

Abstract
Rationale:
Thrombotic microangiopathy (TMA) is a group of clinical syndromes characterized by excessive platelet activation and endothelial injury that leads to acute or chronic microvascular obliteration by intimal mucoid and fibrous thickening, with or without associated thrombi. It frequently involves the kidney but may involve any organ or system at variable frequencies depending on the underlying etiology. Among its numerous causes, drug toxicities and complement regulation abnormalities stand out as some of the most common. A more recently described association is with monoclonal gammopathy. Lung involvement by TMA is infrequent, but has been described in Cobalamin C deficiency and post stem-cell transplantation TMA.
Patient concerns:
This is the case of a patient with smoldering myeloma who received proteasome-inhibitor therapy due to retinopathy and developed acute renal failure within one week of therapy initiation.
Diagnoses:
A renal biopsy showed thrombotic microangiopathy. At the time, mild pulmonary hypertension was also noted and presumed to be idiopathic.
Interventions:
Given the known association of proteasome-inhibitor therapy with thrombotic microangiopathy, Bortezomib was discontinued and dialysis was initiated.
Outcomes:
Drug withdrawal failed to prevent disease progression and development of end-stage renal disease, as well as severe pulmonary hypertension that eventually lead to the patient's death.
Lessons:
To our knowledge, this is the first reported case of pulmonary involvement by TMA associated with monoclonal gammopathy which appears to have been triggered by proteasome-inhibitor therapy. Clinicians should be aware of this possibility to allow for more prompt recognition of pulmonary hypertension as a potential manifestation of monoclonal gammopathy-associated TMA, especially in patients also receiving proteasome-inhibitors, so that treatment aiming to slow disease progression can be instituted.
Keywords: monoclonal gammopathy, proteasome-inhibitor, pulmonary hypertension, smoldering myeloma, thrombotic microangiopathy
1. Introduction
Thrombotic microangiopathies are clinical syndromes characterized by excessive platelet activation and endothelial injury that result in acute and chronic microvascular occlusion.[1] Among its many causes are Shiga-toxin producing bacterial infections, ADAMTS13 deficiency or autoantibodies, complement alternative pathway regulation abnormalities, drug reactions, malignancies, bone marrow transplantation, Cobalamin C deficiency, viral, and bacterial infections.[2] The kidney is often involved; however, any organ or system may be affected. The frequency of extrarenal manifestations may vary according to the underlying etiology, with central nervous system involvement being common in ADAMTS13 deficiency, and renal involvement often seen in complement-mediated TMA. Lung involvement, clinically manifested by pulmonary hypertension, is rare in Complement-mediated TMA but can be seen in TMA secondary to Cobalamin C deficiency[3,4] and stem-cell transplantation.[5] To our knowledge, lung involvement has not been reported in TMA associated with monoclonal gammopathy, nor with proteasome-inhibitor therapy.[1,6]
1.1. Case presentation
The patient was a 53 year-old female who originally presented to an ophthalmologist for blurred vision and was found to have retinal ischemia, cotton wool spots and macular edema, initially attributed to hypertensive retinopathy. Worsening retinal findings eventually led to more extensive workup that revealed a 1.5 g/dL monoclonal protein, immunoglobulin G (IgG) kappa type. She had a negative hypercoagulable panel, normal blood cell counts, elevated erythrocyte sedimentation rate and lactate dehydrogenase, and slightly elevated creatinine (1.1 mg/dL). Total immunoglobulin G was 1176 mg/dL (reference range 700–1600 mg/dL), free light chains (FLC) ratio was abnormal at 28.12 with high free kappa (274 mg/L; reference range 3.3–19.4 mg/L). There was no monoclonal protein in a 24-hour urine collection and no significant proteinuria. Skeletal survey showed no lytic lesions and a bone marrow aspiration and biopsy showed 10%–15% plasma cells. She was diagnosed with smoldering myeloma and plasma cell directed therapy was recommended due to significant vision impairment. One week after starting triple therapy with bortezomib, lenalidomide, and dexamethasone, she presented with acute renal failure (rise in Creatinine from 1.4 to 6.9 mg/dL). Urinalysis showed 1+ protein and greater than five red blood cells per high power field, no casts were seen. Serum albumin was 3.1 g/dL. Hepatitis serologies were negative. Renal ultrasound showed normal-sized kidneys and no evidence of obstruction. A renal biopsy was indicated.
1.2. Renal biopsy
The biopsy contained 11 glomeruli, one of which was globally sclerosed. The remaining glomeruli were shrunken with a bloodless appearance and diffusely wrinkled capillary walls (Figure 1). There was no endocapillary hypercellularity and no glomerular thrombi. Diffuse interstitial edema with focal mild interstitial inflammation were present, along with evidence of acute tubular injury and rare granular casts. No atypical, fractured crystalline eosinophilic casts were seen. Interstitial fibrosis and tubular atrophy were estimated as mild. At least four arterioles were present, all of which showed endothelial swelling, intimal edema and concentric fibroplasia with entrapped red blood cells leading to complete or near-complete luminal obliteration. There were no definite thrombi or fibrinoid necrosis. Congo Red stain for amyloid was negative. Immunofluorescence showed no light chain restriction within casts or in the tubulo-interstitium. Electron microscopy confirmed the absence of amyloid fibrils and showed evidence of endothelial damage and ischemia in the form of subendothelial lucent widening and diffusely wrinkled glomerular basement membranes. Tubules showed intracytoplasmic vacuoles and loss of microvilli consistent with acute tubular injury and no crystals or abnormal lysosomes.
Figure 1.
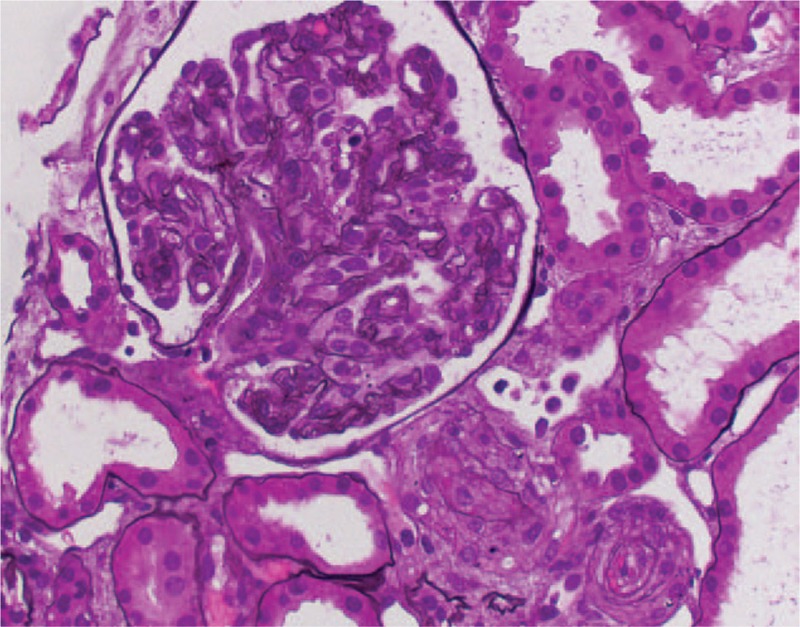
Renal biopsy one week after initiation of bortezomib therapy showing complete arteriolar luminal obliteration by endothelial swelling, fibrous and mucoid intimal thickening, and an ischemic glomerulus with a bloodless appearance. (Jones Silver stain, 400×).
A diagnosis of acute thrombotic microangiopathy was made.
1.3. Follow-up
Given the recent initiation of myeloma therapy including Bortezomib and the known association of proteasome-inhibitor therapy with thrombotic microangiopathy,[1] Bortezomib was discontinued. The patient however did not recover renal function and remained dialysis-dependent ever since. She also experienced progressive decline of vision, with no response to intravitreal anti-VEGF and only partial response to steroids.
She eventually presented to an outside hospital 9 months later with shock that was presumed to be septic in origin. By then, she also had a history of recurrent pericardial effusions, attributed to chronic renal failure, and congestive heart failure with an ejection fraction of 25%. She had severe left ventricular systolic dysfunction and moderate pulmonary hypertension. In retrospect, pulmonary hypertension had been detected at an echocardiogram performed two days after her renal biopsy, at which point she had a normal ejection fraction and had received a diagnosis of primary pulmonary hypertension.
Shortly after presentation, she suffered a cardiac arrest and was successfully resuscitated. At that point, she was transferred to our institution where she was admitted to ICU and broad antibiotic therapy was started even though no clear infection source was identified. Echocardiogram showed moderate pericardial effusion and an enlarged right atrium and right ventricle with tricuspid regurgitation but no evidence of tamponade. Pulmonary artery systolic pressure (PASP) was very high (85.28 mmHg) compared with a previous echocardiogram done 2 months earlier that showed a PASP of 52 mmHg. Left ventricular ejection fraction was estimated at 60%–65%. Pericardiocentesis was arranged and 325 mL of serous fluid were drained; cytopathology and fluid cultures including acid-fast bacilli and fungi were negative.
Given her history of smoldering myeloma, hematology-oncology was consulted. Serum protein electrophoresis showed a 0.66 g/dL monoclonal band in the gamma region, characterized as IgG on serum immunofixation. IgG was 1226 mg/dL (reference range 700–1600 mg/dL). Serum kappa FLC was 340.8 mg/L (reference range 0.33–1.94) with kappa/lambda FLC ratio of 16.15. Skeletal survey showed no lytic lesions. An abdominal fat pad biopsy was negative for amyloid.
Rheumatology was also consulted and as part of the workup, she was found to have low C3 levels (54 mg/dL; reference range 90–180 mg/dL) with normal C4 (19 mg/dL; reference range 10–40 mg/dL). Complement Factor I was normal at 32.9 mcg/mL (reference range 29.3–58.5 mcg/mL) as were Factor H (B1H) levels (176 mcg/mL, reference range 160–412 mcg/mL). ADAMTS13 activity was 17%, ADAMTS13 inhibitor screen was negative. Antiphospholipid antibody panel was negative. Because of peripheral neuropathy, plasma VEGF and anti-myelin associated glycoprotein antibodies were ordered to rule out POEMS syndrome but returned negative.
Over the following days, the patient experienced multiple episodes of lactic acidosis requiring repeated intubations and severe right heart failure due to pulmonary hypertension for which she was initiated on treprostinil. Thirteen days after admission, she suffered another cardiac arrest refractory to resuscitative efforts and a partial (chest-abdomen) autopsy was requested.
Patient's family has provided informed consent. Ethical board review was waived because as a single case study, it did not qualify as human subjects research.
1.4. Autopsy
External examination was unremarkable except for a 2 cm ulcer with fat layer exposure on the dorsal aspect of the left foot (Figure 2). Bilateral serous pleural effusions were present (left 400 mL, right 900 mL). The pericardial sac contained 200 mL of serous fluid and was free of adhesions. The heart weighted 440 g (normal range for females: 148–296 g[8]). Serial sections of the coronary arteries showed no significant atherosclerosis and no thrombi. Concentric left ventricular hypertrophy was noted. The right ventricle was dilated with a wall thickness of 0.5 cm. Lungs were congested weighting each 710 g (normal range for normal BMI women 142–719 g for right lung and 143–518 g for left[9]). The spleen weighted 100 g and showed multiple subcapsular wedge-shaped infarcts. No enlarged lymph nodes were found within the thorax or abdomen. The kidneys were bilaterally atrophic weighting 50 g (right; normal range for normal BMI women 67–261 g[9]) and 60 g (left; normal range for normal BMI women 55–274 g[9]). The liver weighted 1330 g (normal range for normal BMI women 775–1946[9]) and had a nutmeg appearance.
Figure 2.

(A) Ulcer with fat layer exposure on the foot dorsum, similar to reported in skin involvement in atypical hemolytic uremic syndrome.[7](B) Renal parenchyma at autopsy showing severe interstitial fibrosis and tubular atrophy; prominent arterial fibrous intimal thickening (B, Trichrome stain, 100×); arterioles with “onion-skin” changes and ischemic glomeruli (C, PAS stain, 400×). Lung parenchyma at autopsy showing severe fibrous intimal thickening of a large caliber pulmonary artery (D, Jones Silver stain, 200×), medial hypertrophy in medium and small caliber arteries (E, Hematoxylin and eosin, 100×) and prominent obliterative changes in small arterioles (F, Jones Silver stain, 400×).
Microscopic examination confirmed severe renal interstitial fibrosis with mostly shrunken glomeruli and concentric fibrointimal thickening of both small and large caliber arteries (Figure 2B and C). Similar changes were also seen within splenic and pulmonary arteries and arterioles, while large caliber pulmonary arteries showed atherosclerotic changes (Figure 2D–F). The heart showed myocyte hypertrophy, minimal fibrosis and no evidence of amyloid deposition. Liver parenchyma had centrolobular congestion and necrosis, consistent with right-sided heart failure.
2. Discussion
Here, we report the case of a patient with smoldering myeloma for which plasma-cell directed therapy including a proteasome inhibitor (PI) was indicated due to progressively worsening retinopathy. She developed acute renal failure and biopsy-proven renal thrombotic microangiopathy within a week of starting treatment; at which point, in retrospect, pulmonary hypertension was also noted and presumed to represent idiopathic pulmonary hypertension. Bortezomib was discontinued; however the patient did not recover renal function, experienced progressive retinopathy and pulmonary hypertension leading to congestive heart failure and ultimately death.
While both monoclonal gammopathy[6] as well as proteasome inhibitor therapy[1] have been associated with microangiopathic hemolytic anemia and renal thrombotic microangiopathy; to our knowledge, this is the first case of lung involvement in TMA triggered by Bortezomib therapy for monoclonal gammopathy (Table 1). TMA associated with monoclonal gammopathy has been more recently described, and so far, pulmonary involvement has not been reported (Table 2).[6]
Table 1.
Previous case reports or case series of adverse events associated with proteasome-inhibitor therapy.

Table 2.
Previous case reports or case series of TMA associated with monoclonal gammopathy.

It is postulated that monoclonal paraprotein may lead to TMA through Complement alternative pathway dysregulation, however the exact mechanism is not known. It is possible that dimeric light chains may act as an autoantibody against complement Factor H thus interfering with its inhibitory properties. Alternatively, monoclonal immunoglobulins could increase the half-life of alternative pathway C3 convertase by acting as autoantibodies to complement Factor B, akin to a C3 nephritic factor.[6,33,34]
In the case of TMA associated with PI therapy, given the presence of podocytopathy in a few cases (mostly associated with Carfilzomib use),[35] a postulated mechanism of endothelial injury is through decreased VEGF production secondary to decreased NF-kB transcription due to decreased ubiquitination of NF-kB inhibitor IkK because of proteasome inhibition therapy.[13] Finally, direct endothelial damage by the drug has also been invoked.[1]
In our patient, TMA was initially correctly presumed to be secondary to Bortezomib given the temporal association of PI therapy and TMA presentation. However, given the lack of response to drug withdrawal and progressive unremitting TMA, we postulate that Bortezomib may have been only the trigger for activation of Complement alternative pathway dysregulation related to her monoclonal gammopathy, which then remained unchecked. This is supported by the low C3 levels measured 8 months after discontinuation of Bortezomib; and the progressive end-organ-damage due to TMA in spite to prompt discontinuation, which is in strike contrast to most reports of proteasome-inhibitor associated TMA, in which the majority of cases resolved after drug withdrawal[1] (Table 1). The overall outcome of TMA associated with monoclonal gammopathy seems to be worse than that of PI-associated TMA, with up to 50% of patients progressing to end-stage renal disease (ESRD) (Table 2),[6] as was the case with our patient.
Skin lesions can also be a manifestation of Complement-mediated thrombotic microangiopathy, including ulcers with well-delimitated borders, most commonly seen in the lower limbs, similar to those seen in this patient (Figure 2A).[7]
Pulmonary involvement in thrombotic microangiopathy has been reported in adult and pediatric patients with TMA secondary to Cobalamin C deficiency,[3,4] in pediatric patients with TMA post stem cell transplantation.[5] Pulmonary hypertension due to pulmonary artery stenosis was also described in a case of pediatric atypical hemolytic uremic syndrome due to Complement Factor B mutation.[36] Complement-mediated TMA predominantly affects the renal microvasculature; however, extrarenal manifestations including central nervous system, cardiovascular, skin, pulmonary, gastrointestinal, and skeletal muscle involvement can be seen in up to 20% of patients.[37] Importantly, they have been described to occur not only during the disease acute phase but even after renal function is lost, as in our case, likely as a result of chronic over activation/dysregulation of the complement system.[37,38] Cardiovascular complications of PI therapy, including bortezomib-induced heart failure (HF),[22,25] ischemic heart disease and complete heart block[24] have also been reported (Table 1) and appear to be due to a toxic mechanism, as symptoms in most cases occurred only after high cumulative doses.[24,25] This is in contrast to our patient, who only received one cycle of bortezomib-containing chemotherapy and developed HF many months later after drug discontinuation, indicating that her HF was likely secondary to pulmonary hypertension due to chronic complement over activation as opposed to bortezomib toxicity.
Retinal involvement in multiple myeloma can occur in the form of flame-shaped, white hemorrhages, nerve fiber layer infarcts,[39] and macular edema[40] and may be an indication for plasma-cell targeted therapy. The retina can also rarely be involved in systemic TMA, where it can present as cotton wool exudates or multiple retinal detachments.[41–43] In retrospect, it is possible that this patient's retinal manifestations could in at least in part already be a manifestation of monoclonal paraprotein associated Complement dysregulation leading to microvascular injury. Because autopsy was restricted to the thorax and abdomen, histopathological examination of the retina could not be performed to confirm or rule out the presence of TMA.
Cardiac tamponade leading to death has been reported in a patient with HUS and was considered as a possibility in our case.[44] In that regard, she had been assessed multiple times by cardiology, had frequent pericardiocentesis with drainage of up to 325 mL, and in all those occasions, the possibility of tamponade had been ruled out. At postmortem examination, 200 mL of serous fluid were noted in the pericardial space, which were attributed to this patient's chronic pericardial effusions and deemed unlikely to represent cardiac tamponade in her case. Although no anatomical cause of death was identified, patient's death was likely a result of severe pulmonary hypertension leading to cardiogenic shock. This is supported clinically by the very high PASP in the days preceding her death and morphologically by prominent cardiomegaly and right ventricular hypertrophy, and severe pulmonary microvascular obliterative changes (Figure 2D–F).
Our report has limitations that should be acknowledged. First of all, patient care took place at multiple institutions in different states; she only came to our institution during final admission and her diagnosis remained unclear until postmortem examination was completed. Therefore an alternative Complement pathway dysregulation was not thought of as a possible cause for her symptoms and more extensive testing, including functional assays, measurement of Complement activation markers and search for autoantibodies were not performed.[45] Still, we believe our findings are relevant because they show that severe lung involvement eventually leading to death can occur in TMA associated with monoclonal gammopathy, which in our case appears to have been triggered or worsened by proteasome-inhibitor therapy. Clinicians should be aware of this possibility to allow for more prompt recognition of pulmonary hypertension as a potential manifestation of monoclonal gammopathy-associated TMA.
Author contributions
Conceptualization: Clarissa A. Cassol.
Data curation: Clarissa A. Cassol, Michael P.A. Williams, Sophia Rodriguez.
Investigation: Sophia Rodriguez.
Supervision: Clarissa A. Cassol.
Writing – original draft: Clarissa A. Cassol.
Writing – review & editing: Clarissa A. Cassol, Tiffany N. Caza.
Clarissa A. Cassol: 0000-0001-7536-6806.
Footnotes
How to cite this article: Cassol CA, Williams MPA, Caza TN, Rodriguez S. Renal and pulmonary thrombotic microangiopathy triggered by proteasome-inhibitor therapy in patient with smoldering myeloma. Medicine. 2019;98:39(e17148).
Abbreviations: ADAMTS13 = a disintegrin-like and metalloproteinase with thrombospondin type 1 motif, member 13, BMI = body mass index, C3 = complement C3, C4 = complement C4, ESRD = end-stage renal disease, FLC = free light chains, HF = heart failure, HUS = hemolytic uremic syndrome, ICU = intensive care unit, IgG = immunoglobulin G, IkK = IκB kinase, NF-kB = nuclear factor kappa-light-chain-enhancer of activated B cells, PASP = pulmonary artery systolic pressure, PI = proteasome inhibitor, POEMS syndrome = polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes, TMA = thrombotic microangiopathy, VEGF = vascular endothelial growth factor.
The authors of this case report have no conflicts of interest to disclose.
References
- [1].Goodship TH, Cook HT, Fakhouri F, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int 2017;91:539–51. [DOI] [PubMed] [Google Scholar]
- [2].Grangé S, Bekri S, Artaud-Macari E, et al. Adult-onset renal thrombotic microangiopathy and pulmonary arterial hypertension in cobalamin C deficiency. Lancet 2015;386:1011–2. [DOI] [PubMed] [Google Scholar]
- [3].Komhoff M, Roofthooft MT, Westra D, et al. Combined pulmonary hypertension and renal thrombotic microangiopathy in cobalamin C deficiency. Pediatrics 2013;132:e540–4. [DOI] [PubMed] [Google Scholar]
- [4].Jodele S, Hirsch R, Laskin B, et al. Pulmonary arterial hypertension in pediatric patients with hematopoietic stem cell transplant-associated thrombotic microangiopathy. Biol Blood Marrow Transplant 2013;19:202–7. [DOI] [PubMed] [Google Scholar]
- [5].Yui JC, Van Keer J, Weiss BM, et al. Proteasome inhibitor associated thrombotic microangiopathy. Am J Hematol 2016;91:E348–52. [DOI] [PubMed] [Google Scholar]
- [6].Molina DK, DiMaio VJ. Normal organ weights in women: Part I-the heart. Am J Forensic Med Pathol 2015;36:176–81. [DOI] [PubMed] [Google Scholar]
- [7].Ardissino G, Tel F, Testa S, et al. Skin involvement in atypical hemolytic uremic syndrome. Am J Kidney Dis 2014;63:652–5. [DOI] [PubMed] [Google Scholar]
- [8].Molina DK, DiMaio VJ. Normal organ weights in women: Part II-the brain, lungs, liver, spleen, and kidneys. Am J Forensic Med Pathol 2015;36:182–7. [DOI] [PubMed] [Google Scholar]
- [9].Ravindran A, Go RS, Fervenza FC, Sethi S. Thrombotic microangiopathy associated with monoclonal gammopathy. Kidney Int 2017;91:691–8. [DOI] [PubMed] [Google Scholar]
- [10].Chan KL, Filshie R, Nandurkar H, Quach H. Thrombotic microangiopathy complicating bortezomib-based therapy for multiple myeloma. Leuk Lymphoma 2015;56:2185–6. [DOI] [PubMed] [Google Scholar]
- [11].Atallah-Yunes SA, Soe MH. Drug-induced thrombotic microangiopathy due to cumulative toxicity of ixazomib. Case Rep Hematol 2018;2018:7063145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jhaveri KD, Chidella S, Varghese J, et al. Carfilzomib-related acute kidney injury. Clin Adv Hematol Oncol 2013;11:604–5. [PubMed] [Google Scholar]
- [13].Lodhi A, Kumar A, Saqlain MU, Suneja M. Thrombotic microangiopathy associated with proteasome inhibitors. Clin Kidney J 2015;8:632–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mehta N, Saxena A, Niesvizky R. Bortezomib-induced thrombotic thrombocytopaenic purpura. BMJ Case Rep 2012;2012: [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wanchoo R, Khan S, Kolitz JE, Jhaveri KD. Carfilzomib-related acute kidney injury may be prevented by N-acetyl-L-cysteine. J Oncol Pharm Pract 2015;21:313–6. [DOI] [PubMed] [Google Scholar]
- [16].Qaqish I, Schlam IM, Chakkera HA, et al. Carfilzomib: a cause of drug associated thrombotic microangiopathy. Transfus Apher Sci 2016;54:401–4. [DOI] [PubMed] [Google Scholar]
- [17].Salmenniemi U, Remes K. Thrombotic microangiopathy associated with bortezomib treatment in a patient with relapsed multiple myeloma. Hematol Rep 2012;4:e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sullivan MR, Danilov AV, Lansigan F, Dunbar NM. Carfilzomib associated thrombotic microangiopathy initially treated with therapeutic plasma exchange. J Clin Apher 2015;30:308–10. [DOI] [PubMed] [Google Scholar]
- [19].Morita R, Hashino S, Shirai S, et al. Thrombotic microangiopathy after treatment with bortezomib and dexamethasone in a patient with multiple myeloma. Int J Hematol 2008;88:248–50. [DOI] [PubMed] [Google Scholar]
- [20].Moore H, Romeril K. Multiple myeloma presenting with a fever of unknown origin and development of thrombotic thrombocytopenic purpura post-bortezomib. Intern Med J 2011;41:348–50. [DOI] [PubMed] [Google Scholar]
- [21].Hobeika L, Self SE, Velez JC. Renal thrombotic microangiopathy and podocytopathy associated with the use of carfilzomib in a patient with multiple myeloma. BMC Nephrol 2014;15:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Honton B, Despas F, Dumonteil N, et al. Bortezomib and heart failure: case-report and review of the French Pharmacovigilance database. Fundam Clin Pharmacol 2014;28:349–52. [DOI] [PubMed] [Google Scholar]
- [23].Enrico O, Gabriele B, Nadia C, et al. Unexpected cardiotoxicity in haematological bortezomib treated patients. Br J Haematol 2007;138:396–7. [DOI] [PubMed] [Google Scholar]
- [24].Dasanu CA. Complete heart block secondary to bortezomib use in multiple myeloma. J Oncol Pharm Pract 2011;17:282–4. [DOI] [PubMed] [Google Scholar]
- [25].Gupta A, Pandey A, Sethi S. Bortezomib-induced congestive cardiac failure in a patient with multiple myeloma. Cardiovasc Toxicol 2012;12:184–7. [DOI] [PubMed] [Google Scholar]
- [26].Riksen NP, Luken BM, Klasen IS, et al. Antibodies against the CUB1-2 domains of ADAMTS13 in a patient with benign monoclonal gammopathy: no causal relationship. Haematologica 2007;92:e74–6. [DOI] [PubMed] [Google Scholar]
- [27].Cheah CY, Orlowski RZ, Manasanch EE, Oo TH. Thrombotic thrombocytopenic purpura in a patient with lenalidomide-responsive multiple myeloma. Ann Hematol 2015;94:1605–7. [DOI] [PubMed] [Google Scholar]
- [28].Alpay N, Uzun S, Bahat G, et al. Thrombotic thrombocytopenic purpura associated with multiple myeloma. Blood Coagul Fibrinolysis 2008;19:439–41. [DOI] [PubMed] [Google Scholar]
- [29].Koga T, Yamasaki S, Nakamura H, et al. Renal thrombotic microangiopathies/thrombotic thrombocytopenic purpura in a patient with primary Sjogren's syndrome complicated with IgM monoclonal gammopathy of undetermined significance. Rheumatol Int 2013;33:227–30. [DOI] [PubMed] [Google Scholar]
- [30].Yao H, Monge M, Renou M, et al. Thrombotic thrombocytopenic purpura due to anti-ADAMTS13 antibodies in multiple myeloma. Clin Nephrol 2014;81:210–5. [DOI] [PubMed] [Google Scholar]
- [31].Xiao X, Zhong HY, Zhang GS, Deng MY. Thrombotic thrombocytopenic purpura as initial and major presentation of multiple myeloma. J Thromb Thrombolysis 2013;36:422–3. [DOI] [PubMed] [Google Scholar]
- [32].Cheungpasitporn W, Leung N, Sethi S, et al. Refractory atypical hemolytic uremic syndrome with monoclonal gammopathy responsive to bortezomib-based therapy. Clin Nephrol 2015;83:363–9. [DOI] [PubMed] [Google Scholar]
- [33].Jokiranta TS, Solomon A, Pangburn MK, et al. Nephritogenic λ light chain dimer: a unique human miniautoantibody against complement factor H. J Immunol 1999;163:4590–6. [PubMed] [Google Scholar]
- [34].Meri S. Activation of the alternative pathway of complement by monoclonal lambda light chains in membranoproliferative glomerulonephritis. J Exp Med 1992;175:939–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hobeika L, Self SE, Velez JCQJBn. Renal thrombotic microangiopathy and podocytopathy associated with the use of carfilzomib in a patient with multiple myeloma. BMC Nephrol 2014;15:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Azukaitis K, Loirat C, Malina M, et al. Macrovascular involvement in a child with atypical hemolytic uremic syndrome. Pediatr Nephrol 2014;29:1273–7. [DOI] [PubMed] [Google Scholar]
- [37].Hofer J, Rosales A, Fischer C, Giner T. Extra-renal manifestations of complement-mediated thrombotic microangiopathies. Front Pediatr 2014;2:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Noris M, Remuzzi G. Cardiovascular complications in atypical haemolytic uraemic syndrome. Nat Rev Nephrol 2014;10:174–80. [DOI] [PubMed] [Google Scholar]
- [39].Knapp AJ, Gartner S, Henkind P. Multiple myeloma and its ocular manifestations. Surv Ophthalmol 1987;31:343–51. [DOI] [PubMed] [Google Scholar]
- [40].Bernard A. [Macular manifestations of monoclonal dysgammaglobulinemias. Apropos of 3 cases]. J Fr Ophtalmol 1986;9:805–10. [PubMed] [Google Scholar]
- [41].Benson DO, Fitzgibbons JF, Goodnight SH. The visual system in thrombotic thrombocytopenic purpura. Ann Ophthalmol 1980;12:413–7. [PubMed] [Google Scholar]
- [42].Mohsin N, Nooyi C, Jha A, et al. Retinal injury as an early manifestation of posttransplant thrombotic microangiopathy: recovery with plasma exchanges and conversion to sirolimus–case report and review of the literature. Transplant Proc 2007;39:1272–5. [DOI] [PubMed] [Google Scholar]
- [43].Randhawa S, Sharma M. Multifocal serous detachments as the presenting sign of thrombotic thrombocytopenic purpura. Retin Cases Brief Rep 2014;8:295–9. [DOI] [PubMed] [Google Scholar]
- [44].Birk PE, Chakrabarti S, Lacson AG, Ogborn MR. Cardiac tamponade as a terminal event in the hemolytic uremic syndrome in childhood. Pediatr Nephrol 1994;8:754–5. [DOI] [PubMed] [Google Scholar]
- [45].Angioi A, Fervenza FC, Sethi S, et al. Diagnosis of complement alternative pathway disorders. Kidney Int 2016;89:278–88. [DOI] [PubMed] [Google Scholar]