

SUMMARY
Distinct oxygenases and their oxylipin products have been shown to participate in thermogenesis by mediating physiological adaptations required to sustain body temperature. Since the role of the lipoxygenase (LOX) family in cold adaptation remains elusive, we aimed to investigate if, and how, LOX activity is required for cold adaptation, and to identify LOX-derived lipid mediators that could serve as putative cold mimetics with therapeutic potential to combat diabetes. By utilizing mass spectrometry-based lipidomics in mice and humans, we demonstrated that cold and β3-adrenergic stimulation could promote the biosynthesis and release of 12-LOX metabolites from BAT. Moreover, 12-LOX ablation in mouse brown adipocytes impaired glucose uptake and metabolism, resulting in blunted adaptation to the cold in vivo. The cold-induced 12-LOX product 12-HEPE was found to be a batokine that improves glucose metabolism by promoting glucose uptake into adipocytes and skeletal muscle through activation of an insulin-like intracellular signaling pathway.
Keywords: 12-HEPE, obesity, diabetes, brown adipose tissue, fat, thermogenesis, lipokine
eTOC Blurb
Leiria et al. uncover a cold-induced response involving the activation of the enzyme 12-lipoxygenase in brown adipose tissue, which subsequently produces an omega-3 oxylipin 12-HEPE to promote glucose uptake into tissue via an insulin-like intracellular signaling pathway.
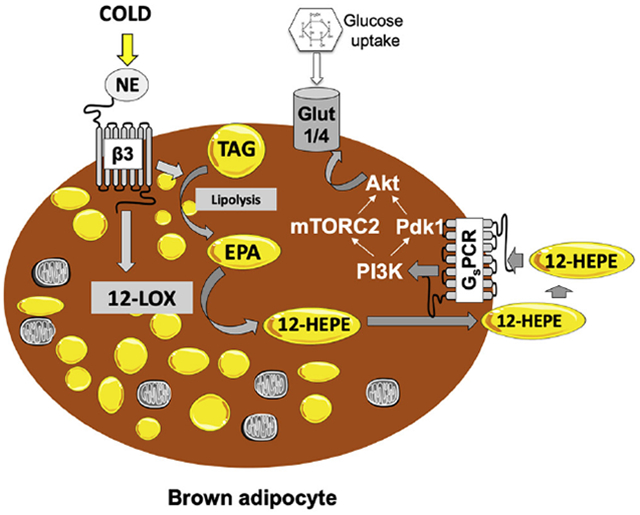
Graphical Abstract
INTRODUCTION
The prevalence of obesity and metabolic syndrome has reached pandemic levels worldwide and has been gradually increasing over the last few decades (GBD 2015 Obesity Collaborators, 2017). Several epidemiologic studies have identified high body mass index (BMI) as a risk factor for many chronic diseases, including cardiovascular disease and diabetes mellitus (Singh et al., 2013; Roberto et al., 2015), illustrating the urgency for identifying novel molecular targets and therapeutic strategies to treat these metabolic disorders. Due to the high capacity of brown adipose tissue (BAT) in consuming glucose and fatty acids to generate heat and expend energy (Cannon and Nedergaard, 2004; Bartelt et al., 2011; Townsend and Tseng, 2014), and the presence of BAT in adult humans (Nedergaard et al., 2007; Cypess et al., 2009; van Marken Lichtenbelt et al., 2009; Virtanen et al., 2009), increasing the amount or activity of functional BAT has been considered an appealing approach for the treatment, or prevention, of obesity and related metabolic diseases.
Cold exposure elevates BAT volume and activity, thus increasing energy expenditure, whole-body insulin sensitivity, and facilitating weight loss in obese humans (van der Lans et al., 2013; Yoneshiro et al., 2013, Hanssen et al., 2015). Furthermore, the second type of thermogenic adipose tissue called beige or brite (brown-in-white) fat is activated by cold or adrenergic stimuli (Frontini and Cinti, 2010). Thermogenic fat serves as a “metabolic sink” by consuming glucose and fatty-acids from the circulation (Frontini and Cinti, 2010; Kajimura et al., 2015). The high metabolic capacity of thermogenic fat is mediated by a tightly controlled transcriptional and post-transcriptional regulatory system (Seale et al., 2008; Chouchani et al., 2015; Lynes et al., 2017; Bartlelt et al., 2018). Although brown/beige fat has attracted attention as a potential target for treating metabolic diseases because of its energy-dissipating function, at least part of its beneficial effect is actually due to its secretory function, and consequent capacity to affect metabolic functions in other tissues such as muscle, white fat, and liver (Stanford et al., 2013; Wang et al., 2014). Thermogenic adipocytes can synthesize and release factors such as peptides, lipids or other metabolites called batokines and can directly or indirectly support thermogenesis (Wang et al., 2015; Chen et al., 2016; Villarroya et al., 2017). These secreted factors may act as endocrine, paracrine, or autocrine agents to regulate a number of physiological functions required for adaptive thermogenesis (Wang et al., 2015; Villarroya et al., 2017). The identification of novel BAT-secreted molecules that may work as cold mimetics to promote substrate metabolism is essential to uncover pathways that can serve as new therapeutic targets for metabolic diseases, including diabetes.
Cold increases the expression and activity of enzymes that catalyze lipid oxidation (Lynes et al., 2017). A cluster of oxidized lipid metabolites called oxylipins are derived from omega-3 or omega-6 polyunsaturated fatty acids (PUFAs) and can regulate a number of metabolic processes, including adipogenesis and fuel utilization (Barquissau et al., 2017). Oxylipins are biosynthesized through the oxidative activity of the cyclooxygenases (COX), lipoxygenases (LOX), or cytochrome P450 epoxygenase (CYP P450), and play well-documented roles in controlling acute inflammation and its resolution (Serhan, 2014; Capdevilla et al., 2015; Barquissau et al., 2017). These lipid mediators can also exert metabolic effects required for non-shivering thermogenesis, such as the formation of beige adipocytes by the COX-derived prostaglandins (Vegiopoulos et al., 2010), or the induction of fatty-acid uptake into BAT and skeletal muscle by the cold/exercise-induced cytochrome P450 epoxygenase/epoxide hydrolase product (±)12,13-dihydroxy-9Z-octadecenoic acid (12,13-diHOME) (Lynes et al., 2017; Stanford et al., 2018). Considering these studies, the role of LOX enzymes for adaptive thermogenesis remains elusive.
In the present study, we revealed a new function for LOX enzymes and their corresponding lipid metabolites in the adaptive thermogenesis. Using lipidomic profiling of humans and mice, we observed that β3-adrenergic stimulation could promote the biosynthesis and release of BAT-derived metabolites from the enzyme 12-LOX. Importantly, one of the 12-LOX omega-3 metabolites, 12-hydroxyeicosapentaenoic acid (12-HEPE), acts as a paracrine/endocrine factor to promote glucose uptake into brown fat and muscle. The ablation of 12-LOX in vitro impairs the glucose uptake capacity of brown adipocytes. Importantly, in vivo CRISPR/Cas9 mediated deletion of 12-LOX, specifically in BAT hampers the animal’s capacity to adapt to cold temperature. These findings uncover a new role for lipoxygenases and their oxylipin products in the regulation of glucose metabolism and adaptive thermogenesis in both mice and humans.
RESULTS
Cold exposure or β3-adrenergic stimulation increase circulating levels of 12-LOX derived lipids in mice and humans
The use of lipidomics technologies, combined with appropriate genetic models, has helped to identify lipid mediators that serve as metabolic messengers to communicate energy status and modulate fuel utilization among tissues (Cao et al., 2008; Liu et al., 2011; Yore et al., 2014; Lynes et al., 2017). Aiming to search for LOX products that reflect the activity of this family of enzymes under cold condition, we used liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) to measure the concentrations of a panel of 88 oxidized lipids in the serum of mice that were housed in cold (5°C) or thermoneutrality (30°C ) for 7 days. Interestingly, we found a coordinated elevation of three 12-LOX metabolites, namely, 12-HEPE, 14-hydroxydocosahexaenoic acid (14-HDHA), and 12-Hydroxyeicosatetraenoic acid (12-HETE) by cold exposure in both male and female C57BL6/J mice (Figures 1A–1D). Omega-3/6 PUFAs, mainly eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and arachidonic acid (AA), can be oxidized by 12-LOX to produce 12-HEPE, 14-HDHA, and 12-HETE, respectively (Hamberg, 1980; Brash, 1999; Serhan et al., 2009; Kuhn et al., 2015) (Figure 1I). Notably, the levels of HEPEs, HDHAs, and HETEs derived from other LOX subtypes, COXs or CYP450 were not modified by cold, except for a reduction in 5-LOX products, 5-HEPE, and 5-HETE (Figures S1A and S1B). Since acute cold exposure activates brown fat glucose and fatty-acid uptake (van Marken Lichtenbelt et al., 2009; Virtanen et al., 2009, Bartelt et al., 2011; Lynes et al., 2017), we sought to measure serum lipid levels after short-term cold exposure and found that the levels of 12-LOX metabolites were elevated after only 1 hour of cold exposure (Figures 1E and 1F). Circulating levels of other HEPEs, HDHAs, and HETEs were not altered, except for a small increase in 9-HEPE, which is a product of non-enzymatic oxidation (Figure S1C). To quantify the circulating concentrations of these lipid mediators, we performed a targeted lipidomics analysis. We found the serum levels of 12-HEPE, 14-HDHA and 12-HETE were around 50, 15, and 25 nMol for male C57BL/6 mice housed for 7 days in cold temperature (5°C) ( Figure S1D).
Figure 1: Cold exposure induces 12-LOX derived lipid secretion into the circulation.

A) Volcano plot of the lipids profiled in serum showing fold change between male C57BL6/J mice exposed to 5°C or 30°C for 7 days, co mpared to the p-value of this comparison.
B) Serum levels of the 12-LOX products 12-HEPE, 14-HDHA and 12-HETE from male mice exposed to 30°C or 5°C for 7 days (n = 5).
C) Volcano plot of the serum lipids from C57BL6/J female mice exposed to 30°C or 5°C for 7 days.
D) Serum levels of the 12-LOX products from female mice exposed to 30°C or 5°C for 7 days (n = 5).
E) Volcano plot of the lipids profiled in serum showing fold change between male C57BL6/J mice exposed to 5°C for 1 hour and mice kept at room temperature (22°C) compared to the p-value of this comparison.
F) Circulating levels of the 12-LOX products from male mice exposed to 5°C or 22°C for 1 hour (n = 5).
G) Alox12 mRNA expression in differentiated WT-1 brown adipocytes stimulated with cyclic AMP (1mM), norepinephrine (1μM) or CL316,243 (1μM) for 4 hours.
H) Western blotting for 12-LOX protein expression in iBAT and ingWAT from mice housed at 5°C or 30°C for 7 days (n = 4).
I) Scheme illustrating the biosynthetic pathways by which 12-HEPE, 14-HDHA and 12-HETE are derived from their polyunsaturated fatty-acid precursors EPA, DHA and AA. For all panels, *P<0.05, unpaired student’s t-test. Data are represented as mean ± S.E.M. See also Figure S1.
All the lipid quantification data were detected using non-targeted lipidomics, thus relative values are shown.
The coordinated regulation of 12-LOX products by cold exposure prompted us to examine whether cold and/or β3-adrenergic activation is capable of inducing the expression of 12-LOX in brown or beige adipocytes. While 12-LOX (Alox12) is known to biosynthesize lipid metabolites such as 12(S)-HEPE, 14(S)-HDHA, and 12(S)-HETE, 15-LOX (Alox15, also known as 12/15-LOX) is able to produce both 12- and 15-LOX products in mice such as 12(S)-HETE and 15(S)-HETE from AA and 15(S)-HEPE from EPA (Brash, 1999; Kuhn et al., 2015). The 12R-LOX enzyme, which is encoded by the Alox12b gene, is responsible for the biosynthesis of the R stereoisomers 12(R)-HETE and 12(R)-HEPE (Brash, 1999; Kuhn et al., 2015). Since brown and beige fat are the primary fat depots activated by cold stimulation, we measured the expression of genes encoding these enzymes in BAT and inguinal white adipose tissue (ingWAT) to determine which lipoxygenase is expressed and could potentially be activated in these tissues. We found negligible levels of mRNA expression of Alox15, Alox15b, and Alox12b (Ct > 32 when measured by qRT-PCR; note that Ct is a relative and not absolute quantification), while Alox12 was expressed at a substantially higher level in BAT and ingWAT (Ct ca. 26-27). These data suggest that the S stereoisomers were the ones being produced under cold. Alox12 mRNA expression was elevated in differentiated brown adipocytes in response to three different adrenergic mimetics, namely, cAMP dibutyrate, norepinephrine, and the β3-adrenergic receptor agonist CL316,243 (Figure 1G). We found that 12-LOX protein expression was significantly higher in both BAT and ingWAT of mice housed in 5°C for 7 days compared to mice kept at 30°C ( Figure 1H), although at the mRNA level, Alox12 mRNA was elevated in BAT, but not in ingWAT (Figure S1E). Alox12 mRNA levels were not altered in either BAT or ingWAT after 1 hour in 5°C (Figure S1E), indicating that upon an acute cold challenge, it is the enzymatic activity, rather than the transcriptional modulation, of 12-LOX that contributes to the production of the metabolites. Furthermore, we found that within the adipose tissue, the cold-induced 12-LOX mRNA expression predominantly occurred in mature adipocytes, but not in stromal vascular fraction SVF (Figure S1F).
Lipolysis in response to acute cold or adrenergic stimulation leads to increased availability of free fatty-acids to serve as substrates for oxidative metabolism, and also contributes to the release of lipid metabolites into the circulation (Haemmerle et al., 2006). To determine whether lipolysis was required for the biosynthesis and secretion of 12-LOX products upon short-term cold exposure, we measured circulating lipid levels in mice with adipose-specific deletion of adipose triglyceride lipase (ATGL) (AdipoqCRE/Atglflox). ATGL is the major lipase that hydrolyzes triglyceride in adipose tissue (Ahmadian et al., 2011). As previously reported, (Ahmadian et al., 2011; Schreiber et al., 2017; Shin et al., 2017), these animals displayed cold intolerance and an increased BAT mass due to brown fat whitening (Figures S1G–S1I). We found that the cold-induced elevation of 12-LOX lipid metabolites was abolished or significantly reduced in the AdipoqCRE/Atglflox mice (Figure S1J), indicating that the activation of the ATGL-dependent lipolytic pathway in adipose tissue is essential for the production of these circulating lipids in response to acute cold challenge.
Elevation of 12-LOX metabolites in the circulation of humans acutely treated with mirabegron
To further test the hypothesis that adrenergic-induced activation of 12-LOX is relevant in humans, we performed a lipidomics analysis in plasma samples from healthy human subjects treated either with a single oral dose of the specific β3-adrenergic agonist mirabegron (200mg), or a placebo. As previously reported by Cypess et al. (2015), these subjects exhibited higher 18F-fluorodeoxyglucose (FDG) uptake in BAT, and elevated energy expenditure after mirabegron treatment. We detected significantly higher levels of 12-HEPE and 14-HDHA in mirabegron treated subjects, while 12-HETE was unchanged (Figure 2A–2B). Importantly, 12-HEPE was the most significantly upregulated lipid metabolite among all the detected lipid species (Figures 2A–2B and S2A–S2C). Furthermore, the circulating levels of these lipids positively correlated with BAT activity measured by FDG uptake (Figures 2C–2E).
Figure 2: Mirabegron treatment increases the secretion of 12-LOX metabolites in human subjects.

A) Volcano plot of the lipids profiled in plasma showing fold change between human subjects treated with a single oral dose of the β3-adrenergic agonist mirabegron (200mg) and subjects treated with placebo compared to the p-value of this comparison, (n = 10-11). p values determined by multiple t-tests.
B) Circulating levels of the 12-LOX derived lipids from human subjects treated with placebo or mirabegron (n = 10-11). **p<0.01, ***p<0.001. Data are represented as paired individual values.
C-E) Spearman correlation between circulating 12-LOX metabolites levels and BAT glucose uptake measured by PET-CT, and expressed as standardized uptake values (SUV), obtained from human subjects treated with placebo or mirabegron. See also Figure S2.
All the lipid quantification data were detected using non-targeted lipidomics, thus relative values are shown.
BAT is a source for cold-induced 12-LOX lipid metabolites
To further assess whether BAT could be the source of the 12-LOX metabolites in response to cold, we measured the circulating oxidized lipid levels in wild-type control mice, and in Myf5CRE/BMPr1aflox mice, which are characterized by impaired BMP signaling in brown fat progenitors leading to a severe paucity of BAT (Schulz et al., 2013). Comparing these mice with the wild-type animals allows for the identification of BAT-secreted factors (Figure 3A). The cold-induced increase in circulating 12-HEPE was completely diminished in the Myf5CRE/Bmpr1aflox group, while 14-HDHA was still induced by cold in both groups, and 12-HETE levels were unchanged (Figure 3B).
Figure 3: 12-HEPE is a cold-inducible lipid that is biosynthesized in BAT.

A) Schematic panel illustrating the experimental design of the lipidomics performed in serum from Bmpr1aflox (WT) and Myf5CRE/Bmpr1aflox (mice with iBAT paucity) male mice exposed to 30°C or 5°C for 2 days.
B) Serum lipid levels in male Bmpr1aflox (WT) and Myf5CRE/Bmpr1aflox mice exposed to 30°C or 5°C (n = 4-5).
C) Schematic panel illustrating the experimental design of the lipidomics performed in iBAT and ingWAT harvested from male C57BL6/J mice exposed to 30°C or 5°C for 7 days.
D) Lipid levels in iBAT and ingWAT from C57BL6/J male mice exposed to 30°C or 5°C for 7 days (n = 5-6). Targeted lipidomics was used for lipid quantification.
E) Schematic panel illustrating the experimental design of the lipidomics performed in conditioned medium incubated with ingWAT or iBAT explants, harvested from C57BL6/J mice exposed to 30°C or 5°C for 7 days.
F) Medium lipid levels in Krebs Ringer buffer after its incubation (1 hour) with iBAT or ingWAT harvested from C57BL6/J male mice exposed to 30°C or 5°C for 7 days.
G) Schematic panel illustrating the experimental design of the lipidomics performed in media conditioned by in vitro differentiated human brown adipocytes stimulated with forskolin or vehicle for 4 hours.
H) 12-HEPE, 14-HDHA and 12-HETE levels in conditioned media of human brown adipocytes stimulated with forskolin or vehicle. *P<0.05, **p<0.01, ***p<0.001. Unpaired Student’s t-test. N=5 per group. Data are represented as mean ± S.E.M. See also Figure S3.
All the lipid quantification data (except for Figure 3D) were detected using non-targeted lipidomics, thus relative values are shown.
Next, targeted lipidomic analyses were performed in BAT and ingWAT of C57BL6/J mice housed at 5°C or 30°C for 7 days (Figure 3C). Although 12-HEPE could be detected in both fat depots, its cold-induced biosynthesis was only observed in the interscapular BAT. In contrast, levels of 14-HDHA were increased by cold in both fat depots, and the levels of 12-HETE were not altered in either tissue (Figure 3D). To validate the capacity of these fat depots to produce lipid metabolites, we dissected BAT and ingWAT from mice exposed to 5°C or 30°C for 7 d ays and incubated the tissue explants in Krebs-Ringer buffer for 1 hour before quantifying lipid concentrations in this buffer. BAT from mice exposed to cold secreted twice as much 12-HEPE as control BAT, while ingWAT secretion was not affected by cold (Figures 3E–3F). Using this assay, we could not detect significant changes of 14-HDHA and 12-HETE release from BAT or ingWAT in response to cold (Figures 3E–3F). Since adipose tissue contains multiple cell types, to determine whether these lipid mediators are directly produced by activated brown adipocytes, we used in vitro differentiated human and murine brown adipocytes treated with sympathomimetics. We found significantly higher levels of 12-HEPE and 14-HDHA in the media of murine brown adipocytes treated with the β-3-adrenergic agonist CL316,243 for 4 hours (Figure S3A–S3C). Importantly, this phenomenon was also observed in human brown adipocytes. As shown in Figure 3G–3H, the levels of all three 12-LOX oxylipins were increased by at least 2-fold in the media of human brown adipocytes treated with forskolin. Altogether, these findings suggest that brown adipocytes are the cellular source producing 12-LOX metabolites in response to cold or β3-adrenergic stimulation in rodents and humans.
12-LOX activity in BAT is required for adaptive thermogenesis
In order to investigate whether 12-LOX activity is required for cold adaptation, we pre-treated C57BL/6 mice with a dual 12- and 12/15-LOX chemical inhibitor (LOXBlock-1) for 30 minutes and then exposed these animals to 5°C for 4 hours. LOXBlock-1 abolished the cold-induced release of 12-LOX products, 12-HEPE and 14-HDHA (Figure 4A), and led to an impairment in cold tolerance, which was prevented by 12(S)-HEPE co-injection (Figures 4B and S4A). We observed similar effects using two other distinct 12-LOX inhibitors (Baicalein and NCTT956) (Figures S4B–S4C). The effect of the 12-LOX inhibitors was not due to changes in the expression of thermogenic genes in BAT because the cold-induced levels of thermogenic mRNA such as Ucp1, Dio2, and Ppargc1a, were not altered between vehicle and LOXBlock-1-treated groups (Figure S4D), suggesting that the 12-LOX lipid mediators may directly and acutely modulate the activity of brown fat, independently of major transcriptional changes.
Figure 4: 12-LOX ablation in brown adipocytes impairs glucose utilization and cold adaptation.

A) Serum lipid levels in C57BL6/J male mice i.p. injected with LOXblock-1 for 15 min or its vehicle DMSO, and then exposed to 5°C for 4h. (n = 6-7).
B) Rectal temperature during cold tolerance test in C57BL6/J male mice i.p. injected with LOXblock-1 or vehicle (DMSO) in the presence or absence of 12(S)-HEPE (200μg/kg via i.p.) and then exposed to 5°C. (n = 5-6).
C) Schematic panel illustrating the strategy for the generation of Ucp1CRE/Alox12 KO mice from the Ucp1CRE and CRISPR/Cas9 knock-in mice.
D) Serum 12-LOX-derived lipid concentrations from the empty vector (EV) mice and Ucp1CRE/Alox12 KO mice, exposed to 22°C or 5°C for 1 hour. (n = 6).
E) Rectal temperature during cold tolerance test in EV and Ucp1CRE/Alox12 KO male mice, by measuring rectal temperature under 5°C (n = 12).
F) Oxygen consumption measured by CLAMS in EV and Ucp1CRE/Alox12 KO mice i.p. injected with norepinephrine (n = 12).
G) Glucose uptake in wild-type, Alox12 KO and 12-LOX re-expressed Alox12 KO differentiated brown adipocytes in vitro.
H) Extracellular acidification rate (ECAR) during the Seahorse glycolysis stress test in Alox12-KO and 12-LOX re-expressed Alox12 KO differentiated brown adipocytes.
I) Oxygen consumption ratio (OCR) during the Seahorse mitochondrial stress test in EV, Alox12-KO and 12-LOX re-expressed Alox12 KO differentiated brown adipocytes.
*p<0.05, **p<0.01, ***p<0.001. Data are represented as mean ± S.E.M. See also Figure S4. The lipid quantification data were detected using non-targeted lipidomics, thus relative values are shown.
To more specifically assess the role of 12-LOX activity in BAT thermogenic capacity and secretory function, we generated brown fat-specific Alox12 knockout mice using a combined Cre-lox and CRISPR/Cas9 technology (Platt et al., 2014). We crossed Cre-dependent Rosa26 Cas9 knockin mice (Rosa26-LSL-Cas9) with Ucp1-Cre animals to generate Ucp1CRE/Cas9 mice (Figure 4C). These mice received bilateral interscapular BAT injections with AAV2/8 expressing either a specific gRNA targeting exon 1 of the Alox12 gene (Figure S4E), or an empty vector (EV) control (Figure 4C), thus generating UCP1CRE-Alox12 KO mice or EV control mice. We confirmed that the level of 12-LOX protein was diminished in BAT, but not in the ingWAT of UCP1CRE-Alox12 KO mice (Figure S4F). UCP1CRE-Alox12 KO mice had no changes in body weight, fasting glucose, or tissue weights when raised at standard room temperature (22°C) ( Figure S4G – S4I). Cold-induced elevation of 12-LOX products in circulation was absent in UCP1CRE-Alox12 KO mice after 1-hour exposure at 5°C ( Figure 4D). Importantly, UCP1CRE-Alox12 KO mice displayed impaired cold adaptation compared to the EV control mice in a cold tolerance test (Figure 4E). In line with these findings, the UCP1CRE-Alox12 KO mice also displayed a significant reduction in whole-body oxygen consumption in response to norepinephrine (NE) stimulation (Figure 4G). Similar to what observed in mice treated with LOXBlock-1-treated mice, the levels of cold-stimulated expression of thermogenic genes were comparable between UCP1CRE-Alox12 KO and EV control mice (Figure S4J).
Given that 12-LOX products positively correlated with glucose uptake into BAT from human subjects treated with mirabegron, we hypothesized that the loss of 12-LOX in brown adipocytes would lead to impaired glucose metabolism and mitochondrial activity in brown adipocytes in a cell-autonomous fashion. To test this hypothesis, we generated Alox12 KO cells by using the CRISPR/Cas9 system and the same gRNA targeting Alox12 that was used for the aforementioned in vivo studies (Figure S4K). Indeed, the capacity of Alox12 KO brown adipocytes to take up glucose was impaired compared to EV control cells, while Alox12 re-expression completely restored basal glucose uptake (Figure 4G). Consistent with these findings, Alox12 deficient brown adipocytes displayed a reduced extracellular acidification rate in a glycolysis stress test which was fully restored by the re-expression of Alox12 (Figure 4H). Next, we investigated whether Alox12 deletion in brown adipocytes could affect mitochondrial respiration. Although basal and uncoupled respiration were unaffected by Alox12 deletion, the Alox12 KO brown adipocytes displayed a severe defect in maximal respiration in response to the mitochondrial uncoupler FCCP (carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone), which was restored by the 12-LOX re-expression (Figure 4I). Although differentiation capacity was unaltered in the 12-LOX KO cells (Figures S4L and S4M), expression of selected thermogenic genes, such as Ucp1 and Cidea, but not Ppargc1a and Dio2, was reduced in the KO cells, which was nominally restored by Alox12 re-expression (Figure S4M). The inability of 12-LOX re-expression in restoring Ucp1 and Cidea expression in the KO cells and the fact that level of Ucp1 mRNA was not altered in BAT of UCP1CRE-Alox12 KO mice suggest that 12-LOX might not be directly involved in the transcriptional regulation of Ucp1 and other thermogenic genes. Therefore, the reduced levels of UCP1 and Cidea in the 12-LOX KO cells might be secondary to other metabolic changes. Taken together, these data indicate that 12-LOX, presumably by generating its downstream lipid metabolites, plays an essential role in BAT-mediated regulation of adaptive thermogenesis and glucose metabolism.
12-HEPE biosynthesis and secretion are repressed in obesity
Since brown fat mass and activity are lower in obese patients and are inversely correlated with BMI in humans (van Marken Lichtenbelt et al., 2009; Cypess et al., 2009), we proceeded to determine the relationship between circulating levels of 12-LOX products and metabolic parameters in a cohort of subjects with a broad distribution of BMI. Interestingly, we found significantly lower levels of the 12-LOX metabolites 12-HEPE, 14-HDHA, and 12-HETE in overweight (BMI = 25-30) and obese subjects (BMI > 30) compared to lean individuals (BMI < 25) (Figures 5A – 5C). Due to the non-linear relationship with the variants, we used the Spearman’s correlation and identified a significant negative correlation between plasma levels of 12-LOX products and BMI (Figures S5A–S5C). Importantly, the levels of these lipids were also negatively correlated with insulin resistance measured by HOMA-IR and circulating leptin levels (Figures 5D – 5I), however, these correlations were dependent on BMI.
Figure 5: Regulation of 12-LOX metabolite levels in human and murine obesity.

A - C) 12-HEPE, 14-HDHA and 12-HETE serum levels in serum samples from lean (BMI<25), overweight (BMI>25, <30) and obese (BMI>30) subjects (n = 60). **P<0.01, ***P<0.001. Data are represented as mean ± S.E.M. See also Figure S4.
D-F) Spearman correlation between the serum levels of 12-LOX products and insulin resistance measured by HOMA-IR index.
G-I) Spearman correlation between the serum levels of 12-LOX products and serum leptin concentration. See also Figure S5.
The lipid quantification data were detected using non-targeted lipidomics, thus relative values are shown.
Because our data suggest that fat depots are potential sources of the circulating 12-LOX metabolites, we measured lipid levels in BAT of diet-induced obese (DIO) mice and control lean mice. We found that the concentration of 12-HEPE was significantly lower in BAT of DIO mice, contrasting with the not significantly altered 14-HDHA and higher 12-HETE levels (Figure S5D).
The 12-LOX derived lipid 12-HEPE is a brown fat-secreted mediator of glucose uptake
Because the levels of 12-HEPE were consistently increased by cold in all of the conditions we tested, and among the 12-LOX metabolites we detected it was the lipid species consistently found to be produced by BAT and repressed by obesity in murine brown fat and human plasma, we sought to determine whether 12-HEPE is able to regulate energy homeostasis and fuel utilization in DIO mice. DIO mice were treated daily with intraperitoneal (i.p.) injections of 12(S)-HEPE (200μg/kg) or PBS for 2 weeks. The i.p. injection increased circulating 12-HEPE levels by 2.5-fold 30 minutes after treatment (Figure S6A). Although no change in body weight gain or fasting glucose was observed (Figures 6A and 6B), 12(S)-HEPE-treated DIO mice displayed a marked improvement in glucose tolerance compared to the vehicle-treated group (Figure 6C). There was no difference in the serum insulin levels before or after glucose injection (Figure S6B), indicating that the effects of 12(S)-HEPE did not rely on the regulation of glucose-induced insulin release. Moreover, 12(S)-HEPE treatment led to an improvement in insulin sensitivity in DIO mice (Figure 6D). Animals treated with 12(S)-HEPE did not show any change in food intake (Figures S6C – S6D), physical activity (Figure S6E), carbon dioxide production (VCO2) or oxygen consumption (VO2) (Figures S6F and S6G). There was a trend for an increase in VO2 and respiratory exchange ratio (Figure S6H). Since the effects of omega-3 PUFAs are associated with their ability to reduce adipose tissue inflammation (Oh et al., 2010; Talukdar et al., 2011, Spite et al., 2014), we examined whether the improved glucose metabolism observed in 12(S)-HEPE-treated DIO mice was mediated by alterations in adipose inflammation. The mRNA expression of pro- and anti-inflammatory genes was not altered by 12(S)-HEPE treatment in ingWAT, perigonadal white adipose tissue (pgWAT) or BAT, with the exception of increased IL-10 expression in pgWAT of 12(S)-HEPE-treated animals (Figure S6I – S6K). We did not observe differences in M1 and M2 macrophage populations in the pgWAT of 12(S)-HEPE treated mice compared to the PBS-treated group by flow cytometry analysis (Figure S6L). Interestingly, BAT weight was increased in DIO mice treated with 12(S)-HEPE compared to control animals (Figure 6E). Although there was no change in the expression of genes involved in the thermogenic program in the BAT (Figure 6F), 12(S)-HEPE treatment increased mRNA expression of the glucose transporter Glut-1 (Figure 6G), the glucose-sensitive transcription factor Chrebp-β and its downstream target fatty-acid synthase (Fasn) (Figure 6H), suggesting that 12(S)-HEPE treatment promotes glucose uptake and utilization in BAT of DIO mice.
Figure 6: 12-HEPE serves as a glucose uptake mediator.

A) Body weight in 12(S)-HEPE and Vehicle treated DIO male mice (n = 7).
B) Fasting serum glucose measured in 12(S)-HEPE and Vehicle treated DIO mice (n = 7).
C) Glucose tolerance test (GTT) performed in 6h fasted 12(S)-HEPE and Vehicle treated DIO mice. (n = 7).
D) Glycemia during the insulin tolerance test (ITT) performed in 6h fasted 12(S)-HEPE and Vehicle treated DIO mice (n = 7).
E) Ratio of tissue weight to body weight calculated for pgWAT, ingWAT, liver and BAT harvested from 12(S)-HEPE and Vehicle treated DIO mice (n = 7).
F-H) mRNA expression measured by qPCR of genes associated with the thermogenic program associated, glucose transport and de novo lipogenesis in BAT from 12(S)-HEPE and Vehicle treated DIO mice (n = 7).
I) Glucose uptake into BAT, ingWAT, pgWAT, quadriceps, gastrocnemius, soleus and liver of C57BL6/J lean mice i.v. injected with 12(S)-HEPE or vehicle, 30 minutes before an [3H]2-deoxyglucose i.v. injection (n = 6-8).
J-N) In vitro glucose uptake into murine brown adipocytes, human brown adipocytes, murine myocytes (C2C12), murine white adipocytes (3T3-F442A cells) and human white adipocytes (hWAT) treated with 12(S)-HEPE or vehicle for 30 minutes before adding [3H]2-deoxyglucose. (n = 4-6 experimental replicates from at least 2 biological replicates). *P<0.05, **P<0.01, ***P<0.001. Data are represented as mean ± S.E.M. See also Figure S6.
To test the hypothesis that 12(S)-HEPE improves glucose homeostasis by promoting glucose uptake into tissues, we measured tissue [3H] 2-deoxyglucose ([3H]2-DG) uptake in lean mice acutely treated with 12(S)-HEPE. 12(S)-HEPE treatment significantly increased [3H]2-DG uptake into BAT and skeletal muscle, but not into pgWAT, ingWAT, or liver (Figure 6I). To examine whether 12(S)-HEPE regulates glucose uptake in a cell-autonomous manner, we measured [3H]2-DG uptake into different cell types in vitro. We found that 12(S)-HEPE was able to increase glucose uptake in differentiated murine and human brown adipocytes as well as in C2C12 myotubes (Figure 6J–6L), indicating that 12(S)-HEPE could directly regulate glucose uptake in adipocytes and muscle cells. Interestingly, while 12(S)-HEPE did not promote 2-DG uptake into WAT in vivo, it could increase glucose uptake into human and mouse white adipocytes differentiated in vitro (Figure 6M and 6N). These data suggest that 12-HEPE functions as a mediator of glucose uptake in adipose tissue and skeletal muscle.
12-HEPE promotes glucose uptake by triggering the PI3K/Akt/Glut pathway in a GsPCR dependent fashion
Since the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/protein kinase B (Akt)/Glut-4 axis is the canonical signaling pathway that regulates glucose uptake, we hypothesized that 12(S)-HEPE activates this pathway to promote glucose uptake into tissues. Akt activation is triggered by the phosphorylation at residues Ser473 and/or Thr308 by the mammalian target of rapamycin complex 2 (mTORC2) and phosphoinositide dependent kinase-1 (Pdk-1), respectively (Sarbassov et al., 2005). Interestingly, we found that 12(S)-HEPE promoted AKT phosphorylation at both Ser473 and Thr308 in murine brown adipocytes (Figures 7A, S7A and S7B), indicating coparticipation of mTORC2 and Pdk1 in the signaling pathway triggered by 12(S)-HEPE. Indeed, in addition to increased mTORC2 phosphorylation at Ser2481, we also observed phosphorylation of the downstream target AS160 at Thr642 in response to 12(S)-HEPE stimulation in brown adipocytes (Figures 7A, S7C and S7D). Importantly, 12(S)-HEPE triggered similar signaling cascade in vivo, since phosphorylation of Akt (Ser473) and mTOR (Ser2481) was increased in BAT of mice acutely injected with 12(S)-HEPE (i.v., 30 min, 200μg/kg) (Figure S7E and S7F). To determine whether the mTORC-PI3K-Akt axis plays an essential role in 12(S)-HEPE-induced glucose uptake, murine brown adipocytes were pre-treated with inhibitors of mTORC1, mTORC1/2, or PI3K, and the glucose uptake in response to 12(S)-HEPE was measured. While the incubation with the mTORC1 inhibitor Rapamycin did not affect glucose uptake, the mTORC1/2 inhibitor Torin1 partially reduced 12(S)-HEPE’s effect on glucose uptake. Pre-treatment with the PI3K inhibitor Wortmannin almost completely abolished this effect (Figure 7B). Furthermore, Glut4 was translocated to the plasma membrane in response to 12(S)-HEPE stimulation in murine brown adipocytes (Figure 7C).
Figure 7: 12-HEPE promotes glucose uptake via a GsPCR/PI3K/AKT axis.

A) Western blotting for phospho-AKT (Ser473 and Th308), Akt, phospho-mTORC2 (Ser2481), mTOR, phospho-AS160 (Th642), and β-tubulin (loading control) in mouse brown adipocytes treated with 12(S)-HEPE or vehicle.
B) In vitro glucose uptake into murine brown adipocytes treated with 12(S)-HEPE or vehicle in the presence or absence of pre-treatment with rapamycin, Torin 1 or Wortmannin.
C) Immunofluorescent staining for Glut-4 in murine brown adipocytes treated with PBS, insulin or 12(S)-HEPE. The white arrows indicate Glut-4 staining in the plasma membrane.
D-F) In vitro glucose uptake into murine brown adipocytes treated with 12(S)-HEPE or vehicle in the presence or absence of pre-treatment with YM254890, Pertussis Toxin or Melittin.
G) In vitro glucose uptake into Scramble (control) or Gnas KD murine brown adipocytes treated with 12(S)-HEPE or vehicle. *P<0.05, **P<0.01, ***P<0.001. Data are represented as mean ± S.E.M. See also Figure S7.
Many lipid mediators exert their effects through stimulation of G-protein coupled receptors (GPCRs). To identify the class of GPCR that mediates the effect of 12(S)-HEPE on promoting glucose uptake, we performed an in vitro glucose uptake assay in murine brown adipocytes pre-treated with inhibitors of 3 subtypes of GPCR, namely Gq/11, Gi, and Gs protein-coupled receptors. Pretreatment with the Gq/11 and Gi antagonists, YM254890 and pertussis toxin, did not change the 12(S)-HEPE effect (Figure 7D–7E); however, the Gs inhibitor melittin, completely blunted the 12(S)-HEPE-induced glucose uptake (Figure 7F), suggesting that 12(S)-HEPE utilizes a GSPCR to trigger downstream signaling. To further validate these findings, we knocked down Gnas, the gene encoding the stimulatory subunit of Gs protein, in brown adipocytes (Figure S7G), and found that 12(S)-HEPE-induced glucose uptake was abolished in the Gnas knock-down cells (Figure 7G). Thus, 12(S)-HEPE exerts the glucose shuttling effect by triggering a GsPCR, which leads to PI3K-mTORC2-Akt activation and Glut4 translocation to the plasma membrane.
DISCUSSION
Oxylipins are oxidized lipids derived from PUFAs that play important roles in health and disease. Using lipidomics analyses, here we demonstrate that the 12-LOX biosynthetic pathway is activated in BAT in response to cold or β-adrenergic stimulation, generating oxylipin mediators that are released into the circulation where they can exert glucose shuttling effect on tissues to support thermogenesis. This effect is mediated through the stimulation of a GsPCR, which triggers an insulin-like signaling pathway that results in Glut4 translocation and glucose uptake into the cell. These data reveal a novel cold-induced 12-LOX-dependent pathway in thermoregulation and fuel utilization.
Among the 12-LOX-derived oxylipins, 12-HETE is the most studied and characterized lipid (Nunemaker et al., 2008; Ma et al., 2010; Cole et al., 2013; Tersey et al., 2014). To date, 12-LOX is described as a potential mediator in the pathogenesis of type 2 diabetes/obesity by means of biosynthesizing 12-HETE, which, leads to impairment of insulin signaling in adipocytes and hinders β-cell function by exerting a pro-inflammatory effect in these tissues (Nunemaker et al., 2008; Ma et al., 2010; Cole et al., 2013; Tersey et al., 2014). 12-LOX whole-body knockout mice are protected against HFD-induced insulin resistance and inflammation (Nunemaker et al., 2008). It is conceivable that under high-fat feeding, lipoxygenase activity might shift towards the arachidonic acid cascade when this substrate is more abundant (Brash et al., 1999; Joshi et al., 2013). Since HFD has an omega-6 to omega-3 ratio approximately 2-fold higher than a regular chow diet, this imbalance results in the increased availability of the pro-inflammatory substrate, which may help to explain the deleterious role of 12-LOX in obesogenic murine models. Here we show that 12-LOX activity, specifically in BAT, has important implications for glucose metabolism and thermoregulation. Deletion of 12-LOX in brown adipocytes in vitro and in vivo results in impaired glucose uptake and glycolysis, as well as cold intolerance. These results suggest that 12-LOX may play tissue-specific roles by producing distinct metabolites, which may explain in part the seemly conflicting results between the previously reported whole body KO mice and the brown fat-specific KO model described in the current study.
Although we demonstrate that brown adipocytes produce 12-HEPE and release it into the circulation, one cannot neglect that other cell types, such as white adipocytes, platelets or myeloid cells, could also contribute to circulating levels of 12-LOX metabolites, since 12-LOX is abundantly expressed in these cell types (Deng et al., 2014; Serhan et al., 2014; Kuhn et al., 2015). However, by combining both serum and tissue lipidomics in normal C57BL/6 mice and a mouse model with BAT paucity with the ex vivo and in vitro studies, we demonstrate that 12-HEPE is a cold-induced 12-LOX metabolite that is produced and released from BAT. Surprisingly, we find that the release of all three 12-LOX products is blunted in the UCP1CRE/Alox12 KO mice in response to an acute cold challenge, suggesting that, in addition to 12-HEPE, 14-HDHA and 12-HETE may also be released from BAT in response to acute cold exposure. Whether such changes are sustained in prolonged cold acclimatization warrants future investigations.
Recently, emerging evidence suggests that both shivering and non-shivering thermogenesis contribute to defending body temperature upon acute cold challenges. First, it is well-known that the UCP1-KO mice are cold intolerant when directly transferred from room temperature to cold environment (Enerbäck et al., 1997). In addition, transgenic mouse models with impaired brown fat activity also display compromised adaptation to acute cold-exposure (Schulz et al., 2013; Ohno et al., 2013). Importantly, data from the current study demonstrate that BAT-derived lipid mediators play an essential role in acute cold tolerance presumably via enhancing non-shivering thermogenesis. Additionally, the data from our AdipoqCRE/Atglflox knockout mice indicate that upon acute cold exposure, lipolysis combined with enhanced 12-LOX activity regulates the secretion of 12-LOX oxylipins into the circulation. Interestingly, it has been recently shown that ATGL-mediated lipolysis in BAT is dispensable for cold adaptation, but adequate nutrient supply is important for proper thermoregulation (Schreiber et al., 2017). It is possible that ATGL-dependent lipolysis in WAT can also contribute to the acute release of 12-LOX metabolites and other lipid substrates for adaptive thermogenesis. This establishes a cross-talk between WAT and BAT via the release of lipid mediators in defending body temperature upon cold challenge.
It has been well established that cold and β3-adrenergic stimulation promote BAT glucose uptake and trigger adaptive thermogenesis (van Marken Lichtenbelt et al., 2009; Virtanen et al., 2009, Cypess et al., 2015). Moreover, the β3-adrenergic-mediated glucose uptake in brown adipocytes is known to also rely on a PI3K-mTORC2-Akt pathway (Albert et al., 2016), which reinforces the link between GsPCR and this signaling pathway in brown fat. The exact mechanism by which glucose supports thermogenesis remains unclear, although it has been shown that BAT glucose uptake and subsequent glycolysis in response to cold are required to compensate for the loss of mitochondrial ATP production due to mitochondrial uncoupling (Vallerand et al., 1990). In addition, glucose is needed for anaplerotic reactions to maintain fatty acid oxidation (Hao et al., 2015), and triggers de novo lipogenesis, which in turn provides fatty acids for UCP1 activation (Sanchez-Gurmaches et al., 2018). In agreement with these findings, 12-LOX protein expression is induced by cold in BAT, and its metabolite 12-HEPE can increase the expression of de novo lipogenesis genes and Glut-1 in BAT, promote Glut-4 translocation to the plasma membrane, and ultimately increase glucose uptake into brown adipocytes. These data provide important mechanistic insights linking cold-induced glucose uptake and adaptive thermogenesis.
Our study reveals a novel role of 12-LOX in adaptive thermogenesis and glucose utilization. We demonstrate that 12-LOX activity in BAT is indispensable for cold tolerance, presumably by biosynthesizing and releasing the orphan lipid mediator 12-HEPE. 12-HEPE then acts as an autocrine and endocrine factor to promote glucose uptake into BAT and skeletal muscle through activation of the PI3K/mTOR/Akt/Glut pathway. Given the non-toxic nature of the lipid, the observed metabolic effects and the mechanisms of action elucidated in our study, 12-HEPE, or 12-HEPE-mimetics, holds great therapeutic potential for reducing hyperglycemia in diabetic patients. Taken together, our data not only reveal a previously unrecognized mechanism coupling lipid oxidation and adaptive thermogenesis, but also provide new therapeutic approaches for individuals with diabetes and other related metabolic disorders.
Limitations of Study
In this study, we have described a mechanism regulating cold adaptation through a coordinated biosynthesis and release of bioactive 12-LOX metabolites from BAT into the circulation. Although we have chosen to study the role of the EPA-derived lipid mediator,12-HEPE, on the regulation of nutrient metabolism, the contributions of the other two metabolites, namely 12-HETE and 14-HDHA, to cold acclimatization and fuel utilization remain to be determined. Furthermore, mechanisms linking 12-HEPE-induced glucose uptake and thermoregulation in brown adipocytes also warrant future investigations. Finally, a thorough pharmacokinetic characterization of 12-HEPE, as well as the identification of its putative receptor, will enhance the understanding of 12-HEPE’s action and contribute to the development of 12-HEPE-based therapies in treating metabolic diseases.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yu-Hua Tseng (yu-hua.tseng@joslin.harvard.edu). This study did not generate new unique reagents.
DATA AND CODE AVAILABILITY
The lipid datasets generated during this study are available at Metabolomics Workbench (URL: www.metabolomicsworkbench.org; DataTrack ID 1749, 1750 and 1751).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice and treatments
All animal procedures were approved by the Institutional Animal Use and Care Committee at Joslin Diabetes Center. The animals utilized in this study were kept at a conventional animal facility at Joslin Diabetes Center. The experiments were not randomized, except the cold tolerance tests, which were performed in a blinded manner. No statistical method was used to predetermine the sample size for the animals. For all of the in vivo studies, in which wild-type mice were used, 11-13-week-old male C57BL/6J mice (Stock no. 000664) were purchased from The Jackson Laboratory. The in vivo experiments are described in the sections below.
For acute BAT activation, mice were either sacrificed as control animals kept at room temperature or individually housed at 4°C for 1 hour, then sacrificed for serum and tissue collection, which were subsequently sent for lipidomics analysis. For chronic cold exposure, C57BL/6J mice were individually housed at 5°C or 30°C for 7 days. Both acute and chronic exposures were done by housing the mice in temperature controlled diurnal incubators (Caron Products & Services Inc.) on a 12 hour light/dark cycle with ad libitum access to food. In all experiments, interscapular BAT, inguinal WAT, and serum were collected after sacrifice. All tissues were snap frozen in liquid nitrogen and stored at −80°C while serum was frozen at −20°C. For the l ipidomic analyses using the ex vivo tissue incubation strategy, interscapular BAT (BAT) and inguinal subcutaneous white adipose tissue (ingWAT) were dissected from 12-week-old male C57BL/6J mice chronically exposed to cold or thermoneutrality (as described above) and incubated at 37°C in 300μl of Krebs-Ringer solution (pH 7.4) for 1 hour, after which the tissue was discarded, and LC-MS/MS was performed on the conditioned Krebs solution.
Transgenic mice carrying floxed alleles of BMP receptor 1A (Bmpr1a) were used to generate conditional gene deletion mouse models by intercrossing with Myf5-driven cre recombinase and compared to cre-negative littermate controls as described previously (Schulz et al, 2013). These mice were used for a 2-day cold exposure experiment, in which their serum was collected and processed for global lipidomics analysis.
The same intercrossing strategy was used to delete Atgl in mouse adipose tissues. Atgl floxed mice were crossed (Stock no. 024278, The Jackson Laboratory), with the AdipoqCRE strain (Stock no. 028020, The Jackson Laboratory). As these animals are cold intolerant, they were used only for the acute (1h) cold exposure experiments.
For the experiments with daily injections of 12(S)-HEPE in high-fat diet induced obese (DIO) mice, 6-week-old C57BL/6J were fed with a high-fat diet containing 60 % kcal fat (Research Diets Stock no. D12492) for 16 weeks prior to treatment and during the course of the experiment. Mice were injected intraperitoneally daily with 200μg/kg body weight of 12(S)-HEPE diluted in PBS solution or with vehicle (PBS). Body weight and food intake were monitored. Serum was collected, and glucose (Infinity® Blood Glucose Meter, US Diagnostics), and insulin (Ultra-Sensitive Mouse Insulin ELISA Kit, Crystal Chem), were measured. All mice were allowed ad libitum access to water and food. To measure the glucose-induced insulin release, we collected additional blood from the tail at the time 0 and 45min after the glucose IP injection (2g/kg) during the glucose tolerance test.
For indirect calorimetry, PBS or 12(S)-HEPE treated DIO mice were individually housed in metabolic cages of a Comprehensive Lab Animal Monitoring System (CLAMS) at room temperature. After a 12h acclimation period, animals were monitored for 24h in order to obtain measurements for the volume of oxygen consumption (VO2), the volume of carbon dioxide production (VCO2) and respiratory exchange ratio (RER), which was calculated as the ratio of total VCO2 produced to total VO2 consumed.
Study with human subjects treated with Mirabegron
Human plasma was acquired from a previously performed clinical trial (Cypess et al., 2015) registered with ClinicalTrials.gov () and had the FDA Investigational New Drug (IND) registration number 116246. It was approved by the Human Studies Institutional Review Boards of Beth Israel Deaconess Medical Center (BIDMC) and Joslin Diabetes Center (JDC). Healthy volunteers were recruited through electronic advertisements and provided written informed consent. The subjects were given a single oral dose of mirabegron, 200 mg. Blood samples were collected 180 minutes after oral dosing. 180 minutes is the corresponding Tmax (time after administration of a drug when the maximum plasma concentration is reached) found for mirabegron in men (Baskin et al., Diabetes 2018).
Study with obese and lean human subjects
A cohort of 55 individuals were selected from the Leipzig biobank (42 women and 13 men) to represent a wide range of BMI (17.5–75.4 kg/m2), categories of lean (BMI < 25 kg/m2; n = 15; 4 male (M)/11 female (F)), overweight (BMI 25.1–29.9 kg/m2; n = 13; 4 M/9 F) or obese (BMI > 30 kg/m2; n = 27; 5 M/22 F) and glucose-metabolism parameters (fasting plasma glucose 3.9–13.4 mmol/liter; fasting plasma insulin 3.8–451 pmol/liter, HOMA-IR: 0.1–25). In the subgroup of lean, all individuals were normal glucose tolerant (NGT), whereas in the overweight subgroup, 10 individuals with NGT and 3 with type 2 diabetes (T2D), and in the obese group, 20 NGT individuals and 7 individuals with T2D were included. Collection of human biomaterial, serum analyses and phenotyping were approved by the ethics committee of the University of Leipzig (approval numbers: 159-12-21052012 and 017-12-23012012), and all individuals gave written informed consent before taking part in the study.
Generation of Alox12 KO cells
To generate a loss of function model for Alox12 in vivo, four different guide RNAs (gRNAs) were designed to target exon 1, 2, or 3 of the mouse Alox12 gene: gRNA1-GGGCCGCTACCGCGTCCGTG (exon 1); gRNA2 – TACCGCGTCCGTGTGGTCAC (exon 1); gRNA3 – GTTTGACTTCGACGTTCCCG (exon 2); gRNA4 – GAAGTATCGAGAAAAGGAAC (exon 4). The gRNAs were cloned into the pSpCas9(BB)-2A-Puro (PX459) V2.0 plasmid (Addgene plasmid #62988). Backbone and Alox12 gRNA were delivered into mouse preadipocytes (DE cells) through the nucleofection method (Nucleofector™ Technology, Lonza). After 6 days of puromycin selection, stable knockdown mouse preadipocytes were established and then used for adipocyte differentiation and functional experiments. Since gRNA number 2 showed the best efficiency in deleting Alox12 in immortalized brown adipocytes (Figure S3D), future in vitro and in vivo studies used this gRNA, which targets exon 1 of the Alox12 gene.
For re-expression of 12-LOX, WT and Alox12 knockdown mouse preadipocytes received GFP or mouse Alox12 cDNA (Myc-DDK-tagged, catalog # MR227302, Origene) through the aforementioned nucleofection method. One day after nucleofection, the cells underwent adipogenic differentiation for 8 days and, once mature, western blot analysis was done in order to confirm the expected modifications in protein expression.
Generation of UCP1CRE/Alox12 KO mice
gRNA 2 targeting exon 1 of the mouse Alox12 gene was cloned into the 1179_pAAV-U6-BbsI-gRNA-CB-EmGFP plasmid (Addgene plasmid #89060). The Alox12 gRNA or empty vector were packaged in AAV2/8 particles and then bilaterally injected into both brown adipose tissue lobes of 10-weeks old UCP1CRE/Cas9 knockin mice, thus generating the Ucp1CRE-Alox12 KO mice or empty vector (EV) mice. For the injection, mice were anesthetized with 2.5% solution of Avertin (15ul/g BW), and a small vertical incision was made between the scapulae. After the injections, the incision area was immediately closed and sutured. The animals were maintained on a heating pad following the surgery until complete recovery, and then were housed individually. There was a 2-week interval between the surgery and the beginning of the experimental procedures in these mice.
For the generation of the Ucp1CRE/Cas9 knockin mice, we crossed the Ucp1cre strain (Stock no. 024670, The Jackson Laboratories) with the homozygous Rosa26-floxed STOP-Cas9 knockin mice (Stock no. 024857, The Jackson Laboratory). Rosa26-floxed STOP-Cas9 knockin mice have a cre recombinase-dependent expression of Cas9. As a result, we obtained 50% UCP1CRE/Cas9 (HET) knockin mice and half WT/Cas9 (HET), which were injected with AAV/Alox12 gRNA and AAV/EV, respectively, through the method described above. The efficiency of the 12-LOX deletion was confirmed through western blots in BAT and ingWAT.
METHOD DETAILS
Preparation of 12(S)-HEPE
12(S)-HEPE was purchased from Cayman Chemical (Michigan, USA). Due to its instability, the preparation was done immediately before the experiments and following the manufacturer’s instructions. 12(S)-HEPE is supplied as a solution in ethanol. To change the solvent, the ethanol was evaporated under a gentle stream of nitrogen and the resulting neat oil was immediately dissolved in PBS, pH 7.2. The aqueous solution was used immediately after reconstitution.
Cold tolerance tests
For cold tolerance tests, mice were intraperitoneally injected with 50μl of the 12-LOX inhibitors, Bacalein (50mg/kg), NCTT956 (50mg/kg), LOXBlock-1 (50mg/kg), or DMSO, 15 minutes before placing the mice at 5°C. In another set of experiments, LOXBlock-1 was co-injected with 200 μg/kg of 12(S)-HEPE 15 minutes before starting the cold exposure. Rectal temperature measurements were done using an aRET-3 rectal probe (Physitemp). Bacalein and LOXBlock1 dosages were chosen based on previous publications (Yigitkanli et al., 2013; Sahu et al., 2015; Shi et al., 2018). Since NCTT956 has not been published yet for in vivo application, we decided to adopt the same dosage used for the other two inhibitors.
For the interscapular temperature, telemeters recording both temperature and activity (TA11TA-F10; Data Sciences International) were implanted under the skin in the intrascapular space of anesthetized 12-week old C57Bl/6J mice. Mice were maintained on a heating pad following the surgery and then housed individually. Mice were allowed at least 48 hours for recovery before experiments. Temperature and activity data were collected every 5 minutes.
In vivo thermogenic capacity through indirect calorimetry
Mice were injected intraperitoneally with Pentobarbital (65 mg/kg). Basal energy expenditure was measured for 30 minutes before stimulation with Norepinephrine (Sigma, 1 mg/kg in 0.9 % w/v NaCl). Energy expenditure was measured using a CLAMS. After norepinephrine injection, we monitored VO2 for the following 30 minutes.
Glucose and Insulin tolerance tests
For the glucose tolerance test (GTT), animals were fasted for 6 hours (7AM to 1PM) with free access to drinking water. A baseline blood sample was collected from the tail of fully conscious mice, followed by i.p. injection of dextrose (2.0 g/kg body weight), and blood was taken from the tail at 15, 30, 45, 60, 90, and 120 minutes after injection. For the insulin tolerance test (ITT), animals were fasted for 6 hours (7AM to 1PM) with free access to drinking water. Baseline blood samples were collected from the tail of fully conscious mice. Insulin (1 U/kg body weight) (Humulin®; Eli Lilly) was administered by i.p. injection, and blood samples were taken from the tail at 15, 30, 45, and 60 minutes after injection. Glucose concentrations were determined from blood using an Infinity® Blood Glucose Meter (US Diagnostics).
In vivo Glucose uptake
Glucose uptake in vivo was measured as previously described (Bonadonna et al., 1993). Briefly, 12-week-old C57BL6/J mice were fasted for 5 hours (7AM to 11AM) and then retro-orbitally injected with 12(S)HEPE (200ug/Kg) or vehicle (PBS). Mice were anesthetized 15 minutes after the treatments with sodium pentobarbital (85–100 mg/kg mouse body weight, i.p. injection). After 15 minutes, blood was taken from the tail to assess basal glucose concentrations and background radioactivity levels. Then, mice were injected with [3H]2-deoxyglucose-6-P (Perkin Elmer Life and Analytical Science, Waltham, MA) at 0.33 μCi /g body weight, administered via the retro-orbital sinus, and blood samples were taken after 5, 10, 15 and 25 minutes for the determination of glucose and [3H] levels. After the last blood draw, animals were sacrificed by cervical dislocation, and BAT, perigonadal and subcutaneous WAT, liver, soleus, quadriceps and gastrocnemius were harvested and immediately frozen in liquid nitrogen. Accumulation of [3H]2-deoxyglucose-6-P was assessed in tissues using a perchloric acid/Ba(OH)2/ZnSO4 precipitation procedure modified from previous work (Ferré et al., 1985).
Signaling Lipidomics
Tissue samples were homogenized in 0.1x PBS in Omni homogenizing tubes with ceramic beads at 4°C. Aliquots of 100 μL serum or 1mg protein from homogenized tissue (measured by BCA) were taken, depending on the experiment. A mixture of deuterium-labeled internal standards was added to each aliquot, followed by 3x volume of sample of cold methanol (MeOH). Samples were vortexed for 5 minutes and stored at −20 °C overnight. Cold samples were centrifuged at 14,000g for 10 minutes, and the supernatant was then transferred to a new tube and 3 mL of acidified H2O (pH 3.5) was added to each sample prior to C18 SPE. The methyl formate fractions were collected, dried under nitrogen, and reconstituted in 50 μL MeOH:H2O (1:1, by vol). Samples were transferred to 0.5 mL tubes and centrifuged at 20,000g at 4 °C for 10 minutes. (35ul) of supernatant was transferred to LC–MS/MS vials for analysis using the BERG LC–MS/MS mediator lipidomics platform. Separation of signaling lipids was performed on an Ekspert MicroLC 200 system (Eksigent Technologies) with a Synergi™ Fusion-RP capillary C18 column (150 × 0.5 mm, 4 μm; Phenomenex Inc., Torrance, CA, USA) heated to 40°C. A sample volume of 11 μL was injected at a flow rate of 20 μL/min. Lipids were separated using mobile phases A (100 % H2O, 0.1 % acetic acid) and B (100 % MeOH, 0.1 % acetic acid) with a gradient starting at 60% B for 0.5 min, steadily increasing to 80% B by 5 min, reaching 95% B by 9 minutes, holding for 1 minute, and then decreasing to 60% B by 12 minutes. MS analysis was performed on a SCIEX TripleTOF® 5600+ system using the HR-MRM strategy consisting of a TOF MS experiment looped with multiple MS/MS experiments. MS spectra were acquired in high-resolution mode (>30,000) using a 100-ms accumulation time per spectrum. Full-scan MS/MS was acquired in high sensitivity mode, with an accumulation time optimized per cycle. Collision energy was set using rolling collision energy with a spread of 15V. The identity of a component was confirmed using PeakView® software (SCIEX), and quantification was performed using MultiQuant™ software (SCIEX). C18SPE cartridges were purchased from Biotage. All solvents were of HPLC or LC-MS/MS grade and were acquired from Sigma-Aldrich, Fisher Scientific, or VWR International.
Global lipidomics was also performed in cell-conditioned media samples. Differentiated human brown and white adipocytes were treated with vehicle (ethanol) or forskolin (10μM). After that, 100 μL of the medium was collected and immediately snapfrozen in liquid nitrogen. In a separate experimental setting, differentiated murine brown adipocytes were stimulated with the β3-adrenergic receptor agonist CL316,243 (1 μM) or its vehicle (PBS), for 4 hours, and after this period 100 μL of the medium was collected and immediately snap-frozen. Lipidomic analysis of these samples was performed detailed above.
Targeted Lipidomics
Targeted LC-MS/MS analysis was done in serum, BAT and ingWAT collected from C57BL6/J mice housed in cold or thermoneutral temperature, and in BAT collected from either lean or DIO mice kept in room temperature. We also detected circulating levels of 12-HEPE at time 0 (zero) and 30 min after 12(S)-HEPE i.p. injections. After collection, the tissues were then minced in ice-cold methanol containing an internal deuterium-labeled standard (d8–5-HETE) used to assess extraction recovery and aid quantification. For media and serum samples, 2-3 volumes of ice-cold methanol containing d8–5-HETE was added and the samples were stored at −80C prior to extraction. Following centrifugation (3,000 rpm), the supernatants were subjected to solid phase extraction and LC-MS/MS analysis, as described in detail in Dalli et al., 2018. Briefly, samples were acidified (pH 3.5) and lipid mediators were extracted by C18 column chromatography with elution in methyl formate, which was then evaporated under a steady stream of N2 gas. The samples were resuspended in methanol:water (50:50) and injected into a high-performance liquid chromatograph (HPLC, Shimadzu) coupled to a QTrap5500 mass spectrometer (AB Sciex) that was operated in negative ionization mode. Lipid mediators were identified using retention time, specific multiple reaction monitoring (MRM) transitions, and diagnostic MS/MS fragmentation spectra, as compared with authentic standards. The abundance of individual lipid mediators was determined by normalizing to extraction recovery of the internal d8–5-HETE and extrapolation to calibration curves for external standards using synthetic 12-HEPE, 14-HDHA, and 12-HETE (Cayman Chemical).
In vitro glucose uptake
All the in vitro glucose uptake assays were performed in differentiated cells, regardless of the cell type. WT-1 and 3T3-F442A cells were differentiated for 9 days according to a standard adipogenic differentiation protocol. Human brown (hBAT) and white adipocytes hWAT were cultured and differentiated for 12 days as previously described (Xue et al., 2015). C2C12 cells were differentiated for 7 days in a DMEM-high glucose with 1% horse serum. Both human and murine brown and white adipocyte cell lines were serum starved in no-glucose DMEM (Gibco; catalog# 11966025), for 4 hours before the treatments. The mature C2C12 cells were serum starved overnight (18 hours) with low-glucose DMEM (Gibco; catalog# 11885084), before the treatments. The cells were treated with PBS, 12(S)-HEPE (0.01, 0.1 and 1μM) or insulin (1μM) for 30 minutes before starting the glucose transport procedure. Cells were washed once with HEPES Buffered Saline (HBS), which was then completely aspirated. Then, 300μl of transport solution (TS) containing [3H]2-deoxyglucose (0.5 uCi/ml) and 2-deoxyglucose (5mM) diluted in 20mM HBS solution was added. Cells were incubated in this solution for 5 minutes at room temperature, which was then quickly aspirated. An ice-cold stop solution (0.9% Saline) was added and washed, before the addition of 0.5 ml of a 0.05M NaOH solution to the wells. Cells were scraped and homogenized, and the homogenate transferred to fresh scintillation tubes (0.35ml), where it was vigorously mixed with 4ml of liquid scintillation cocktail (CytoScint™- ES Liquid Scintillation Cocktail, MP Biomedicals) before scintillation counting. Protein levels were determined by BCA assay in the remaining homogenate for the normalization of values.
Gnas was transiently knocked down in brown adipocytes in order to generate Gnas KD cells and then confirm the requirement of the GsPCR for the 12(S)-HEPE mediated glucose uptake. Differentiated brown adipocytes were transfected with 50nM of Gnas siRNA (Dharmacon®) or with Scrambled control using PolyJet. After 2 days, we proceeded as described above for wild type cells.
Glut-4 translocation
WT-1 cells were seeded in a chamber cell culture slide and underwent differentiation for 9 days in the same way we described for the in vitro glucose uptake experiments. After serum starvation in no-glucose DMEM for 4 hours, differentiated WT-1 cells were treated with PBS, 1 μM insulin, or 1 μM 12-HEPE. 30 minutes later, cells were fixed with 4% paraformaldehyde, followed by incubation in blocking buffer (2.5% BSA, 0.3% Triton X-100 in PBS) at 4°C overnight. Glut-4 protein was stained by anti-Glut-4 antibody (1:500, Millipore) followed by anti-rabbit IgG secondary antibody conjugated with Alexa Fluor 488 (1:1000). 5 ug/ml DAPI solution was used for nucleus staining. The images were captured using a confocal microscope (Zeiss LSM 700).
mRNA Expression
Total RNA was extracted from tissue with Trizol and purified using a spin column kit (Zymo Research). RNA (500 ng-1 μg) was reverse transcribed with a high-capacity complementary DNA (cDNA) reverse transcription kit (Applied Biosystems). Real-time PCR was performed in mouse tissues starting with 10 ng of cDNA and forward and reverse oligonucleotide primers (300 nM each) in a final volume of 10 μl with SYBR green PCR Master Mix (Life Technologies). Fluorescence was determined and analyzed in an ABI Prism 7900 sequence detection system (Applied Biosystems). Acidic ribosomal phosphoprotein P0 (ARBP) expression was used to normalize gene expression.
Protein expression
WT-1 cells were differentiated into mature brown adipocytes according to a standard adipogenic differentiation protocol (Xue et al., 2015). Cells were serum-starved in no-glucose DMEM for 4 hours and then treated with 12(S)-HEPE (1μM) for 5, 15, 30 and 45 minutes or treated with Insulin (1μM), as a positive control for 15 minutes. After that, cells were scraped from tissue-culture plates into RIPA buffer (Boston BioProducts Inc, Ashland, MA) supplemented with protease and proteinase inhibitors cocktails (Sigma-Aldrich, Dallas, TX) and further homogenized for protein detection. Protein concentrations were determined by using the Pierce BCA kit (Life Technologies), according to the manufacturer’s instructions. For immunoblots, lysates were diluted into Laemmli buffer and boiled and then loaded onto 10% Tris gels for SDS–PAGE. After complete separation of the proteins, they were transferred onto a PVDF membrane (Amersham Biosciences), blocked in western blocking buffer (Roche), and primary antibodies were applied in blocking buffer overnight at 4°C. After washing 4x for 15 min with TBS-T, secondary antibodies were applied for 1 h in blocking buffer. Membranes were washed again 3x times for 15 min in TBS-T and developed using chemiluminescence (Thermo Fisher). To quantify the bands in scanned immunoblots, regions of interest of identical size were drawn in each lane at the same molecular weight, and integrated pixel density was measured using ImageJ software. β-tubulin was used as an endogenous control for normalization. For the western blots in tissues in response to 12(S)-HEPE, we anesthetized 12 weeks old C57BL6/J mice with Avertin (375mg/kg), opened the visceral cavity and injected 12(S)-HEPE (200μg/kg, 50μl) or PBS via the vena cava. After 10 minutes, mice were sacrificed, and tissues were collected for protein extraction and western blotting. Protein lysates were stored at −80°C until further use. The data are expressed as the average normalized value for each lane, with the error bars representing S.E.M.
Cellular bioenergetic profile
Preadipocytes were seeded onto gelatin-coated Seahorse Plates and differentiated according to standard protocols. Cells were serum-starved for 1 hour prior to the beginning of the experiment. The extracellular acidification rate (ECAR) was monitored in a Seahorse XF24 instrument using the standard glycolysis stress test protocol of 3 minutes mix, 2 minutes wait, and 3 minutes measure. For the glycolysis stress test, cells were challenged with Glucose (25 mM), Oligomycin (2μM), and 2-deoxyglucose (100 mM), allowing 4 measurements after each injection. For the oxygen consumption ratio (OCR) measurements, cells were stimulated with Oligomycin (2μM), FCCP (2μM) and antimycin A (2μM), also allowing 4 measurements after each injection.
For the normalization of respiration to protein content, cells were lysed in RIPA buffer and protein concentration was measured using the Pierce BCA kit (Thermo Fisher).
Oil Red O staining
Cells were washed twice with PBS and fixed with 10% buffered formalin for 30 minutes at room temperature. Cells were then stained with a filtered Oil Red O solution (0.5% Oil Red O in isopropyl alcohol) for 2 hours at room temperature, washed several times with distilled water, and then visualized.
Flow cytometry
SVF was obtained from pgWAT by treatment with 2 mg/mL collagenase (Sigma) for 45 minutes at 37°C. The isolated SVF was resuspended in cold Hank’s balanced salt solution (HBSS) with 2% fetal bovine serum (FBS). Cells were incubated with CD45-PE-Cy7 (eBiosciences), F4/80-APC-Cy7 (BioLegend), CD206-Alex647 (Serotec, Inc.) and CD11c-PE (BD Pharmingen) antibodies for 30 minutes in HBSS containing 2% FBS on ice and then washed and resuspended in solution with Sytox Blue (Thermo Scientific). Cells were analyzed on a BD FACS Aria cell sorter after selection by forward scatter and side scatter, followed by exclusion of dead cells with Sytox Blue staining, and analyzed for cell-surface markers. M1 or M2 macrophages were identified as F4/80+/CD11c+/CD206- or F4/80+/CD11c-/CD206+ cells, respectively. The data are shown as the percentage of M1 and M2 macrophages.
QUANTIFICATION AND STATISTICAL ANALYSIS
No statistical method was used to predetermine sample size. The experiments were not blinded, except the cold tolerance tests. All statistics were calculated using Microsoft Excel and GraphPad Prism. Unpaired Student’s t-test was done to compare only 2 groups, and paired Student’s t-test was used for 2 paired-groups experiments. One-Way ANOVA followed by a Tukey’s post-hoc test was done when comparing more than 3 groups. Correlations were established based on Spearman correlation tests, and the Spearman correlation coefficient was provided and the p-value calculated. P<0.05 was adopted as significant.
Supplementary Material
KEY RESOURCE TABLE
| Reagent | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Phospho-Akt (Ser473) | Cell Signaling Technology | Cat# 9271 |
| Phospho-Akt (Thr308) | Cell Signaling Technology | Cat# 13038 |
| Akt | Cell Signaling Technology | Cat# 4685 |
| Phospho-mTOR (Ser2481) | Cell Signaling Technology | Cat# 2974 |
| mTOR (7C10) | Cell Signaling Technology | Cat# 2983 |
| Phospho-AS160 (Thr642) | Cell Signaling Technology | Cat# 8881 |
| Beta-tubulin | Cell Signaling Technology | Cat# 2146 |
| Actin | Millipore | Cat# MAB1501R |
| 12-LO Antibody (C5) | Santa Cruz Biotechnology | Cat# sc-365194 |
| CD45-PE-Cy7 | eBiosciences | Cat# 25-0451-82 |
| F4/80-APC-Cy7 | BioLegend | Cat# 123118 |
| CD206-Alex647 | BioRad | Cat# MCA2235A647 |
| CD11c-PE | BD Pharmingen | Cat# 561044 |
| Chemicals, peptides and recombinant protein and recombinant proteins | ||
| 12(S)-HEPE | Cayman Chemicals | Cat# 32550 |
| Insulin (adipocytes differentiation) | Sigma Aldrich | Cat# I6634 |
| Insulin (human, for glucose uptake) | Sigma Aldrich | Cat# I9278 |
| Insulin (Humulin, for ITT)) | Elly Lilli | N/A |
| NCTT956 | Sigma Aldrich | Cat# SML0499 |
| Norepinephrine | Sigma Aldrich | Cat# A0937 |
| Rosiglitazone | Sigma Aldrich | Cat# R2408 |
| Indomethacin | Sigma Aldrich | Cat# I7378 |
| Dexamethasone | Sigma Aldrich | Cat# D4902 |
| IBMX | Sigma Aldrich | Cat# I5879 |
| Rotenone | Sigma Aldrich | Cat# R8875 |
| Antimycin A | Sigma Aldrich | Cat# A8674 |
| FCCP | Sigma Aldrich | Cat# C2920 |
| Oligomycin | Millipore | Cat# 495455 |
| ZnSO4 | Sigma Aldrich | Cat# Z2876 |
| Ba(OH)2 | Sigma Aldrich | Cat# B4059 |
| Perchloric acid | Sigma Aldrich | Cat# 244252 |
| Pertussis toxin | Sigma Aldrich | Cat# P7208 |
| Bacalein | Tocris | Cat# 1761 |
| Melittin | Tocris | Cat# 1193 |
| CL316,243 | Tocris | Cat# 1499 |
| YM254890 | FujiFilm Wako Chemicals | Cat# 257-00631 |
| LOXBlock-1 | Donated by Epigen Biosciences | N/A |
| [3H]2-deoxyglucose | Perkin Elmer | Cat# 760562 |
| Critical Commercial Assays | ||
| Insulin Kit | Cristal Chemicals | Cat# 90080 |
| Cell Line Nucleofector kit | Lonza | Cat# VVCA-1003 |
| Pierce BCA Protein Assay kit | Thermo Fisher | Cat# 23225 |
| RNA extraction kit | Zymo Research | Cat# R2052 |
| Sybr Green Master Mix | Thermo Fisher | Cat# A25778 |
| cDNA Reverse Transcription Kit | Thermo Fisher | Cat# 4368813 |
| Oligonucleotides | ||
| Primers for qPCR | See Table S1 – this paper | N/A |
| Primers for gRNAs | See Method Details – this paper | N/A |
| Gnas siRNA | Dharmacon | Cat# R040646-00-0005 |
| Control siRNA | Dharmacon | Cat# D-001810-10-05 |
| Recombinant DNA | ||
| Alox 12 Myc-DDK-tagged | Origene | Cat# MR227302 |
| 1179_pAAV-U6-BbsI-gRNA-CB-EmGFP | Addgene | Cat# 89060 |
| pSpCas9(BB)-2A-Puro (PX459) V2.0 | Addgene | Cat# 62988 |
| Experimental Models: Cell Lines | ||
| WT-1 cells | Developed in Tseng lab | Tseng et al., 2004 |
| hWA A41 | Developed in Tseng lab | Xue et al., 2015; Kriszt et al., 2017 |
| hBA A41 | Developed in Tseng lab | Xue et al., 2015; Kriszt et al., 2017 |
| C2C12 | ATCC | Cat# CRL-1772 |
| 3T3-F442A | Sigma | Cat# 00070654 |
| Experimental Models: Organisms/Strains | ||
| C57BL6/J | Jackson Laboratories | Cat# 000664 |
| Atgl FLOX | Jackson Laboratories | Cat# 024278 |
| Cas9 knockin mice | Jackson Laboratories | Cat# 024857 |
| Myf5+ CRE | Jackson Laboratories | Cat# 007893 |
| STOCK Bmpr1atm2.1Bhr/Mmnc | MMRRC | Cat# 030469-UNC |
| Ucp1-CRE | Jackson Laboratories | Cat# 024670 |
| Adiponectin-CRE | Jackson Laboratories | Cat# 028020 |
| Datasets | ||
| Global Lipidomics Datasets | www.metabolomicsworkbench.org | DataTrack ID 1749, 1750 and 1751 |
| Algorithms and Software | ||
| GraphPad Prizm | GraphPad Software, San Diego, CA | Versions 7 and 8 |
| ImageJ | NIH | https://fiii.sc/ |
Highlights.
Cold exposure increases 12-LOX activity in brown adipose tissue
12-LOX activity in BAT contributes to cold adaptation
12-HEPE is a cold-induced and BAT-secreted oxylipin
12-HEPE promotes glucose uptake in vivo and in vitro
Context and Significance.
Brown adipose tissue (BAT) maintains body temperature in response to cold by burning blood glucose and fatty-acids. Understanding how BAT clears blood glucose is of importance for developing new therapeutic approaches to combat diabetes. In this study, the authors find that cold stress activates the enzyme 12-lipoxygenase (12-LOX) in BAT, which produces lipids that help maintaining body temperature. Deletion of 12-LOX from mouse BAT inhibits production of 12-HEPE, a 12-LOX-derived lipid that can shuttle glucose into tissues and improve glucose tolerance in obese mice.
ACKNOWLEDGEMENTS
This work was supported in part by US National Institutes of Health (NIH) grants R01DK077097 and R01DK102898 (to Y.-H.T.), R01HL106173 and P01GM095467 (to M.S.), R01DK099511 (to L.J.G.), and P30DK036836 (to Joslin Diabetes Center’s Diabetes Research Center) from the National Institute of Diabetes and Digestive and Kidney Diseases, and by US Army Medical Research grant W81XWH-17-1-0428 (to Y.-H.T.). L.O.L. was supported by American Diabetes Association post-doctoral fellowship (1-16-PDF-063) and by the grant São Paulo Research Foundation (FAPESP) grant 2017/02684. M.D.L was supported by NIH grants T32DK007260, F32DK102320 and K01DK111714. K.v.L. was supported by the grant R21NS087165. A.B. was supported by a Deutsche Forschungsgemeinschaft Research Fellowship (BA 4925/1-1) and the Deutsches Zentrum für Herz-Kreislauf-Forschung. B.E.S. is supported by an NRSA from the NIH (HL136044). We thank A. Clermont, A. Dean, and K. Longval of the Joslin Diabetes Center Physiology core. We also would like to thank Dr. Fabio Tucci of Epigen Biosciences, San Diego, who synthesized LOXBlock-1. We apologize to colleagues whose work we could not cite due to space limitations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
E.Y.C., V. B., N.R.N. and M.A.K. are employees of BERG.
REFERENCES
- Ahmadian M, Abbott MJ, Tang T, Hudak CS, Kim Y, Bruss M, Hellerstein MK, Lee HY, Samuel VT, Shulman GI, et al. (2011). Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 13, 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert V, Svensson K, Shimobayashi M, Colombi M, Munoz S, Jimenez V, Handschin C, Bosch F and Hall MN (2016). mTORC2 sustains thermogenesis via Akt-induced glucose uptake and glycolysis in brown adipose tissue. EMBO Mol. Med. 8, 232–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barquissau V, Ghandour RA, Ailhaud G, Klingenspor M, Langin D, Amri EZ, and Pisani DF (2017). Control of adipogenesis by oxylipins, GPCRs and PPARs. Biochimie 136, 3–11. [DOI] [PubMed] [Google Scholar]
- Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, Kaul MG, Tromsdorf UI, Weller H, Waurisch C, et al. (2011). Brown ad- ipose tissue activity controls triglyceride clearance. Nat. Med 17, 200–205. [DOI] [PubMed] [Google Scholar]
- Bartelt A, Widenmaier SB, Schlein C, Johann K, Goncalves RLS, Eguchi K, Fischer AW, Parlakgül G, Snyder NA, Nguyen TB, et al. (2018). Brown adipose tissue thermogenic adaptation requires Nrf1-mediated proteasomal activity. Nat. Med 24, 292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskin AS, Linderman JD, Brychta RJ, McGehee S, Anflick-Chames E, Cero C, Johnson JW, O’Mara AE, Fletcher LA, Leitner BP, et al. (2018). Regulation of Human Adipose Tissue Activation, Gallbladder Size, and Bile Acid Metabolism by a β3-Adrenergic Receptor Agonist. Diabetes 67, 2113–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brash AR. (1999). Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J. Biol. Chem 274, 23679–23682. [DOI] [PubMed] [Google Scholar]
- Bonadonna RC, Del Prato S, Saccomani MP, Bonora E, Gulli G, Ferrannini E, Bier D, Cobelli C, DeFronzo RA (1993). Transmembrane glucose transport in skeletal muscle of patients with non-insulin-dependent diabetes. J. Clin. Invest 92, 486–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B, and Nedergaard J (2004). Brown adipose tissue: function and physiolgical significance. Physiol. Rev 84, 277–359. [DOI] [PubMed] [Google Scholar]
- Cao H, Gerhold K, Mayers JR, Wiest MM, Watkins SM, and Hotamisligil GS (2008). Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell 134, 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capdevila JH, Wang W, Falck JR (2015). Arachidonic acid monooxygenase: Genetic and biochemical approaches to physiological/pathophysiological relevance. Prostaglandins Other Lipid Mediat. 120, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Buyel JJ, Hanssen MJ, Siegel F, Pan R, Naumann J, Schell M, van der Lans A, Schlein C, Froehlich H, et al. (2016). Exosomal microRNA miR-92a concentration in serum reflects human brown fat activity. Nat. Commun 7, 11420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Kazak L, Jedrychowski MP, Lu GZ, Erickson BK, Szpyt J, Pierce KA, Laznik-Bogoslavski D, Vetrivelan R, Clish CB, et al. (2016). Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 532, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole BK, Lieb DC, Dobrian AD, and Nadler JL (2013). 12- and 15-lipoxygenases in adipose tissue inflammation. Prostaglandins Other Lipid Mediat. 104-105, 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng Y-H, Doria A, et al. (2009). Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med 360, 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cypess AM, Weiner LS, Roberts-Toler C, Franquet Elía E, Kessler SH, Kahn PA, English J, Chatman K, Trauger SA, Doria A, and Kolodny GM (2015). Activation of human brown adipose tissue by a β3-adrenergic receptor agonist. Cell Metab. 21, 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalli J, Colas RA, Walker ME, Serhan CN (2018). Lipid Mediator Metabolomics Via LC-MS/MS Profiling and Analysis. Methods Mol. Biol. 1730, 59–72. [DOI] [PubMed] [Google Scholar]
- Deng B, Wang CW, Arnardottir HH, Li Y, Cheng CY, Dalli J, Serhan CN Maresin biosynthesis and identification of maresin 2, a new anti-inflammatory and pro-resolving mediator from human macrophages. PLoS One 9, e102362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enerbäck S, Jacobsson A, Simpson EM, Guerra C, Yamashita H, Harper ME, Kozak LP (1997). Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature 387, 90–94. [DOI] [PubMed] [Google Scholar]
- Ferré P, Leturque A, Burnol AF, Penicaud L, Girard J (1985). A method to quantify glucose utilization in vivo in skeletal muscle and white adipose tissue of the anaesthetized rat. Biochem. J 228, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frontini A, and Cinti S (2010). Distribution and development of brown adipocytes in the murine and human adipose organ. Cell Metab. 11, 253–6. [DOI] [PubMed] [Google Scholar]
- GBD 2015 Obesity Collaborators, Afshin A, Forouzanfar MH, Reitsma MB, Sur P, Estep K, Lee A, Marczak L, Mokdad AH, Moradi-Lakeh M, et al. (2017). Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med 377, 13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder S, et al. (2006). Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 312, 134–737. [DOI] [PubMed] [Google Scholar]
- Hamberg M (1980). Transformations of 5,8,11,14,17-eicosapentaenoic acid in human platelets. Biochim. Biophys. Acta 618, 389–98. [DOI] [PubMed] [Google Scholar]
- Hanssen MJ, Hoeks J, Brans B, van der Lans AA, Schaart G, van den Driessche JJ, Jörgensen JA, Boekschoten MV, Hesselink MK, Havekes B, et al. (2015). Short-term cold acclimation improves insulin sensitivity in patients with type 2 diabetes mellitus. Nat. Med 21, 863–865. [DOI] [PubMed] [Google Scholar]
- Hao Q, Yadav R, Basse AL, Petersen S, Sonne SB, Rasmussen S, Zhu Q, Lu Z, Wang J, Audouze K, et al. (2015). Transcriptome profiling of brown adipose tissue during cold exposure reveals extensive regulation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab 308, E380–E392. [DOI] [PubMed] [Google Scholar]
- Joshi N, Hoobler EK, Perry S, Diaz G, Fox B, and Holman TR (2013). Kinetic and structural investigations into the allosteric and pH effect on the substrate specificity of human epithelial 15-lipoxygenase-2. Biochemistry 52, 8026–8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Spiegelman BM, and Seale P (2015). Brown and Beige Fat: Physiological Roles beyond Heat Generation. Cell Metab. 22, 546–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriszt R, Arai S, Itoh H, Lee MH, Goralczyk AG, Ang XM, Cypess AM, White AP, Shamsi F, Xue R, et al. (2017). Optical visualisation of thermogenesis in stimulated single-cell brown adipocytes. Sci. Rep 7, 1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn H, Banthiya S, and van Leyen K (2015). Mammalian lipoxygenases and their biological relevance. Biochim. Biophys. Acta 1851, 308–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Brown JD, Stanya KJ, Homan E, Leidl M, Inouye K, Bhargava P, Gangl MR, Dai L, Hatano B, et al. (2013). A diurnal serum lipid integrates hepatic lipogenesis and peripheral fatty acid use. Nature 502, 550–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynes MD, Leiria LO, Lundh M, Bartelt A, Shamsi F, Huang TL, Takahashi H, Hirshman MF, Schlein C, Lee A, et al. (2017). The cold-induced lipokine 12,13-diHOME promotes fatty acid transport into brown adipose tissue. Nat. Med 23, 631–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma K, Nunemaker CS, Wu R, Chakrabarti SK, Taylor-Fishwick DA, and Nadler JL (2010). 12-Lipoxygenase Products Reduce Insulin Secretion and beta-Cell Viability in Human Islets. J. Clin. Endocrinol. Metab 95, 881–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard J, Bengtsson T, and Cannon B (2007). Unexpected evidence for active brown adipose tissue in adult humans. Am. J. Physiol. Endocrinol. Metab 293, E444–E452. [DOI] [PubMed] [Google Scholar]
- Nunemaker CS, Chen M, Pei H, Kimble SD, Keller SR, Carter JD, Yang Z, Smith KM, Wu R, Bevard MH, et al. (2008). 12-Lipoxygenase-knockout mice are resistant to inflammatory effects of obesity induced by Western diet. Am. J. Physiol. Endocrinol. Metab 295, E1065–E1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, and Olefsky JM (2010). GPR120 is an omega-3 fatty acid receptor mediating potent antiinflammatory and insulin-sensitizing effects. Cell 142, 687–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno H, Shinoda K, Ohyama K, Sharp LZ, Kajimura S. (2013). EHMT1 controls brown adipose cell fate and thermogenesis through the PRDM16 complex. Nature 504, 163–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, et al. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto CA, Swinburn B, Hawkes C, Huang TT, Costa SA, Ashe M, Zwicker Cawley, J H, and Brownell KD. (2015). Patchy progress on obesity prevention: emerging examples, entrenched barriers, and new thinking. Lancet 385, 2400–2409. [DOI] [PubMed] [Google Scholar]
- Sahu BD, Mahesh Kumar J, Sistla R (2015). Baicalein, a bioflavonoid, prevents cisplatin-induced acute kidney injury by up-regulating antioxidant defenses and down-regulating the MAPKs and NF-κB pathways. PLoS One. 10, e0134139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Gurmaches J, Tang Y, Jespersen NZ, Wallace M, Martinez Calejman C, Gujja S, Li H, Edwards YJK, Wolfrum C, Metallo CM, et al. (2018). Brown fat AKT2 is a cold-induced kinase that stimulates ChREBP-mediated de novo lipogenesis to optimize fuel storage and thermogenesis. Cell Metab. 27, 195–209.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, and Sabatini DM (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- Schulz TJ, Huang P, Huang TL, Xue R, McDougall LE, Townsend KL, Cypess AM, Mishina Y, Gussoni E, and Tseng YH (2013). Brown-fat paucity due to impaired BMP signalling induces compensatory browning of white fat. Nature 495, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber R, Diwoky C, Schoiswohl G, Feiler U, Wongsiriroj N, Abdellatif M, Kolb D, Hoeks J, Kershaw EE, Sedej S, et al. (2017). Cold-induced thermogenesis depends on ATGL-mediated lipolysis in cardiac muscle, but not brown adipose tissue. Cell Metab. 26, 753–763.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scime A, Devarakonda S, Conroe HM, Erdjument-Bromage H et al. (2008). PRDM16 controls a brown fat/skeletal muscle switch. Nature 454, 961–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN (2014). Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014 510, 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Yang R, Martinod K, Kasuga K, Pillai PS, Porter TF, Oh SF, and Spite M. (2009). Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med 206, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Hao Z, Zhang S, Wei M, Lu B, Wang Z, Ji L (2018). Baicalein and baicalin alleviate acetaminophen-induced liver injury by activating Nrf2 antioxidative pathway: The involvement of ERK1/2 and PKC. Biochem. Pharmacol 150, 9–23. [DOI] [PubMed] [Google Scholar]
- Shin H, Ma Y, Chanturiya T, Cao Q, Wang Y, Kadegowda AKG, Jackson R, Rumore D, Xue B, Shi H, et al. (2017). Lipolysis in Brown Adipocytes Is Not Essential for Cold-Induced Thermogenesis in Mice. Cell Metab. 26, 764–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh GM, Danaei G, Farzadfar F, Stevens GA, Woodward M, Wormser D, Kaptoge S, Whitlock G, Qiao Q, Lewington S, et al. (2013). The age-specific quantitative effects of metabolic risk factors on cardiovascular diseases and diabetes: a pooled analysis. PLoS One 8, e65174–e65174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spite M, Clària J, and Serhan CN (2014). Resolvins, specialized proresolving lipid mediators, and their potential roles in metabolic diseases. Cell Metab. 19, 21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford KI, Lynes MD, Takahashi H, Baer LA, Arts PJ, May FJ, Lehnig AC, Middelbeek RJW, Richard JJ, So K, et al. (2018). 12,13-diHOME: An Exercise-Induced Lipokine that Increases Skeletal Muscle Fatty Acid Uptake. Cell Metab. 27, 1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford KI, Middelbeek RJW, Townsend KL, An D, Nygaard EB, Hitchcox KM, Markan KR, Nakano K, Hirshman MF, Tseng Y-H, et al. (2013). Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J. Clin. Invest 123, 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukdar S, Olefsky JM, and Osborn O (2011). Targeting GPR120 and other fatty acid-sensing GPCRs ameliorates insulin resistance and inflammatory diseases. Trends Pharmacol. Sci. 32, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tersey SA, Maier B, Nishiki Y, Maganti AV, Nadler JL, and Mirmira RG (2014). 12-lipoxygenase promotes obesity-induced oxidative stress in pancreatic islets. Mol. Cell. Biol 34, 3735–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend KL, and Tseng YH (2014). Brown fat fuel utilization and thermogenesis. Trends Endocrinol. Metab. 25, 168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng YH, Kriauciunas KM, Kokkotou E, Kahn CR (2004). Differential roles of insulin receptor substrates in brown adipocyte differentiation. Mol. Cell. Biol 24, 1918–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallerand AL, Pérusse F, and Bukowiecki LJ (1990). Stimulatory effects of cold exposure and cold acclimation on glucose uptake in rat peripheral tissues. Am. J. Physiol 259, R1043–R1049. [DOI] [PubMed] [Google Scholar]
- van der Lans AA, Hoeks J, Brans B, Vijgen GH, Visser MG, Vosselman MJ, Hansen J, Jorgensen JA, Wu J, Mottaghy FM et al. (2013). Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J. Clin. Invest 123, 3395–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JMAFL, Kemerink GJ, Bouvy ND, Schrauwen P, and Teule GJJ (2009). Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med 360, 1500–1508. [DOI] [PubMed] [Google Scholar]
- Vegiopoulos A, Müller-Decker K, Strzoda D, Schmitt I, Chichelnitskiy E, Ostertag A, Berriel-Diaz M, Rozman J, Hrabe de Angelis M, Nüsing RM, et al. (2010). Cyclooxygenase-2 controls energy homeostasis in mice by de novo recruitment of brown adipocytes. Science 328, 1158–1161. [DOI] [PubMed] [Google Scholar]
- Villarroya F, Cereijo R, Villarroya J, and Giralt M (2017). Brown adipose tissue as a secretory organ. Nat. Rev. Endocrinol 13, 26–35. [DOI] [PubMed] [Google Scholar]
- Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, Taittonen M, Laine J, Savisto N-J, Enerback S, et al. (2009). Functional brown adipose tissue in healthy adults. N. Engl. J. Med 360, 1518–1525. [DOI] [PubMed] [Google Scholar]
- Wang GX, Zhao XY, and Lin JD (2015). The brown fat secretome: metabolic functions beyond thermogenesis. Trends Endocrinol Metab. 26, 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GX, Zhao XY, Meng ZX, Kern M, Dietrich A, Chen Z, Cozacov Z, Zhou D, Okunade AL, Su X, et al. (2014). The brown fat-enriched secreted factor Nrg4 preserves metabolic homeostasis through attenuation of hepatic lipogenesis. Nat Med. 20, 1436–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GX, Zhao XY, Meng ZX, Kern M, Dietrich A, Chen Z, Cozacov Z, Zhou D, Wecksler AT, Jacquot C, et al. (2009). Mechanistic investigations of human reticulocyte 15- and platelet 12-lipoxygenases with arachidonic acid. Biochemistry 48, 6259–6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue R, Lynes MD, Dreyfuss JM, Shamsi F, Schulz TJ, Zhang H, Huang TL, Townsend KL, Li Y, Takahashi H, et al. (2015). Clonal analyses and gene profiling identify genetic biomarkers of the thermogenic potential of human brown and white preadipocytes. Nat. Med 21, 760–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yigitkanli K, Pekcec A, Karatas H, Pallast S, Mandeville E, Joshi N, Smirnova N, Gazaryan I, Ratan RR, Witztum JL, et al. (2013). Inhibition of 12/15-lipoxygenase as therapeutic strategy to treat stroke. Ann Neurol. 73, 129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneshiro T, Aita S, Matsushita M, Kayahara T, Kameya T, Kawai Y, Iwanaga T, and Saito M (2013). Recruited brown adipose tissue as an antiobesity agent in humans. J. Clin. Invest 123, 3404–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yore MM, Syed I, Moraes-Vieira PM, Zhang T, Herman MA, Homan EA, Patel RT, Lee J, Chen S, Peroni OD, et al. (2014). Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell 159, 318–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The lipid datasets generated during this study are available at Metabolomics Workbench (URL: www.metabolomicsworkbench.org; DataTrack ID 1749, 1750 and 1751).