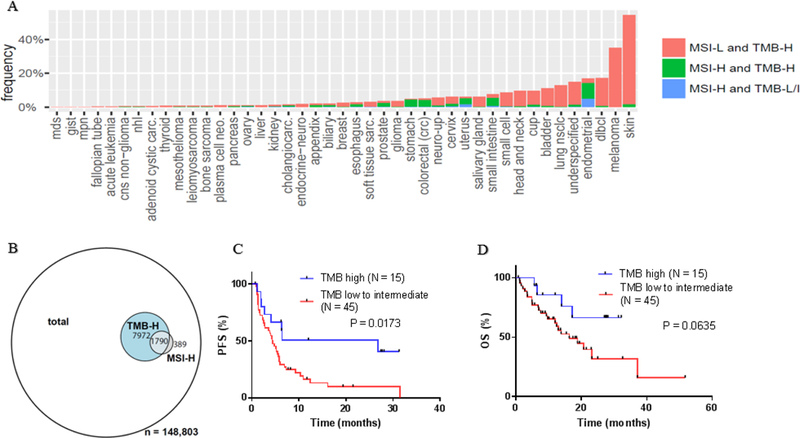
A) Distribution of MSI status and mutational burden amongst various tumor histologies (N = 148,803). B) Of148,803 total patients (with 2179 being MSI-H and 9762 being TMB-H), 389 patients are MSI-H and TMB-Low or TMB-intermediate; (ii) 1790 patients are MSI-H and TMB-H; and (iii) 7972 patients are MSS and TMB-H. C) Median PFS for MS-stable tumors dichotomized by TMB. The median PFS for TMB-high tumors compared to TMB-Low to -Intermediate tumors was 26.8 months vs. 4.3 months. (P = 0.0173, HR 0.42 [95% CI 0.22–0.77]). D) Mediant OS for MS-stable tumors dichotomized by TMB. The median OS for TMB-high tumors compared to TMB-Low to -Intermediate tumors was not reached vs. 16.3 months (median follow up of 17.2 months) (P = 0.0635, HR 0.4581 [95% CI 0.20–1.0]). PFS and OS were calculated by the method of Kaplan and Meier (P values by log-rank test)
The UCSD data set consisted of 60 patients who were all MSI stable. Fifteen patients (25%) had TMB-high tumors. Histologies that were TMB-low to -intermediate included non-small cell lung cancer (N = 13), melanoma (N = 12), head and neck cancer (N = 7), bladder cancer (N = 4), sarcoma (N = 3), breat cancer (N = 2), glioblastoma (N = 1), cervical cancer (N = 1), ovarian cancer (N = 1), and adrenal cancer (N = 1) (Supplemental Table S1). Histologies that were TMB-high included melanoma (N = 6), bladder cancer (N = 2), cutaneous squamous cell carcinoma (N =2), glioblastoma (N = 1), breast cancer (N = 1), basal cell carcinoma (N = 1), esophageal carcinoma (N = 1), and prostate cancer (N = 1) (Supplemental Table S1). All patients were treated with either PD-1/L1 or CTLA4 checkpoint blockade (some received a combination of these agents) (Supplemental Tables S1 and S2). Seventeen of 45 (38%) of TMB-Low to -Intermediate patients were treated with combination therapy (Supplemental Table S2) whereas 5 of 15 (33%) TMB-high patients were treated with combination therapy (Supplemental Table S2).
The benefit rate (stable disease ≥6 months/partial and complete remission (SD ≥6 months/PR/CR)) for MS-stable/TMB-high versus MS-stable/TMB-Low to –Intermediate patients was 10/15 (75%) vs. 17/54 (38%) (P = 0.0734, odds ratio (OR) 3.29 [95% confidence interval (CI) 0.91–10.29]). The median PFS for MS-stable/TMB-high versus MS-stable/TMB-Low/TMB-Intermediate tumors was 26.8 months vs. 4.3 months (P = 0.0173, hazard ratio (HR) 0.42 [95% CI 0.22–0.77]); median OS, not reached (median follow up, 17.2 months vs. 16.3 months (P = 0.0635, HR 0.4581 [95% CI 0.20–1.0])) (Fig. 1).
Discussion:
Our data suggested that MS-stable/TMB-high tumors have significantly longer median PFS (26.8 months vs. 4.3 months (P = 0.0173)) after checkpoint blockade than MS-stable/TMB-Low/Intermediate tumors. Furthermore, MS-stable/TMB-high characterized a subgroup of cancers considerably larger than the MSI-high subset (7,972/148,803 versus 2,179/148,803 patients). Although the salutary effects of checkpoint blockade for MSI-high tumors wasclear (8), and most MSI-high tumors were TMB-high, about ~18% of malignancies that were MSI-high, were not TMB-high. It would be worth ascertaining if patients with MSI-high but lower TMBs (a subset of patients to small to assess in our current study) respond less well to immunotherapy.
The limitations of our study included the relatively small sample size and the retrospective analysis. Furthermore, our patients were not all treated with the same therapy. Due to the limited number of patients included in our analysis, we were unable to perform a multivariate analysis to assess for potential confounding factors such as heterogeneity of treatments (different agents, combinations vs. monotherapies) and tumor types that may have influenced outcomes. PD-L1 expression was not available for many patients and was not included in the analysis. PD-L1 expression and TMB are not significantly correlated within most cancer subtypes (14). Even so, our data showed that significant subgroups of patients have MS-stable/TMB-high tumors and these individuals appear to respond favorably to immunotherapy.
MS-stable/TMB-high characterized a subgroup of cancers that was larger than the MSI-high subset. However, the optimal cutoff between TMB low and high remains to be defined. It is currently unknown whether individual cutoffs for specific tumor types or a universal cutoff point for all tumors should be adopted (15). TMB-high patients, regardless of MSI status, respond to checkpoint blockade, and FDA-approval of checkpoint inhibitors based off TMB status may be warranted (7,8). This will greatly expand the population of cancer patients who could receive checkpoint blockade. The current observations underscore the importance of prospective clinical trials evaluating the utility of TMB in diverse tumors treated with checkpoint blockade.
Supplementary Material
Acknowledgments
Competing interests: Aaron Goodman receives speaking fees from Seattle Genetics and consulting fees from Jazz Pharmaceuticals. Ethan Sokol and Garrett Frampton are employees of Foundation Medicine and Roche shareholders. Razelle Kurzrock receives research funding from Genentech, Incyte, Merck, Serono, Pfizer, Sequenom, Foundation Medicine, and Guardant, as well as consultant fees from Loxo, X Biotech, NeoMed and Actuate Therapeutics, speaker fees from Roche, and an ownership interest in IDbyDNA and Curematch Inc
Funding: Funded in part by grant: P30 CA023100 (RK) and the Joan and Irwin Jacobs fund
Footnotes
Declarations
Ethics approval and consent to participate: This study was performed in accordance with UCSD Institutional Review Board guidelines for data analysis () and for any investigational treatments for which patients provided consent. Approval for the Foundation Medicine data analysis was obtained from the Western International Review Board (Protocol number 20152817).
Consent for publication: The authors give consent for publication.
Availability of data and material: All of the data has been provided in the supplementary tables.
Authors’ information: Not applicable
Bibliography
- 1.Patel SP, Kurzrock R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol Cancer Ther 2015;14:847–56. [DOI] [PubMed] [Google Scholar]
- 2.Goodman A, Patel SP, Kurzrock R. PD-1-PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat Rev Clin Oncol 2017;14:203–20. [DOI] [PubMed] [Google Scholar]
- 3.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hellmann MD, Ciuleanu T-E, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. New England Journal of Medicine 2018;378:2093–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol Cancer Ther 2017;16:2598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miao D, Margolis CA, Vokes NI, Liu D, Taylor-Weiner A, Wankowicz SM, et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat Genet 2018;50:1271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 2015;372:2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med 2017;377:2500–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cortes-Ciriano I, Lee S, Park W-Y, Kim T-M, Park PJ. A molecular portrait of microsatellite instability across multiple cancers. Nat Commun [Internet] 2017. [cited 2018 Aug 20];8 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5467167/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Medicine 2017;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31:1023–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Halll M, Gowen K, Sanford E. Evaluation of microsatellite instability (MSI) status in 11,573 diverse solid tumors using comprehensive genomic profiling (CGP). J Clin Oncol 2016;34(15):1523. [Google Scholar]
- 14.Yarchoan M, Albacker LA, Hopkins AC, Montesion M, Murugesan K, Vithayathil TT, et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samstein RM, Lee C-H, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 2019;51:202–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.