

Abstract
High-affinity binding of antibodies provides for increased specificity and usually higher effector functions in vivo. This goal, well documented in cancer immunotherapy, is very relevant to vaccines as well, and has particularly significant application toward glycan antigens. The inability to elicit high-affinity antibodies has limited potential applications of glycan-based immunogens, giving rise to insufficient population coverage due to low titers and short duration of protection. That such vaccines have achieved widespread use in spite of these shortcomings highlights the surpassing importance of glycans as prophylactic immunological targets. New advances in the combination of synthetic chemistry, bioconjugation, and mechanistic immunology offer the possibility to vastly expand the number of potential molecular targets in cancer and infectious diseases by opening a wider world of carbohydrate structures to immunological recognition and high-affinity response.
Introduction
A series of papers by N.A. Mitchison in the European Journal of Immunology in 1971 first dissected the recognition of haptens and developed the principle of hapten-carrier immunogenicity to elicit specific antibodies against non-natural small molecules[1–5]. According to this design, the hapten represents the antibody epitope and is attached to a carrier protein that is seen by helper T cells to provide bystander maturation and mutagenesis to the hapten-specific B cells. This scheme also highlights the key inherent limitation of glycan haptens: their inability to bind to MHC class II molecules and elicit specific T helper responses.
From the perspective of structural diversity, uniqueness, and disease relevance, glycans have long been recognized as excellent potential antigens for the development of anti-cancer and anti-infectious vaccines. Many cancer cells re-express unique surface glycans, while microbes, including pathogenic fungi and bacteria, are covered with glycans not found in eukaryotic cells.[6] An effective tetravalent anti-Streptococcus pneumoniae (SP) vaccine was first developed in 1945 using purified polysaccharides[7], but it was subsequently rarely used. More than 25 years were required to see the deployment of a 14-valent and then a 23-valent version of this vaccine (PneumoVax), which has significantly impacted the prevalence of pneumonia and meningitis caused by this microbe.
Shortly thereafter came polysaccharide-based vaccines against Haemophilus influenzae type B (HiB, 1977), Neisseria meningitidis (NM, 1981), and Salmonella typhi, the agent of typhoid fever (ST, 1994). In all cases, effectiveness was shown in adults but not young children or the elderly, and protection was achieved typically for periods of only two or three years. Mitchison’s “conjugate vaccine” concept was then employed to develop the next generation of these vaccines, for HiB in 1987[8], SP in 2000[9], ST in 2001 [10], and NM in 2005[11 ]. (For the purposes of this discussion, we define “conjugate vaccines” as those in which the desired antigenic molecule, or collection of molecules, is attached to a “carrier,” usually a protein.) The translation of basic science to the commercial stage of these vaccines took 20-30 years: if we want to see a third generation of glycan-targeting vaccines, the sooner we implement changes, the better.
Why should changes be introduced for vaccines that have shown strong efficacy and led to a very efficient control of the diseases they target? The medical and financial (~$4.0 billion in annual sales) success of the two main commercial glycan-based streptococcal vaccines should not hide their shortcomings. As with the landmark vaccines mentioned above, the ability of Prevnar and PneumoVax to protect the very young, the old, and the immunocompromised is marginal, and the duration of protection very limited, often not exceeding 2-3 years, especially for some of the serotypes[12]. The same type of polysaccharide-based approach has repeatedly failed to produce vaccines against microbes such as Staphylococcus aureus, one of the most important emerging antibiotic-resistant pathogens[13]. An analysis of these shortcomings of current conjugate vaccines identifies affinity maturation of antibodies as the primary issue. While current approaches promote some cell-cell interactions supportive of affinity maturation, bystander CD4 T cell help is usually suboptimal as it targets primarily anti-protein B cells and not the rare anti-glycan B cell. Efficient B cell maturation and productive mutagenesis require intimate CD4 T cell-B cell interactions, of which CD40-CD40 ligand pair association is the most important[14], along with a high local concentration of cytokines such as IL-4 and IL-13. In this context, the limitations of bystander T cell help to B cells include the production of low- to medium-affinity antibodies and very little expansion of the memory B cell compartment. Consequently, these vaccines have short duration and high-affinity anti-glycan antibodies against SP, NM, ST, and HiB have never been isolated. However, the clinical and commercial success of these vaccines, and the heavy investment that vaccine development requires, have stopped most R&D on conjugate vaccines.
We discuss here the potential for new approaches aimed at increasing the T cell dependence of the anti-glycan response in order to promote the production of high-affinity antibodies. We regard this as the best route to the development of a third generation of anti-glycan based vaccines that will open a far larger range of disease-relevant molecular targets to the power of the prophylactic immune response.
The fundamental issue: no MHC class II binding for glycans
Apart from reports involving zwitterionic polysaccharides, there is to date no example of glycans binding to MHC class II molecules[15], making this class of molecules T cell-independent and not capable of driving directly the generation of high-affinity antibodies. However, the immune system is very well suited to recognize glycans, especially those repetitively displayed as on the surfaces of bacteria, capsular polysaccharides being the best example. This function is assigned to lectins and IgMs. Lectin recognition is usually of low affinity unless part of the adjacent protein displaying the sugar is also part of the epitope[16]; nevertheless, the interactions are sufficient for microbe uptake and destruction. IgMs, the first effector molecules of adaptive immunity, use multivalency to compensate for the low affinity and low specificity of binding to individual glycans. This enables fast sampling (off-rates) of non-optimal recognition events, and makes IgMs the most efficient innate defense system in the mammalian arsenal. For instance, the hyper-IgM syndrome, in which patients have mutations that preclude immunoglobulin switch, is rarely revealed by a severe bacterial infection but rather by pulmonary infections with an unusual fungus, Pneumocystis carinii, also encountered in AIDS patients[17].
In addition to their essential role in protecting us from pathological microbes, IgMs display glycan specificity in three other contexts. The first is the rapid emergence after birth of anti-blood group antibodies directed at the A and B glycans of red blood cells. Titers are usually low, the switch to IgG not efficient, and the sequences of both heavy and light chains show few mutations from germline[18]. Secondly, all of us produce large quantities of antibodies against the αGal epitope (Galα1-3Galβ1-4GlcNac), a motif recognized as foreign because of the loss of α1-3-galactosyl transferase function in humans and Old World apes[19]. Surprisingly, these antibodies become problematic only in instances where a massive immunogenic challenge is applied, as for blood group incompatibility during transfusion or antibody transfer to the newborn in multiparous women, or xenogenic organ transplantation. A third example of anti-glycan IgM effect is in the pathological context of Guillain-Barre syndrome, in which the recognition of the ganglioside-like sugar moiety of the Campylobacter jejuni polysaccharide leads to immunological attack of axons[20].
From a molecular perspective, these examples illustrate that the adaptive immune system is fully equipped to detect a range of glycans. So, the essential problem in the robust and general production of high-affinity anti-glycan antibodies is to provide B cells sufficient help to undergo full maturation and somatic mutagenesis. In an ideal and most efficient scenario, to recruit help, B cells recognize the antigen through their idiotypic receptors and internalize it upon recognition to produce a series of antigen-derived peptides. These peptides bind to MHC class II and are then presented at high density at the surface of that same B cell to CD4 T helper cells. As B cell survival and proliferation in the germinal center is proportional to the amount of peptide they can present to cognate CD4 T cells, the probability of generating a high-affinity anti-glycan clone is dependent on the efficient execution of all the steps leading to peptide presentation. This series of steps is governed by some simple principles that are not optimally fulfilled by current conjugate vaccines, as discussed below.
A practical issue for conjugate vaccines: natural glycans
The manufacturing of conjugate vaccines remains a difficult exercise. Because the glycans used in these vaccines are from natural sources, each of the pathogens targeted by the vaccine must be cultured in very large quantities. Then, polysaccharides have to be purified and coupled to a carrier protein. In addition to the obvious difficulties of handling large quantities of hazardous organisms, even the best-established protocols for the purification of microbial glycans leaves immunogenic contaminants in varying quantities. Furthermore, traditional methods of coupling of the complex sugars to the carrier do not control for the sites of coupling, their heterogeneity, and the accessibility of the glycan epitope after coupling; consequently, antigen amounts and quality are not well quantified. It is very difficult to implement good quality control procedures in this process[21]. As a result, it is certain that each sugar is presented in multiple different ways, and that significant variability exists between batches. These problems are especially important for complex vaccines in which many glycan epitopes are targeted.
We believe that the reliance on natural polysaccharide sources has resulted in more complex formulations than necessary. In contrast to conformational epitopes of proteins, the structural motifs encrypted in bacterial glycans are reasonably simple and modular. The target polysaccharides are very often composed of many repeats of short oligosaccharides, which should be the focus of our attention. For example, the unique molecular signatures of the 13 polysaccharides targeted by Prevnar 13 can be represented by six or seven di- and trisaccharides (Figure 1), vastly reducing the complexity of the system. Thus, a trisaccharide motif, Gal-α1,3-Glu-α1,3-Rha, uniquely identifies serotypes 6A and 6B, and only disaccharides are required to distinguish serotypes 6A, 6B, 18C, and 19A (Glu-α1,3-Rha) from serotypes 18C and 23F (Glu-β1,4-Gal-P-glycerol). The successful development of vaccines using such simplified glycan antigens requires us to demonstrate serotype specificity, which will be greatly aided by driving maturation against them to achieve high-affinity antibodies.
Figure 1:
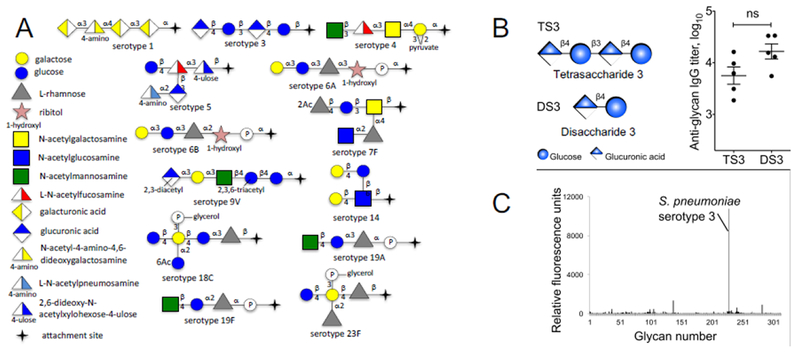
A: Schematic representation of the 13 serotypes of Streptococcus pneumoniae retained in Prevnar 13. This cartoon illustrates some of the redundant motifs that are shared by multiple serotypes, e.g. terminal disaccharide of 6A and 6B, or 19A and 19F. B-C: A tetrasaccharide and a disaccharide unit from the serotype 3 of Streptococcus pneumoniae were compared head to head in mouse vaccination. Serum titers after two injections (day 0 and day 42) were nearly identical between the two forms of the glycan, and specificity of the anti-disaccharide sera was perfect (C, tested on a glycan microarray of 312 microbial sugars), indicating that affinity and specificity were kept when using a minimal glycan unit.
This reductionist strategy both requires and makes practical the production of target glycans by synthetic chemistry. The synthesis of complex carbohydrates remains challenging, and is largely impossible for polysaccharides. However, without minimizing the level of sophistication required, it is fair to say that most carbohydrates of four sugars or fewer are accessible with full control of stereochemistry by competent practitioners using current methods. Synthetic sugars offer absolute control of linker design and of overall purity, characterized by high-resolution mass spectrometry and nuclear magnetic resonance. As show in the example presented in Figure 1, this approach has been successful in our hands for several glycans in the family of SP capsular antigens and we have now managed to reduce the complexity of the sugar from tetra- to disaccharide (Figure 1)[22]. Furthermore, it has been shown by others that synthetic glycan-based conjugates can be co-formulated with existing commercial vaccines, expanding their coverage[23].
A neglected component: carrier design
A variety of carrier proteins have been employed in conjugate vaccine design, with the most popular being inactivated recombinant diptheria toxin (CRM197), recombinant tetanus toxoid, keyhole limpet hemocyanin, bovine serum albumin, and several types of virus-like particles; only the toxoids have so far reached the clinic[24]. While many labs have their favorites, all are chosen for their apparent immunogenicity and are rarely compared head-to-head, nor are design features often explored to optimize the immune response.
Consider, for example, the widely used CRM197. It suffers from being large (535 amino acids) and containing no less than 40 primary amines, most of which are likely to be addressed in coupling reactions to attach the antigen of interest[25]. The resulting highly heterogeneous conjugates are unlikely to produce a focused and optimized immune response. To circumvent this issue, we have employed the self-assembled virus-like particle derived from the 132-amino-acid Qβ phage capsid protein[22,26]. When recombinantly expressed, 180 copies of this protein assemble in high yields into 30-nm diameter icosahedral particles. Such carriers have many advantages: optimal size for lymphatic transport and dendritic cell uptake, high stability toward chemical modification and extended storage, tailorable amino acid sequences to introduce unique conjugation sites (including unnatural amino acids) and peptide insertions and extensions, and easy GMP-level production in large quantities[27].
We have taken advantage of the availability of synthetic glycan epitopes bearing different linkages to explore the effects of different attachment chemistries and linker lengths on the immune response of Qβ-based glycan immunogens. The ligation chemistry determines to a large degree the specificity of the positions on the carrier protein to which glycans are appended. The standard approach is crosslinking to lysine -NH2 groups with activated acyl linkers. In this case, the attachment site can be rendered homogeneous by mutating away all lysines but one. The use of other methods allows the investigator to achieve site-specific functionalization, such as by introducing an artificial cysteine or unnatural amino acid at a chosen position. The latter approach also provides the minimum possible length for the linkage between peptide and sugar, other than that provided by enzymatic O- or N-glycosylation. We believe short linkers to be important for the recognition of glycopeptides by CD4 T cells to increase T cell help. Other than chain length, “natural” connectivity is not required to present a glycan moiety in such a way as to achieve a strong anti-glycan immune response[28], showing that nature has not chosen to be very sensitive to the molecular nature of the connection. Another important parameter is the display valency: how many copies of the desired antigen are attached to the display platform. We have observed optimal ranges that provide a sufficient concentration of the relevant glycopeptide while avoiding inhibition of B cell maturation which otherwise favors IgM and T-independent isotypes like natural polysaccharides do[22].
A successful combination of these features has been realized in our approach to anti-SP3 and anti-SP14 vaccines. These are composed of synthetic tetrasaccharide antigens, attached by a short linker to at least half of 180 identical peptide sites that are repeated on the surface of a virus-like particle. Alternatives in each of the display parameters resulted in diminished performance, whereas the optimized design gave a well-characterized immunogen seen by both B and T cells, and supporting a strong expansion of T follicular helper cells to provide high-affinity IgG molecules, an outcome not reported for any other strategy[22]. Most critical appears to be focusing the immune response on a single glycopeptide, allowing for consistent presentation of the epitope in sufficient amounts over sufficient time. We note, for example, that increased antigen dosage has been shown to be sufficient in elderly people to improve flu vaccine protection[29], and that similar results have been reported for SP vaccination[9]. It also allows for co-dependent optimization of the peptide sequence for MHC binding, a parameter that we will turn to in future studies. This approach, while highly focused on the glycopeptide of interest with the objective to reduce the complexity of the antigen, retains the remaining of the carrier protein and its ability to support additional T cell help.
Adjuvants: a new frontier
When pathogens invade us, they provide a collection of molecules that stimulate a series of innate “pattern recognition” receptors that are essential to initiate and potentiate immune responses. For each microbe, a minimum of two molecules with this type of “adjuvant” property is usually recognized: one surface molecule such as lipopolysaccharide (LPS) for Gram-negative bacteria and peptidoglycan for Grampositive bacteria, and one internal molecule, usually a component of the bacterial genome. In all instances where natural primary infections confer efficient protection against secondary challenges, the adjuvant effect is strong. Inactivated or killed whole microbial vaccines which have proven efficacious are all based on this property. Thus, one is well advised to include one or more adjuvants in subunit or conjugate vaccine candidates; we believe this to be absolutely required for glycan-focused targets[30]. However, regulatory approval of adjuvants has been slow due to their frequent induction of side effects such as pain and soreness at the site of injection, short duration flu-like symptoms, and mild headache.
Alum, the only currently approved adjuvant used for anti-glycan vaccines, is unfortunately weak and biases the immune response towards a Th2 phenotype, not the Th1 polarization that would help B cells efficiently. The addition of more sophisticated adjuvants to anti-SP vaccines has been extensively explored but successful reports are limited to some CpG DNA sequences capable of engaging TLR9[31]. For anti-SP formulations, we have combined the effects of bacterial RNA packaged by the recombinant Qβ virus-like particle with strong agonism of NKT cells provided by a synthetic galactosyl ceramide derivative[32], an adjuvant that has shown efficacy in humans[33]. The NKT cell is the main provider of IL4 at the initiation of B cell immune responses[34] and IFNγ later, thereby contributing to affinity maturation of the dendritic cell, recruitment of CD4 T cells, and the direct cell contact-mediated help to B cells. These functions are essential for high titers and quality of anti-microbial antibodies.
The dream of a synthetic microbial vaccine
We contend that the consistent generation of high-affinity anti-glycan antibodies, and therefore effective anti-glycan vaccines, relies not on a dramatically new concept but a series of small additive modifications: synthetic short glycans, limited sites of attachment of the glycan to polyvalent carriers, glycopeptides capable of MHC binding and recognizable by CD4 T cells, more efficacious adjuvants, and platforms with optimal uptake and delivery to the draining lymph nodes. These improvements combine to produce “synthetic microbes” in terms of size, shape, repetitive antigenic display, and presence of adjuvants, constructed with tools of synthetic biology and chemistry (Figure 2). These components, needed to drive protective immune responses, are all tailorable to a wide variety of glycans or other types of antigens. We nurture the hope that this approach represents a viable solution to outbreaks of new SP serotypes, influenza, or other mutating pathogens.
Figure 2:
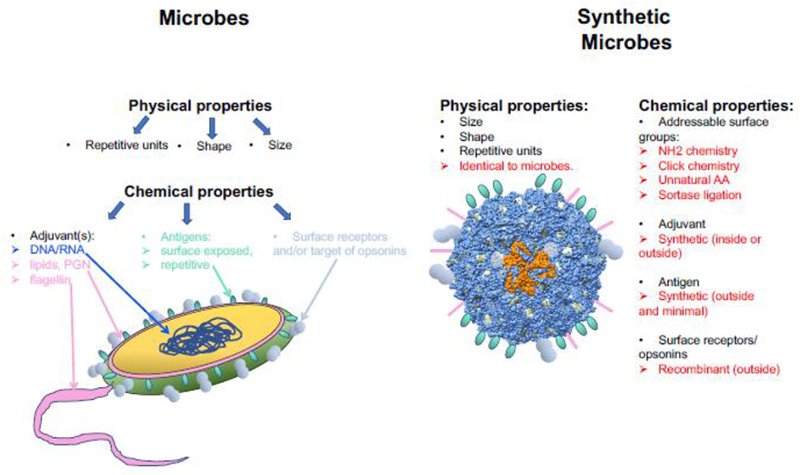
Basic principles defining the concept of “synthetic microbes” as the next generation of vaccines. The physical properties of microbes are directly integrated in the design by using a VLP that is recombinant phage. The collection of chemical properties of real microbes is “copied” for the synthetic microbe by designing the subunit of the VLP to have multiple addressable surface groups, either natural (free NH2-of side chains), or engineered (cysteines, unnatural amino acids, modifiable tags). Each of these groups is then used to couple the synthetic antigens and adjuvants, as well as the recombinant proteins used to promote uptake by B cells and/or dendritic cells. The same platform can be configured for any type of viral, fungal, or bacterial target, by simply changing the synthetic antigen. The model of the synthetic microbe was built with Chimera[37] using the 3 dimensional structure of Qβ (pdb 1QBE)[38].
High-affinity anti-glycan antibodies also offer significant promise for the fields of diagnosis and passive immunotherapy. For example, serotype-specific reagents for SP, Staphylococcus aureus, and a variety of other pathogens do not currently exist and would be extraordinarily useful, as would monoclonal anti-blood group antibodies usable for blood typing. Humanized high-affinity anti-glycan antibodies would be immediately useful in acute infections not amenable to vaccination, and play a similar (but safer and potentially more effective) role as some anti-sera have in diphteria and tetanus. Even more importantly, anti-tumor glycan antibodies could be used to arm CAR T cells in solid tumors, as recently reported[35]. In the field of HIV and HIV vaccine, antibodies directed at the glycan shield of the virus have been reported[36]; however, because these glycans are normal mammalian N-linked sugars, their targeting with high affinity antibodies elicited by vaccination might be problematic.
Conclusions
We believe that methods are now available to develop a new generation of anti-glycan based vaccines and immunotherapies based on the ability to elicit high-affinity anti-glycan antibodies. With renewed interest and augmented capabilities, immunization against carbohydrate antigens should finally be able to help the immunocompromised and the elderly, two patient populations that have so far been poorly served by current glycan-focused vaccines.
Highlights.
High-affinity anti-glycan antibodies can now be produced by using synthetic immunogen and adjuvants that specifically help B cell maturation and increase somatic hypermutation. Re-design of current glycan-based vaccines would help protect immunocompromised as well as elderly patients who do not respond to current commercial conjugate vaccines. Additionally, the same new approaches could be used to target unique glycans of cancer cells and numerous infectious microbes with surface-exposed glycans.
Acknowledgements:
This work was supported by the National Institute of Health, grant AI139748 to PBS, MGF, and LT.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Boak JL, Mitchison NA, Pattisson PH: The carrier effect in the secondary response to hapten-protein conjugates. 3. The anatomical distribution of helper cells and antibody-forming-cell-precursors. Eur J Immunol 1971, 1:63–65. [DOI] [PubMed] [Google Scholar]
- 2.Britton S, Mitchison NA, Rajewsky K: The carrier effect in the secondary response to hapten-protein conjugates. IV. Uptake of antigen in vitro and failure to obtain cooperative induction in vitro. Eur J Immunol 1971, 1:65–68. [DOI] [PubMed] [Google Scholar]
- 3.Mitchison NA: The carrier effect in the secondary response to hapten-protein conjugates. V. Use of antilymphocyte serum to deplete animals of helper cells. Eur J Immunol 1971, 1:68–75. [DOI] [PubMed] [Google Scholar]
- 4.Mitchison NA: The carrier effect in the secondary response to hapten-protein conjugates. II. Cellular cooperation. Eur J Immunol 1971, 1:18–27. [DOI] [PubMed] [Google Scholar]
- 5.Mitchison NA: The carrier effect in the secondary response to hapten-protein conjugates. I. Measurement of the effect with transferred cells and objections to the local environment hypothesis. Eur J Immunol 1971, 1:10–17. [DOI] [PubMed] [Google Scholar]
- 6.Astronomo RD, Burton DR: Carbohydrate vaccines: developing sweet solutions to sticky situations? Nat Rev Drug Discov 2010, 9:308–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grabenstein JD, Klugman KP: A century of pneumococcal vaccination research in humans. Clin Microbiol Infect 2012, 18 Suppl 5:15–24. [DOI] [PubMed] [Google Scholar]
- 8.Zarei AE, Almehdar HA, Redwan EM: Hib Vaccines: Past, Present, and Future Perspectives. J Immunol Res 2016, 2016:7203587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poolman JT, Peeters CC, van den Dobbelsteen GP: The history of pneumococcal conjugate vaccine development: dose selection. Expert Rev Vaccines 2013, 12:1379–1394. [DOI] [PubMed] [Google Scholar]
- 10.D’Amelio E, Salemi S, D’Amelio R: Anti-Infectious Human Vaccination in Historical Perspective. Int Rev Immunol 2016, 35:260–290. [DOI] [PubMed] [Google Scholar]
- 11.Dull PM, McIntosh ED: Meningococcal vaccine development--from glycoconjugates against MenACWY to proteins against MenB--potential for broad protection against meningococcal disease. Vaccine 2012, 30 Suppl 2:B18–25. [DOI] [PubMed] [Google Scholar]
- 12.Artz AS, Ershler WB, Longo DL: Pneumococcal vaccination and revaccination of older adults. Clin Microbiol Rev 2003, 16:308–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fattom AI, Horwith G, Fuller S, Propst M, Naso R: Development of StaphVAX, a polysaccharide conjugate vaccine against S. aureus infection: from the lab bench to phase III clinical trials. Vaccine 2004, 22:880–887. [DOI] [PubMed] [Google Scholar]
- 14.Kawabe T, Naka T, Yoshida K, Tanaka T, Fujiwara H, Suematsu S, Yoshida N, Kishimoto T, Kikutani H: The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity 1994, 1:167–178. [DOI] [PubMed] [Google Scholar]
- 15.Cobb BA, Wang Q, Tzianabos AO, Kasper DL: Polysaccharide processing and presentation by the MHCII pathway. Cell 2004, 117:677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagae M, Yamaguchi Y: Sugar recognition and protein-protein interaction of mammalian lectins conferring diverse functions. Curr Opin Struct Biol 2015, 34:108–115. [DOI] [PubMed] [Google Scholar]
- 17.Leven EA, Maffucci P, Ochs HD, Scholl PR, Buckley RH, Fuleihan RL, Geha RS, Cunningham CK, Bonilla FA, Conley ME, et al. : Hyper IgM Syndrome: a Report from the USIDNET Registry. J Clin Immunol 2016, 36:490–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaneko M, Kato Y, Horiuchi H, Osawa M: Molecular characterization of a human monoclonal antibody to B antigen in ABO blood type. Immunol Lett 2003, 86:45–51. [DOI] [PubMed] [Google Scholar]
- 19.Galili U: Anti-Gal: an abundant human natural antibody of multiple pathogeneses and clinical benefits. Immunology 2013, 140:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu RK, Usuki S, Ariga T: Ganglioside molecular mimicry and its pathological roles in Guillain-Barre syndrome and related diseases. Infect Immun 2006, 74:6517–6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu Y, Yan M, Lasanajak Y, Smith DF, Song X: Large scale preparation of high mannose and paucimannose N-glycans from soybean proteins by oxidative release of natural glycans (ORNG). Carbohydr Res 2018, 464:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]; *A strategy and workflow to purify and characterize glycans from natural sources.
- 22.Polonskaya Z, Deng S, Sarkar A, Kain L, Comellas-Aragones M, McKay CS, Kaczanowska K, Holt M, McBride R, Palomo V, et al. : T cells control the generation of nanomolar-affinity anti-glycan antibodies. J Clin Invest 2017, 127:1491–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]; *The first report of high affinity anti-Streptococcus pneumoniae antibodies, a new approach for their production, and a 3D structure of one of these antibodies.
- 23.Kaplonek P, Khan N, Reppe K, Schumann B, Emmadi M, Lisboa MP, Xu FF, Calow ADJ, Parameswarappa SG, Witzenrath M, et al. : Improving vaccines against Streptococcus pneumoniae using synthetic glycans. Proc Natl Acad Sci U S A 2018, 115:13353–13358. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Describes the addition of new serotypes to exisiting vaccines using synthetic chemistry.
- 24.Pichichero ME: Protein carriers of conjugate vaccines: characteristics, development, and clinical trials. Hum Vaccin Immunother 2013, 9:2505–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shinefield HR: Overview of the development and current use of CRM(197) conjugate vaccines for pediatric use. Vaccine 2010, 28:4335–4339. [DOI] [PubMed] [Google Scholar]
- 26.Yin Z, Comellas-Aragones M, Chowdhury S, Bentley P, Kaczanowska K, Benmohamed L, Gildersleeve JC, Finn MG, Huang X: Boosting immunity to small tumor-associated carbohydrates with bacteriophage qbeta capsids. ACS Chem Biol 2013, 8:1253–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Braun M, Jandus C, Maurer P, Hammann-Haenni A, Schwarz K, Bachmann MF, Speiser DE, Romero P: Virus-like particles induce robust human T-helper cell responses. European journal of immunology 2012, 42:330–340. [DOI] [PubMed] [Google Scholar]
- 28.Speir JA, Abdel-Motal UM, Jondal M, Wilson IA: Crystal structure of an MHC class I presented glycopeptide that generates carbohydrate-specific CTL. Immunity 1999, 10:51–61. [DOI] [PubMed] [Google Scholar]
- 29.DiazGranados CA, Dunning AJ, Kimmel M, Kirby D, Treanor J, Collins A, Pollak R, Christoff J, Earl J, Landolfi V, et al. : Efficacy of high-dose versus standard-dose influenza vaccine in older adults. N Engl J Med 2014, 371:635–645. [DOI] [PubMed] [Google Scholar]
- 30.Sogaard OS: The clinical use of adjuvants in pneumococcal vaccination: current status and future perspectives. Hum Vaccin 2011, 7:276–280. [DOI] [PubMed] [Google Scholar]
- 31.Sogaard OS, Lohse N, Harboe ZB, Offersen R, Bukh AR, Davis HL, Schonheyder HC, Ostergaard L: Improving the immunogenicity of pneumococcal conjugate vaccine in HIV-infected adults with a toll-like receptor 9 agonist adjuvant: a randomized, controlled trial. Clin Infect Dis 2010, 51:42–50. [DOI] [PubMed] [Google Scholar]
- 32.Savage PB, Teyton L, Bendelac A: Glycolipids for natural killer T cells. Chemical Society reviews 2006, 35:771–779. [DOI] [PubMed] [Google Scholar]
- 33.Tefit JN, Crabe S, Orlandini B, Nell H, Bendelac A, Deng S, Savage PB, Teyton L, Serra V: Efficacy of ABX196, a new NKT agonist, in prophylactic human vaccination. Vaccine 2014, 32:6138–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaya M, Barral P, Burbage M, Aggarwal S, Montaner B, Warren Navia A, Aid M, Tsui C, Maldonado P, Nair U, et al. : Initiation of Antiviral B Cell Immunity Relies on Innate Signals from Spatially Positioned NKT Cells. Cell 2018, 172:517–533 e520. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Demonstrates that NKT cells are the main producers of IL-4 supporting B cell maturation at the initiation of anti-viral B cell responses.
- 35.Steentoft C, Migliorini D, King TR, Mandel U, June CH, Posey AD Jr.: Glycan-directed CAR-T cells. Glycobiology 2018, 28:656–669. [DOI] [PubMed] [Google Scholar]; *First report of the use of anti-glycan antibodies to produce engineered CAR T cells in cancer
- 36.Doores KJ, Burton DR: Variable loop glycan dependency of the broad and potent HIV-1-neutralizing antibodies PG9 and PG16. J Virol 2010, 84:10510–10521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE: UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004, 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 38.Golmohammadi R, Fridborg K, Bundule M, Valegard K, Liljas L: The crystal structure of bacteriophage Q beta at 3.5 A resolution. Structure 1996, 4:543–554. [DOI] [PubMed] [Google Scholar]