

Abstract
Purpose
We evaluated clinical and genetic features enriched in patients with multiple Mendelian conditions to determine which patients are more likely to have multiple potentially relevant genetic findings (MPRF).
Methods
Results of the first 7698 patients who underwent exome sequencing at Ambry Genetics were reviewed. Clinical and genetic features were examined and degree of phenotypic overlap between the genetic diagnoses was evaluated.
Results
Among patients referred for exome sequencing, 2% had MPRF. MPRF were more common in patients from consanguineous families and patients with greater clinical complexity. The difference in average number of organ systems affected is small: 4.3 (multiple findings) vs. 3.9 (single finding) and may not be distinguished in clinic.
Conclusion
Patients with multiple genetic diagnoses had a slightly higher number of organ systems affected than patients with single genetic diagnoses, largely because the comorbid conditions affected overlapping organ systems. Exome testing may be beneficial for all cases with multiple organ systems affected. The identification of multiple relevant genetic findings in 2% of exome patients highlights the utility of a comprehensive molecular workup and updated interpretation of existing genomic data; a single definitive molecular diagnosis from analysis of a limited number of genes may not be the end of a diagnostic odyssey.
Keywords: comorbidity, distinct vs. overlapping phenotypes, multiple genetic diseases, multilocus genomic variation, diagnostic exome sequencing
INTRODUCTION
Traditionally in clinical genetics, identifying the correct diagnosis in a patient requires abstracting from specific phenotypes to recognize a pattern of a Mendelian condition. Additional clinical features that do not fit into the pattern of this genetic etiology might be a phenotype expansion, but they may also indicate an additional diagnosis.1,2 The clinical distinction between an expanded phenotype and comorbidity can be especially difficult when the phenotypes of the dual diagnoses interact to produce a more complex phenotype.
Since the completion of the Human Genome Project, rapid advances in technology such as diagnostic exome sequencing (DES) or genome sequencing have given medical professionals an extensive ability to interrogate their patients’ DNA. This ability to examine all genes in an unbiased manner can be very useful for providing the correct single or multiple diagnosis(es). If clinicians could identify which patients are more likely to have multiple Mendelian conditions, they might be able to prioritize DES testing over single-gene or panel testing for these patients. Thus we asked which clinical indications for testing are enriched in patients who receive multiple potentially relevant findings, and therefore would be more likely to benefit from DES. We also examined other characteristics such as inheritance patterns and asked if patients with more organ systems reported as affected were more likely to have multiple findings. We reviewed the results from the first seven years of DES at Ambry Genetics to clarify what types of patients tend to receive multiple diagnoses.
Previous studies have examined the influence of clinical complexity on diagnostic results: Trujillano et al. concluded that more clinically complex patients have a nonsignificant trend toward a higher rate of single positive findings.3 Karaca et al. found that many cases of presumed phenotypic expansion are actually patients with multiple Mendelian conditions.2 Posey et al. used Human Phenotype Ontology (HPO) terms to determine the degree of phenotypic overlap of individual patients’ dual diagnoses.4 To our knowledge, however, this study is the first to compare clinical complexity between patients with single and multiple potentially relevant findings. The data suggest that more clinically complex patients have a slightly higher rate of multiple diagnoses but that additional diagnoses often have overlapping clinical features.
MATERIALS AND METHODS
The indications for testing and results of the first 7698 consecutive patients who underwent DES at Ambry Genetics were reviewed. Some of these patients have been reported previously.5–8 Clinicians were encouraged to refer all first-degree relatives and other informative family members for testing. Solutions Institutional Review Board determined the study to be exempt from the Office for Human Research Protections Regulations for the Protection of Human Subjects (45 CFR 46) under category 4. Retrospective data analysis of anonymized data exempted the study from the requirement of receiving consent from patients. Patients’ clinical and testing histories, along with pedigrees, provided by referring physicians, were reviewed and summarized for each case by a team of genetic counselors at Ambry Genetics. The diagnostic alterations identified in the current study are/will be available in the ClinVar repository (https://www.ncbi.nlm.nih.gov/clinvar/).
In step 1, patients who had multiple potentially relevant findings (MPRF) were identified. For these patients, at least one relevant finding needs to be interpreted as a positive (Pos) or likely positive (LPos) result, i.e., pathogenic (P) or likely pathogenic (VLP) variants in a gene with good phenotypic match (biallelic P or VLP for recessive Mendelian conditions). Additional findings for step 1 had more relaxed criteria: the second finding could be either Pos, LPos, or uncertain, i.e., variants of uncertain significance (VUS); the gene could have an uncertain phenotypic match; or heterozygous P or VLP identified in a gene with highly specific clinical correlation and generally autosomal recessive inheritance. Variants were classified according to Ambry’s clinical variant classification scheme,9,10 which incorporates the American College of Medical Genetics and Genomics (ACMG) variant classification recommendations.11 Clinical indications for testing were examined in this group of 153 de-identified patients with MPRF. Organ system involvement was determined by clinician-submitted check boxes on the test requisition form; cases with zero selected were manually reviewed. In step 2, a subgroup of MPRF was identified: 33 patients had multiple genetic diagnoses (MGD), i.e., P/VLP findings in at least two genes with significant clinical correlation. The level of phenotypic overlap between the diagnoses was scored independently by five exome analysts. Finally, reclassifications were considered including proactive reclassification due to newly published gene–disease relationship information as described previously,8 reanalysis due to clinician request, and re-evaluation of variants. Variants are always fully re-evaluated if they are encountered in a new case or during case reanalysis, or systematically reassessed when there is better understanding of the mutational mechanism of a gene and/or release of new population databases. All reclassification reports received through the end of April 2018 were reviewed as part of this study. All discussed variants are considered primary findings related to clinical findings submitted with the exome sequencing order and are not considered secondary findings.
Statistical analysis
A chi-squared test was used for comparison between trios and nontrios of overall diagnostic rate, rates of MPRF and single genetic diagnosis (SGD), rates of uncertain findings, and rates of de novo findings. Chi-squared test was also used to compare rates of MPRF versus SGD by mode of inheritance (autosomal dominant [AD], autosomal recessive [AR], X-linked, mitochondrial), sex (male, female), consanguinity (declared, denied), and mode of inheritance within each sex. One-way analysis of variance (ANOVA) was used to compare average number of affected organ systems as in Fig. 3a, p < 0.0001, and Tukey post hoc is the p value reported in the results.
Fig. 3.
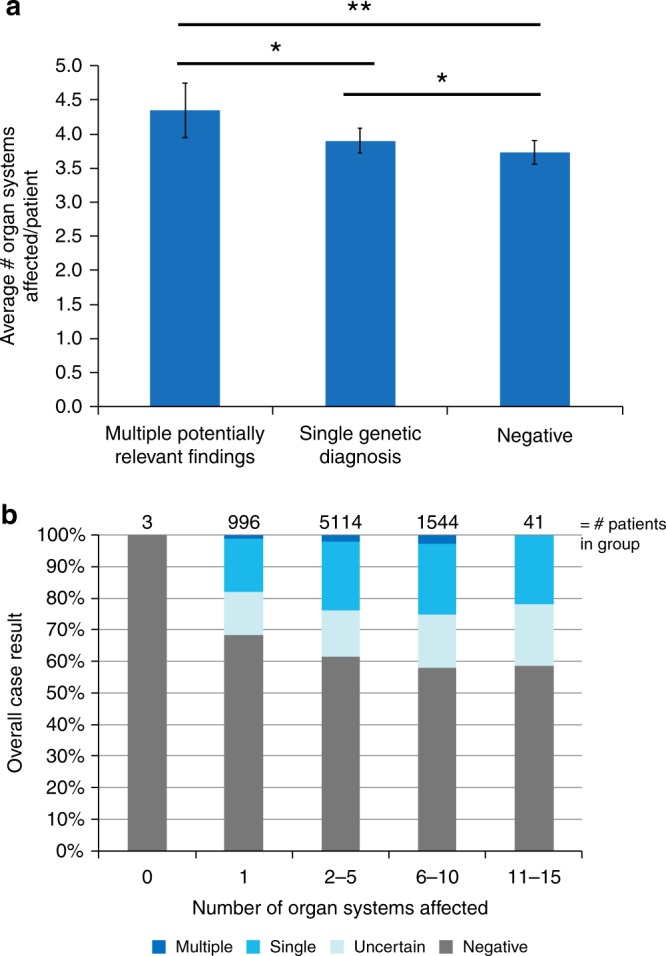
Relationship of clinical complexity to number of genetic diagnoses. (a) Patients with more organ systems affected received more genetic diagnoses. Shown is mean +/- s.e.m. (b) Histogram shows the diagnostic rate for patients with the number of organ systems indicated. Cases with 6–10 organ systems affected had the highest rate of single findings (22.5%) and of multiple potentially relevant findings (2.7%).
RESULTS
Among 7698 patients who underwent DES at Ambry Genetics, 1792 patients (23%, Fig. 1 and Supp Table 1) received at least one definitive genetic diagnosis, i.e., at least one variant that was either VLP or P in a gene with significant clinical correlation with a characterized Mendelian condition. A separate group of patients had an uncertain overall result: 15% (1160 patients) had a finding of uncertain significance in either a characterized or a novel candidate gene. Lastly, 62% (4746 patients) had a negative result. Within the cohort of patients who received at least one positive genetic diagnosis, 1639 patients had a SGD and 153 patients (8.5% of patients with positive reports) had MPRF. These patients with MPRF comprise the data set for step 1 and make up 2.0% of the total cohort of patients tested. Multiple potentially relevant findings were reported in two genes for 142 patients and three genes for 11 patients. In step 2, at least 2 definitive genetic diagnoses were identified in 33 patients within the group of MPRF, meaning that about 1.8% of patients with positive results actually had MGD.
Fig. 1.

Diagnostic rates of first 7698 cases submitted for diagnostic exome sequencing. Cases that simultaneously had two potentially relevant findings were included in the multiple potentially relevant findings (MPRF) group.
Step 1: clinical and genetic features of patients with multiple potentially relevant findings
Trios
Parent–proband trios are well established to have a higher diagnostic rate in DES than nontrios, a difference generally explained by a higher rate of uncertain findings among nontrios.6,12–15 In this study, 25% (1382/5471) of trios received a genetic diagnosis compared with 18% of nontrios (410/2227, p < 0.00001). Of all diagnosed trios, 7.9% had MPRF while 92.1% had SGD. Diagnosed patients from nontrios had similar rates: 10.7% MPRF and 89.3% SGD (Fig. 2a, p = 0.086). We evaluated the proportion of MRPF that were uncertain findings in trios versus nontrios since a benefit of trio sequencing is the ability to rule out VUS that were inherited from unaffected parents.16 The rate of uncertain findings among trios with multiple findings (6.0%, 83/1382) was similar to that of nontrios with multiple findings (9.0%, 37/410, p = 0.28). Inherited findings were more common in cases with MPRF than with SGD. Of all AD findings reported in trios SGD had 11% inherited findings while MPRF had 34% inherited. This could be attributed to inclusion of uncertain findings in MPRF, as inherited findings are more likely to be VUS. However, inherited findings were also more common in cases with MGD than with SGD, which is a comparison of only likely positive and positive findings between groups (MGD had 32% inherited variants). This difference also remains when accounting for the reported affected status of parents: in the subset of trios with two unaffected parents (848 trios) there was also a significantly higher proportion of inherited AD findings in patients with MPRF than in SGD (p < 0.0001). These data show that second findings are more often inherited, even when both parents are reportedly unaffected.
Fig. 2.

Clinical features of cases that had multiple relevant findings. (a) The proportion of solved cases that had multiple potentially relevant findings was not significantly different between trios and nontrios. Both trios and nontrios had about 2% rate of multiple positive or likely positive results. Nontrios had a trend toward more uncertain second results, but this difference was not significant. (b) The origin of autosomal dominant (AD) findings in trios. Cases with multiple potentially relevant findings had a higher proportion of inherited variants than did cases with single findings. (c) Inheritance patterns of genetic diagnoses. (d) Males had a slightly higher proportion of multiple relevant findings than single findings, compared with females. (e) Cases from consanguineous families had significantly more multiple potentially relevant findings (MPRF) than those from nonconsanguineous families. AR autosomal recessive, LP likely pathogenic, MGD multiple genetic diagnoses, P pathogenic, XL X-linked.
Mode of inheritance
The distribution of inheritance patterns for patients with SGD was very similar to the distribution for patients with MPRF (Fig. 2c) and to all patients (data not shown). There was no significant difference in distribution of mode of inheritance of variants between patients that had SGD versus MPRF (Fig. 2c, p = 0.063).
Sex
The patient’s sex can affect diagnostic rate as X-hemizygosity in males can reveal X-linked molecular diagnoses and some neurodevelopmental Mendelian conditions are more penetrant or more severe in males.17–19 In this cohort, 56% (4347) of patients were male. The percentage of solved cases who received MPRF was slightly but significantly different between males and females (Fig. 2d). Of patients with SGD, 55% were male while of patients with MPRF, 63% were male (p = 0.177). There was no significant difference in mode of inheritance between the sexes (p = 0.21).
Consanguinity
Fig. 2e shows the percent of reported findings in patients with MPRF. A family history of consanguinity was reported for 296 patients. Of all diagnosed patients from consanguineous families, 22% received MPRF. In contrast, only 8% of diagnosed patients from nonconsanguineous families had MPRF (p < 0.00001). For patients from consanguineous families, homozygous findings in genes associated with recessive Mendelian conditions were more common. They comprised 41% of all results for consanguineous families but only 23% of all results for nonconsanguineous families.
Organ systems
Patients with MPRF, similar to all patients undergoing testing, predominantly had childhood onset indications for testing, with neurologic, musculoskeletal, and craniofacial organ systems most commonly affected in order of decreasing frequency. Gastrointestinal and ophthalmologic clinical features were, respectively, fourth and fifth most common in the whole group but ended up as fifth and fourth most common in the cohort of patients with MPRF. Patients who received MPRF had complex clinical presentations involving an average of 4.3 organ systems per patient (n = 153). This was significantly different from the average of 3.9 organ systems per patient with a SGD (n = 1639, Fig. 3a mean +/- s.e.m., MPRF vs. SGD p < 0.05), and from the average 3.7 organ systems per patient with a negative result (n = 4746, MPRF vs. negative p < 0.01). Patients with a SGD also had more organ systems affected on average than patients with a negative result (SGD vs. negative p < 0.05). Visualized another way, MPRF were identified in 1.1% of patients with 1 reported organ system affected, 2.0% of patients with 2–5 organ systems affected, and 2.7% of patients with 6–10 organ systems affected (Fig. 3b).
Step 2: phenotypic overlap between multiple diagnoses
Organ system involvement
In step 2, the amount of phenotypic overlap between the diagnoses for 33 patients with MGD was evaluated (Table 1). For one patient (3%), the two relevant findings could explain the same symptom in the patient, icthyosis vulgaris. In 42% (14 patients), each diagnosis could be responsible for some unique clinical features, but there was sufficient symptom overlap that the molecular diagnoses appear intertwined. For 55% (18 patients), the diagnoses explained completely separate clinical features, suggesting the presence of two different Mendelian conditions in the same patient (see Supp Table 2 for information regarding the MPRF cohort).
Table 1.
Phenotypic overlap in patients with MGD
| Gene | Zygosity | Alteration(s) pathogenicity | Genetic diagnosis | Indication for testing | |
|---|---|---|---|---|---|
| Completely overlapping | |||||
| 1 | FLG | AD | P | Ichthyosis vulgaris, MIM 146700 | Allergy/Immune/Infections; Derm; Pulmonary |
| SLC27A4 | AR | P/P | Ichthyosis prematurity syndrome, MIM 608649 | ||
| Partially overlapping | |||||
| 4 | DNA2 | AD | P | Progressive external ophthalmoplegia, MIM 615156 | Neuro |
| POLG | AR | P/P | Mitochondrial recessive ataxia syndrome, MIM 607459 | ||
| 5 | HNRNPU | AD | P (dn) | Early infantile epileptic encephalopathy, MIM 617391 | Audiologic; Cardio; Craniofacial; Endocrine; MusSkel; Neuro |
| SMC1A | XLR | VLP | Cornelia de Lange syndrome, MIM 300590 | ||
| 6 | FOXG1 | AD | P (dn) | Rett syndrome, MIM 613454 | GI; Neuro; Pulmonary |
| KAT6B | AD | P (dn) | SBBYSS syndrome, MIM 603736 | ||
| 7a | ETFDH | AR | P (homoz) | Glutaric acidemia, MIM 231680 | MusSkel |
| HEXA | AR | VLP (homoz) | Tay–Sachs disease, MIM 272800 | ||
| 8 | CYP21A2 | AR | P/P | Congenital adrenal hyperplasia, MIM 201910 | Endocrine; Neuro |
| PTEN | AD | VLP | Macrocephaly/autism syndrome, MIM 605309 | ||
| SCN8A | AD | P (dn) | Intellectual developmental disorder, MIM 614306 | ||
| 13 | PTEN | AD | VLP (dn) | Macrocephaly/autism syndrome, MIM 605309 | GI; GU; MusSkel; Neuro |
| ANKRD11 | AD | P | KBG syndrome, MIM 148050 | ||
| 14 | SATB2 | AD | P (dn) | Glass syndrome, MIM 612323 | Cardio; Craniofacial; Neuro; Ophthalmologic |
| CAMTA1 | AD | VLP | Cerebellar ataxia with intellectual disability, MIM 614756 | ||
| 15 | PDE6A | AR | P (homoz) | Retinitis pigmentosa, MIM 613810 | MusSkel; Neuro; Ophthalmalogic |
| SH3TC2 | AR | P (homoz) | Charcot–Marie–Tooth, MIM 601596 | ||
| SLC26A2 | AR | P (homoz) | Multiple epiphyseal dysplasia, MIM 226900 | ||
| 17 | SLC6A1 | AD | P | Myoclonic–atonic epilepsy, MIM 616421 | Neuro; Ophthalmalogic |
| TCF20 | AD | P | Neurodevelopmental abnormalities MIM 603107 | ||
| 21 | PTEN | AD | VLP (dn) | Macrocephaly/autism syndrome, MIM 605309 | Cardio; Craniofacial; MusSkel; Neuro; Pulmonary |
| SON | AD | P (dn) | ZTTK syndrome, MIM 617140 | ||
| 24 | CLTC | AD | P (dn) | Intellectual developmental disorder 56, MIM 617854 | Craniofacial; Hematologic; Metabolic |
| DNMT3A | AD | P (dn) | Tatton–Brown–Rahman syndrome, MIM 615879 | ||
| 26 | NLGN3 | XLR | P | NLGN3 neurodevelopmental disorder, gene MIM 300336 | Craniofacial; Derm; Neuro |
| CAMK2B | AD | VLP (dn) | Intellectual developmental disorder 54, MIM 617799 | ||
| 27 | DNMT3A | AD | P (dn) | Tatton–Brown–Rahman syndrome, MIM 615879 | Craniofacial; Derm; Endocrine; MusSkel; Neuro; Oncologic; Ophthalmalogic |
| ATP1A2 | AD | VLP (dn) | Familial hemiplegic migraine 2, MIM 602481 | ||
| 32 | ARSA | AR | P/P | Metachromatic leukodystrophy, MIM 250100 | Cardio; Craniofacial; GI; Neuro |
| KMT2D | AD | P (dn) | Kabuki syndrome 1, MIM 147920 | ||
| Completely separate | |||||
| 2 | PKD1 | AD | P | Polycystic kidney disease, MIM 173900 | Allergy/Immune/Infections; Derm; GI; Neuro; Ophthalmologic; Renal |
| ANK2 | AD | P (dn) | ANK2 -related autism (no MIM number) | ||
| 3 | ANO3 | AD | VLP (dn) | Dystonia, MIM 615034 | GI; Neuro; Ophthalmologic |
| NALCN | AR | P/P | Hypotonia, psychomotor retardation, MIM 615419 | ||
| 9 | WDR45 | XLD | P (dn) | Neurodegeneration with brain iron accumulation, MIM 300894 | Cardio; Craniofacial; Neuro; Ophthalmologic |
| NOTCH1 | AD | P | Aortic valve disease MIM 109730 | ||
| 10 | ANKRD11 | AD | P (dn) | KBG syndrome, MIM 148050 | Craniofacial; GI; MusSkel; Neuro |
| GAA | AR | P/P | Glycogen storage disease II, MIM 232300 | ||
| 11 | CRB1 | AR | P/P | Retinitis pigmentosa-12, MIM 600105 | Dental; Ophthalmologic |
| WDR72 | AR | P (homoz) | Amelogenesis imperfecta, type IIA3, MIM 613211 | ||
| 12a | DDX11 | AR | P (homoz) | Warsaw breakage syndrome, MIM 613398 | Audiologic; Craniofacial; Hematologic; Derm; MusSkel; Neuro |
| ITK | AR | P (homoz) | Lymphoproliferative syndrome 1, MIM 613011 | ||
| 16 | LRP5 | AR | P/P | Exudative vitreoretinopathy, MIM 601813 | Cardio; GI; Metabolic; MusSkel; Neuro; Renal |
| MT-ND5 | Mito | P | Leigh syndrome, MIM 256000 | ||
| 18 | ATRX | XLR | P | ɑ-thalassemia/intellectual disability syndrome, MIM 301040 | Craniofacial; GI; GU; Neuro, Ophthalmalogic |
| G6PD | XLR | P | Hemolytic anemia, G6PD deficient (favism), MIM 300908 | ||
| 19 | INSL3 | AD | VLP | Cryptorchidism, MIM 219050 | Cardio; Craniofacial; GI; GU; MusSkel; Neuro; Ophthalmalogic |
| ZSWIM6 | AD | P | Frontonasal dysostosis, MIM 603671 | ||
| 20 | ADNP | AD | VLP (dn) | Helsmoortel–van der Aa syndrome, MIM 615873 | Neuro; Renal |
| PKD1 | AD | P | Polycystic kidney disease, MIM 173900 | ||
| 22a | CYP21A2 | AR | P (homoz) | Congenital adrenal hyperplasia, MIM 201910 |
Childhood onset: GU; Endocrine; MusSkel Adult onset: Derm; Endocrine; GI; MusSkel; Neuro; Oncologic; Ophthalmalogic |
| SPAST | AD | VLP | Spastic paraplegia, MIM 182601 | ||
| 23 | PPM1D | AD | P (dn) | Intellectual developmental disorder, MIM 617450 | Audiologic; Craniofacial; Dental; Derm; GI; Metabolic; MusSkel; Neuro |
| WNT10A | AD | P | Tooth agenesis, selective, MIM 150400 | ||
| 25a | GNB5 | AR | P (homoz) | Neurodevelopmental disorder, cardiac arrhythmia, MIM 617173 | Cardio; Pulmonary |
| LRRC6 | AR | P (homoz) | Primary ciliary dyskinesia 19, MIM 614935 | ||
| 28 | PPM1D | AD | VLP (dn) | Intellectual developmental disorder, MIM 617450 | Audiologic; Cardio; Craniofacial; Endocrine; GI; GU; MusSkel; Neuro; Ophthalmalogic; Renal |
| ELN | AD | P | Supravalvar aortic stenosis, MIM 185500 | ||
| 29 | HBB | AR | P/P | Sickle cell anemia, MIM 603903 | Cardio; Craniofacial; Hematologic; MusSkel; Neuro; Ophthalmalogic |
| FOXP1 | AD | P | Intellectual developmental disorder, MIM 613670 | ||
| 30a | CFTR | AR | P/P | Cystic fibrosis, MIM 219700 | Cardio; MusSkel; Neuro; Ophthalmalogic; Pulmonary |
| CNTNAP2 | AR | P (homoz) | CNTNAP2 neurodevelopmental disorder, MIM 610042 | ||
| 31 | CHD7 | AD | P | CHARGE syndrome, MIM 214800 | Derm; MusSkel; Neuro; Ophthalmalogic |
| NF1 | AD | P | Neurofibromatosis type 1, MIM 162200 | ||
| 33a | TYR | AR | P (homoz) | Oculocutaneous albinism type IA, MIM 203100 | Cardio; Craniofacial; Derm; GI; Neuro; Ophthalmalogic |
| VPS13B | AR | P (homoz) | Cohen syndrome, MIM 216550 | ||
For the group of patients with multiple genetic diagnoses (MGD), the phenotypic overlap, or degree that multiple diagnoses could explain the same symptoms in a proband, was scored by exome analysts. Genetic diagnoses that affect only one organ system in that patient at the time of diagnosis are in bold. All indications for testing are childhood onset unless otherwise noted.
AD autosomal dominant, AR autosomal recessive, Derm dermatologic, dn de novo, GI gastrointestinal, GU genitourinary, homoz homozygous, MusSkel musculoskeletal, P pathogenic variant, VLP variant likely pathogenic.
aFamilies who reported consanguinity closer than second cousins.
Copy-number variants
Copy-number variants (CNVs) were not clinically validated for the exome analysis pipeline used here. Therefore, the data presented in this study specifically reflect the detection rate of DES for single-nucleotide substitutions and small indels. To estimate the number of patients with multiple diagnoses including CNVs, testing results were added from orthogonal methods such as karyotype (n = 88) or microarray (n = 1066), either performed at Ambry or recorded in the clinic notes. A total of 44 patients had CNVs that were considered pathogenic or likely pathogenic in addition to a Pos/LPos finding from DES (Supplemental Table 1). In total, 195 patients (2.5% of the whole cohort) had multiple potentially relevant findings including CNVs. This calculation is likely to underestimate the true percentage of patients with dual genetic diagnoses because not all patients included in the study had karyotype or microarray and many patients who are found to have a clinically relevant CNV do not proceed to DES.
Reclassifications
Each molecular diagnosis is based on current knowledge of the patient’s clinical features and the known condition characteristics at the time of reporting. Therefore updated clinical information and new literature that affects variant or gene characterization can lead to result reclassification. All reclassification reports issued to MPRF patients were reviewed, including some with multiple simultaneous changes to results, e.g., one finding was added and another was removed in the same report. More than half (15/24) of patients who received reclassification reports had a new genetic diagnosis: 9 due to discovery of a new genetic etiologies (ZBTB18, DDX3X, GNAO1, BPTF, NACC1, SIN3A, KMT2B, GNB5, CAMK2B), 5 due to new clinical information (ATP7B, HADHA, PAX9*, CLCN1*, BASP1; *denotes same patient), 1 due to finding an allelic de novo variant in the same novel gene in a subsequent internal patient (NR2F1), and 1 due to discovery that the presence of significant numbers of heterozygotes in healthy controls in gnomAD was actually of somatic origin in DNMT3A (discussed in Carlston et al.)20 Five were upgrades from an uncertain to a positive overall result: four due to characterization of a candidate gene (MTOR, ETV6, HNRNPK, TRAK1), and one due to RNA studies establishing pathogenicity of an alteration (ADNP). One quarter of the reclassification reports (6/24) downgraded an uncertain potential relevant finding to negative, removing an initial result: in one patient the clinical validity of the gene SRI was refuted. Additionally, for about 3.8% (6/159) of patients who initially had MPRF, it was possible to rule out at least one initial finding with updated clinical information (OPHN1, MIB1), the release of bigger control population databases with better representation of rare ethnic groups (TTN and ASPM), and family segregation data (ALG13).
Case example
Detection of MPRF in a proband may have implications for the rest of the family because these Mendelian conditions are expected to segregate independently. For example, a deceased 3-month-old girl was reported to have global developmental delay, dysmorphic features, hypotonia with slight flexion contractures of the elbows, bilateral complete retinal detachment consistent with persistent fetal vasculature, coronary artery fistula, and cardiomyopathy, which was considered the cause of death. A head ultrasound revealed a dysplastic corpus callosum and colpocephaly. She had 11 pairs of ribs, a handlebar shaped clavicle, dysphagia, growth retardation, and microcephaly. DES was performed as previously described.5 Both the patient and her partially affected sister were found to harbor compound heterozygous alterations in the gene LRP5 (gene MIM 603506): a paternally inherited pathogenic variant c.2718_2721DELTATG (p.M907TFS*52), and a maternally inherited likely pathogenic variant c.1709G>A (p.R570Q). LRP5 is associated with familial exudative vitreoretinopathy (FEVR) and with various forms of osteopetrosis. These alterations could explain the retinal detachment, but would leave unexplained the patient’s skeletal dysplasia, hypotonia, cardiomyopathy, renal abnormalities, elevated BUN/creatinine ratio, and dysmorphic features. Mitochondrial sequencing revealed a pathogenic alteration m.13513G>A p.D393N in mitochondrial gene ND5 (MT-ND5, MIM 516005), with a heteroplasmy level of about 72% in blood. This variant is of maternal origin and the mother apparently has a low variant load (~6% in blood and saliva). This second diagnosis of Leigh disease can explain most of the proband’s growth retardation and neurodevelopmental features, but skeletal dysplasia is still unexplained by this diagnosis.
For the family of this proband, obtaining two separate diagnoses was particularly important. The proband’s sister also has exudative vitreoretinopathy, Chiari malformation, and was described as clumsy. The sister harbored the compound heterozygous LRP5 variants but the ND5 alteration was not detected in buccal swab or blood, suggesting a better prognosis than the proband’s. In addition, a brother was identified with 23% heteroplasmy on buccal swab and 27% on blood.
DISCUSSION
Our results show that DES can identify multiple genetic diagnoses in many patients, as 2.1% of the whole cohort and 8.9% of solved cases had MPRF (Fig. 1). Including CNVs detected by orthogonal methods, 2.6% of cases submitted for DES have more than one molecular diagnosis, which is comparable with the 1.6% in Yang et al.,15 4.0% in Yavarna et al.,21 0.92% reported in Retterer et al.,22 1.0% in Balci et al.,23 and 1.4% reported in Posey et al.4 The ability to compare rates between laboratories is hindered by slightly different protocols for variant evaluation, reporting criteria, and potentially different rates of reclassifications. Despite these limitations, most studies have consistently reported a small proportion of patients with multiple Mendelian conditions.
Patients who underwent trio exome analysis did not have a significantly different rate of MPRF than patients submitted without an informative trio (Fig. 2a). Trio sequencing allows prioritization of de novo variants and relevant heterozygous alterations in trios with a SGD were overwhelmingly de novo (89%) rather than inherited (11%, Fig. 2b). Interestingly, trios with MPRF had a higher proportion of inherited variants (66% de novo, 34% inherited) and trios with MGD had a similar proportion of inherited variants (68% de novo, 32% inherited). Stated another way, second findings were more often inherited than single findings. This may be because Mendelian conditions due to inherited variants may be milder than conditions due to de novo variants,24 and families with milder phenotypic presentations may not be recommended for DES until a second, more severe condition arises in the proband. One prediction from these data is that trios including partially affected family members may receive multiple results: one that explains the inherited phenotype and a separate result that explains the clinical features unique to the proband. Surprisingly this pattern remained even when analysis was limited to trios with two reportedly healthy unaffected parents. Inherited findings from parents who were marked as unaffected were Mendelian conditions associated with variable expressivity (e.g., PTEN, RYR1), reduced penetrance (e.g., CACNA1A, CAMTA1), and imprinting (e.g., KCNK9). In a few cases, the parent had a Mendelian condition (e.g., PKD1, NOTCH1) that was considered irrelevant to the indication for testing in the proband so the parent was marked as unaffected (for the neurodevelopmental disorder).
No mode of inheritance was significantly enriched in patients with MPRF compared with patients with SGD (Fig. 2c), but males had a slightly higher proportion of MPRF than females (Fig. 2d). It was initially suspected that males may have a higher rate of MPRF due to X-hemizygosity, but surprisingly no mode of inheritance was enriched in either sex. A family history of consanguinity correlated most strongly with having MPRF (Fig. 2e) in agreement with other reports.4,21–23 As expected, homozygous findings in genes associated with recessive molecular diagnoses were enriched in patients from consanguineous families. Additionally, patients from consanguineous families still have a risk of Mendelian conditions due to de novo variants.22,25 This higher rate of MPRF suggests that pretest counseling for families with a history of consanguinity should optimally include disclosures that multiple molecular diagnoses may be found.
A previous exome cohort study suggested that patients with more clinical features are more likely to have multiple Mendelian conditions but it did not find a significant difference between groups.1 In the present cohort, patients with more clinically complex phenotypic presentation had a slightly higher proportion of MPRF (Fig. 3a). Although there was a statistically significant difference between the number of organ systems affected in each group of patients, an average difference of 0.5 organ systems affected between patients with MPRF and SGD may not be clinically significant. As most patients referred for DES have clinically complex presentations, those found to have MPRF may appear in clinic to be similarly affected as patients with SGD. Clinically complex patients are also more likely to have any positive result, with the highest diagnostic rates of both SGD (22.5%) and of MPRF (2.7%) in the group of patients with 6–10 organ systems affected (Fig. 3b). These trends also suggest that thorough patient phenotyping and communication with the diagnostic laboratory increase the chance of diagnosing patients through DES. Further, clinicians are likely to stop testing once a single diagnosis is obtained on panel and or single-gene testing, potentially resulting in missed diagnoses. DES is uniquely positioned to pick up multiple diagnoses and is therefore a good first-tier strategy for helping these patients.
How can patients with multiple molecular conditions have an average number of affected organ systems so similar (i.e., 4.4 vs. 3.9 organ systems affected) to patients with only one Mendelian condition? One possibility is that the two Mendelian conditions affect the same organ systems, causing overlapping clinical features. Partial overlap of phenotypes from multiple Mendelian conditions can appear to be a phenotype expansion of a single condition or it can indicate the presence of multiple Mendelian conditions.2 In organs affected by multiple Mendelian conditions, the molecular pathways may interact to produce an oligogenic phenotype. In the present cohort, about half of the patients who received multiple diagnoses had some clinical features that could be explained by either diagnosis, suggesting that the comorbid conditions cause overlapping phenotypes. Additionally many of the nonoverlapping diagnoses presented in only one organ system in the proband at the time of diagnosis, such as PKD1 (renal), ANK2 (neurological), NOTCH1 (cardiovascular), LRP5 (ophthalmologic), INSL3 (genitourinary), WNT10A and WDR72 (dental), ELN (cardiovascular), HBB (hematologic), CFTR (pulmonary), NF1 (dermatologic), and TYR (dermatologic). Therefore additional conditions do not double the number of organ systems involved for two reasons: both conditions affect the same organ system, and at least one condition affects only one organ system.
New information about gene–disease relationships is published continuously, so reanalyzing previous patients can help identify new diagnoses and can increase diagnostic yield.8,26 On the other hand, better defining the phenotypic spectrum of a specific Mendelian condition can allow elimination of a competing diagnosis. We reviewed reclassification reports to determine both how often new diagnoses were added to previous findings and how often initial diagnoses were ruled out. Because fewer than 4% of reclassification reports for MPRF patients removed a previous diagnosis, it appears that the majority of the time (more than 96%) MPRF likely reflect true coexisting Mendelian conditions in the proband. In a similar vein, a patient’s phenotypic presentation may diverge from the course expected for the initial genetic diagnosis and indicate an additional Mendelian condition in the patient, as recently reported.27 Emergence of new clinical features is an excellent time to reanalyze previous exome data, whether the patient already has a diagnosis or not. This may lead to identification of an (additional) genetic diagnosis that was initially ruled out as clinically irrelevant.
The ability to simultaneously identify multiple genetic etiologies in a single patient dramatically demonstrates the utility of diagnostic exome sequencing. Detection of multiple competing relevant findings may require discussion between the diagnostic laboratory and the clinic to determine if one diagnosis fits better than the other, or if the patient in fact has multiple conditions. It is likely that the presence of multiple Mendelian conditions in a single patient may have implications in the clinic. For instance, a patient with oligogenic traits may respond differently to a treatment and therefore might require additional check-ins. Still unanswered is whether multiple Mendelian conditions that affect the same organ systems can have an additive effect on symptom severity. For instance, do patients with pathogenic alterations in multiple genes in the same pathway have more severe phenotype than patients with a single alteration in that pathway, as was suggested by Bayram et al.?28 How often might blended phenotypes from multiple Mendelian conditions mask a true diagnosis? Answering these questions will require collaboration on a large scale and quantification of symptom severity, but may reveal whether specific clinical presentations are truly oligogenic. Finally, the data suggest that a diagnosis that explains only some of the patient’s clinical features might actually be only a portion of the diagnostic answer.
Supplementary information
Acknowledgements
We are grateful to the patients and their families for their participation and to their physicians and genetic counselors for providing samples and clinical histories.
Disclosure
E.D.S., K.R., K.B., S.A.S., J.M.H., D.N.S., B.W., M.R., J.H., W.A., K.D.F.H. and S.T. are full-time, salaried employees of Ambry Genetics. Diagnostic exome sequencing is one of the publicly available tests offered by Ambry Genetics. The other authors declare no conflicts of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41436-019-0477-2) contains supplementary material, which is available to authorized users.
References
- 1.Chong JX, Buckingham KJ, Jhangiani SN, et al. The genetic basis of Mendelian phenotype: discoveries, challenges, and opportunities. Am J Hum Genet. 2015;97:199–215. doi: 10.1016/j.ajhg.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karaca E, Posey JE, Coban Akdemir Z, et al. Phenotypic expansion illuminates multilocus pathogenic variation. Genet Med. 2018;20:1528–1537. doi: 10.1038/gim.2018.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trujillano D, Bertoli-Avella AM, Kumar Kandaswamy K, et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur J Hum Genet. 2017;25:176–182. doi: 10.1038/ejhg.2016.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Posey JE, Harel T, Liu P, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med. 2017;376:21–31. doi: 10.1056/NEJMoa1516767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farwell Hagman KD, Shinde DN, Mroske C, et al. Candidate-gene criteria for clinical reporting: diagnostic exome sequencing identifies altered candidate genes among 8% of patients with undiagnosed diseases. Genet Med. 2017;19:224–235. doi: 10.1038/gim.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farwell KD, Shahmirzadi L, El-Khechen D, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med. 2015;17:578–586. doi: 10.1038/gim.2014.154. [DOI] [PubMed] [Google Scholar]
- 7.Shahmirzadi L, Chao EC, Palmaer E, Parra MC, Tang S, Gonzalez KD. Patient decisions for disclosure of secondary findings among the first 200 individuals undergoing clinical diagnostic exome sequencing. Genet Med. 2014;16:395–399. doi: 10.1038/gim.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith ED, Radtke K, Rossi M, et al. Classification of genes: standardized clinical validity assessment of gene-disease associations aids diagnostic exome analysis and eclassifications. Hum Mutat. 2017;38:600–608. doi: 10.1002/humu.23183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laduca H, Stuenkel AJ, Dolinsky JS, et al. Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genet Med. 2014;16:830–837. doi: 10.1038/gim.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pesaran T, Karam R, Huether R, et al. Beyond DNA: an integrated and functional approach for classifying germline variants in breast cancer genes. Int J Breast Cancer. 2016;2016:2469523. doi: 10.1155/2016/2469523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Atwal PS, Brennan ML, Cox R, et al. Clinical whole-exome sequencing: are we there yet? Genet Med. 2014;16:717–719. doi: 10.1038/gim.2014.10. [DOI] [PubMed] [Google Scholar]
- 13.Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med. 2013;369:1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee H, Deignan JL, Dorrani N, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312:1880–1887. doi: 10.1001/jama.2014.14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sawyer SL, Hartley T, Dyment DA, et al. Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin Genet. 2016;89:275–284. doi: 10.1111/cge.12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacquemont S, Coe BP, Hersch M, et al. A higher mutational burden in females supports a “female protective model” in neurodevelopmental disorders. Am J Hum Genet. 2014;94:415–425. doi: 10.1016/j.ajhg.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;9:114. doi: 10.1038/nature21062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilfert AB, Sulovari A, Turner TN, Coe BP, Eichler EE. Recurrent de novo mutations in neurodevelopmental disorders: properties and clinical implications. Genome Med. 2017;9:101. doi: 10.1186/s13073-017-0498-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carlston CM, O’Donnell-Luria AH, Underhill HR, et al. Pathogenic ASXL1 somatic variants in reference databases complicate germline variant interpretation for Bohring-Opitz syndrome. Hum Mutat. 2017;38:517–523. doi: 10.1002/humu.23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yavarna T, Al-Dewik N, Al-Mureikhi M, et al. High diagnostic yield of clinical exome sequencing in Middle Eastern patients with Mendelian disorders. Hum Genet. 2015;134:967–980. doi: 10.1007/s00439-015-1575-0. [DOI] [PubMed] [Google Scholar]
- 22.Retterer K, Juusola J, Cho MT, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2015;18:696–704. doi: 10.1038/gim.2015.148. [DOI] [PubMed] [Google Scholar]
- 23.Balci TB, Hartley T, Xi Y, et al. Debunking Occam’s razor: diagnosing multiple genetic diseases in families by whole-exome sequencing. Clin Genet. 2017;92:281–289. doi: 10.1111/cge.12987. [DOI] [PubMed] [Google Scholar]
- 24.Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet. 2012;13:565–575. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]
- 25.Powis Z, Farwell KD, Alamillo CL, Tang S. Diagnostic exome sequencing for patients with a family history of consanguinity: over 38% of positive results are not autosomal recessive pattern. J Hum Genet. 2016;61:173–175. doi: 10.1038/jhg.2015.125. [DOI] [PubMed] [Google Scholar]
- 26.Wenger AM, Guturu H, Bernstein JA, Bejerano G. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med. 2017;19:209–214. doi: 10.1038/gim.2016.88. [DOI] [PubMed] [Google Scholar]
- 27.Jehee FS, de Oliveria VT, Gurgel-Giannetti J, et al. Dual molecular diagnosis contributes to atypical Prader-Willi phenotype in monozygotic twins. Am J Med Genet A. 2017;173:2451–2455. doi: 10.1002/ajmg.a.38315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bayram Y, Karaca E, Coban Akdemir Z, et al. Molecular etiology of arthrogryposis in multiple families of mostly Turkish origin. J Clin Invest. 2016;126:762–778. doi: 10.1172/JCI84457. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.