

Abstract
Purpose:
RET is an emerging oncogenic target showing promise in phase I/II clinical trials. An understudied aspect of RET-driven cancers is the extent to which co-occurring genomic alterations exist and how they may impact prognosis or therapeutic response.
Experimental Design:
Somatic activating RET alterations were identified among 32,989 consecutive patients with metastatic solid tumors tested with a clinical cell-free circulating tumor DNA (cfDNA) assay. This comprehensive next-generation sequencing (NGS) assay evaluates single-nucleotide variants, and select indels, fusions, and copy number gains in 68–73 clinically relevant cancer genes.
Results:
A total of 176 somatic activating RET alterations were detected in 170 patients (143 fusions and 33 missense mutations). Patients had non-small cell lung (NSCLC, n = 125), colorectal (n = 15), breast (n = 8), thyroid (n = 8), or other (n = 14) cancers. Alterations in other oncogenic signaling pathway genes were frequently identified in RET-positive samples and varied by specific RET fusion gene partner. RET fusions involving partners other than KIF5B were enriched for alterations in MAPK pathway genes and other bona fide oncogenic drivers of NSCLC, particularly EGFR. Molecular and clinical data revealed that these variants emerged later in the genomic evolution of the tumor as mechanisms of resistance to EGFR tyrosine kinase inhibitors.
Conclusions:
In the largest cancer cohort with somatic activating RET alterations, we describe novel co-occurrences of oncogenic signaling pathway aberrations. We find that KIF5B-RET fusions are highly specific for NSCLC. In our study, only non-KIF5B-RET fusions contributed to anti-EGFR therapy resistance. Knowledge of specific RET fusion gene partner may have clinical significance.
Introduction
The RET proto-oncogene encodes a receptor tyrosine kinase (RTK) and is an emerging target for cancer therapy (1). Next-generation sequencing (NGS) studies have identified RET alterations in approximately 2% of diverse solid tumors; however, not all are clearly oncogenic (2, 3). Gain-of-function amino acid substitutions and genomic rearrangements producing chimeric fusion proteins can cause ligand-independent constitutive activation of RET. The most commonly described oncogenic RET alterations are in thyroid and non-small cell lung cancers (NSCLC). RET rearrangements have been reported in 20%–40% of papillary thyroid cancer (PTC), 1%–2% of NSCLC (particularly in adenocarcinoma histology with minimal tobacco exposure), and occasionally in a variety of other cancer types (4–10). RET M918T, a hotspot mutation within the tyrosine kinase domain, is the most common somatic molecular event in sporadic medullary thyroid cancer (MTC), found in 43%–71% of cases, and is associated with poor prognosis (11–13). A subset of patients with MTC have germline RET missense mutations. Responses of these tumors to RET inhibition has been reported (14). The prevalence of oncogenic alterations in RET is not well described in other cancer types.
Another understudied aspect of RET-driven cancers is the extent to which co-occurring genomic alterations exist and how they may impact prognosis or therapeutic response. Advanced-stage EGFR- or KRAS-driven lung cancers commonly harbor co-occurring mutations in pathways that impact tumor biology and/or response to targeted therapy, chemotherapy, or immune checkpoint inhibition (15–19). Tissue NGS compendia often include data on early stage and/or untreated patients, so the landscape of tumors with RET aberrations in advanced, treated cancers is not well described. A tissue-based NGS study found that >80% of tumors harboring a RET alteration had coexisting alterations, most commonly in TP53, cell-cycle-associated genes, the PI3K pathway, MAPK effectors, or other tyrosine kinase families (3). However, this study included variants not clearly known to be oncogenic, germline alterations, and treatment history was largely unavailable. In the setting of treatment-naïve NSCLC, oncogenic drivers, including RET fusions, are considered to be mutually exclusive. In contrast, two recent cell-free circulating tumor DNA (cfDNA) studies of patients with NSCLC harboring EGFR or ALK driver mutations found frequent co-occurrence of multiple oncogenic divers in the setting of acquired resistance to targeted therapy (15, 19). RET fusions have been reported as an acquired resistance mechanism to EGFR tyrosine kinase inhibitors (TKI) in approximately 5% of osimertinib-resistant biopsies (20–23).
Several FDA-approved multikinase inhibitors (e.g., vandeta-nib, cabozantinib, lenvatinib, sunitinib, alectinib, sorafenib, ponatinib, nintedanib, regorafenib) have activity against RET and are approved for thyroid cancer, renal cell carcinoma, leukemia, gastrointestinal stromal tumors, colorectal cancer, and/or hepatocellular carcinoma; however, none require identification of a RET alteration or other biomarker for patient selection. While these agents are nonspecific, they have been investigated in RET-driven NSCLC with response rates as high or higher than with chemotherapy, although lower than that of other oncogene-targeted therapies in NSCLC such as those for EGFR, ALK, ROS1, and BRAF V600E (24–26). Gatekeeper RET V804M/L mutations may be acquired after targeting of RET fusions with vandetanib or cabozantinib; however, their prevalence in a clinical setting is not known (27–30). More selective RET inhibitors are showing promise in early-phase clinical trials in patients with RET-driven advanced solid tumors and also have activity against the V804 gatekeeper mutation (NCT03157128, NCT03037385, NCT01877811; refs. 29, 31–33). There is limited evidence suggesting that specific drugs may have differential anti-RET activity depending on the upstream fusion partner. In a phase II study of vandetinib in advanced NSCLC, 5 of 6 patients with a CCDC6-RET fusion achieved an objective response compared with 2 of 10 patients with a KIF5B-RET fusion (34). In a phase I/Ib study of the VEGFR-sparing multikinase inhibitor, RXDX-105 in NSCLC, none of the 20 patients with a KIF5B-RET fusion responded, whereas 6 of 9 patients with non-KIF5B RET fusions responded (35). However, a difference in response rate by fusion partner was not observed in a larger retrospective study evaluating several multikinase inhibitors in NSCLC or in two recent trials evaluating selective RET inhibitors (LOXO-292 and BLU-667) across multiple cancer types (24, 31, 33). Given limited data, the clinical impact of the fusion partner is not clear.
In this study, we evaluated genomic sequencing data from nearly 33,000 patients with diverse advanced cancers tested with a validated comprehensive NGS cfDNA assay, Guardant360 (G360; Guardant Health). We exploit the ability of cfDNA NGS to differentiate germline from somatic RET alterations and truncal clonal drivers from acquired subclonal variants (36, 37). cfDNA analysis can provide information on genomic alterations shed from multiple metastatic lesions, thereby capturing tumor heterogeneity that may not be appreciable in the primary tumor or from biopsy of a single metastatic site. We estimate the frequency of RET and co-occurring alterations, focusing only on somatic alterations predicted to be functionally relevant.
Methods
Consecutive patients with at least one somatic RET-activating alteration detected on the G360 cfDNA assay were identified from the Guardant Health deidentified database (Guardant Health). Patients were tested between February 2015 and July 2017 and had stage III or IV solid tumors. Only samples with evidence of tumor DNA present (e.g., at least one alteration was present) were included to determine prevalence estimates. Germline alterations were filtered out in this study using a method previously described that differentiates germline from somatic mutations based on the relative variant allele fraction and position of known germline single-nucleotide polymorphisms (36). Patients were included if at least one sample contained a somatic RET alteration predicted to be oncogenic, which included RET rearrangements and mis-sense alterations leading to single amino acid substitutions that have been previously characterized as causing aberrant RET activation. For co-occurring alterations, synonymous alterations and variants of uncertain significance were excluded so that only variants known or predicted to be functionally significant were included. Curation resources include COSMIC, cBioPortal for Cancer Genomics, UniProt, Integrative Genomics Viewer, and literature cataloged by PubMed and the International Agency for Research on Cancer database. Co-occurrence of other alterations was determined on a per patient basis by summarizing all unique alterations present from all samples available for review. Pathway-level prevalence estimates were also determined on a per patient basis, by counting patients with at least one alteration affecting each pathway (patients with multiple alterations in the same pathway were counted once). A list of genes included in each pathway can be found in Supplementary Figs. S3 and S4. Prevalence estimates accounted for the most extensive panel used. Clinical information (cancer type, age at testing, sex, records on treatment history, and/or tissue results) was taken from test request forms and confirmed by the ordering clinician where possible. This research was conducted in accordance with recognized ethical guidelines (e.g., Declaration of Helsinki, CIOMS, Belmont Report, U.S. Common Rule) with a waiver of patient consent under an Institutional Review Board–approved protocol for the generation of deidentified datasets for research purposes.
G360 is a 68–73-gene CLIA-certified, College of American Pathologists-accredited, New York State Department of Health–approved clinical cfDNA NGS test with analytic and clinical validation reported (37,38). cfDNA isolation and analytic methods were performed as previously described. Briefly, extracted cfDNA is subjected to paired-end NGS on an Illumnia NextSeq 500 and/or HiSeq 2500 (Illumina, Inc, average read depth 15,000×) following generation of sequencing libraries using nonrandom oligonucleotide adapters and hybrid capture enrichment (IDT, Inc and Aligent Technologies, Inc). Sequencing reads were mapped to the hg19/GRCh37 human reference sequence and were evaluated for SNVs in 68–73 clinically relevant cancer genes as well as small insertions/deletions (indels), gene rearrangements/fusions, and copy number amplification (CNA) in a subset of genes using a proprietary bioinformatics pipeline that reconstructs the original double-stranded cfDNA molecules. The critical regions of RET are sequenced (exons 10, 11, 13, 15, and 16; exons 9, 12, and 14 are also sequenced on the 73-gene panel). Fusions known to be biologically important are reported. The reportable range for SNVs, indels, fusions, and CNAs is > 0.04%, > 0.02%, > 0.04%, and > 2.12 copies, respectively, with a >99.9999% per-position analytic specificity (38).
In plasma samples from patients with advanced cancer, the majority of cfDNA is typically germline-derived and only a small fraction is tumor-derived. The variant allele fraction (VAF) for a given mutation is the total number of cfDNA molecules harboring the mutation divided by the total number of unique cfDNA molecules at that position. The median VAF using this assay is 0.46% (38). Clonality assessment was based on modeling that considers the relative timing of point mutations and amplifications in a sample and adjusts for mutations on amplified genes as described previously (37). Relative VAF is the copy number–adjusted VAF normalized to the highest VAF in the sample. For the purpose of this study, clonal alterations were those with relative clonality >0.9. The cutoff of >0.9 was chosen as it has been shown to recapitulate mutual exclusivity of truncal oncogenic drivers in NSCLC. Notably, these are likely conservative estimates of clonal alterations as truncal drivers can have clonality <0.9(37). Comparisons between RET-positive and negative cases as well as proportions with co-occurring alterations were performed using two-sided Fisher exact tests. Comparison of median VAFs was done by Kruskal-Wallis test.
Results
Prevalence of activating RET alterations in cfDNA of patients with advanced cancer
Among 32,989 consecutive patients with diverse stage III-IV solid tumors, 176 distinct somatic RET alterations predicted to be oncogenic were detected in the cfDNA of 170 (0.5%) patients (patients with multiple samples containing the same RET alteration were only counted once). Another 529 patients who had only variants of uncertain significance, inactivating, or synonymous alterations in RET were not further evaluated in this study (a list of recurrent RET VUS can be found in Supplementary Fig. S1).
Oncogenic RET alterations included 143 in-frame fusions found in 141 patients and 33 single-nucleotide variants (SNV) resulting in an amino acid substitution found in 29 patients. RET fusions were most prevalent among patients with NSCLC, thyroid cancer, or colorectal cancer (Table 1). Seven different fusion partners (KIF5B, CCDC6, NCOA4, TRIM24, TRIM33, ERC1, APAF1) were observed. The most common fusion partner was KIF5B, which was only observed in NSCLC (n = 75) or cancer of unknown primary (CUP, n = 2). Twenty-five different breakpoint combinations were observed, >95% of which involved intron 11 of RET, most commonly fused with intron 15 of KIF5B or intron 1 of CCDC6 (Fig. 1A; ref. 39). One patient with colorectal cancer had two RET fusions involving different partners (CCDC6 and NCOA4), and one patient with NSCLC had KIF5B-RET fusions involving two distinct breakpoints.
Table 1.
Prevalence of oncogenic RET alterations detected in the cfDNA of patients with advanced solid tumors, by tumor type, and RET alteration type (including subcategories of RET fusion or SNVs)
| Alteration, number of unique patients (%) |
NSCLC (n = 14,639) |
Colorectal (n = 3,059) |
Breast (n = 3,921) |
Thyroid (n = 137) |
Othera (n = 11,233) |
Total (n = 32,989) |
|---|---|---|---|---|---|---|
| Patients with any RET fusion | 116 (0.79) | 14 (0.46)b | 2 (0.05) | 1 (0.73) | 8 (0.07) | 141 (0.4) |
| KIF5B-RET | 75 | 0 | 0 | 0 | 2c | 77 |
| CCDC6-RET | 31 | 6 | 2 | 1 | 3 | 43 |
| NCOA4-RET | 6 | 8 | 0 | 0 | 0 | 14 |
| Otherd | 4 | 1 | 0 | 0 | 3 | 8 |
| Patients with any RET SNV | 9 (0.06) | 1 (0.03) | 6 (0.15) | 7 (5.1)e | 6 (0.05)f | 29 (0.09) |
| M918T | 0 | 0 | 3 | 5 | 0 | 8 |
| Other TKD | 5 | 0 | 1 | 3 | 2 | 11 |
| CRD | 4 | 1 | 2 | 1 | 3 | 13 |
| Other | 0 | 0 | 0 | 0 | 1 | 1 |
Abbreviations: CRD, cysteine-rich domain; TKD, tyrosine kinase domain.
Other cancer types with RET fusion included CUP (n = 4), small-cell lung (n = 1), esophageal (n = 1), head and neck (n = 1), mesothelioma (n = 1); or with RET SNV included atypical pulmonary carcinoid (n = 1), gastric (n = 1), bladder (n = 1), prostate (n = 1), pancreatic neuroendocrine (n = 1), and sarcoma (n = 1).
One patient with colorectal cancer had two RET fusions (CCDC6-RET + NCOA4-RET).
Both patients with KIF5B-RETfusions without NSCLC had unknown primary cancers.
Other fusion partners included ERC1 (n = 3), TRIM24 (n = 2), TRIM33 (n = 2), and AFAP1 (n = 1).
One patient with MTC had 3 TKD alts.
One patient with atypical pulmonary carcinoid had 3 CRD alts.
Figure 1.
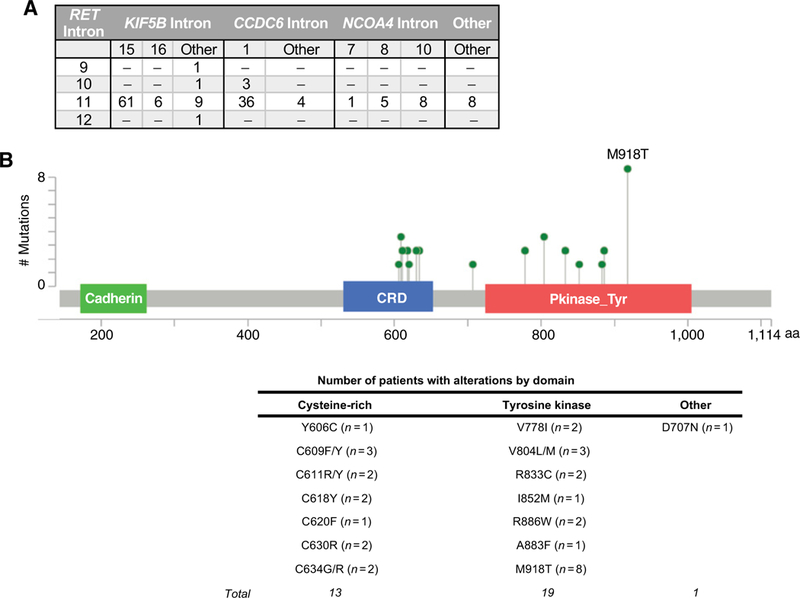
Somatic oncogenic RET alterations detected by NGS of cfDNA from patients with advanced solid tumors. A, Distribution of breakpoints detected among samples containing RET rearrangements (n = 24 unique fusions involving 7 different gene partners in 156 samples from 141 patients). B, Distribution of characterized SNVs in the coding region of RET(n = 19 unique alterations in 34 samples from 29 patients). Counts are unique patients. CRD, cysteine-rich domain.
RET SNVs were most commonly identified among patients with thyroid or breast cancer, but were found in a range of other cancer types including NSCLC (Table 1; Fig. 1B; Supplementary Table S1). The most common alterations were located in the tyrosine kinase domain (n = 19 patients, 8 of whom had M918T) or affected cysteine residues in the extracellular domain (n = 12 patients). One patient with gastric cancer had D707N, which affects a caspase cleavage site within the cytoplasmic domain of RET and has been shown to inhibit apoptosis in preclinical studies (40, 41). Two patients had multiple RET SNVs. One was a patient with MTC harboring M918T, V804M, and V804L alterations who had been treated with several TKIs in an investigational setting. The other patient had an atypical lung carcinoid harboring C611R, C618Y, and C620F alterations (treatment history unknown).
Co-occurrence with other oncogenic alterations
A total of 210 cfDNA samples were available from the 170 RET-positive patients from which to analyze the prevalence of co-occurring alterations and relative clonality. The alterations present in patients with multiple samples were summarized to avoid duplicate counting. Only characterized nonsynonymous alterations known or predicted to have a functionally relevant impact were included (e.g., alterations resulting in enhanced oncogene signaling or loss-of-function mutations in tumor suppressor genes). Patients were evaluated for co-occurring alterations in 72 (n = 62 patients), 69 (n = 75), or 67 other genes (n = 33), based on the assay version available at the time (Supplementary Fig. S2).
A total of 141 patients (82.9%) had 473 additional alterations besides RET affecting 52 genes, 291 of which were unique variants. The prevalence of co-occurring alterations was similar between samples that had a RET fusion and those with a RET SNVs. The median number of additional alterations per patient was 3 (range 1–25), including copy number amplifications (CNA). The most common alterations were in TP53 (n = 99/ 170 patients, 58.2%), other receptor tyrosine kinases (RTK, n = 57/170, 33.5%), or genes involved in the cell cycle (n = 48/170, 28.2%), MAPK (n = 46/170, 27.1%), and PI3K (n = 27/170, 15.9%) pathways (Supplementary Fig. S3). The proportion of patients with at least one co-occurring alteration affecting various oncogenic pathways differed on the basis of the RET alteration type (Fig. 2).
Figure 2.
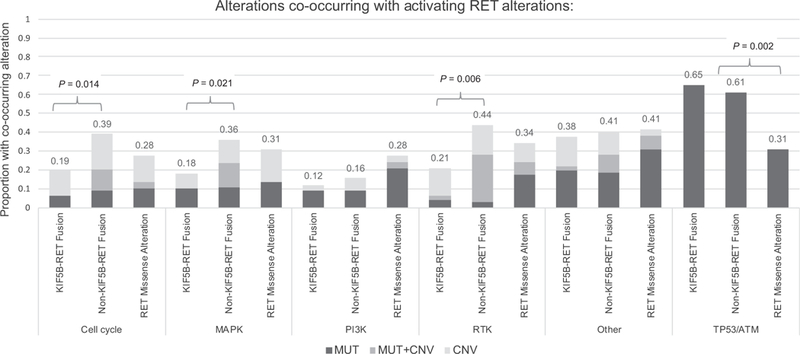
Co-occurring somatic genomic alterations detected in the cfDNA of 170 patients with advanced solid tumors harboring an activating RET mutation, by pathway and RET alteration type (KIF5B-RET fusion, fusion involving partner other than KIF5B, and RET missense SNVs). Proportions are calculated by counting the number of patients with at least one sample containing an alteration of one or more genes in the pathway (see also Supplementary Fig. S3).
Other RTK alterations co-occurred with RET mutations only in patients with CUP, colorectal cancer, or NSCLC. In contrast to previous tissue-based studies, RET fusions were found in samples positive for other driver mutations in NSCLC and/or with MAPK pathway alterations (3). Alterations in at least one MAPK pathway gene were observed with RET fusions in 37 of 141 (26.2%) patients with either breast cancer, colorectal cancer, NSCLC, or CUP.
Given the cfDNA testing population is primarily comprised of patients with advanced and typically pretreated cancers, we hypothesized this novel observation was due to selection of RET and/or MAPK pathway alterations as a mechanism of therapy resistance. Across all RET-positive samples, approximately 30% of the 197 oncogenic RET and 2% of the 42 non-CNA MAPK pathway alterations detected were clonal (clonality >0.9) based on a validated copy number-adjusted clonality assessment (see Methods). This suggested that a proportion of RET variants and the majority of the MAPK pathway variants detected in this series emerged later in the genomic evolution of the tumor. We next examined the contribution of RET as a driver or resistance mutation by evaluating relative clonality and patterns of co-occurring alterations. We focused on NSCLC for which the most genomic and clinical data were available and because patterns of therapy resistance are relatively well-described.
RET fusions in NSCLC may arise as a mechanism of resistance to EGFR tyrosine kinase inhibitors
Among the 125 RET-positive patients with NSCLC, 103 (82.4%) had at least one additional functionally relevant somatic alteration (Fig. 3A). Twenty-three patients had another classic driver mutation detected in at least one cfDNA sample though the proportion differed significantly by RET mutation type (Fig. 3B). Only two of the75 (2.7%) patients with a KIF5B-RET fusion had other NSCLC drivers including one with KRAS K117N and one with EGFR R776H, which has been shown to confer constitutive ligand-independent activation of EGFR in vitro but is not well-described in patients (42, 43). In both cases, the KIF5B-RET fusion had higher clonality (1 vs. 0.47 in the KRAS-positive case and 0.54 vs. 0.05 in the EGFR-positive case), suggesting that the RET fusion was likely a primary driver in both cases. We also cannot rule out the possibility that the RET/KRAS-positive case could have had an additional primary cancer or clonal hematopoiesis given a lack of detailed clinical history. Overall the median clonality of KIF5B-RET fusions in NSCLC was 0.55 (95% CI: 0.47–0.8) and in all KIF5B-RET-positive samples, the mutations that did have higher clonality were in tumor suppressor genes or were VUS or synonymous alterations (Fig. 3C). These observations are consistent with the KIF5B-RET fusions being a primary NSCLC driver.
Figure 3.
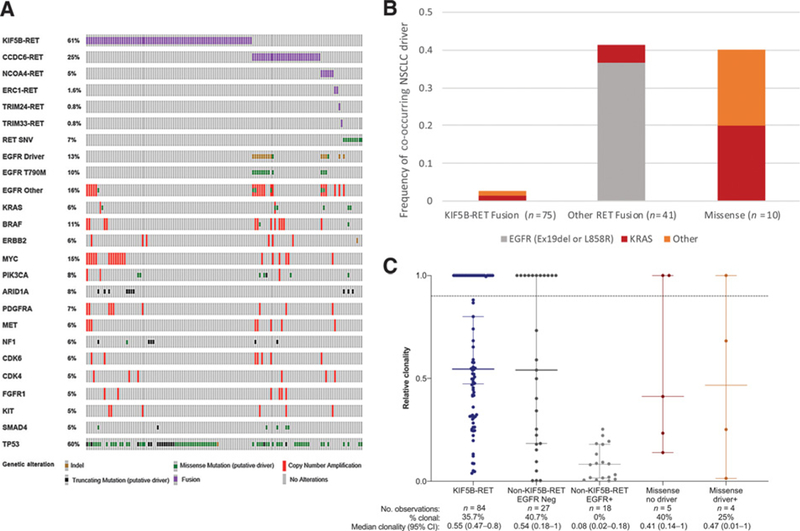
Co-occurring somatic alterations detected by NGS of cfDNA from 125 patients with advanced NSCLC harboring an oncogenic RET mutation. A, Oncoprint of somatic genomic alterations present in at least 5% of samples. The first four rows indicate RET rearrangements by fusion partner, and the fifth row represents RET oncogenic SNVs. B, Co-occurrence of oncogenic RET mutations with mutations in classic NSCLC driver genes: EGFR exon 19 deletion or L858R (gray), KRAS (red), or other (orange). Other includes EGFR R776H, EGFR exon 20 insertion, ERBB2 exon 20 insertion (n = 1 patient each). One patient with EGFR-positive NSCLC also had an ALK rearrangement and another had BRAF V600E in the setting of EGFR TKI resistance (see Fig. 4). No cases of ROS1 rearrangements, MET exon 14 skipping, or ERBB2-activating SNVs were observed. C, Relative clonality of oncogenic RET alterations (see Methods). Solid horizontal lines indicate median and 95% confidence intervals, and dashed line indicates clonality >0.9. Medians and proportions are calculated on a per alteration basis across all samples.
In contrast, other NSCLC driver mutations were commonly observed in samples containing RET SNVs and RET fusions involving partners other than KIF5B. Among the 9 patients with NSCLC and RET SNVs, 4 (44%) had classic driver mutations, including two with KRAS alterations, 1 with an EGFR exon 20 insertion, and 1 with an ERBB2 exon 20 insertion. Median clonality of RET SNVs was similar in samples containing another driver mutation as those without another driver (0.47 vs. 0.41, respectively; Fig. 3C). The RET SNVs in patients with NSCLC affected codon 609 (n = 2), 778 (n = 2), 630, 833, 886, 606, and 804 (n = 1 each). Cumulatively, our data are inconclusive as to whether RET SNVs have a strong role as drivers of NSCLC.
Seventeen of the 41 (41.4%) patients with a non-KIF5B RET fusion had one or more samples positive for either a deletion of EGFR exon 19 (n = 13) or an L858R mutation (n = 2) or a KRAS G12V or K117N alteration (n = 1 each). The median clonality of the non-KIF5B-RET fusion among EGFR-positive samples was significantly lower than those without EGFR mutations (0.08 vs. 0.53, P <0.0001; Fig. 3C). Consistent with this observation, the RET fusion appeared to be arising as a mechanism of resistance to EGFR TKIs in all 15 patients (Fig. 4). Twelve ofthe 15 (80%) RET/EGFR+ cases also had the EGFR T790 gatekeeper mutation detected, three of whom also had EGFR C797S, which has been shown to arise as a mechanism of resistance to osimertinib (44). CTNNB1 exon 3 alterations were found in 3 of the 15 patients. While CTNNB1 mutations are enriched in EGFR-mutant NSCLC, particularly clones that have acquired T790M, this frequency is higher than what has been reported in other EGFR T790M-positive NSCLC cfDNA cohorts (15). In total, 14 of 15 RET/EGFR-positive patients’ tumors harbored at least one, and typically multiple, known genomic mechanisms of resistance to EGFR TKIs (45). While serial sampling and pre-EGFRTKI tissue RET status were not available for most of the cases to confirm whether RET was acquired over time, the copy number-adjusted EGFR driver to RET fusion variant allele fraction (VAF) ratio was consistent with the RET fusion arising later in tumorigenesis in all 15 cases (Fig. 5A).
Figure 4.
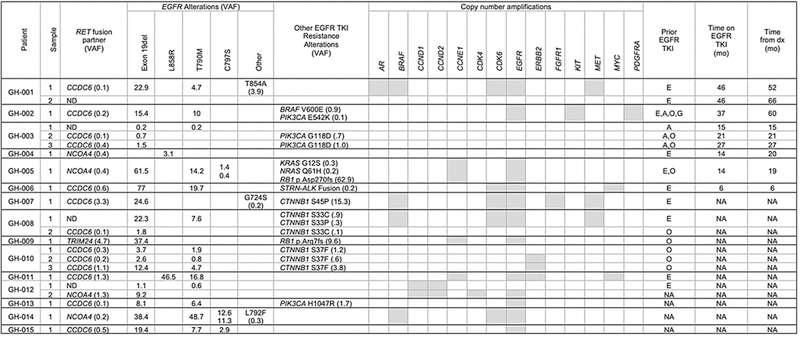
Landscape of genomic alterations found in the cfDNA of 15 patients with both an EGFR driver mutation (exon 19 deletion or L858R) and a RET fusion. The variant allele frequency (VAF) of detected alterations in each of the 22 samples from the 15 patients is shown for fusions, SNVs, and indels. Samples with copy number amplification are shaded gray. Only select alterations that are known mechanisms of resistance to anti-EGFR therapy are shown for readability. Common tumor suppressor alterations (e.g., NF1, TP53) and other SNVs (BRCA2 C3155S in patient 5, and MLH1 E433Q and JAK2 V617F in patient 12) are not shown. Patients GH-001, GH-003, GH-008, GH-010, and GH-012 had multiple samples collected; in some, the RET rearrangement was not detected in all samples. Exposure to EGFR tyrosine kinase inhibitors (TKI) prior to sample collection is indicated in the last three columns. Patients GH-001 through GH-006 had detailed history available, including order of therapies, duration of therapy in months (mo), and other molecular testing history (see Fig. 5B).Treatment history of the other patients was taken from test request forms when available. A, afatinib; E, erlotinib; G, gefitinib; NA, not available; ND, not detected; O, osimertinib.
Figure 5.
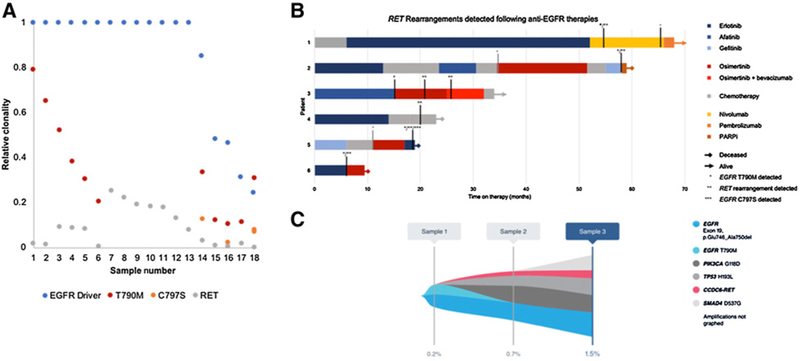
Landscape of genomic alterations found in the cfDNA of 15 patients with both an EGFR driver mutation (exon 19 deletion or L858R) and a RET fusion. A, Relative clonality of the EGFR driver mutation and RET fusion compared with on-target EGFR TKI mechanisms of resistance (T790M and C797S) among the 18 samples with RET fusion detected. B, Order and duration of systemic therapies among the 6 patients with both an EGFR driver mutation (exon 19 deletion or L858R) and a RET rearrangement detected in cfDNA and detailed clinical history available. EGFR TKIs are shown in shades of blue or red, chemotherapy (various) in gray, and other therapies in orange. Patients still living are indicated with an arrow, and time of death is indicated with a diamond. Vertical black lines indicate timing of the Guardant360 blood draw relative to treatment timing, and vertical gray lines indicate timing of other liquid biopsy or tissue molecular tests. The first detection of EGFR T790, the RET fusion, or EGFR C797S is indicated with a single, double, or tripleasterisk, respectively. C, Tumor response map for patient GH-003 showing suppression of EGFR T790M between sample 1 and sample2while the patient was on osimertinib, but emergence of RET fusion and alterations in PIK3CAand TP53.Treatment between samples2and 3 included addition of bevacizumab to osimertinib with relativelystable VAF of the RET fusion, PIK3CA, and TP53 alterations, but emergence of a SMAD4 alteration.
Furthermore, 11 of the 15 patients were indicated on test request forms as having prior exposure to EGFR TKIs and more detailed treatment and molecular testing information was available for patients GH-001toGH-006(Fig. 5B). These 6 patients all had lung adenocarcinoma at a median age of diagnosis of 55 years (range 41–65). Five had EGFR exon 19 deletion and one had L858R detected in tissue either prior to first-line therapy (patients GH-002-GH-006) or shortly after starting first-line chemotherapy (patient GH-001). Only one patient (GH-006) had evaluation of RET in tissue collected prior to EGFR TKI exposure that was RET negative by NGS. All 6 patients had been treated with at least one EGFR TKI prior to collection of the cfDNA assay that detected the RET fusion. The median time on TKI prior to detection of the RET fusion in cfDNA was 17.5 months (range 6–46 months). The RET fusion was detected following therapy with erlotinib in 3 patients, and osimertinib and first-or second-generation TKIs in the other 3 patients. The RET fusion was found without (n = 1), concurrent with (n = 2), and subsequent to (n = 3) the first detection of EGFR T790M. Three patients (patients GH-001, GH-002, and GH-003) had multiple cfDNA NGS assays. In two patients (GH-002 and GH-003), EGFR T790M, but not the RET fusion, was detected on the first sample that was drawn following progression on erlotinib and/or afatinib. The RET fusion was detected on a subsequent sample following progression on osimertinib. These cases are consistent with the RET fusion arising as an acquired mechanism of resistance.
Patients GH-002, GH-004, and GH-005 died shortly after the detection of the RET fusion without a change in therapy. Patient GH-001 had tissue “hotspot” testing for T790M that was negative upon progression after 46 months of treatment with erlotinib. The patient was placed on nivolumab and had stabilization of disease. Two months after initiating nivolumab, the patient had G360 testing that revealed 5 EGFR alterations (exon 19 deletion, T790M, L777M, T854A, and amplification), a CCDC6-RETfusion and amplifications of MET, BRAF, AR, and CDK6. The patient continued on nivolumab and another G360 was drawn after 12 months that showed resolution of the EGFR and RET alterations. The patient was switched to pembrolizumab due to side effects and continues to have stable disease 2 months after starting pembrolizumab.
In patient GH-003, G360 revealed EGFR exon 19 deletion and T790M following progression on afatinib (15-month treatment duration). Therapy was switched to osimertinib; however, after 7 months, the patient experienced disease progression. A second G360 at that timepoint identified the EGFR exon 19 deletion, the RET fusion as well as alterations in TP53 and PIK3CA, but not EGFR T790M, implicating the RET fusion as a mechanism of resistance independent of EGFR T790M (Fig. 5C). The patient was continued on osimertinib for another 10 months with the addition of bevacizumab. On progression, a third G360 revealed the same EGFR exon 19 deletion, the RET fusion, TP53, and PIK3CA alterations as well as a new alteration in SMAD4; however, EGFR T790M was not detected. The patient was placed on chemotherapy and had a partial response according to RECIST 1.1 criteria.
In patient GH-006, following progression on erlotinib, a G360 test revealed an exon 19 deletion, T790M, and EGFR amplification in addition to the RET fusion, an ALK rearrangement, a TP53 alteration, and amplifications of MYC and CDK6. Therapy was switched to osimertinib. The patient had a mixed response, progressed after 3.5 months of therapy, and died shortly thereafter.
Across the entire dataset, the prevalence of RET fusions among all patients with NSCLC with an EGFR exon 19 deletion was higher than that of patients with L858R (0.8% vs. 0.2%, P = 0.04) and higher among patients with coexisting T790M and/or C797S versus those with EGFR drivers without T790M or C797S (1.1% and 4.6%, respectively, vs. 0.6%). Among patients with NSCLC with treatment history information submitted at the time of sample collection, the prevalence of a RET fusion was higher following exposure to osimertinib (9/184 samples, 4.9%) than first-or second-generation EGFR TKIs (13/1,627 samples, 0.8%, P = 0.0001).
Discussion
This is the largest series to date describing the genomic features of advanced cancers harboring activating somatic RET alterations. Somatic activating RET alterations are found in cfDNA of approximately 1 in 200 patients in a wide range of solid tumor types. They are potentially targeteable as demonstrated by the successful treatment of cfDNA NGS-detected RET fusion driver alterations in NSCLC (25). In nearly 33,000 patients, KIF5B-RET fusions were only identified in patients with NSCLC (n = 75) or CUP (n = 2), suggesting that the presence of a KIF5B-RET fusion may be pathognomonic for a NSCLC diagnosis. If confirmed in a larger CUP cohort, this finding may help determine patients with CUP with NSCLC primary tumors.
The detection of both clonal and acquired subclonal RET alterations, including notation of the fusion partner for rearrangements, builds upon previous findings that a plasma-based cfDNA NGS assay may be useful. The quantitative nature of variant allele fractions (VAF) in cfDNA permits the assessment of the relative burden of various alterations contributing to tumor progression (15, 37). The highest VAF alterations are generally the earliest events in tumorigenesis while alterations that arise later in tumorigenesis tend to have a VAF that is lower than these truncal events.
In a study of 4,871 diverse tumor types tested with a 182-or 236-gene tissue NGS panel, 88 (1.3%) had a RET alteration and similar to this study, most (81%) had coexisting alterations (although all here are non-VUS alterations; ref. 3). In tissue, RET fusions did not co-occur with alterations in MAPK effectors and co-occurred with alterations in other tyrosine kinase family genes in only 7.4% of cases. In contrast, in this cfDNA cohort, RET fusions co-occurred with alterations in these pathways in 26.2% and 33.3% of patients, respectively.
Given that the cfDNA assay used in this study has been extensively validated with high specificity, the differences in co-occurring alterations between plasma and tissue are likely due to biological factors and we provide evidence that this observation is likely a result of some RET fusions and MAPK pathway alterations arising as a mechanism of therapy resistance (38). The cfDNA testing population is comprised of patients with advanced cancers and who are often treated with one or more systemic therapies prior to sample collection, whereas tissue compendia may contain early-stage tumors that have not been exposed to systemic therapy. Furthermore, cfDNA overcomes challenges of intra-and inter-tumor heterogeneity. The frequent presence of subclonal resistance alterations has previously been shown to account for major differences in molecular profiles identified using cfDNA NGS in advanced generally pretreated cohorts compared with generally early-stage, treatment-naive tissue cohorts (37). The differences between tissue and cfDNA genomic landscapes highlight the value of reassessing tumor genomic status in patients with advanced cancer, particularly following treatment with a targeted agent. As activation of RET promotes downstream pathways including RAS/MAPK, JAK/STAT, and PI3K/AKT, inhibiting RET in the setting of a co-occurring MAPK or other downstream pathway may be less efficacious (4, 46, 47).
The majority of cfDNA-detected MAPK and other RTK alterations were identified in patients with NSCLC or colorectal cancer who may have received EGFR-directed therapy prior to cfDNA collection and acquired alterations in these pathways as a mechanism of therapy resistance (48, 49). Indeed, NSCLCs harboring subclonal RET fusions and coexisting EGFR driver mutations with available treatment histories all had exposure to first-, second-, and/or third-generation EGFR TKIs prior to cfDNA sample collection. Additional work in a larger cohort is needed to better understand the contribution of RET to therapy resistance or as a driver in other cancer types, particularly in colorectal cancer and breast cancer that are commonly treated with targeted therapies.
Interestingly, only acquired subclonal non-KIF5B-RET fusions (particularly with CCDC6 or NCOA4 as the fusion partner) arise at progression in NSCLC, whereas the more common gene partner is KIF5B when RET fusions are the clonal oncogenic driver. The different frequency of fusion partners seen as drivers versus acquired mechanisms of resistance in NSCLC has been reported (19). The dominant partner in ALK fusion drivers is EML4 (>95% of cases), whereas EML4 is the partner in only approximately 45% of EGFR TKI-resistant cases. However, the fact that no KIF5B-RET fusion arose as a mechanism of resistance in this series suggests emergent KIF5B-RET fusions may not be tolerated in the presence of anti-EGFR TKI and/or that loss of CCDC6 or NCOA4 affords a particular survival advantage to tumors exposed to an EGFR TKI. In a Drosophila model, KIF5B-RET fusions (particularly the motor domain that is unique to KIF5B relative to the other fusion partners) are highly reliant on EGFR signaling to promote enhanced cell growth, more so than CCDC6-RET or NCOA4-RET fusions (50). There may be important cell biology differences based on the RET fusion partner. Future studies of RET should involve assays such as NGS that can distinguish between fusion partners.
While non-KIF5B-RET fusions appear to be a rare mechanism of EGFR TKI resistance, they may be more likely to emerge in more advanced disease given their frequent co-occurrence with multiple mechanisms of EGFR TKI resistance. Our data also suggest that acquired RET fusions may be more common following exposure to osimertinib than to first-or second-generation EGFR TKIs. While our observation that 4.9% of NSCLCs with prior osimertinib exposure had a RET fusion should be interpreted with caution given the limited available clinical history, Piotrowska and colleagues recently found RET fusions in a similar proportion (2/41, 4.9%) of confirmed osimertinib-resistant biopsies (23). The cfDNA results of patient GH-003 suggest that during therapy with osimertinib, the RET fusion arose concurrent with suppression of T790M indicating that RET fusions may arise in subclones separate from other mechanisms of resistance. Piotrowska and colleagues reported two patients with similar findings where the T790M resistance clone was successfully suppressed with osimertinib while the RET fusion was detected at progression. In these two cases, combination therapy with osimertinib and BLU-667 (a novel selective RET inhibitor) was well-tolerated and led to rapid radiographic responses. Given the recent FDA approval of osimertinib for first-line treatment of EGFR-driven NSCLC, RET fusions may be a relevant factor for treatment selection at progression. The frequency and diversity of coexisting altered oncogenic pathways suggests that comprehensive genomic profiling may be needed in patients progressing on EGFRTKIs to appreciate the entire resistance profile and that optimal targeting of patients with acquired RET alterations may require customized combination strategies. Detection of targetable resistance mutations using a comprehensive cfDNA assay may facilitate enrollment in clinical trials as well as serve as biomarkers for therapeutic efficacy.
This study is a retrospective review of genomic findings from a cohort of patients with clinically ordered cfDNA testing and complete treatment history and follow-up data are not available for most of the cohort. Even with this limitation, this cohort offers a snapshot of advanced cancer genomics encountered in a “real-world” setting. As patients for this study were selected on the basis of having a positive RET alteration in cfDNA, the overall sensitivity of plasma detection of RET alterations cannot be assessed. In addition, the cfDNA assay used only reports RET fusions with partners known to be biologically significant. However, as has been previously reported by Zill and colleagues, we found the prevalence of intronic breakpoints largely recapitulates what has been described in tissue, which provides validation of the cfDNA NGS assay from a biological perspective (37).
In summary, cfDNA NGS testing may be beneficial at identifying less common, but potentially targetable alterations in RET as well as multiple resistance mechanisms that may be present in different tumor populations. This study provides additional evidence that advanced-stage cancers, particularly those having progressed on targeted therapy, may be driven by multiple oncogenic pathways, which may not be apparent from tissue taken at initial diagnosis. The clinical impact of these findings, such as targeting acquired RET fusions in NSCLCs resistant to anti-EGFR therapy, requires further investigation. As cfDNA testing provides a noninvasive method of capturing tumor heterogeneity and can evaluate the dynamics of response to therapy, it may provide a useful platform for further studying the impact of clonal and subclonal mutations on tumor progression as well as inform studies of rational combination therapies.
Supplementary Material
Translational Relevance.
Cell-free circulating tumor DNA next-generation sequencing testing may be beneficial at identifying less common, but potentially targetable alterations such as activating RET alterations as well as providing an overview of multiple resistance mechanisms that may be present in different tumor populations. This study provides additional evidence to a growing body of literature that advanced stage cancers, particularly those having progressed on targeted therapy, maybe driven by multiple oncogenic pathways that may not be apparent from tissue taken at initial cancer diagnosis or from single lesions at the time of progression.
Acknowledgments
T.A. Rich has ownership interests (including patents) at Guardant Health. K. L. Reckamp is a consultant/advisory board member for and reports receiving commercial research support from Guardant. Y.K. Chae reports receiving speakers bureau honoraria from Genentech, AstraZeneca, Guardant Health, and Lilly Oncology; is a consultant/advisory board member for Foundation Medicine, Guardant Health, Takeda, Genentech, and AstraZeneca; and reports receiving commercial research grants from Biodesix, Freenome, Lexent Bio, Abbvie, and Bristol-Myers Squibb. R.C. Doebele has ownership interests (including patents) at Rain Therapeutics; is a consultant/advisory board member for Rain Therapeutics, Genentech/Roche, Bayer, and Takeda/Millenium; and has licensing fees for a patent with Abbott. W.T. Iams is a consultant/advisory board member for Outcome Insights and has done clinical trial planning conference travel for EMD Serono. R.B. Lanman is an employee of Biolase; has ownership interests (including patents) at Guardant Health, Biolase, and Forward Medical; and is a consultant/advisory board member for Forward Medical. J.W. Reiss is a consultant/advisory board member for Loxo Oncology. T.E. Stinchcombe is a consultant/advisory board member for Takeda, AstraZeneca, Genetech/Roche and G1 Therapeutics. V. Subbiah reports receiving commercial research support from LOXO Oncology, Blueprint Medicines, Exelixis, Takeda, Roche, Novartis, Bayer, GlaxoSmithKline, Nanocarrier, Vegenics, Celgene, Northwest Biother-apeutics, Berghealth, Incyte, Fujifilm, Pharmamar, D3, Pfizer, Multivir, Amgen, Abbvie, Alfasigma, Agensys, Boston Biomedical, Idera Pharma, and Inhibrx. S.R. Fairclough has ownership interests (including patents) at Guardant Health. J. Yen has ownership interests (including patents) at Guardant Health.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed by the other authors.
Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Ettinger DS, Wood DE, Aisner DL, Akerley W, Bauman J, Bharat A, et al. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): non-small cell lung cancer, version 3.2019 [Internet]; 2019. Available from: www.nccn.org.
- 2.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23:703–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kato S, Subbiah V, Marchlik E, Elkin SK, Carter JL, Kurzrock R. RET aberrations in diverse cancers: next-generation sequencing of 4,871 Patients. Clin Cancer Res 2017;23:1988–97. [DOI] [PubMed] [Google Scholar]
- 4.Mulligan LM. RET revisited: expanding the oncogenic portfolio. Nat Rev Cancer 2014;14:173–86. [DOI] [PubMed] [Google Scholar]
- 5.Ciampi R, Nikiforov YE. RET/PTC rearrangements and BRAF mutations in thyroid tumorigenesis. Endocrinology 2007;148:936–41. [DOI] [PubMed] [Google Scholar]
- 6.Wang R, Hu H, Pan Y, Li Y, Ye T, Li C, et al. RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J Clin Oncol 2012;30:4352–9. [DOI] [PubMed] [Google Scholar]
- 7.Drilon A, Wang L, Arcila ME, Balasubramanian S, Greenbowe JR, Ross JS, et al. Broad, hybrid capture-based next-generation sequencing identifies actionable genomic alterations in lung adenocarcinomas otherwise negative for such alterations by other genomic testing approaches. Clin Cancer Res 2015;21:3631–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamatani K, Eguchi H, Ito R, Mukai M, Takahashi K, Taga M, et al. RET/PTC rearrangements preferentially occurred in papillary thyroid cancer among atomic bomb survivors exposed to high radiation dose. Cancer Res 2008; 68:7176–82. [DOI] [PubMed] [Google Scholar]
- 9.Marsh DJ, Learoyd DL, Andrew SD, Krishnan L, Pojer R, Richardson AL, et al. Somatic mutations in the RET proto-oncogene in sporadic medullary thyroid carcinoma. Clin Endocrinol 1996;44:249–57. [DOI] [PubMed] [Google Scholar]
- 10.Pietrantonio F, Di Nicolantonio F, Schrock AB, Lee J, Morano F, Fucà G, et al. RET fusions in a small subset of advanced colorectal cancers at risk of being neglected. Ann Oncol 2018;29:1391–401. [DOI] [PubMed] [Google Scholar]
- 11.Moura MM, Cavaco BM, Pinto AE, Domingues R, Santos JR, Cid MO, et al. Correlation of RET somatic mutations with clinicopathological features in sporadic medullary thyroid carcinomas. Br J Cancer 2009;100:1777–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elisei R, Cosci B, Romei C, Bottici V, Renzini G, Molinaro E, et al. Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study. J Clin Endocrinol Metab 2008; 93:682–7. [DOI] [PubMed] [Google Scholar]
- 13.Dvorakova S, Vaclavikova E, Sykorova V, Vcelak J, Novak Z, Duskova J, et al. Somatic mutations in the RET proto-oncogene in sporadic medullary thyroid carcinomas. Mol Cell Endocrinol 2008;284:21–7. [DOI] [PubMed] [Google Scholar]
- 14.Wirth LJ, Cabanillas ME, Sherman EJ, Solomon B, LeBoulleux S,Robinson B, et al. Clinical activity of LOXO-292, a highly selective RET inhibitor, in patients with RET-altered thyroid cancers. Thyroid 2018;28:1.29334341 [Google Scholar]
- 15.Blakely CM, Watkins TBK, Wu W, Gini B, Chabon JJ, McCoach CE, et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet 2017;49: 1693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arbour KC, Jordan E, Kim HR, Dienstag J, Yu HA, Sanchez-Vega F, et al. Effects of co-occurring genomic alterations on outcomes in patients with KRAS-Mutant non-small cell lung cancer. Clin Cancer Res 2018;24: 334–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov 2015;5:860–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov 2018;8:822–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCoach CE, Blakely CM, Banks KC, Levy B, Chue BM, Raymond VM, et al. Clinical utility of cell-free DNA for the detection of ALK fusions and genomic mechanisms of ALK inhibitor resistance in non-small cell lung cancer. Clin Cancer Res 2018;24:2758–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ko R, Kenmotsu H, Serizawa M, Koh Y, Wakuda K, Ono A, et al. Frequency of EGFR T790M mutation and multimutational profiles of rebiopsy samples from non-small cell lung cancer developing acquired resistance to EGFR tyrosine kinase inhibitors in Japanese patients. BMC Cancer 2016; 16:864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCoach CE, Le AT, Gowan K,Jones K, Schubert L, Doak A, et al. Resistance mechanisms to targeted therapies in ROS1 + and ALK+ non-small cell lung cancer. Clin Cancer Res 2018;24:3334–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klempner SJ, Bazhenova LA, Braiteh FS, Nikolinakos PG, Gowen K, Cervantes CM, et al. Emergence of RET rearrangement co-existing with activated EGFR mutation in EGFR-mutated NSCLC patients who had progressed on first-or second-generation EGFRTKI. Lung Cancer 2015; 89:357–9. [DOI] [PubMed] [Google Scholar]
- 23.Piotrowska Z, Isozaki H, Lennerz JK, Gainor JF, Lennes IT, Zhu VW, et al. Landscape of acquired resistance to osimertinib in EGFR-mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU-667 for acquired RET fusion. Cancer Discov 2018; 8:1529–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gautschi O, Milia J, Filleron T, Wolf J, Carbone DP, Owen D, et al. Targeting RET in patients with RET-rearranged lung cancers: results from the global, multicenter RET registry. J Clin Oncol 2017;35:1403–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarfaty M, Moore A, Neiman V, Dudnik E, Ilouze M, Gottfried M, et al. RET fusion lung carcinoma: response to therapy and clinical features in a case series of 14 patients. Clin Lung Cancer 2017;18:e223–32. [DOI] [PubMed] [Google Scholar]
- 26.Aisner DL, Sholl LM, Berry L, Rossi M, Chen H, Fujimoto J, et al. The impact of smoking and TP53 mutations in lung adenocarcinoma patients with targetable mutations-the Lung Cancer Mutation Consortium (LCMC2). Clin Cancer Res 2018;24:1038–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moccia M, Liu Q, Guida T, Federico G, Brescia A, Zhao Z, et al. Identification of novel small molecule inhibitors of oncogenic RET kinase. PLoS ONE 2015;10:e0128364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carlomagno F, Guida T, Anaganti S, Vecchio G, Fusco A, Ryan AJ, et al. Disease associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene 2004;23: 6056–63. [DOI] [PubMed] [Google Scholar]
- 29.Subbiah V, Velcheti V, Tuch BB, Ebata K, Busaidy NL, Cabanillas ME, et al. Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol 2018;29:1869–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dagogo-Jack I, Stevens SE, Lin JJ, Nagy R, Ferris L, Shaw AT, et al. Emergence of a RETV804M gatekeeper mutation during treatment with vandetanib in RET-rearranged NSCLC. J Thorac Oncol 2018;13:e226–7. [DOI] [PubMed] [Google Scholar]
- 31.Subbiah V, Gainor JF, Rahal R, Brubaker JD, Kim JL, Maynard M, et al. Precision targeted therapy with BLU-667 for RET-driven cancers. Cancer Discov 2018;8:836–49. [DOI] [PubMed] [Google Scholar]
- 32.Drilon AE, Liu S, Doebele R, Rodriguez C, Fakih M, Reckamp KL, et al. LBA19A phase 1b study of RXDX-105, a VEGFR-sparing potent RET inhibitor, in RETi-naïve patients with RET fusion-positive NSCLC. Ann Oncol 2017;28:mdx440.012. [Google Scholar]
- 33.Drilon AE, Subbiah V, Oxnard GR, Bauer TM, Velcheti V, Lakhani NJ, et al. A phase 1 study ofLOXO-292, a potent and highly selective RET inhibitor, in patients with RET-altered cancers. J Clin Oncol 36:15s, 2018. (suppl; abstr 102). [Google Scholar]
- 34.Yoh K, Seto T, Satouchi M, Nishio M, Yamamoto N, Murakami H, et al. Vandetanib in patients with previously treated RET-rearranged advanced non-small-cell lung cancer (LURET): an open-label, multicentre phase 2 trial. Lancet Respir Med 2017;5:42–50. [DOI] [PubMed] [Google Scholar]
- 35.Drilon A, Siqing F, Patel MR, Fakih M, Wang D, Olszanski AJ, et al. A phase I/Ib trial of the VEGFR-sparing multikinase RET inhibitor RXDX-105. Cancer Discov 2019;9:384–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu Y, Alden RS, Odegaard JI, Fairclough SR, Chen R, Heng J, et al. Discrimination of germline EGFR T790M mutations in plasma cell-free DNA allows study of prevalence across 31,414 cancer patients. Clin Cancer Res 2017;23:7351–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zill OA, Banks KC, Fairclough SR, Mortimer SA, Vowles JV, Mokhtari R, et al. The landscape of actionable genomic alterations in cell-free circulating tumor DNA from 21,807 advanced cancer patients. Clin Cancer Res 2018;24:3528–38. [DOI] [PubMed] [Google Scholar]
- 38.Odegaard JI, Vincent JJ, Mortimer S, Vowles JV, Ulrich BC, Banks KC, et al. Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue-and plasma-based methodologies. Clin Cancer Res 2018;24:3539–49. [DOI] [PubMed] [Google Scholar]
- 39.Mizukami T, Shiraishi K, Shimada Y, Ogiwara H, Tsuta K, Ichikawa H, et al. Molecular mechanisms underlying oncogenic RET fusion in lung adenocarcinoma. J Thorac Oncol 2014;9:622–30. [DOI] [PubMed] [Google Scholar]
- 40.Cañibano C, Rodriguez NL, Saez C, Tovar S, Garcia-Lavandeira M, Borrello MG, et al. The dependence receptor Ret induces apoptosis in somatotrophs through a Pit-1/p53 pathway, preventing tumor growth. EMBO J 2007;26: 2015–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bordeaux MC, Forcet C, Granger L, Corset V, Bidaud C, Billaud M, et al. The RET proto-oncogene induces apoptosis: a novel mechanism for Hirsch-sprung disease. EMBO J 2000;19:4056–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruan Z, Kannan N. Mechanistic insights into R776H mediated activation of epidermal growth factor receptor kinase. Biochemistry 2015;54:4216–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Noesel J, van der Ven WH, van Os TAM, Kunst PWA, Weegenaar J, Reinten RJA, et al. Activating germline R776H mutation in the epidermal growth factor receptor associated with lung cancer with squamous differentiation. J Clin Oncol 2013;31:e161–4. [DOI] [PubMed] [Google Scholar]
- 44.Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFRT790M. Nat Med 2015;21: 560–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol 2014;11: 473–81. [DOI] [PubMed] [Google Scholar]
- 46.Alberti L, Carniti C, Miranda C, Roccato E, Pierotti MA. RET and NTRK1 proto-oncogenes in human diseases. J Cell Physiol 2003;195:168–86. [DOI] [PubMed] [Google Scholar]
- 47.Phay JE, Shah MH. Targeting RET receptor tyrosine kinase activation in cancer. Clin Cancer Res 2010;16:5936–41. [DOI] [PubMed] [Google Scholar]
- 48.Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun 2016;7:11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pietrantonio F, Vernieri C, Siravegna G, Mennitto A, Berenato R, Perrone F, et al. Heterogeneity of acquired resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer. Clin Cancer Res 2017;23:2414–22. [DOI] [PubMed] [Google Scholar]
- 50.Das TK, Cagan RL. KIF5B-RET oncoprotein signals through a multi-kinase signaling hub. Cell Rep 2017;20:2368–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.