

Abstract
Preeclampsia is characterized by new onset hypertension and fetal growth restriction and is associated with aberrant activation of the innate immune complement system and stressed or ischemic placenta. Previous studies have suggested a role for both endothelin and complement system activation products in new onset hypertension in pregnancy, but inter-relationships of the pathways are unclear. We hypothesized that complement activation following placental ischemia stimulates the endothelin pathway to cause hypertension and impair fetal growth. The Reduced Uterine Perfusion Pressure (RUPP) model results in hypertension and fetal growth restriction in a pregnant rat due to placental ischemia caused by mechanical obstruction of blood flow to uterus and placenta. The effect of inhibitor of complement activation soluble Complement Receptor 1 (sCR1) and endothelin A receptor (ETA) antagonist atrasentan on hypertension, fetal weight, complement activation (systemic circulating C3a and local C3 placental deposition) and endothelin [circulating endothelin and message for preproendothelin (PPE), ETA and endothelin B receptor (ETB in placenta] in the RUPP rat model were determined. Following placental ischemia, sCR1 attenuated hypertension but increased message for PPE and ETA in placenta, suggesting complement activation causes hypertension via an endothelin independent pathway. With ETA antagonism the placental ischemia-induced increase in circulating C3a was unaffected despite inhibition of hypertension, indicating systemic C3a alone is not sufficient. In normal pregnancy, inhibiting complement activation increased plasma endothelin but not placental PPE message. Atrasentan treatment increased fetal weight, circulating endothelin and placental ETA message, and unexpectedly increased local complement activation in placenta (C3 deposition) but not C3a in circulation, suggesting endothelin controls local placental complement activation in normal pregnancy. Atrasentan also significantly decreased message for endogenous complement regulators Crry and CD55 in placenta and kidney in normal pregnancy. Results of our study indicate that complement/endothelin interactions differ in pregnancies complicated with placental ischemia vs normal pregnancy, as well as locally vs systemically. These data clearly illustrate the complex interplay between complement and endothelin indicating that perturbations of either pathway may affect pregnancy outcomes.
Keywords: complement, endothelin, placental ischemia, pregnancy
Graphical Abstract
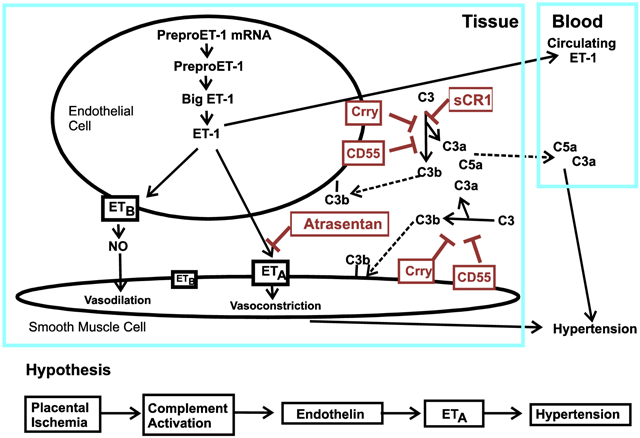
1. Introduction
Control of the immune system is very important in maintaining a normal pregnancy. Consequently, dysregulation of the immune system can play a role in the pathogenesis of adverse pregnancy outcomes including pregnancy-induced hypertension and fetal growth restriction. Both adaptive and innate immune mechanisms have been implicated in abnormal spiral artery remodeling in the developing placenta, resulting in placental ischemia, hypertension and intrauterine growth restriction. Our studies focus on events following placental ischemia, where exacerbated immune activation has a demonstrated role in the pathophysiology. In particular, our studies are designed to define inter-relationships of the innate immune complement system and the endothelin system in normal pregnancy in the rat, as well as in the stressed placenta following third trimester placental ischemia using the Reduced Uterine Perfusion Pressure (RUPP) model to cause gestational hypertension and fetal growth restriction.
The complement system is composed of more than 30 different proteins operating extracellularly and intracellularly to protect from invaders, both in concert with the adaptive immune response as well as independent of cellular and humoral immunity [1]. The complement system is essential for a normal pregnancy and for protection of both mother and child from infection. The innate immune complement system is activated in a normal pregnancy [2, 3], and numerous studies have demonstrated more pronounced activation in preeclamptic pregnancies [4], with excessive activation of the complement system having deleterious effects. In fact, a case study provided evidence that treatment of a preeclamptic pregnancy with antibody to complement component C5 prolonged the pregnancy by more than 2 weeks, indicating the clinical relevance of excessive complement activation in the pathology of preeclampsia [5].
Chronic placental ischemia in the RUPP model in the rat results in increased blood pressure in the mother, fetal growth restriction and excessive complement activation as indicated by generation of C3a [6, 7] with increased C3 deposition in placenta [8]. Inhibiting complement activation with soluble Complement Receptor 1 (sCR1), as well as antagonism of C3a or C5a receptors, attenuates placental ischemia-induced hypertension in the RUPP rat model [6, 7] but does not reverse fetal growth restriction. In addition to complement system activation, placental ischemia in the RUPP model results in activation of endothelin 1 (ET-1) pathway [9] with clinical evidence to indicate that endothelin is increased in preeclampsia [10]. ET-1 is produced by the endothelium abluminally and acts on the endothelin A receptor (ETA) in smooth muscle to cause vasoconstriction [11]. It can also act via the endothelin B receptor (ETB) to cause vasodilation and/or to clear endothelin, minimizing entry into the circulation. Evidence suggests that ETB on smooth muscle plays a minor role in blood pressure regulation [12] with the most significant contribution from ETA. In a rat model of placental ischemia-induced hypertension, antagonism of ETA with the selective ETA antagonist, atrasentan, prevents the increase in blood pressure [13, 14], but does not reverse the intrauterine growth restriction. An ET-1 dependent mechanism is also responsible for hypertension in the pregnant rat induced by TNF [15], sFlt-1 [16] and autoantibodies to the angiotensin receptor [17], providing supportive evidence that ET-1 is a central downstream mediator of placental ischemia-induced hypertension [18]. However, not all mediators of placental ischemia-induced hypertension are ET-1 dependent as data indicates that IL-17 causes hypertension in pregnancy but acts independently of ET-1 [19]. Whether complement activation leads to placental ischemia-induced hypertension by an endothelin dependent mechanism is unknown. Though complement activation has been linked with endothelin activation in a number of other experimental model systems [20-24], connections of the two pathways have not been investigated following placental ischemia.
Multiple interventions in the RUPP model result in attenuation of the hypertension, but without favorable effects on fetal growth. In our previous studies using sCR1 to inhibit complement activation, we noted that sCR1 treatment exacerbated the placental ischemia-induced fetal growth restriction, suggesting a role for complement activation in maintaining normal fetal growth following placental ischemia. In addition, evidence indicates that endothelin acting through the ETA receptor results in fetal growth restriction following hypoxia in the pregnant rat [25, 26]. In the present study, we hypothesized that complement activation following placental ischemia results in activation of endothelin pathway to cause hypertension and impact fetal growth. To test this hypothesis, we determined if an inhibitor of complement activation (soluble complement receptor 1; sCR1) that attenuates placental ischemia-induced hypertension would affect changes in the ET-1 system following placental ischemia, or whether ETA antagonist atrasentan altered placental ischemia-induced complement activation systemically or locally. Our results reveal important interactions between the endothelin and complement systems that may impact fetal growth and pregnancy outcomes, with different interactions identified following placental ischemia compared to normal pregnancy, either locally or systemically.
2. Materials and Methods
2.1. Reduced Uterine Perfusion Pressure (RUPP) procedure
The RUPP procedure was used as a model of placental ischemia-induced hypertension in the third trimester pregnant rat as previously described [6-8, 27]. All studies in animals were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health, and were approved by the University of Minnesota Institutional Animal Care and Use Committee. Timed pregnant Sprague Dawley dams (Crl:CD IGS, Charles River Laboratories, Raleigh NC) were anesthetized with isoflurane on gestation day (GD)14 with the date of vaginal plug designated as 0. A ventral midline incision was made, the lower abdominal aorta isolated, and a sterile silver clip (0.203 mm inner diameter) placed around the aorta above the iliac bifurcation. Both right and left uterine arcades were also clipped at the ovarian end, directly before the first segmental artery, using a silver clip (0.100 mm inner diameter) to prevent compensatory blood flow to the placenta. For the comparison group, a Sham surgery differing only in absence of the clips was also conducted. On GD18, the carotid artery was cannulated under isoflurane anesthesia using a 25% dextrose lock solution in sterile pyrogen free saline to maintain cannula patency. On GD19, mean arterial pressure (MAP) was measured from the arterial catheter in an unanesthetized restrained rat. Following measurement of blood pressure, the serum, plasma and tissues were collected in necropsy. The uterus was exteriorized, the total number of viable and resorbed pups counted and the pups and placentae weighed. From the right horn, select placenta in atrasentan experiments were frozen in OCT for immunohistochemistry and measurement of C3 deposition. From the left horn, select placenta were flash frozen in liquid nitrogen for isolation of RNA for qRT PCR.
2.2. Experimental Design and Treatments
sCR1.
We previously reported that daily intravenous treatment with 15 mg/kg sCR1 from GD14 to 18 significantly attenuated placental ischemia-induced hypertension [6]. sCR1 (Celldex Therapeutics, Inc., Needham, MA) is a soluble form of the endogenous complement regulator CR1 (CD35) with demonstrated ability to inhibit complement activation at the C3 and C5 convertases of the complement pathways, and demonstrated efficacy in numerous rat models of autoimmune and inflammatory diseases. Flash frozen placenta and plasma archived from those published studies were used to determine if sCR1 treatment significantly altered message for preproendothelin (PPE), ETA or ETB or plasma endothelin. Control injections in Sham and RUPP animals consisted of daily intravenous treatment with saline (Veh).
Atrasentan.
Atrasentan hydrochloride was purchased from ChemScene (CAS No 195733-43-8; Monmouth Junction, NJ) and was 99.64% pure by LCMS. Rats were provided with either 50 or 75 ug/ml atrasentan in drinking water beginning GD13 until necropsy. Total water consumption was measured over the 6 day period and body weight monitored daily. Rats were randomly assigned to one of six experimental groups based on surgical procedure and water treatment: 1) RUPP surgery with water ad lib (RUPP Water, n=8); 2) RUPP surgery with 50 ug/ml atrasentan in water (RUPP 50 ug/ml atrasentan, n=6); 3) RUPP surgery with 75 ug/ml atrasentan in water (RUPP 75 ug/ml atrasentan, n=6); 4) sham surgery with water ad lib (Sham Water, n=10); 5) sham surgery with 50 ug/ml atrasentan in water (Sham 50 ug/ml atrasentan, n=7); 5) sham surgery with 75 ug/ml atrasentan in water (Sham 75 ug/ml atrasentan, n=5).
2.3. Complement System measurements
C3a.
The circulating complement activation product C3a was measured in serum by Western blot as previously described [6] using the IgG fraction of a rabbit polyclonal antibody to the 9 carboxy terminal amino acids of rat C3a (Research Genetics, Huntsville, AL) as primary antibody. Modifications included use of NuPAGE Novex 10% Bis-Tris gels with MES SDS Running buffer and the secondary antibody IRDye 800CW Goat anti-Rabbit IgG (H+L) at 1/10,000 dilution and LiCor Odyssey Fc for imaging. A standard curve of rat serum activated by yeast was included on each gel. Relative amounts of C3a were expressed as C3a units/ul based on signal intensity of 1 ul of standard pool of rat serum activated by yeast.
CH50.
Total hemolytic complement activity was determined as previously described [28]. Briefly, the assay involves serial dilution of serum and incubation with antibody-coated sheep red blood cells for one hour at 37 C. The antibody coated cells are lysed by complement, releasing hemoglobin. The inverse dilution of serum that results in 50% hemolysis of sensitized sheep erythrocytes (CH50) was determined in the presence or absence of atrasentan. CH50 is a general measure of the overall function of the classical pathway of complement activation.
Immunohistochemistry for C3 and IgM.
Placenta were frozen in OCT freezing medium and 8 um sections cut and placed on slides for immunohistochemistry as previously described [8] using polyclonal goat anti-rat C3 (MP Biomedical 55713; Santa Ana, CA) and appropriate isotype control antibody followed by appropriate secondary antibodies. This polyclonal antibody will detect C3 and its fragments but cannot distinguish between C3b or C3c/d to provide an indication of the acute vs chronic nature of the complement activation. The images were scored by two blinded observers in comparison to the isotype control as negative (0), weakly positive (1), positive (2) or strongly positive (3).
2.4. qRT PCR
RNA isolation and cDNA synthesis from flash frozen tissues was as previously described [8], Primers for assessment of rat Vascular endothelial growth factor A (VEGF) were obtained from Bio-Rad (Prime PCR SYBR Green Assay; Vegfa, Rat) and reaction efficiency confirmed. For all other primers, expected product size was confirmed by gel electrophoresis, and qRT PCR products were sequenced to verify that the product was of the expected size and sequence for each primer set (University of Minnesota Genomics Center). Equal primer efficiencies for β-actin and each of the targets were validated. Real time PCR reactions for all primers were performed in duplicate and the Delta-delta Ct method of relative quantification was used to determine fold change in mRNA expression compared to β-actin with the change in Sham animals with plain drinking water defined as 1. Primers for rat ETA and ETB receptors were as published [29], For other targets, custom primers were obtained from Integrated DNA technologies (Coralville, Iowa). For rat complement regulators Crry, CD55 and rat β-actin we used previously published and validated primers [8], Primers for preproendothelin (PPE) were; forward 5-GAACTCCGAGCCCAAAGTACCATG-3, reverse 5-TTAGTTTTCTTCCCTCCACCAGCTG-3. In addition, selected experiments used previously published primers for PPE [30] to confirm that qRT PCR results with either primer set were equivalent.
2.5. Endothelin measurements
Endothelin-1 was quantitated in EDTA plasma using R&D Endothelin Quantikine ELISA kit (DET100, Minneapolis, MN).
2.6. Effect of atrasentan on cells in culture
IEC-6 (Intestinal epithelial cells) cells were obtained from ATCC and cultured as described by ATCC and previously published [31]. RBE4 cells were a kind gift from Dr. Les Drewes and were cultured as originally described by Roux et al [32]. Atrasentan at 0, 10, 20 and 40 ug/ml was added to the culture media and cells cultured in duplicate for 24 hours. Cells were harvested and RNA isolated for qRT PCR as for tissues. Values presented represent the mean± standard error for 4-5 separate experiments (each with duplicates) and are expressed as the change from control media (0 atrasentan), paired for the same cell date. With the analysis paired for cell date, all values in media alone are defined as 1.
2.7. Statistical analysis
Data were expressed as mean ± standard error of the mean. Differences were considered significant when p<0.05; p values 0.05-0.1 were considered trends. Two way ANOVA was conducted to determine if either the surgery (RUPP vs Sham) or the treatment (atrasentan vs water; sCR1 vs Vehicle) had a significant effect. In addition, post hoc individual contrasts using JMP and SAS software (SAS Institute, Cary, NC) were considered. For atrasentan treatment, the following comparisons were considered: RUPP water vs Sham water; RUPP water vs RUPP atrasentan 50; RUPP water vs RUPP atrasentan 75; Sham water vs Sham atrasentan 50; Sham water vs Sham atrasentan 75. For sCR1 or saline (Veh) treatment, the following comparisons were considered: Sham Veh vs RUPP Veh; Sham Veh vs Sham sCR1; RUPP Veh vs RUPP sCR1; Sham sCR1 vs RUPP sCR1.
3. Results
3.1. Effect of inhibiting complement activation on placental ischemia-induced events
Our previous studies in the RUPP model demonstrated that inhibiting complement activation with sCR1 significantly attenuated placental ischemia induced hypertension [6] and did not reverse RUPP-induced fetal growth restriction. In our previous study, a comparison of Sham Veh to RUPP Veh demonstrated a statistically significant increase in both mean arterial pressure and C3a. A more comprehensive analysis of the fetal and placental weights from that study are shown in Fig 1A. A significant difference between Sham Veh and RUPP Veh was not detected with an individual contrast for fetal and placental weight. However, as expected in the model, ANOVA indicated a significant main effect with a decrease in fetal and placental weights with RUPP surgery (**p<0.05). Strikingly, post hoc comparisons showed both fetal and placental weight were significantly lower in RUPP animals treated with sCR1 compared to vehicle, suggesting a role for complement activation differs in normal vs ischemic pregnancy. To determine if inhibiting complement activation altered indicators of the endothelin pathway activation (PPE, ETA and ETB receptor message; plasma endothelin), archived plasma and placenta from Sham and RUPP animals treated with 15 mg/kg sCR1 or Veh were evaluated [6]. Synthesis of the potent 21 amino acid peptide endothelin in endothelial cells is a 3 step process starting with translation of preproendothelin (PPE) mRNA into preproendothelin followed by sequential cleavage of PPE by furin and endothelin converting enzymes into Big ET-1 and ET-1, respectively. ET-1 released abluminally from the endothelial cell acts on the local ETA receptor on smooth muscle to cause vasoconstriction. Previous studies by others have demonstrated marked increase in PPE message in kidney and placenta with RUPP-induced increases in blood pressure in the rat [13, 29, 33, 34], no increase in circulating ET-1, with a decrease in ETB but not ETA reported in aorta [29]. Evaluating message for placental preproendothelin, no change was evident in normal pregnant Sham animals treated with sCR1, nor comparing Sham Veh to RUPP Veh animals (Fig 1B). However, following placental ischemia, sCR1 treatment surprisingly increased message for placental PPE and ETA (Fig. 1B) with no changes noted in ETB (data not shown). This increased PPE and ETA message in placenta paralleled the decreases in fetal and placental weight seen in RUPP animals treated with sCR1 (Fig 1A). Given these changes, we also evaluated circulating endothelin (Fig 1C) with increased circulating endothelin noted in Sham animals treated with sCR1. These data suggest that following placental ischemia, complement activation is important in the hypertension [6] as well as in favoring normal placental and fetal growth and counteracting placental ischemia-induced fetal growth restriction.
Fig. 1.

Effect of inhibitor of complement activation, sCR1, on average fetal and placental weight and the endothelin system. Animals were treated daily with 15 mg/kg sCR 1 or saline (Veh) iv from GD14-18 and plasma endothelin measured by ELISA in plasma collected from the abdominal aorta at GD19. mRNA was isolated from GD19 placenta and the Delta delta Ct method of relative quantification was used to determine fold change in mRNA expression compared to ß actin with change in Sham Veh defined as 1. **p<0.05 for main surgery effect by ANOVA, *p<0.05 for indicated post hoc comparisons. Values represent the mean ± SE in Veh (n=10-20) or sCR1 treated animals (n=5-11). A. RUPP surgery significantly decreased fetal and placental weight and sCR1 exacerbated the effect following placental ischemia. B. sCR1 treatment increased PPE and ETA message following placental ischemia. C. Post hoc comparisons demonstrated a significant increase in plasma endothelin in Sham animals treated with sCR1.
3.2. Effect of inhibiting ETA receptor on placental ischemia induced events
Pregnant dams were treated with 2 different concentrations of atrasentan in drinking water, 50 or 75 ug/ml. The delivered dose of atrasentan was estimated by measuring total water consumption over the 6 days (GD13 to 19; average ml of water or mg atrasentan consumed/day) and daily monitoring of body weight. The highest and lowest body weight recorded over the six day period were used to estimate the range of delivered dose of atrasentan to each animal (mg atrasentan consumed/highest body weight in kg/day to mg atrasentan consumed/lowest body weight in kg/day). Because of RUPP surgery and fetal resorptions, RUPP animals weigh less and also consume slightly less water than Sham animals (152.0±6.4 vs 125.2±6.6 ml water/kg/day; p<0.05). The amount of water consumed did not differ when adding atrasentan to the water. Animals receiving 50 ug/ml atrasentan in drinking water received 5.4 mg/kg/day at a minimum (estimated range 5.4-5.7 mg/kg/day). Animals drinking 75 ug/ml atrasentan received 7 mg/kg/day atrasentan at a minimum (estimated range 7.0-9.2 mg/kg/day). Previous studies of others reported a dose of 5 mg/kg/day delivered in drinking water attenuated placental ischemia-induced hypertension in the rat RUPP model [13].
As seen in Fig 2A, RUPP surgery significantly increased mean arterial pressure (MAP) that was attenuated by the ETA antagonist atrasentan as previously reported by others. Atrasentan significantly reduced MAP in Sham animals as well. Also consistent with previous reports, RUPP surgery did not increase circulating ET-1 concentrations in animals with no atrasentan in drinking water, and atrasentan treatment significantly increased circulating plasma endothelin [35]. Considering fetal and placental weight of the treatment groups (Fig 2B), ANOVA demonstrated a significant RUPP surgery effect with a decrease in both fetal and placental weights. Atrasentan also significantly increased fetal weight in Sham animals, similarly to a previous report [36].
Fig. 2.

ETA antagonist atrasentan inhibits placental ischemia induced increase in mean arterial pressure, increases circulating endothelin and increases ETA message. Animals received drinking water ad lib either with or without 50 or 75 ug/ml atrasentan from GD13-19 and mean arterial pressure and average fetal and placental weight determined on GD19. Plasma endothelin was measured by ELISA in plasma collected from the abdominal aorta at GD19. mRNA was isolated from GD19 placenta and the Delta delta Ct method of relative quantification was used to determine fold change in mRNA expression compared to ß actin with change in Sham Water defined as 1. **p<0.05 for main surgery effect by ANOVA, *p<0.05 for indicated post hoc comparisons. Values represent the mean ± SE of mean arterial pressure or average fetal weight measured GD19 in water (n=7-10) or atrasentan treated animals (n=5-7). A. The increase in mean arterial pressure in RUPP animals was decreased by treatment with atrasentan. Atrasentan also significantly decreased the mean arterial pressure in Sham animals. B. RUPP surgery significantly decreased average fetal and placental weight as determined by ANOVA analysis. Post hoc comparison indicated increased fetal weight in Sham animals. C. By ANOVA RUPP surgery did not significantly increase PPE message in placenta. Post hoc comparisons indicated that PPE message was significantly increased in RUPP animals treated with atrasentan. For ETA., ANOVA analysis demonstrated a significant surgery and treatment effect, with increased ETA in Sham animals detected post hoc.
As seen in Fig. 2C, no significant change in PPE message was detected in placenta comparing RUPP vs Sham animals with no atrasentan in the drinking water. Post hoc analysis demonstrated a significant increase in PPE message in placenta of RUPP animals treated with the higher concentration of atrasentan. ETA message significantly increased with RUPP surgery (**p<0.05 by ANOVA) and post hoc analysis revealed a significant atrasentan effect as well in the Sham. No changes in ETB in placenta was noted (data not shown). In addition, PPE message in kidney cortex did not change with RUPP surgery (data not shown). We also used PPE primers published by Santiago-Font [30] and were unable to detect any changes in PPE message in placenta comparing RUPP to Sham animals, indicating that a lack of detectable change was not due to different primers.
3.3. Effect of ETA antagonism on complement activation systemically and locally
Our previous studies demonstrated that placental ischemia increased complement activation product C3a in circulation, as well as local complement deposition of C3b in the placenta (C3 deposition) and IgM deposition [8]. In addition, sCR1 significantly inhibited the increased C3a in the circulation coincident with attenuation of placental ischemia-induced hypertension. As seen in Fig. 3, placental ischemia increased circulating complement activation product C3a, with no significant change due to atrasentan treatment. Unexpectedly, atrasentan treatment of Sham animals resulted in increased C3 deposition in placenta (Fig. 3), a method that detects both C3 and C3b. This is the first demonstration of an effect of ETA antagonism on local placental complement activation in normal pregnancy. This increased complement deposition in placenta in Sham animals treated with atrasentan was not associated with adverse effects on fetal and placental growth in Sham animals (Fig 2B).
Fig. 3.

Effect of the ETA antagonist atrasentan on complement activation. Animals were treated with either 50 or 75 ug/ml atrasentan in drinking water from GD13-19 and placenta, kidney cortex and serum obtained at GD19. Values represent the mean ± SE in water (n=7-12) or atrasentan treated animals (n=5-6). Serum C3a was determined by Western blot with units of C3a relative to a standard pool of yeast activated rat serum as described in Methods. C3 deposition in GD19 placenta was determined by immunohistochemistry with staining graded by two blinded observers from 0 to 3, negative to strongly positive vs isotype control. **p<0.05 for main surgery effect. *p<0.05 for indicated comparisons. The RUPP-induced increase in C3a was not significantly altered by atrasentan. Atrasentan treatment significantly increased C3 deposition in Sham animals, and C3 deposition was significantly increased comparing Sham Water to RUPP Water animals.
Uncontrolled and excessive complement activation can result in depletion of complement components because activation exceeds new synthesis of complement proteins. Total hemolytic complement activity (CH50) of rat serum in the 6 treatment groups depicted in Fig 3 did not differ (data not shown), indicating that complement components were not being depleted in RUPP vs Sham. Treatment of normal rat serum with atrasentan in vitro also did not affect total hemolytic complement activity (data not shown).
3.4. Effect of ETA antagonism on complement regulators in normal pregnancy
In normal pregnant Sham animals, 50 ug/ml atrasentan had no effect on MAP or circulating C3a, yet still significantly increased circulating endothelin and C3 deposition in placenta (Fig 2A and Fig 3). Increased complement activation can be due to increased activation of the pathway(s) and/or a decrease in endogenous integral membrane complement regulators that protect our own cells. Membrane bound regulators Crry and CD55 control C3 activation in rodents. We considered that increased C3 deposition in the placenta of Sham normal pregnant animals treated with atrasentan (Fig. 3) was due to a reduction in expression of complement regulators Crry or CD55. As seen in Fig. 4A, atrasentan treatment in vivo significantly decreased the message for Crry and CD55 in placenta and kidney, with different doses resulting in inhibition in the two tissues. These data suggest that in normal pregnancy, endothelin maintains adequate complement regulators to prevent excessive complement activation locally. When ETA is blocked, complement regulators may decline and excessive complement deposition occurs.
Fig 4.

Effect of the ETA antagonist atrasentan on message for complement regulators. A. Animals were treated with either 50 or 75 ug/ml atrasentan in drinking water from GD13-19 and placenta and kidney cortex obtained at GD19. Values represent the mean ± SE in water (n=7-12) or atrasentan treated animals (n=5-6). Atrasentan treatment in vivo significantly decreased message for complement regulators Crry and CD55 in placenta and kidney cortex of Sham animals. B. IEC-6 cells were treated with either 0, 10, 20 or 40 ug/ml atrasentan in media for 24 hours and the change in message for complement regulators Crry and CD55 determined in cells harvested after 24 hours of treatment. mRNA was isolated from cells and the Delta-delta Ct method of relative quantification used to determine fold change in mRNA expression compared to ß actin with change in media alone defined as 1. Values represent the mean ± SE of the change from media alone in 4-5 different experiments. *p<0.05 for indicated comparisons.
To further assess whether atrasentan directly affects complement regulators at the cell level, we assessed the effect of atrasentan in vitro on cultured rat cells, using a rat intestinal epithelial cell line (IEC-6) and rat brain endothelial cell line (RBE4). As seen in Fig 4B, incubation of IEC-6 cells with 10-40 ug/ml atrasentan for 24 hours resulted in a significant increase in message for the regulators. In RBE4 cells, no significant changes in CD55 and Crry were noted (data not shown). Comparing message for regulators indicated that RBE4 cells had 2.6 times higher expression of CD55 and 1.8 times higher Crry than IEC cells.
An imbalance in angiogenic (vascular endothelial growth factor; VEGF) and anti-angiogenic (s-Flt-1) factors has been implicated in the pathophysiology of placental ischemia-induced hypertension. VEGF has also been demonstrated to regulate local inhibitory effect of the complement regulator Factor H in the eye and kidney [37]. Podocytes produce VEGF that crosses the basement membrane and maintains the fenestrated epithelium. Disruption of VEGF in kidney decreases local Factor H and other complement regulators, leading to increased complement deposition in kidney. Thus, we hypothesized that the decrease in regulators in placenta following atrasentan treatment was associated with decreased message for VEGF in placenta. However, evaluation of VEGF message in placenta of Sham animals revealed no significant change with atrasentan treatment (data not shown). The concentration of VEGF in plasma and placental extracts could not be measured accurately since it was below the level of reliable detection by rat ELISA (R&D VEGF Quantikine kit for rat VEGF, RRV00, Minneapolis, MN), consistent with published limitations regarding measurement of rat VEGF [38] by commercial ELISA. sFlt-1 was not measured in this study, but our previous study [6] demonstrated no increase in sFlt-1 with RUPP surgery with concentrations of sFlt-1 being measured near the level of reliable detection.
4. Discussion
The role of endothelin in preeclampsia has not been thoroughly tested, though evidence clearly indicates that endothelin is increased in plasma of women with preeclampsia compared to a normal pregnancy, particularly in women with severe preeclampsia and proteinuria [10]. Using animal models, ETA is of demonstrated importance in placental ischemia-induced hypertension in the rat RUPP model, as previously published [13] and confirmed in the present study. However, direct therapeutic modulation of the endothelin pathway during pregnancy is contraindicated due to the potential of adverse effects on fetal development [39]. Thus, it is increasingly important to understand interactions of the endothelin pathway and other mediator pathways activated following placental ischemia. Increased activation of one such pathway, the complement system, is evident in preeclampsia compared to normal pregnancy [4]. Our previous studies in the RUPP model demonstrated that inhibiting complement activation attenuates placental ischemia-induced hypertension attesting to its importance in the mechanism leading to hypertension [6]. We simplistically hypothesized that complement activation results in activation of the endothelin pathway with ETA activation leading to placental ischemia-induced hypertension. If our hypothesis was correct, placental ischemia would cause increased complement activation in the presence of ETA antagonism as noted. Placental ischemia would also result in a decrease in PPE message in the presence of an inhibitor of complement activation. However, PPE message increased followed sCR1 treatment, suggesting that hypertension induced by placental ischemia-induced complement activation was independent of the endothelin system. Thus, a complex interplay between endothelin and complement system activation is operating to result in placental ischemia-induced hypertension.
Results of our current study also demonstrate a previously unrecognized role for endothelin in normal pregnancy; endothelin via the ETA receptor influences the level of local placental C3 deposition in normal pregnancy (Fig 3). This endothelin influence in the presence of ETA blockade in the placenta could be due to increased circulating endothelin acting through ETB or due to a decrease in local placental complement regulators or both. Thus, we evaluated both ETB and complement regulators following atrasentan treatment in the placenta. Clearly, placental complement regulators were decreased in vivo in the presence of ETA antagonism which is consistent with increased placental C3 deposition. We extended these studies to cultured cells and found that intestinal epithelial cells up-regulated CD55 and Crry message with atrasentan treatment, but no change was detected in RBE-4 cells. The placenta is composed of numerous cell types including trophoblasts, endothelial cells, and multiple immune cells, with evidence for endothelin receptors on each. Thus, effect of endothelin antagonism on complement regulators differs with the cell type and the tissue, so complex in vivo effects cannot be attributed solely to a direct effect on a single cell type.
In normal pregnancy, ETA antagonism increased complement activation in the placenta, but not the circulation. If complement activation is important for the blood pressure increase following placental ischemia, why doesn’t blood pressure increase with atrasentan treatment in the Sham animals? Our previously published studies suggest it is the circulating complement activation products C3a and/or C5a acting in the maternal vasculature to increase the blood pressure. The evidence for this lies in our previous studies where C3a and C5a receptor antagonists attenuated placental ischemia-induced hypertension and/or increased heart rate and C5a receptor antagonists reversed endothelial dysfunction in mesentery of rats following placental ischemia, suggesting that C3a/C5a generation and endothelial function is critical. Taken together, these data indicate that the site and extent of complement activation is important in determining whether complement activation is positively or negatively impacting pregnancy outcomes. In our study, placental C3 deposition following atrasentan treatment in normal pregnancy did not adversely affect blood pressure or fetal weight. However, in conditions of placental ischemia, with increases in circulating C3a and C3b deposition, fetal and placental weight are decreased with inhibition of complement activation exacerbating the reduced fetal weight (Fig 1A).
In our study, sCR1 is used as a tool to assess the role of complement activation. sCR1 (TP10, CDX1135) was one of the first complement drugs to reach clinical trials but its development has been halted in lieu of more promising and smaller molecules targeted to endothelial cells in transplantation to inhibit complement activation [40]. The effectiveness of sCR1 in inhibiting complement activation in the pregnant rat is evidenced by significant decrease of circulating C3a following placental ischemia, as well as a significant reduction in the CH50 (Lillegard et al 2013) at doses that have been used in previous studies in the rat.
Limitations of this study primarily lie in the use of an animal model to mimic a pregnancy disorder manifested by a variety of symptoms with widely varying severity and timeline. The RUPP model is a model of hypertension in pregnancy with fetal growth restriction due to placental insufficiency induced mechanically. The RUPP model most closely mimics early-onset preeclampsia (symptoms present <34 weeks of pregnancy) rather than the late-onset (>34 weeks of pregnancy) type that is not usually associated with fetal growth restriction or placental ischemic injury [41]. Despite the many similarities between the RUPP model and early onset preeclampsia, it is clearly not a spontaneous model of preeclampsia. Preeclampsia is far more complex and varied than simply placental ischemia, with a variety of genetic and individual risk factors contributing to the preeclampsia pathology. Another limitation is placental structure. While the rat and human share a hemochorial placental structure, differences in trophoblast invasion and placental structure limit translation of findings in a rat placenta to the human condition. A comparison of rat and human placenta by Soares et al [42] indicate that both human and rat undergo similar spiral artery remodeling but the uterine and vascular structure clearly differ. Hence, our studies focus on the events that occur after placental ischemia. Moreover, any mechanistic findings in rat need to be grounded in findings in human preeclamptic placenta. Another limitation of this study is the limited evidence of the importance of endothelin in human preeclampsia. Our studies of endothelin in placenta do not localize changes in PPE message to placental regions where changes in turbulence and flow due to ischemia may significantly alter a role for endothelin in different placental regions.
Our studies clearly confirmed the effectiveness of an ETA antagonist on placental ischemia induced hypertension, and atrasentan clearly increased circulating endothelin as expected from previous studies. Previous studies have also seen increases in PPE message in the placenta, but not circulating endothelin following placental ischemia, presumably because endothelin is released abluminally from the endothelial cells and does not readily enter the circulation. However, we were unable to detect changes in PPE message in placenta or kidney that have been previously reported following placental ischemia [13, 29, 33, 34, 43]. Besides validating our PCR product, we also used published primers [34] and were still unable to detect any change in PPE message in RUPP compared to Sham animals. Our published studies have consistently used Sprague Dawley rats from Charles River comparing a Sham surgery to RUPP surgery, whereas the majority of studies published compare RUPP surgery on Harlan Sprague Dawley rats to a normal pregnant animal with no surgery. As the surgery itself alters the immune response, appropriate comparisons are critical, but Sprague Dawley rats also appear to differ based on the supplier and strain (CD vs Sasco). A significant body of literature has demonstrated mechanistic differences in cardiovascular responses in rats from these two different suppliers [44-47], but neither has an obvious advantage for modeling pregnancy disorders.
Atrasentan is a highly selective ETA antagonist with minimal ETB antagonistic activity; a 28,000 fold higher affinity for ETA than ETB [48]. In our study, consistent with results of others, plasma endothelin did not increase following placental ischemia in the rat RUPP model, and atrasentan itself significantly increased circulating endothelin in both RUPP and Sham animals. In experimental animals following treatment with endothelin antagonists, particularly ETB antagonists [35] but also ETA antagonists [49], increased plasma endothelin has been reported. With ETB antagonists, the increase of endothelin in plasma may be due to blockade of ETB and impaired clearance of endothelin. Mazzuca reported a decrease in ETB receptor in aorta following placental ischemia with a concomitant increase in endothelin ETA vasoconstriction [29]. Atrasentan treatment may also result in partial ETA receptor internalization [50], potentially changing the ETA/ETB ratio and the ultimate effect of endothelin in placenta or vasculature. Opgenorth [35] suggested that a feedback mechanism increased PPE message following ETA receptor blockade. Our studies measuring PPE message also suggested it increases with atrasentan treatment in placenta of RUPP animals.
Both complement activation and endothelin production have been reported in attacks of hereditary angioedema due to C1-inhibitor deficiency [23] as well as in endotoxin induced systemic inflammation in healthy volunteers [24]. In addition, activation of both systems has been documented in mouse models of glaucoma [21, 22] and heart failure [20]. Literature indicating that complement activation recruits the endothelin pathway is quite limited. In a study of nonocclusive mesenteric ischemia using partial aortic occlusion in the rat, a C5a receptor antagonist prevented the increase in plasma endothelin along with increased mean arterial pressure, heart rate and cardiac output resulting from mesenteric hypoperfusion [51]. In a hemorrhagic model of shock in rat, preventing complement activation using sCR1 prevented the observed increase in endothelin [52]. In addition, C5b-9 causes increased endothelin production by glomerular epithelial cells [53]. None of these studies determined the effect of complement inhibition on endothelin production or message in a control animal or in pregnancy.
Our results reveal a complex interaction of the endothelin and complement system in pregnancy with significant downregulation of normal complement regulators in vivo in placenta following ETA receptor antagonism. Thus, our data suggest that activation of the ETA receptor in vivo is important in maintaining optimal levels of complement regulators to protect tissues from excessive complement activation during normal pregnancy. With blockade of the ETA receptor, excessive tissue complement activation may occur. Our data also suggest that inhibition of complement activation may compromise control of the endothelin pathway. Thus, therapeutic strategies for pregnancy-induced hypertension should optimally target upstream events leading to activation of the complement and endothelin systems to minimize unintended complications of therapy. Our data provide evidence for a strikingly important role of endothelin and the ETA receptor in maintaining adequate complement regulators in placenta to prevent excessive and potentially damaging complement activation.
HIGHLIGHTS.
Following placental ischemia:
Complement activation and ETA mediate placental ischemia induced-hypertension
Complement inhibition elevates PPE mRNA, suggesting complement activation does not cause hypertension by an endothelin dependent pathway.
With ETA antagonism, high plasma C3a is not sufficient to cause hypertension
In normal pregnancy:
Placental ETA maintains complement regulators to prevent excess complement activation
Complement regulates endothelin systemically, but not locally in placenta.
Acknowledgements
The authors acknowledge Dr. Ronald Regal, Department of Mathematics and Statistics, University of Minnesota Duluth for statistical analysis of the data. In addition, Dr. Henry Marsh, Celldex Therapeutics, Inc. (Needham, MA) is acknowledged for providing the sCR1 used in published studies that provided the archived samples from sCR1 treated animals.
Funding
This work was supported by the National Institutes of Health National Heart Lung and Blood Institute (Grant R15-HL109843 to J.F.R., J.S.G., and S.D.F), American Heart Association (17GRNT33650049 to J.F.R., S.D.F.), University of Minnesota OVPR Research Grant in Aid (J.F.R.) and the Institutional Development Award (IDeA) from the National Institute of General Medical Sciences (Grant P20GM103418 to S.D.F).
Nonstandard Abbreviations:
- ET-1
endothelin-1
- ETA
Endothelin A receptor
- ETB
Endothelin B receptor
- GD
gestation day
- PPE
preproendothelin
- RUPP
reduced uterine perfusion pressure
- sCR1
soluble complement receptor 1
- VEGF
vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures or Conflict of Interest
The authors have no conflicts to report
References
- [1].Kolev M, Le Friec G, Kemper C, Complement - tapping into new sites and effector systems, Nat Rev Immunol, 14 (2014) 811–820. [DOI] [PubMed] [Google Scholar]
- [2].Regal JF, Burwick RM, Fleming SD, The Complement System and Preeclampsia, Curr Hypertens Rep, 19 (2017) 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Derzsy Z, Prohaszka Z, Rigo J Jr., Fust G, Molvarec A, Activation of the complement system in normal pregnancy and preeclampsia, Mol Immunol, 47 (2010) 1500–1506. [DOI] [PubMed] [Google Scholar]
- [4].Regal JF, Gilbert JS, Burwick RM, The complement system and adverse pregnancy outcomes, Mol Immunol, 67 (2015) 56–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Burwick RM, Feinberg BB, Eculizumab for the treatment of preeclampsia/HELLP syndrome, Placenta, 34 (2013) 201–203. [DOI] [PubMed] [Google Scholar]
- [6].Lillegard KE, Johnson AC, Lojovich SJ, Bauer AJ, Marsh HC, Gilbert JS, Regal JF, Complement activation is critical for placental ischemia-induced hypertension in the rat, Mol Immunol, 56 (2013) 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lillegard KE, Loeks-Johnson AC, Opacich JW, Peterson JM, Bauer AJ, Elmquist BJ, Regal RR, Gilbert JS, Regal JF, Differential effects of complement activation products c3a and c5a on cardiovascular function in hypertensive pregnant rats, J Pharmacol Exp Ther, 351 (2014) 344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Regal JF, Strehlke ME, Peterson JM, Wing CR, Parker JE, Nieto NF, Bemis LT, Gilbert JS, Fleming SD, Role of IgM and angiotensin II Type I receptor autoantibodies in local complement activation in placental ischemia-induced hypertension in the rat, Mol Immunol, 78 (2016) 38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].LaMarca B, Cornelius D, Wallace K, Elucidating immune mechanisms causing hypertension during pregnancy, Physiology (Bethesda), 28 (2013) 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lu YP, Hasan AA, Zeng S, Hocher B, Plasma ET-1 Concentrations Are Elevated in Pregnant Women with Hypertension -Meta-Analysis of Clinical Studies, Kidney Blood Press Res, 42 (2017) 654–663. [DOI] [PubMed] [Google Scholar]
- [11].Davenport AP, Hyndman KA, Dhaun N, Southan C, Kohan DE, Pollock JS, Pollock DM, Webb DJ, Maguire JJ, Endothelin, Pharmacological reviews, 68 (2016) 357–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Miller E, Czopek A, Duthie KM, Kirkby NS, van de Putte EE, Christen S, Kimmitt RA, Moorhouse R, Castellan RF, Kotelevtsev YV, Kuc RE, Davenport AP, Dhaun N, Webb DJ, Hadoke PW, Smooth Muscle Endothelin B Receptors Regulate Blood Pressure but Not Vascular Function or Neointimal Remodeling, Hypertension, 69 (2017) 275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Alexander BT, Rinewalt AN, Cockrell KL, Massey MB, Bennett WA, Granger JP, Endothelin type a receptor blockade attenuates the hypertension in response to chronic reductions in uterine perfusion pressure, Hypertension, 37 (2001) 485–489. [DOI] [PubMed] [Google Scholar]
- [14].LaMarca B, Wallukat G, Llinas M, Herse F, Dechend R, Granger JP, Autoantibodies to the angiotensin type I receptor in response to placental ischemia and tumor necrosis factor alpha in pregnant rats, Hypertension, 52 (2008) 1168–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].LaMarca B, Speed J, Fournier L, Babcock SA, Berry H, Cockrell K, Granger JP, Hypertension in response to chronic reductions in uterine perfusion in pregnant rats: effect of tumor necrosis factor-alpha blockade, Hypertension, 52 (2008) 1161–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Murphy SR, LaMarca BB, Cockrell K, Granger JP, Role of endothelin in mediating soluble fms-like tyrosine kinase 1-induced hypertension in pregnant rats, Hypertension, 55 (2010) 394–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].LaMarca B, Parrish M, Ray LF, Murphy SR, Roberts L, Glover P, Wallukat G, Wenzel K, Cockrell K, Martin JN Jr., Ryan MJ, Dechend R, Hypertension in response to autoantibodies to the angiotensin II type I receptor (AT1-AA) in pregnant rats: role of endothelin-1, Hypertension, 54 (2009) 905–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].George EM, Granger JP, Linking placental ischemia and hypertension in preeclampsia: role of endothelin 1, Hypertension, 60 (2012) 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Cornelius DC, Wallace K, Kiprono L, Dhillon P, Moseley J, LaMarca B, Endothelin-1 is not a Mechanism of IL-17 Induced Hypertension during Pregnancy, Medical journal of obstetrics and gynecology, 1 (2013). [PMC free article] [PubMed] [Google Scholar]
- [20].Gombos T, Forhecz Z, Pozsonyi Z, Szeplaki G, Kunde J, Fust G, Janoskuti L, Karadi I, Prohaszka Z, Complement anaphylatoxin C3a as a novel independent prognostic marker in heart failure, Clin Res Cardiol, 101 (2012) 607–615. [DOI] [PubMed] [Google Scholar]
- [21].Howell GR, Macalinao DG, Sousa GL, Walden M, Soto I, Kneeland SC, Barbay JM, King BL, Marchant JK, Hibbs M, Stevens B, Barres BA, Clark AF, Libby RT, John SW, Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma, J Clin Invest, 121 (2011) 1429–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Howell GR, MacNicoll KH, Braine CE, Soto I, Macalinao DG, Sousa GL, John SW, Combinatorial targeting of early pathways profoundly inhibits neurodegeneration in a mouse model of glaucoma, Neurobiol Dis, 71 (2014) 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kajdacsi E, Jani PK, Csuka D, Varga L, Prohaszka Z, Farkas H, Cervenak L, Novel Vasoregulatory Aspects of Hereditary Angioedema: the Role of Arginine Vasopressin, Adrenomedullin and Endothelin-1, Journal of clinical immunology, 36 (2016) 160–170. [DOI] [PubMed] [Google Scholar]
- [24].Soop A, Albert J, Weitzberg E, Bengtsson A, Lundberg JO, Sollevi A, Complement activation, endothelin-1 and neuropeptide Y in relation to the cardiovascular response to endotoxin-induced systemic inflammation in healthy volunteers, Acta Anaesthesiol Scand, 48 (2004) 74–81. [DOI] [PubMed] [Google Scholar]
- [25].Thaete LG, Neerhof MG, Caplan MS, Endothelin receptor A antagonism prevents hypoxia-induced intrauterine growth restriction in the rat, Am J Obstet Gynecol, 176 (1997) 73–76. [DOI] [PubMed] [Google Scholar]
- [26].Zhou J, Xiao D, Hu Y, Wang Z, Paradis A, Mata-Greenwood E, Zhang L, Gestational hypoxia induces preeclampsia-like symptoms via heightened endothelin-1 signaling in pregnant rats, Hypertension, 62 (2013) 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Regal JF, Lillegard KE, Bauer AJ, Elmquist BJ, Loeks-Johnson AC, Gilbert JS, Neutrophil Depletion Attenuates Placental Ischemia-Induced Hypertension in the Rat, PloS one, 10 (2015)e0132063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Larsen CP, Regal RR, Regal JF, Trimellitic anhydride-induced allergic response in the guinea pig lung involves antibody-dependent and -independent complement system activation, J Pharmacol Exp Ther, 296 (2001) 284–292. [PubMed] [Google Scholar]
- [29].Mazzuca MQ, Li W, Reslan OM, Yu P, Mata KM, Khalil RA, Downregulation of microvascular endothelial type B endothelin receptor is a central vascular mechanism in hypertensive pregnancy, Hypertension, 64 (2014) 632–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Santiago-Font JA, Amaral LM, Faulkner JL, Ibrahim T, Vaka VR, Cunningham MW Jr., LaMarca BD, Serelaxin improves the pathophysiology of placental ischemia in the Reduced Uterine Perfusion Pressure (RUPP) rat model of Preeclampsia, Am J Physiol Regul Integr Comp Physiol, DOI 10.1152/ajpregu.00192.2016(2016) ajpregu 00192 02016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Quaroni A, Wands J, Trelstad RL, Isselbacher KJ, Epithelioid cell cultures from rat small intestine. Characterization by morphologic and immunologic criteria, J Cell Biol, 80 (1979) 248–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Roux F, Couraud PO, Rat brain endothelial cell lines for the study of blood-brain barrier permeability and transport functions, Cell Mol Neurobiol, 25 (2005) 41–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].George EM, Cockrell K, Aranay M, Csongradi E, Stec DE, Granger JP, Induction of heme oxygenase 1 attenuates placental ischemia-induced hypertension, Hypertension, 57 (2011) 941–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Santiago-Font JA, Amaral LM, Faulkner J, Ibrahim T, Vaka VR, Cunningham MW, LaMarca B, Serelaxin improves the pathophysiology of placental ischemia in the reduced uterine perfusion pressure rat model of preeclampsia, Am J Physiol Regul Integr Comp Physiol, 311 (2016) R1158–R1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Opgenorth TJ, Wessale JL, Dixon DB, Adler AL, Calzadilla SV, Padley RJ, Wu-Wong JR, Effects of endothelin receptor antagonists on the plasma immunoreactive endothelin-1 level, Journal of cardiovascular pharmacology, 36 (2000) S292–296. [DOI] [PubMed] [Google Scholar]
- [36].Thaete LG, Dewey ER, Neerhof MG, Endothelin and the regulation of uterine and placental perfusion in hypoxia-induced fetal growth restriction, Journal of the Society for Gynecologic Investigation, 11 (2004) 16–21. [DOI] [PubMed] [Google Scholar]
- [37].Keir LS, Firth R, Aponik L, Feitelberg D, Sakimoto S, Aguilar E, Welsh GI, Richards A, Usui Y, Satchell SC, Kuzmuk V, Coward RJ, Goult J, Bull KR, Sharma R, Bharti K, Westenskow PD, Michael IP, Saleem MA, Friedlander M, VEGF regulates local inhibitory complement proteins in the eye and kidney, J Clin Invest, 127 (2017) 199–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Weissgerber TL, McConico A, Knudsen BE, Butters KA, Hayman SR, White WM, Milic N, Miller VM, Garovic VD, Methodological differences account for inconsistencies in reported free VEGF concentrations in pregnant rats, Am J Physiol Regul Integr Comp Physiol, 306 (2014) R796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Luscher TF, Barton M, Endothelins and endothelin receptor antagonists: therapeutic considerations for a novel class of cardiovascular drugs, Circulation, 102 (2000) 2434–2440. [DOI] [PubMed] [Google Scholar]
- [40].Ricklin D, Mastellos DC, Reis ES, Lambris JD, The renaissance of complement therapeutics, Nature reviews. Nephrology, 14 (2018) 26–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Stanek J, Histological Features of Shallow Placental Implantation Unify Early-Onset and Late-Onset Preeclampsia, Pediatr Dev Pathol, 22 (2019) 112–122. [DOI] [PubMed] [Google Scholar]
- [42].Soares MJ, Chakraborty D, Karim Rumi MA, Konno T, Renaud SJ, Rat placentation: an experimental model for investigating the hemochorial maternal-fetal interface, Placenta, 33 (2012) 233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Harmon A, Cornelius D, Amaral L, Paige A, Herse F, Ibrahim T, Wallukat G, Faulkner J, Moseley J, Dechend R, LaMarca B, IL-10 supplementation increases Tregs and decreases hypertension in the RUPP rat model of preeclampsia, Hypertension in pregnancy : official journal of the International Society for the Study of Hypertension in Pregnancy, 34 (2015) 291–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Buhimschi IA, Shi SQ, Saade GR, Garfield RE, Marked variation in responses to longterm nitric oxide inhibition during pregnancy in outbred rats from two different colonies, Am J Obstet Gynecol, 184 (2001) 686–693. [DOI] [PubMed] [Google Scholar]
- [45].Griffin K, Polichnowski A, Licea-Vargas H, Picken M, Long J, Williamson G, Bidani A, Large BP-dependent and -independent differences in susceptibility to nephropathy after nitric oxide inhibition in Sprague-Dawley rats from two major suppliers, American journal of physiology. Renal physiology, 302 (2012) F173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Pollock DM, Rekito A, Hypertensive response to chronic NO synthase inhibition is different in Sprague-Dawley rats from two suppliers, The American journal of physiology, 275 (1998)R1719–1723. [DOI] [PubMed] [Google Scholar]
- [47].Wehrwein EA, Yoshimoto M, Guzman P, Shah A, Kreulen DL, Osborn JW, Role of cardiac sympathetic nerves in blood pressure regulation, Auton Neurosci, 183 (2014) 30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Luo K, Thaete LG, Neerhof MG, Endothelin Receptor A Antagonism and Fetal Growth in Endothelial Nitric Oxide Synthase Gene Knockout Maternal and Fetal Mice, Reprod Sci, 23 (2016) 1028–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].LaMarca BB, Cockrell K, Sullivan E, Bennett W, Granger JP, Role of endothelin in mediating tumor necrosis factor-induced hypertension in pregnant rats, Hypertension, 46 (2005) 82–86. [DOI] [PubMed] [Google Scholar]
- [50].Chiou WJ, Wessale JL, von Geldern T, Opgenorth TJ, Wu-Wong JR, ‘Irreversible’ endothelin-1 binding does not prohibit ABT-627 from reversing endothelin-1-induced effects, Journal of cardiovascular pharmacology, 36 (2000) S48–52. [DOI] [PubMed] [Google Scholar]
- [51].Erces D, Nogrady M, Varga G, Szucs S, Meszaros AT, Fischer-Szatmari T, Cao C, Okada N, Okada H, Boros M, Kaszaki J, Complement C5a inhibition improves late hemodynamic and inflammatory changes in a rat model of nonocclusive mesenteric ischemia, Surgery, 159 (2016) 960–971. [DOI] [PubMed] [Google Scholar]
- [52].Chen D, Song MQ, Liu YJ, Xue YK, Cheng P, Zheng H, Chen LB, Inhibition of complement C3 might rescue vascular hyporeactivity in a conscious hemorrhagic shock rat model, Microvasc Res, 105 (2016) 23–29. [DOI] [PubMed] [Google Scholar]
- [53].Cybulsky AV, Stewart DJ, Cybulsky MI, Glomerular epithelial cells produce endothelin-1, J Am Soc Nephrol, 3 (1993) 1398–1404. [DOI] [PubMed] [Google Scholar]