

Abstract
Increased de novo synthesis of fatty acids is implicated in the pathogenesis of human prostate cancer, but a safe and effective clinical inhibitor of this metabolic pathway is still lacking. We have shown previously that leelamine (LLM) suppresses transcriptional activity of androgen receptor, which is known to regulate fatty acid synthesis. Therefore, the present study was designed to investigate the effect of LLM on fatty acid synthesis. Exposure of 22Rv1, LNCaP, and PC-3 prostate cancer cells, but not RWPE-1 normal prostate epithelial cell line, to LLM resulted in a decrease in intracellular levels of neutral lipids or total free fatty acids. LLM was superior to another fatty acid synthesis inhibitor (Cerulenin) for suppression of total free fatty acid levels. LLM treatment downregulated protein and/or mRNA expression of key fatty acid synthesis enzymes including ATP citrate lyase, acetyl-CoA carboxylase 1, fatty acid synthase, and sterol regulatory element-binding protein 1 (SREBP1) in each cell line. Consistent with these in vitro findings, we also observed a significant decrease in ATP citrate lyase and SREBP1 protein expression as well as number of neutral lipid droplets in vivo in 22Rv1 tumor sections of LLM-treated mice when compared to that of controls. LLM-mediated suppression of intracellular levels of total free fatty acids and neutral lipids was partly attenuated by overexpression of SREBP1. In conclusion, these results indicate that LLM is a novel inhibitor of SREBP1-regulated fatty acid/lipid synthesis in prostate cancer cells that is not affected by androgen receptor status.
Keywords: Leelamine, Fatty Acids, Prostate Cancer
Introduction
Prostate cancer continues to be a major health concern for American men reflected by more than 29,000 deaths from this disease every year (1). Metabolic adaptation is now considered one of the hallmarks of different cancers (2). The de novo synthesis of fatty acids, which is very low in normal prostate tissue, is elevated in cancerous prostate (3, 4). The fatty acid synthesis starts with ATP citrate lyase (ACLY)-mediated conversion of citrate to acetyl-CoA that is then metabolized to malonyl-CoA in a reaction catalyzed by the rate limiting enzyme acetyl-CoA carboxylase 1 (ACC1) (3, 4). Fatty acid synthase (FASN) complex facilitates the terminal steps in the fatty acid synthetic pathway, and is responsible for generation of saturated fatty acids including 16-carbon palmitic acid, 14-carbon myristic acid, and 18-carbon stearic acid (3, 4). The palmitic acid is the predominant de novo synthesized saturated fatty acid. The saturated fatty acids are then modified by desaturases and elongases to insert double bonds and increase carbon chain length, respectively, to produce complex fatty acids (3, 4).
The fatty acids and the metabolic intermediates of this pathway are utilized by rapidly dividing cancer cells for energy production (mitochondrial β-oxidation of fatty acids), cell signaling (e.g., palmitoylation of oncogenes) or incorporation into complex lipids for manintenance of the membrane integrity (3–6). Studies have shown that β-oxidation of fatty acids is the dominant bioenergetic pathway in prostate cancer (5). A role for increased fatty acid synthesis in the pathogenesis of human prostate cancer is substantiated by the studies showing: (a) overexpression of mRNA and/or protein levels of key fatty acid synthesis enzyme(s) in early (prostatic intraepithelial neoplasia) and/or advanced stages (adenocarcinoma and metastatic disease) of the disease when compared to normal prostate (7, 8); (b) emergence of prostatic intraepithelial neoplasia lesions in FASN transgenic mice (9); and (c) a decrease in prostate cancer cell proliferation upon silencing of the ACC1 and FASN genes by their RNA interference (3, 4, 10). Furthermore, levels of saturated as well as monounsaturated fatty acids in the blood were suggested to be the risk factors for prostate cancer (11). In a very recent case-control study (2291 cases and 2661 controls), circulating levels of fatty acids and other complex lipids were strongly associated with prostate cancer (12).
Androgen receptor (AR), which is essential for normal male reproductive function and activated after binding with androgens (e.g., dihydrotestosterone), plays an important role in prostate cancer progression (13, 14). Studies have implicated AR signaling in regulation of fatty acid synthesis in prostate cancer. For example, exposure of LNCaP cells to a synthetic androgen (R1881) resulted in increased lipid synthesis as well as induction of ACLY, ACC1, and FASN protein expression (15). Moreover, AR was shown to be a regulator of the expression of sterol regulatory element binding protein 1 (SREBP1), which is a master regulator of fatty acid synthesis (16, 17). Androgens stimulated lipogenic gene expression in prostate cancer cells by activation of the SREBP1 (18). Interestingly, published studies have also demonstrated overexpression of SREBP1 protein in prostate cancer tissue arrays in comparison with normal/benign prostate tissue (19). Only 20% of the normal/benign prostate cells exhibited SREBP1 protein positivity that increased to 71% in high-grade prostate cancers (19). Another study showed overexpression of SREBP1 mRNA in prostate cancer when compared to the matched normal prostate (20). Therefore, suppression of the AR-SREBP1 signaling axis represents a promising therapeutic strategy for inhibition of fatty acid synthesis in prostate cancer.
We have shown previously that leelamine (LLM), a small molecule phytochemical present in the bark of pine tree, inhibits expression and transcriptional activity of AR in cultured human prostate cancer cells (LNCaP, C4–2B, and 22Rv1) in vitro and in 22Rv1 xenograft in vivo (21). These effects were observed at plasma achievable doses of LLM based on pharmacokinetic studies in mice (22). The present study was designed to investigate the effect of LLM on fatty acid/lipid synthesis using a castration-resistant (22Rv1) and an androgen-responsive (LNCaP) prostate cancer cells. An AR-independent cell line (PC-3) was included to determine the possibility of AR-independent effect of LLM on fatty acid synthesis, whereas an immortalized normal human prostate epithelial cell line (RWPE-1) was used to test for cancer cell selectivity. The 22Rv1 xenograft tissues/sections from control and LLM-treated mice (21) were used to determine in vivo relevance of the cellular results.
Materials and Methods
Ethics statement
Xenograft tissues from control and LLM-treated mice from our previously published study (21), which was approved by the University of Pittsburgh Animal Care and Use Committee, were used for western blotting, immunohistochemistry, and staining for neutral lipid droplets. Mice were treated intraperitoneally with either 100 µL vehicle (control group; n=6) or 9.1 mg LLM/kg body weight (LLM group, n=6) 5 times/week (21). A solution consisting of 10% ethanol, 10% dimethyl sulfoxide (DMSO), 30% Kolliphor EL (Sigma-Aldrich, St. Louis, MO), and 50% phosphate-buffered saline (PBS) was the vehicle for LLM (21). A portion of tumor tissues and vital organs from control and LLM-treated mice were fixed in 10% neutral buffered formalin for haematoxylin and eosin staining or immunohistochemistry.
Reagents
LLM (purity ≥ 98%) was purchased from the Cayman Chemical Company. Fetal bovine serum, antibiotic mixture, and other cell culture reagents were purchased from Life Technologies-ThermoFisher Scientific. The RPMI 1640 media was purchased from Mediatech. Anti-ACC1 (cat. #4190) and anti-FASN (cat. #3180) antibodies were purchased from Cell Signaling Technology. The anti-ACLY (cat. #ab40793) and anti-SREBP1 (cat. #ab28481) antibodies were purchased from Abcam. Kits for determination of total free fatty acids (cat. #MAK044) and anti-ACC1 antibody for immunohistochemistry (cat. #SAB4501396) were purchased from Sigma-Aldrich.
Cell lines
The LNCaP, 22Rv1, PC-3, and RWPE-1 cell lines were purchased from the American Type Culture Collection, and maintained as recommended by the supplier. The LNCaP, 22Rv1, and PC-3 cells were more recently authenticated by us in 2017 by short tandem repeat profiling by IDEXX BioResearch (Columbia, MO). Each cell line was found to be of human origin and devoid of inter-species contamination. The RWPE-1 cell line was not authenticated by us. The PC-3 and 22Rv1 cells were stably transfected with empty pcDNA3.1 vector (hereafter abbreviated as EV) or SREBP1 plasmid (abbreviated as SREBP1). Stably transfected cells were selected by culture in the presence of 800 µg/mL for PC-3 and 600 µg/mL for 22Rv1 of G418.
Staining for neutral lipids by BODIPY staining
The 22Rv1 (3×104), LNCaP (1.5×104), PC-3 (2×104), and RWPE-1 (2×104), cells were plated on coverslips in 24-well plates. After overnight incubation, cells were exposed to ethanol (vehicle for LLM), LLM or Etomoxir {ethyl 2-[6-(4-chlorophenoxy)hexyl]oxirane-2-carboxylate} for 24 hours. The cells were then fixed with 2% paraformaldehyde for 1 hour, permeabilized with 0.5% Triton X-100 for 5 minutes, and treated with 0.5 µg/mL BODIPY for 30 minutes at room temperature. The cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) for 5 minutes at room temperature, mounted, and examined under a Nikon confocal microscope. Number of lipid droplets/cell was quantified using ImageJ software.
Oil Red O staining
22Rv1 (3×104), LNCaP (2×104) or PC-3 (2×104) cells were plated in 24-well plates. Following overnight incubation, the cells were treated with ethanol or LLM (2.5 and 5 µmol/L) for 24 hours, and then fixed with 10% formalin at room temperature for 30 minutes. The cells were washed with 60% isopropanol followed by staining with Oil Red O solution for 30 minutes. The cells were washed with water and images were acquired under a Leica microscope equipped with a digital camera. After imaging, Oil red O was eluted using 100% isopropanol and the absorbance was measured at 492 nm using 100% isopropanol as a blank.
Quantitation of total free fatty acids level
A colorimetric assay kit was used to measure intracellular levels of total free fatty acids (C8 and longer) by following the manufacturer’s protocol. 22Rv1 (5×105) or LNCaP (3.5×105) cells were plated in 6-cm culture dishes, allowed to attach by overnight incubation, and then treated with ethanol (control) or the indicated doses of LLM, Cerulenin {(2R,3S)-3-[(4E,7E)-nona-4,7-dienoyl]oxirane-2-carboxamide} or Etomoxir for 24 hours. The cells were sonicated in 200 µL of 1% Triton X-100 in chloroform. Homogenate was centrifuge at 14,000 rpm for 10 minutes, and organic phase was collected and dried. Dried lipids were dissolved in fatty acid buffer and used for the colorimetric assay.
Cell proliferation assay
LNCaP cells (750 cells/well) or 22Rv1 cells (1,000 cells/well) were seeded in 96-well plates and allowed to attach. The attached cells were treated with ethanol (control) or the indicated doses of LLM, Etomoxir or Cerulenin for 24, 48 or 72 hours. Subsequently, 20 µL of manufacturer supplied colour development reagent was added to each well and the plates were incubated at 37°C for 2 hours. Absorbance was measured at 492 nm.
Western blotting
LNCaP (3.5×105) or 22Rv1 (5×105) cells were plated in 6-cm dishes, allowed to attach, and then treated with ethanol (control) or desired concentrations of LLM for 12 or 24 hours. Other details of western blotting using cell lysates and tissue supernatants have been described by us previously (21, 23, 24).
Real-time reverse transcription polymerase chain reaction (RT-PCR)
The effect of LLM treatment on mRNA levels of ACLY, ACC1, and FASN was determined by RT-PCR. Briefly, total RNA from control and LLM-treated cells was isolated using RNeasy kit. Aliquot containing 2 µg RNA was used for cDNA synthesis. The primers for the RT-PCR included: ACLY- forward: 5’- GAAGCTGACCTTGCTGAACC-3’; reverse: 5’- TGCCTCCAATGATGAGGAT-3’; ACC1- forward: 5’-ACCACCAATGCCAAAGTAGC-3’; reverse: 5’-CTGCAGGTTCTCAATGCAAA-3’; FASN- forward: 5’-CTGGCTCAGCACCTCTATCC-3’; reverse: 5’-CAGGTTGTCCCTGTGATCCT-3’; and glyceraldehyde 3-phosphate dehydrogenase (GAPDH)- forward: 5’-GGACCTGACCTGCCGTCTAGAA-3’; reverse: 5’-GGTGTCGCTGTTGAAG TCAGAG-3’. The PCR conditions were as follows: 95°C for 10 minutes followed by 40 cycles at 95°C for 15 seconds, 60°C for 1 minute, and 72°C for 30 seconds. Relative gene expression levels were calculated using the method of Livak and Schmittgen (25).
Immunohistochemistry
The 22Rv1 xenograft sections of 4–5 μm thickness from control (n=6) and LLM-treated mice (n=5) were de-paraffinized, hydrated, washed with PBS, and exposed to antigen retrieval buffer solution (pH 6.0) for 20 minutes followed by treatment with 0.3% hydrogen peroxide in methanol for 20 minutes at room temperature. Sections were then treated with blocking buffer for 1 hour followed by incubation with the desired primary antibody, including anti-ACLY (1:100 dilution), anti-ACC1 (1:50 dilution), anti-FASN (1:200 dilution), and anti-SREBP1 (1:100 dilution) overnight in humidified chambers at 4°C. Sections were washed with PBS and incubated with horseradish peroxidase-conjugated secondary antibody (1:200 dilution) for 1 hour at room temperature. Colour was developed by incubation with 3,3’-diaminobenzidine tetrahydrochloride. The sections were counterstained with haematoxylin. Stained sections were examined under a Leica microscope. At least five non-overlapping representative images were captured from each section and analysed using Aperio ImageScope v9.1 software. The H-score was calculated as described by us previously (24).
Visualization and quantitation of neutral lipid droplets in 22Rv1 xenograft sections
Neutral lipid droplets in 22Rv1 tumor sections were stained by BODIPY. The 22Rv1 xenograft sections (4–5 µm) were de-paraffinized, hydrated, treated at room temperature with 25 μg/mL BODIPY for 1 hour, washed with PBS, and mounted in medium containing DAPI. The sections were examined under a Nikon confocal microscope. Number of neutral lipid droplets/cell was determined using ImageJ software from at least 50 cells/high-power field from five non-overlapping areas of each section.
Immunofluorescence microscopy
22Rv1 (3×104), LNCaP (2×104), and PC-3 (3×104) cells were plated on coverslips in 24-well plates. After overnight incubation, the cells were treated with ethanol (control) or LLM (2.5 and 5 µmol/L) for 24 hours. The cells were then fixed and permeabilized with 2% paraformaldehyde and 0.5% Triton X-100, respectively. After blocking with PBS containing 0.5% bovine serum albumin and 0.15% glycine, the cells were treated with anti-SREBP1 antibody (1:500 dilution) overnight at 4°C. The cells were then treated with Alexa Fluor 488-conjugated rabbit secondary antibody (1:1000 dilution) for 1 hour at room temperature in the dark. The cells were counterstained with DAPI (nuclear stain) for 5 minutes at room temperature in the dark prior to microscopic examination. The cells were examined under a Nikon confocal microscope. Corrected total cell fluorescence (CTCF) was determined using ImageJ software and these values were normalized by cell number.
Analysis of RNA-seq expression profile in The Cancer Genome Atlas (TCGA)
Correlation of SREBP1 (SREBF1) or AR expression with that of ACLY, ACC1 (ACACA) or FASN in prostate tumors (n=497) and normal prostate tissues (n=52) in TCGA dataset was determined using University of California Santa Cruz Xena Browser (http://xena.ucsc.edu/public-hubs/). The correlation coefficient and statistical significance was determined by Pearson’s test.
Statistical analysis
Statistical tests were performed using GraphPad Prism software (version 7.02). One-way analysis of variance (ANOVA) followed by Dunnett’s test or Bonferroni’s test was used for dose-response and multiple comparisons, respectively. Student’s t-test was used for two sample comparisons.
Results
LLM treatment decreased neutral lipid accumulation in prostate cancer cells
Neutral lipid droplets in 22Rv1 cells following treatment with LLM or Etomoxir can be visualized in Fig. 1A. Data from similar experiments using LNCaP, PC-3, and RWPE-1 cells can be found in the Supplementary Fig. S1. Etomoxir was included in this assay as a positive control. Etomoxir is an irreversible inhibitor of carnitine palmitoyltransferase-1 and prevents the formation of acyl carnitines, which is a critical step in the transport of fatty acyl chains from the cytosol into the mitochondrial intermembrane space. Etomoxir is known to cause accumulation of lipid droplets in cancer cells (26). The average number of lipid droplets/cell was decreased significantly in a dose-dependent manner upon 24-hour treatment of 22Rv1, LNCaP, and PC-3 cells with 2.5 and 5 µmol/L of LLM when compared to corresponding solvent controls (Fig. 1B). On the other hand, LLM treatment failed to decrease number of neutral lipid droplets/cell in RWPE-1 cells. The basal staining for neutral lipid droplets/cell was markedly higher in prostate cancer cells than in RWPE-1 cells (Fig. 1B). These results indicated that AR was not involved in suppression of neutral lipid droplets by LLM treatment.
Figure 1.
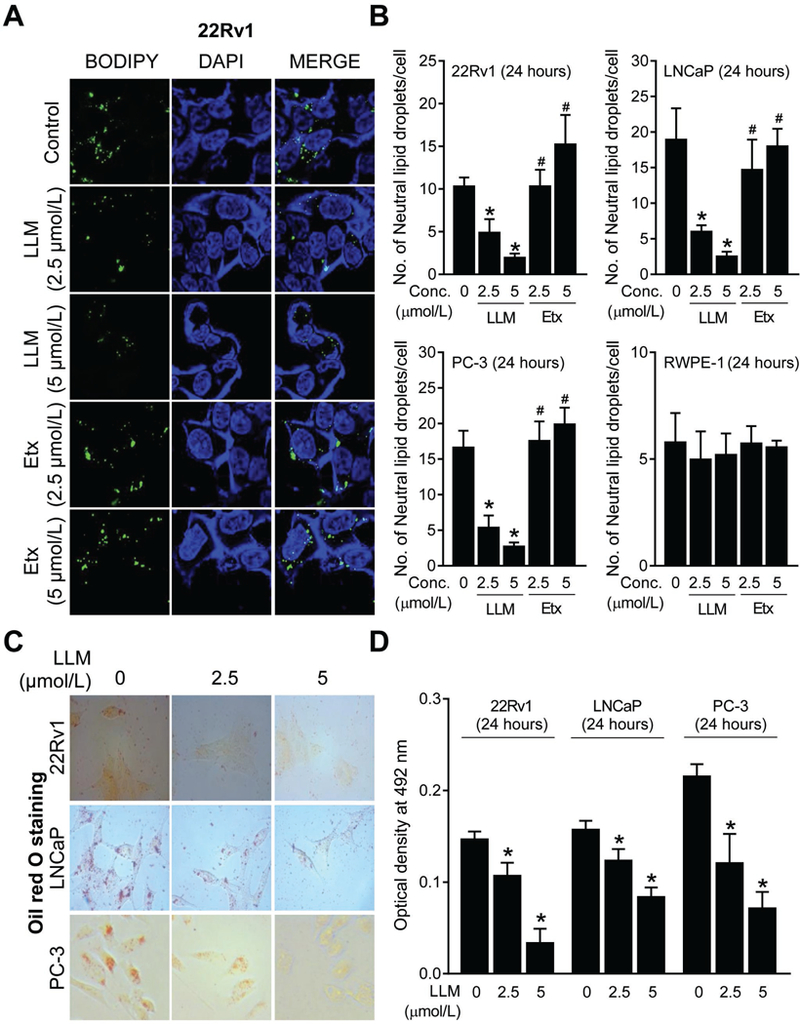
Leelamine (LLM) treatment decreased number of neutral lipid droplets in prostate cancer cells. A, Representative confocal microscopy images for BODIPY staining depicting neutral lipid droplets in 22Rv1 cells after 24-hour treatment with ethanol (control) or 2.5 and 5 µmol/L of LLM or Etomoxir (Etx; positive control). B, Quantitation of the number of neutral lipid droplets/cell in control and LLM-treated 22Rv1, LNCaP, PC-3, and RWPE-1 cells (24-hour treatment). Results shown are mean ± SD (n=3). Significantly different (P<0.05) *compared with ethanol-treated control, and #between LLM and Etx group by one-way ANOVA followed by Bonferroni’s test. C, Oil red O staining in 22Rv1, LNCaP, and PC-3 cells after 24 hours of treatment with ethanol (control) or the indicated doses of LLM. D, Quantitation of Oil Red O colour intensity through measurement of absorbance at 492 nm. Results are shown as mean ± SD (n=3). *Significantly different (P<0.05) compared with corresponding ethanol-treated control cells by one-way ANOVA followed by Dunnett’s test. Consistent results were observed in independent replicate experiments.
Oil red O is another probe for visualization of neutral lipid droplets (27). As shown in Fig. 1C, the Oil Red O-positive neutral lipid droplets were lower in the LLM-treated 22Rv1, LNCaP, and PC-3 cells when compared with the corresponding solvent-treated control cells. Measurement of the absorbance for Oil Red O confirmed statistically significant decrease in the levels of neutral lipids in LLM-treated cells when compared to the corresponding solvent-treated control cells (Fig. 1D). These results indicated a decrease in lipid accumulation upon exposure of prostate cancer cells to LLM and this effect was not observed in RWPE-1 cells.
LLM was a superior inhibitor of total free fatty acid levels than Cerulenin
Next, we compared the effect of LLM treatment on intracellular levels of total free fatty acids with Cerulenin, which is an inhibitor of FASN (28). The levels of total free fatty acids were dose-dependently decreased upon 24-hour treatment of 22Rv1 cells with LLM when compared to the control (Fig. 2A). Similar results were observed with LLM in the LNCaP cell line (Fig. 2A). At equimolar concentrations, Cerulenin or Etomoxir was unable to decrease the levels of total free fatty acids in 22Rv1 and LNCaP cells (Fig. 2A). LLM was also superior to both Cerulenin and Etomoxir for inhibition of 22Rv1 and LNCaP cell proliferation (Fig. 2B). These results indicated that LLM was superior to Cerulenin for inhibition of total free fatty acid levels.
Figure 2.
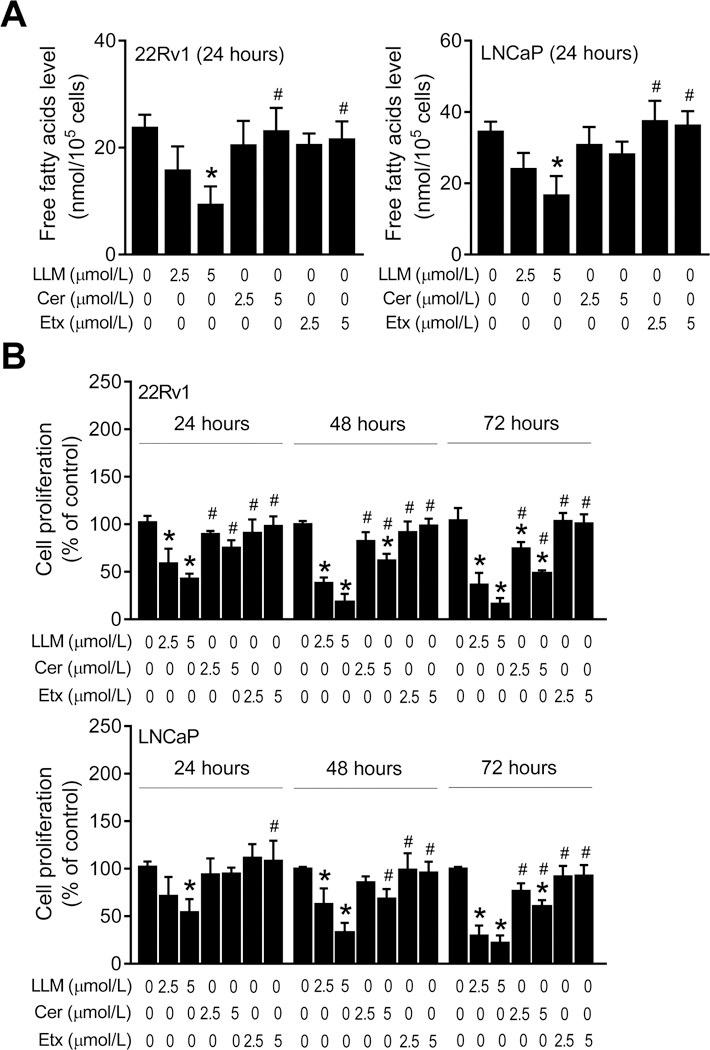
Leelamine (LLM) was superior to Cerulenin for inhibition of intracellular levels of total free fatty acids and cell proliferation. A, Effects of LLM, Cerulenin (Cer) or Etomoxir (Etx) treatments (24-hour treatment) on intracellular levels of total free fatty acids in 22Rv1 and LNCaP cells. B, Effects of LLM, Cer or Etx treatments (24-, 48- or 72-hour treatment) on proliferation of 22Rv1 and LNCaP cells. Each experiment was repeated twice in triplicate, and representative data from one such experiment is shown as mean ± SD (n=3). Significantly different (P<0.05) *compared with ethanol-treated control, and #between LLM and Cer or Etx groups by one-way ANOVA followed by Bonferroni’s test.
Effect of LLM treatment on expression of fatty acid synthesis enzymes
Because LLM treatment decreased intracellular levels of total free fatty acids (Fig. 2A), we proceeded to determine its effect on expression of ACLY, ACC1, and FASN proteins by western blotting (Fig. 3A). The level of each protein was lower in LLM-treated 22Rv1 and LNCaP cells at both 12-hour and 24-hour time points when compared to corresponding vehicle-treated control cells (Fig. 3A). LLM treatment also resulted in a statistically significant decrease in ACLY, ACC1, and FASN mRNA in both cell lines (Fig. 3B). These results indicated transcriptional suppression of ACLY, ACC1, and FASN expression upon LLM treatment in both 22Rv1 and LNCaP cells.
Figure 3.
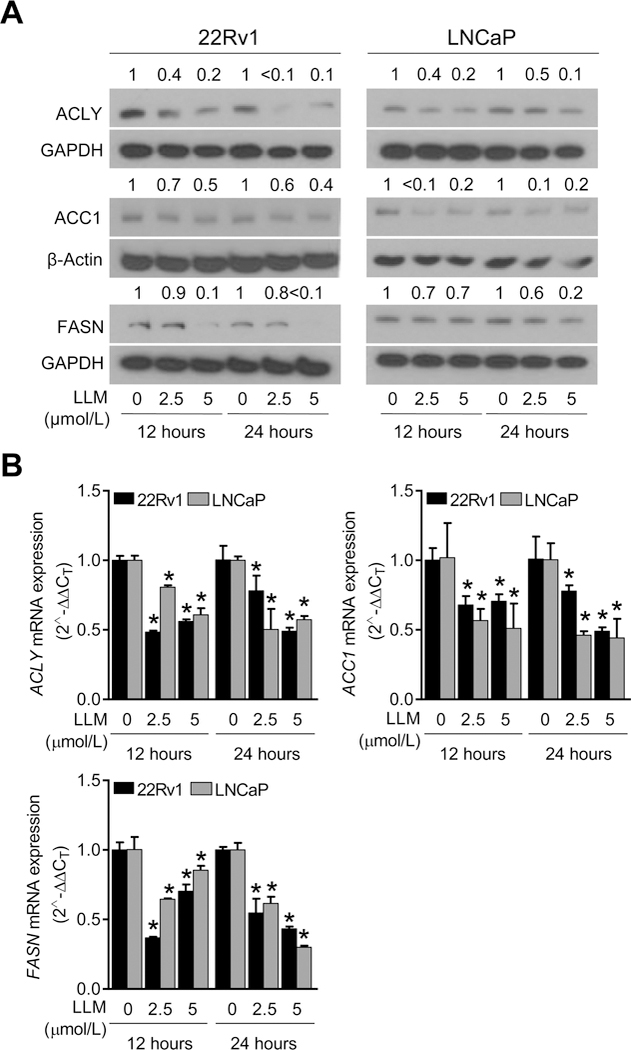
Leelamine (LLM) treatment downregulated expression of fatty acid metabolism proteins in prostate cancer cells. A, Immunoblotting for ATP citrate lyase (ACLY), acetyl-CoA carboxylase 1 (ACC1), and fatty acid synthase (FASN) proteins using lysates from 22Rv1 and LNCaP cells after 12-hour or 24-hour treatment with ethanol or the indicated concentrations of LLM. The blots were probed with anti-β-Actin or anti-GAPDH antibody to correct for differences in protein loading. The numbers above bands represent quantitation of the protein level changes relative to corresponding ethanol-treated control. B, RT-PCR data showing effect of LLM treatment (12-hour or 24-hour treatment) on mRNA levels of ACLY, ACC1, and FASN. Results shown are mean ± SD (n=3). *Significantly different (P<0.05) compared with corresponding ethanol-treated control by one-way ANOVA followed by Dunnett’s test. Similar results were observed in replicate experiments.
In vivo effect of LLM administration on expression of ACLY, ACC1, and FASN proteins
Fig. 4A shows the results of western blotting for ACLY, ACC1, and FASN protein expression using 22Rv1 xenograft supernatants from control and LLM-treated mice. Densitometric quantitation of the ACLY immunoreactive band revealed a 66% decrease in its protein level in tumors of LLM-treated mice in comparison with control (Fig. 4B). The expression levels of ACC1 and FASN proteins were also lower in 22Rv1 xenografts from LLM-treated mice (n=5) when compared to those of controls (n=6), but the difference was insignificant due to large variability and a small sample size (Fig. 4B). The results of western blotting were confirmed by immunohistochemistry. Fig. 4C shows immunohistochemical staining for ACLY, ACC1, and FASN proteins in a representative 22Rv1 xenograft section of the control group and the LLM treatment group. Consistent with the results of western blotting, immunohistochemistry revealed a significant decrease in expression of ACLY protein in 22Rv1 xenograft sections of LLM-treated mice when compared to control (Fig. 4D). Neutral lipid droplets in a representative xenograft section of the control mouse and that of a LLM-treated mouse can be seen in Fig. 4E. In comparison with control, the average number of lipid droplets/cell was lower by about 44% in 22Rv1 tumor sections of LLM-treated group (Fig. 4F). These results provided in vivo evidence for LLM-mediated downregulation of ACLY protein and neutral lipid accumulation in 22Rv1 xenografts.
Figure 4.
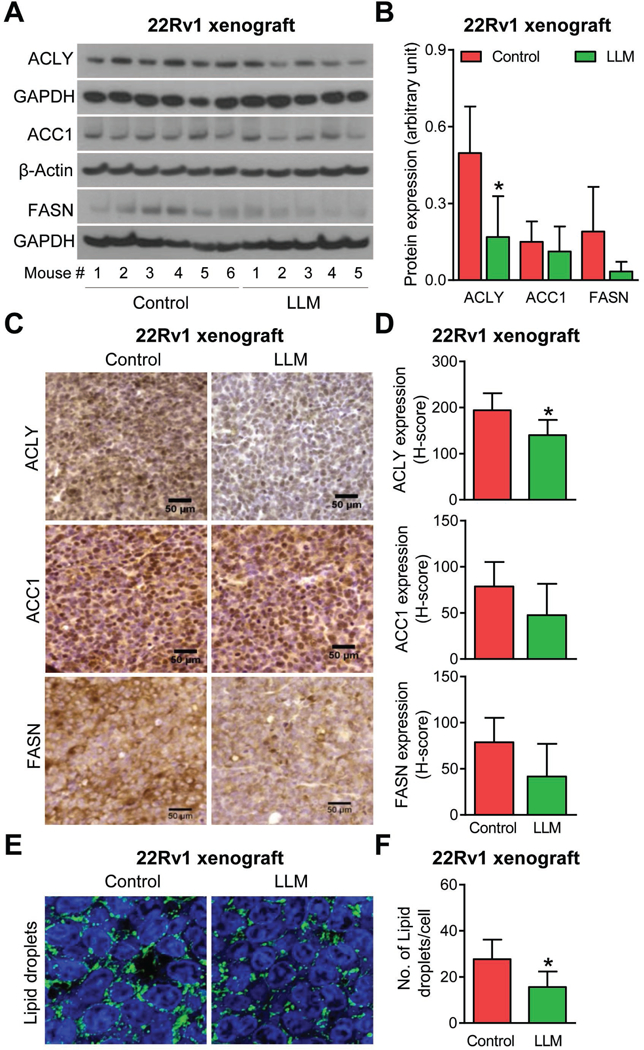
Leelamine (LLM) administration downregulated protein level of ATP citrate lyase (ACLY) and suppressed accumulation of neutral lipid droplets in 22Rv1 xenografts in vivo. A, Western blotting for ACLY, acetyl-CoA carboxylase 1 (ACC1), and fatty acid synthase (FASN) proteins using supernatants from 22Rv1 xenografts from control (n=6) and LLM-treated mice (n=5). B, Densitometric quantitation of the ACLY, ACC1 and FASN expression in 22Rv1 xenografts. The results shown are mean ± SD. *Statistically significant (P<0.05) compared with control by Student’s t-test. C, Immunohistochemistry for ACLY, ACC1, and FASN protein expression in a representative 22Rv1 xenograft section of the control mouse and that of LLM-treated mouse (×400 magnification, scale bar= 50 μm). D, Quantitation of ACLY, ACC1, and FASN expression (H-score). The results shown are mean ± SD. *Statistically significant (P<0.05) compared with control by Student’s t-test. E, Representative confocal microscopy images for BODIPY staining depicting neutral lipid droplets in a representative 22Rv1 xenograft section of a control mouse and that of a LLM-treated mouse. F, Quantitation of the number of lipid droplets/cell in the tumors of control- and LLM-treated mice. Results shown are mean ± SD (n=5–6). *Statistically significant (P<0.05) compared with control by Student’s t-test.
LLM treatment downregulated SREBP1 protein expression in vitro and in vivo
Next, we determined the effect of LLM treatment on levels of SREBP1 protein, which is known to regulate expression of several fatty acid synthesis enzyme proteins (17). Exposure of 22Rv1, LNCaP, and PC-3 cells to LLM resulted in a dose-dependent and statistically significant decrease in SREBP1 protein level as revealed by immunocytochemistry and quantitation of CTCF (Fig. 5A, B). Fig. 5C shows immunohistochemical staining for SREBP1 protein in a representative 22Rv1 xenograft of the control mouse and that of the LLM-treated mouse. The average H-score for SREBP1 protein expression was lower by about 52% in the 22Rv1 xenografts of the LLM treatment group when compared to the controls (Fig. 5D). Histology of vital organs (Supplementary Fig. S2A) or wet weight of vital organs (Supplementary Fig. S2B) was not affected by LLM treatment. These results indicated downregulation of SREBP1 protein by LLM treatment in vivo without any toxicity.
Figure 5.
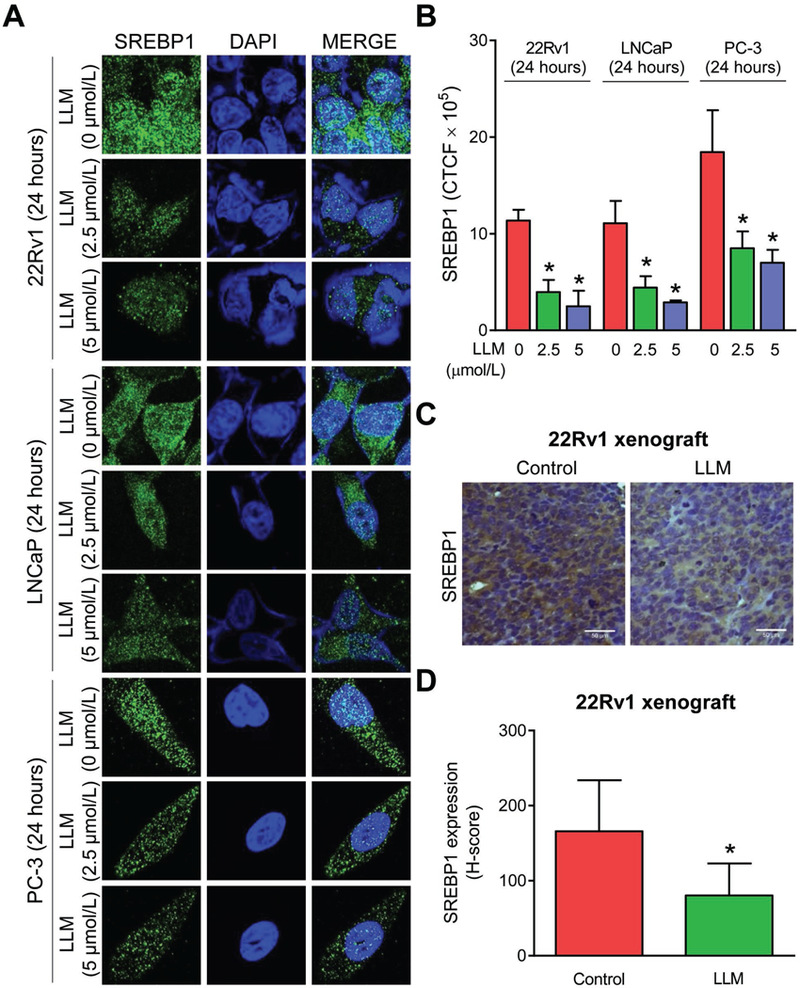
LLM treatment downregulated SREBP1 protein expression in prostate cancer cells in vitro and in 22Rv1 xenografts in vivo. A, Confocal microscopy images depicting expression of SREBP1 protein in 22Rv1, LNCaP, and PC-3 cells after 24-hour treatment with ethanol (control) or 2.5 or 5 µmol/L of LLM. B, Quantitation of corrected total cell fluorescence (CTCF) for SREBP1 protein expression using ImageJ software. Results shown are mean ± SD (n=3). *Significantly different (P<0.05) compared with corresponding ethanol-treated control by one-way ANOVA followed by Dunnett’s test. Comparable results were observed in replicate experiments. C, Immunohistochemistry for SREBP1 protein in a representative 22Rv1 xenograft section of control mouse and that of a LLM-treated mouse (×400 magnification, scale bar= 50 μm). D, Quantitation of SREBP1 expression (H-score). The results shown are mean ± SD (n=6 for control and n=5 for LLM treatment group). *Significantly different compared with control by Student’s t-test.
SREBP1 overexpression attenuated LLM-mediated inhibition of lipid accumulation
Immunoblotting confirmed overexpression of SREBP1 in stably transfected PC-3 and 22Rv1 cells in relation to empty vector transfected control cells (Fig. 6A). Overexpression of SREBP1 resulted in relatively higher level of lipid accumulation in PC-3 cells when compared to empty vector transfected PC-3 cells (Fig. 6B). Moreover, SREBP1 overexpression conferred partial protection against LLM-mediated inhibition of intracellular levels of neutral lipids in PC-3 cells (Fig. 6C) and total free fatty acids in both PC-3 and 22Rv1 cells (Fig. 6D). Collectively, these results indicated a role for SREBP1 in LLM-mediated inhibition of fatty acid synthesis as summarized by a mechanistic model in Fig. 6E.
Figure 6.
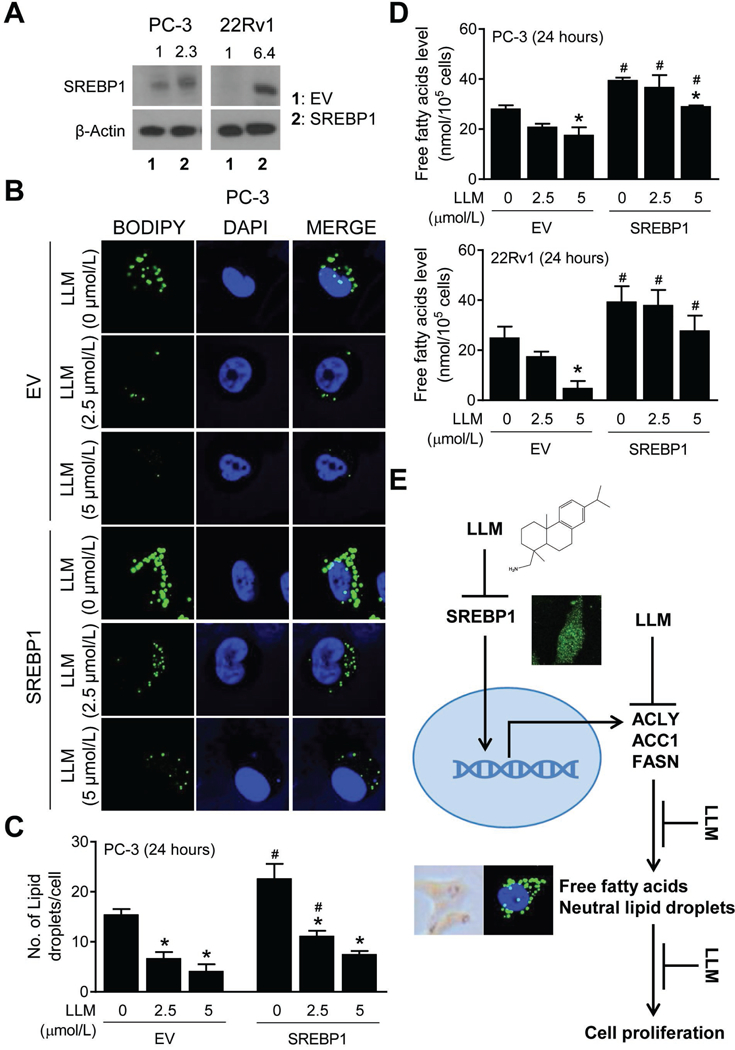
Sterol regulatory element binding protein 1 (SREBP1) overexpression partly attenuated leelamine (LLM)-mediated inhibition of accumulation of total free fatty acids and neutral lipids. A, Western blot for SREBP1 using lysates from empty vector transfected control cells (EV) and SREBP1 overexpressing cells (SREBP1). B, BODIPY staining in EV and SREBP1 (PC-3) cells after 24-hour treatment with ethanol or the indicated doses of LLM. C, Quantitation of number of neutral lipid droplets/cell. The results shown are mean ± SD (n=3). *Significantly different (P<0.05) compared with corresponding ethanol-treated control, and #between EV and SREBP1 cells by one-way ANOVA followed by Bonferroni’s test. D, Quantitation of intracellular levels of total free fatty acids in EV and SREBP1 cells (PC-3 and 22Rv1) after 24-hour treatment with ethanol or the indicated doses of LLM. Results shown are mean ± SD (n=2–3). Significantly different (P<0.05) *compared with corresponding ethanol-treated control, and #between EV and SREBP1 cells by one-way ANOVA followed by Bonferroni’s test. E, A mechanistic model showing LLM-mediated inhibition of fatty acid synthesis in prostate cancer cells.
Correlation between expression of SREBP1 (SREBF1) or AR with that of ACLY, ACC1 (ACACA), and FASN in prostate cancer and normal prostate
Analysis of the RNA-seq data in TCGA dataset revealed a statistically significant positive correlation between expression of SREBP1 with that of ACLY, ACACA, and FASN in prostate cancers (Supplementary Fig. S3). A similar positive correlation was observed for ACACA and FASN in normal prostate tissue (Supplementary Fig. S4). Like SREBP1, expression of AR positively correlated with that of ACLY, ACACA, and FASN in prostate cancers (Supplementary Fig. S3). The positive correlation with AR expression was only observed for ACACA in normal prostate. We suspect that the discrepancy in results between prostate tumor and normal tissues is likely due to smaller sample size (n=52) for the later cohort. These results indicated a role for SREBP1 axis in regulation of fatty acid synthesis enzymes in human prostate cancers.
Discussion
The present study demonstrates suppression of neutral lipids/fatty acids accumulation by LLM treatment in prostate cancer cells in vitro and in vivo. In cancer cells, the fatty acids are used not only for membrane biogenesis but also for energy production through mitochondrial β-oxidation as well as post-translation modification (e.g., palmitoylation) of oncogenic proteins (3–6). Fatty acids can also be stored in neutral lipids such as triglycerides and sterol esters in lipid droplets. Increased number of lipid droplets has been observed in cancer cells (29). Even though the role of lipid droplet accumulation in pathogenesis or progression of cancer is not fully understood, one study showed cholesteryl ester accumulation in clinical high-grade and metastatic human prostate cancer tissues, and its depletion in prostate cancer cells resulted in suppression of cell proliferation and migration in vitro and tumor xenograft growth in vivo (30). Interestingly, cholesteryl ester accumulation was not observed in normal prostate or benign prostatic hyperplasia tissues (30). LLM-mediated cell death in melanoma cells was associated with inhibition of intracellular cholesterol transport (31). Thus, it is reasonable to postulate that inhibition of accumulation of neutral lipids/fatty acids may play an important role in anti-cancer activity of LLM.
This study shows that LLM is a superior inhibitor of not only cellular proliferation but also intracellular levels of total free fatty acids, at least in LNCaP and 22Rv1 cells than Cerulenin. The mechanism underlying fatty acid level suppression by LLM is likely different from that of Cerulenin, which inhibits FASN activity by irreversible covalent modification of a cysteine sulfhydryl in β-keto-acyl-ACP synthase moiety of the complex (28, 32). Covalent modification of proteins is not expected for LLM because it is not an electrophile. In this context, however, we have reported previously that non-covalent interactions of LLM with certain amino acids residues (e.g., Y739) in the ligand-binding domain of AR is functionally important with regards to inhibition of the transcriptional activity of AR (21). It remains to be determined whether LLM non-covalently interacts with amino acids in the active sites of fatty acid synthesis enzyme proteins. Downregulation of key fatty acid synthesis enzyme proteins like ACLY is likely responsible for the decrease in total free fatty acids following treatment with this agent.
We found that the fatty acid synthesis inhibition by LLM in 22Rv1 and LNCaP cells is accompanied by downregulation of mRNA and protein levels of ACLY, ACC1, and FASN. However, the expression of only ACLY protein is decreased significantly in vivo in 22Rv1 tumor xenografts of LLM-treated mice when compared to those of controls as revealed by western blotting and confirmed by immunohistochemistry. These results indicate that ACLY is a critical target of fatty acid synthesis inhibition by LLM treatment. A few other studies have also examined the role of ACLY in prostate cancer cells. Stable knockdown of ACLY in PC3M prostate cancer cell line was accompanied by reduced growth rate, G0-G1 cell cycle arrest, and apoptosis induction when grown under reduced-lipid conditions (33). The effect of LLM on cell cycle progression is not known but its treatment induces apoptosis in prostate cancer cells (21). In another study, pharmacological inhibition of ACLY by an investigational drug resulted in sensitization of castration-resistant prostate cancer cells to enzalutamide, a clinically used AR antagonist (34). Inactivation of ACLY was shown to be responsible for growth inhibitory and proapoptotic effect of a dietary phytochemical cucurbitacin B (35). These observations raise the questions of whether LLM can sensitize castration-resistant prostate cancer cells like 22Rv1 to enzalutamide or whether apoptosis induction by LLM is partly dependent on its ability to downregulate ACLY expression. Further studies are needed to systematically explore these possibilities. Studies are also needed to test whether fatty acid synthesis inhibition by LLM is augmented by knockdown of ACLY, ACC1 and/or FASN.
The present study shows a statistically significant decrease in expression of SREBP1 protein level following LLM treatment not only in cultured prostate cancer cells but also in the 22Rv1 xenografts in vivo. SREBP1 is known to regulate expression of fatty acid synthesis proteins (17, 36). A cross-talk between AR and SREBP1 signaling axis has also been demonstrated recently in prostate cancer cells (16). AR along with mammalian target of rapamycin mTOR is recruited at the promoter region of SREBP1 and its expression is induced upon treatment with a synthetic androgen R1881 in LNCaP cells (16). Cleavage and nuclear localization of SREBP1 is increased by dual activation of the AR and mTOR pathways (16). Analysis of the RNA-seq data in TCGA reveals a significant positive correlation between expression of SREBP1 as well as AR with that of ACLY, ACACA, and FASN in prostate cancers (present study). In this study, we show partial abrogation of LLM-mediated inhibition of accumulation of total free fatty acids and neutral lipids by overexpression of SREBP1 that led to the conceptualization of mechanistic model summarized in Fig. 6E. Interestingly, LLM treatment causes a decrease in levels of lipids in PC-3 cell line, which lacks AR expression. These results indicate that AR is not involved in fatty acid synthesis inhibition by LLM.
In conclusion, the present study demonstrates that LLM treatment inhibits fatty acid synthesis in prostate cancer cells by suppressing SREBP1-ACLY expression. Thus, circulating levels of total free fatty acids/neutral lipids and/or tissue levels of ACLY or SREBP1 may be considered as biomarkers of LLM in future clinical investigations. The present article represents the second thorough study on mechanistic investigation of LLM in prostate cancers. Additional studies are necessary for clinical advancement of LLM for possible therapy of human prostate cancer. For example, evaluation of the anti-tumor effect of LLM and enzalutamide in a panel of castration-resistant prostate cancer cell lines and their xenografts could provide a novel combination regimen for the treatment of enzalutamide resistant tumors. In this context, it is important to highlight that a subset of patients is inherently resistant to enzalutamide due to expression of constitutively active splice variants of AR like AR-V7 (37).
Supplementary Material
Acknowledgment
This work was supported in part by the grant RO1 CA225716-01A1 and RO1 CA101753-14 awarded by the National Cancer Institute (S.V. Singh). This research used the Tissue and Research Pathology Facility supported in part by the Cancer Center Support Grant from the National Cancer Institute (P30 CA047904).
Footnotes
Conflict of interest: The authors do not have any conflict of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 3.Suburu J, Chen YQ. Lipids and prostate cancer. Prostaglandins Other Lipid Mediat 2012;98:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zadra G, Photopoulos C, Loda M. The fat side of prostate cancer. Biochim Biophys Acta 2013;1831:1518–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Y Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis 2006;9:230–4. [DOI] [PubMed] [Google Scholar]
- 6.Resh MD. Palmitoylation of proteins in cancer. Biochem Soc Trans 2017;45:409–16. [DOI] [PubMed] [Google Scholar]
- 7.Rossi S, Graner E, Febbo P, Weinstein L, Bhattacharya N, Onody T, et al. Fatty acid synthase expression defines distinct molecular signatures in prostate cancer. Mol Cancer Res 2003;1:707–15. [PubMed] [Google Scholar]
- 8.Wu X, Daniels G, Lee P, Monaco ME. Lipid metabolism in prostate cancer. Am J Clin Exp Urol 2014;2:111–20. [PMC free article] [PubMed] [Google Scholar]
- 9.Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, et al. Fatty acid synthase: a metabolic enzyme and candidate oncogene in prostate cancer. J Natl Cancer Inst 2009;101:519–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brusselmans K, De Schrijver E, Verhoeven G, Swinnen JV. RNA interference-mediated silencing of the acetyl-CoA-carboxylase-α gene induces growth inhibition and apoptosis of prostate cancer cells. Cancer Res 2005;65:6719–25. [DOI] [PubMed] [Google Scholar]
- 11.Chavarro JE, Kenfield SA, Stampfer MJ, Loda M, Campos H, Sesso HD, et al. Blood levels of saturated and monounsaturated fatty acids as markers of de novo lipogenesis and risk of prostate cancer. Am J Epidemiol 2013;178:1246–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adams C, Richmond RC, Santos Ferreira DL, Spiller W, Tan VY, Zheng J, et al. Circulating metabolic biomarkers of screen-detected prostate cancer in the ProtecT study. Cancer Epidemiol Biomarkers Prev 2018;28:208–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shand RL, Gelmann EP. Molecular biology of prostate-cancer pathogenesis. Curr Opin Urol 2006;16:123–31. [DOI] [PubMed] [Google Scholar]
- 14.Tan MHE, Li J, Xu HE, Melcher K, Yong EL. Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacol Sinica 2015;36:3–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moon JS, Jin WJ, Kwak JH, Kim HJ, Yun MJ, Kim JW, et al. Androgen stimulates glycolysis for de novo lipid synthesis by increasing the activities of hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2 in prostate cancer cells. Biochem J 2011;433:225–33. [DOI] [PubMed] [Google Scholar]
- 16.Audet-Walsh É, Vernier M, Yee T, Laflamme C, Li S, Chen Y, et al. SREBF1 activity is regulated by an AR/mTOR nuclear axis in prostate cancer. Mol Cancer Res 2018;16:1396–405. [DOI] [PubMed] [Google Scholar]
- 17.Shao W, Espenshade PJ. Expanding roles for SREBP in metabolism. Cell Metab 2012;16:414–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heemers H, Maes B, Foufelle F, Heyns W, Verhoeven G, Swinnen JV. Androgens stimulate lipogenic gene expression in prostate cancer cells by activation of the sterol regulatory element-binding protein cleavage activating protein/sterol regulatory element-binding protein pathway. Mol Endocrinol 2001;15:1817–28. [DOI] [PubMed] [Google Scholar]
- 19.Huang WC, Li X, Liu J, Lin J, Chung LW. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Mol Cancer Res 2012;10:133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Malley J, Kumar R, Kuzmin AN, Pliss A, Yadav N, Balachandar S, et al. Lipid quantification by Raman microspectroscopy as a potential biomarker in prostate cancer. Cancer Lett 2017;397:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh KB, Ji X, Singh SV. Therapeutic Potential of Leelamine, a Novel Inhibitor of Androgen Receptor and Castration-Resistant Prostate Cancer. Mol Cancer Ther 2018;17:2079–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song M, Lee D, Lee T, Lee S. Determination of leelamine in mouse plasma by LC-MS/MS and its pharmacokinetics. J Chromatogr B Analyt Technol Biomed Life Sci 2013;931:170–3. [DOI] [PubMed] [Google Scholar]
- 23.Xiao D, Srivastava SK, Lew KL, Zeng Y, Hershberger P, Johnson CS, et al. Allyl isothiocyanate, a constituent of cruciferous vegetables, inhibits proliferation of human prostate cancer cells by causing G2/M arrest and inducing apoptosis. Carcinogenesis 2003;24:891–7. [DOI] [PubMed] [Google Scholar]
- 24.Hahm ER, Lee J, Kim SH, Sehrawat A, Arlotti JA, Shiva SS, et al. Metabolic alterations in mammary cancer prevention by withaferin A in a clinically relevant mouse model. J Natl Cancer Inst 2013;105:1111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT Method. Methods 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 26.Zirath H, Frenzel A, Oliynyk G, Segerström L, Westermark UK, Larsson K, et al. MYC inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc Natl Acad Sci USA 2013;110:10258–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mehlem A, Hagberg CE, Muhl L, Eriksson U, Falkevall A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat Protoc 2013;8:1149–54. [DOI] [PubMed] [Google Scholar]
- 28.Funabashi H, Kawaguchi A, Tomoda H, Omura S, Okuda S, Iwasaki S. Binding site of cerulenin in fatty acid synthetase. J Biochem 1989;105:751–5. [DOI] [PubMed] [Google Scholar]
- 29.Bozza PT, Viola JP. Lipid droplets in inflammation and cancer. Prostaglandins Leukot Essent Fatty Acids 2010;82:243–50. [DOI] [PubMed] [Google Scholar]
- 30.Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab 2014;19:393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuzu OF, Gowda R, Sharma A, Robertson GP. Leelamine mediates cancer cell death through inhibition of intracellular cholesterol transport. Mol Cancer Ther 2014;13:1690–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Angeles TS, Hudkins RL. Recent advances in targeting the fatty acid biosynthetic pathway using fatty acid synthase inhibitors. Expert Opin Drug Discov 2016;11:1187–99. [DOI] [PubMed] [Google Scholar]
- 33.Zaidi N, Royaux I, Swinnen JV, Smans K. ATP citrate lyase knockdown induces growth arrest and apoptosis through different cell- and environment-dependent mechanisms. Mol Cancer Ther 2012;11:1925–35. [DOI] [PubMed] [Google Scholar]
- 34.Shah S, Carriveau WJ, Li J, Campbell SL, Kopinski PK, Lim HW, et al. Targeting ACLY sensitizes castration-resistant prostate cancer cells to AR antagonism by impinging on an ACLY-AMPK-AR feedback mechanism. Oncotarget 2016;7:43713–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao Y, Islam MS, Tian J, Lui VW, Xiao D. Inactivation of ATP citrate lyase by Cucurbitacin B: A bioactive compound from cucumber, inhibits prostate cancer growth. Cancer Lett 2014;349:15–25. [DOI] [PubMed] [Google Scholar]
- 36.Galbraith L, Leung HY, Ahmad I. Lipid pathway deregulation in advanced prostate cancer. Pharmacol Res 2018;131:177–84. [DOI] [PubMed] [Google Scholar]
- 37.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014;371:1028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.