

Abstract
Treatment with nitisinone (NTBC) has brought about a drastic improvement in the treatment and prognosis of hereditary tyrosinemia type I (HT1). We conducted a retrospective observational multicentric study in Spanish HT1 patients treated with NTBC to assess clinical and biochemical long-term evolution.
We evaluated 52 patients, 7 adults and 45 children, treated with NTBC considering: age at diagnosis, diagnosis by clinical symptoms, or by newborn screening (NBS); phenotype (acute/subacute/chronic), mutational analysis; symptoms at diagnosis and clinical course; biochemical markers; doses of NTBC; treatment adherence; anthropometric evolution; and neurocognitive outcome.
The average follow-up period was 6.1 ± 4.9 and 10.6 ± 5.4 years in patients with early and late diagnosis respectively. All patients received NTBC from diagnosis with an average dose of 0.82 mg/kg/d. All NBS-patients (n = 8) were asymptomatic at diagnosis except 1 case with acute liver failure, and all remain free of liver and renal disease in follow-up. Liver and renal affectation was markedly more frequent at diagnosis in patients with late diagnosis (P < .001 and .03, respectively), with ulterior positive hepatic and renal course in 86.4% and 93.2% of no-NBS patients, although 1 patient with good metabolic control developed hepatocarcinoma.
Despite a satisfactory global nutritional evolution, 46.1% of patients showed overweight/obesity. Interestingly lower body mass index was observed in patients with good dietary adherence (20.40 ± 4.43 vs 24.30 ± 6.10; P = .08) and those with good pharmacological adherence (21.19 ± 4.68 vs 28.58 ± 213.79).
intellectual quotient was ≥85 in all NBS- and 68.75% of late diagnosis cases evaluated, 15% of which need pedagogical support, and 6.8% (3/44) showed school failure.
Among the 12 variants identified in fumarylacetoacetate hydrolase gene, 1 of them novel (H63D), the most prevalent in Spanish population is c.554–1 G>T.
After NTBC treatment a reduction in tyrosine and alpha-fetoprotein levels was observed in all the study groups, significant for alpha-fetoprotein in no NBS-group (P = .03), especially in subacute/chronic forms (P = .018).
This series confirms that NTBC treatment had clearly improved the prognosis and quality of life of HT1 patients, but it also shows frequent cognitive dysfunctions and learning difficulties in medium-term follow-up, and, in a novel way, a high percentage of overweight/obesity.
Keywords: nephrocalcinosis, phenotype, severe liver dysfunction, tubulopathy, tyrosine
1. Introduction
Hereditary tyrosinemia type 1 (HT1, OMIM 276700) is a severe autosomal recessive disorder due to deficiency of fumarylacetoacetate hydrolase (FAH, EC 3.7.1.2), the last enzyme in tyrosine (Tyr) degradation pathway. Due to this defect, toxic metabolites including succinylacetone (SA), maleylacetoacetate, and fumarylacetoacetate, are formed. These products cause severe liver dysfunction, renal tubulopathy, porphyria like syndrome, cardiomyopathy, and hepatocellular carcinoma (HCC) early in life.[1,2] Acute (presenting before 6 months of age), subacute (presenting between 6 and 12 months) and chronic (presenting after 1 year) forms of the disease have been described.[1] The age at onset of symptoms broadly correlates with severity.[3] The HT1 frequency worldwide is about 1 in 100,000 individuals, but a higher incidence is observed in some regions such as Turkey, Quebec; and India.[4] The FAH gene has been cloned and mapped in human chromosome 15q, and 100 FAH mutations have been reported so far.[4]
Neonatal mass screening is conducted in many countries around the world and is essential to guarantee early diagnosis and treatment during the neonatal period. Tyr as a screening parameter is not appropriate due to the high rate of false-positive, related to transient neonatal tyrosinemia, and false-negative results; SA is the screening parameter of choice.[5,6]
Until the early 1990s, the only strategies available to manage the symptoms of HT1 were protein-restricted diets (usually low in phenylalanine, methionine, and Tyr) and liver transplantation. In 1992, an alternative treatment, (2-(2-nitro-trifluoromethylbenzoyl) 1, 3-cyclohexanedione, also known as nitisinone or NTBC, was introduced.[7] This treatment inhibits the upstream enzyme 4-hydroxyphenylpyruvate dioxygenase, thereby preventing the formation of the toxic metabolites fumarylacetoacetate and succinylacetoacetate (Fig. 1). NTBC treatment strongly increases plasma Tyr concentrations, so this therapy should be complementary to diet restricted in Tyr and its precursor phenylalanine (Phe) and should not be administered alone.[8,9] Liver dysfunction is now controlled in >90% of patients, and extrahepatic manifestations have been abolished or greatly improved with combined treatment.[10] It is essential an early start of NTBC therapy to avoid sequelae; ideally in the neonatal period.[1,11,12] Eye pain, eye itching/conjunctivitis, corneal crystals, and thrombocytopenia are the most common side effects reported[6] and are mostly related to high Tyr levels and reversible when Tyr levels decrease by strict dietary adherence. The risk of HCC has markedly diminished after the introduction of NTBC, although its occurrence has been also described in some HT1 subjects under NTBC therapy.[1,13,14] Neurocognitive deficits could also represent a problem in long-term management.[2,15,16]
Figure 1.
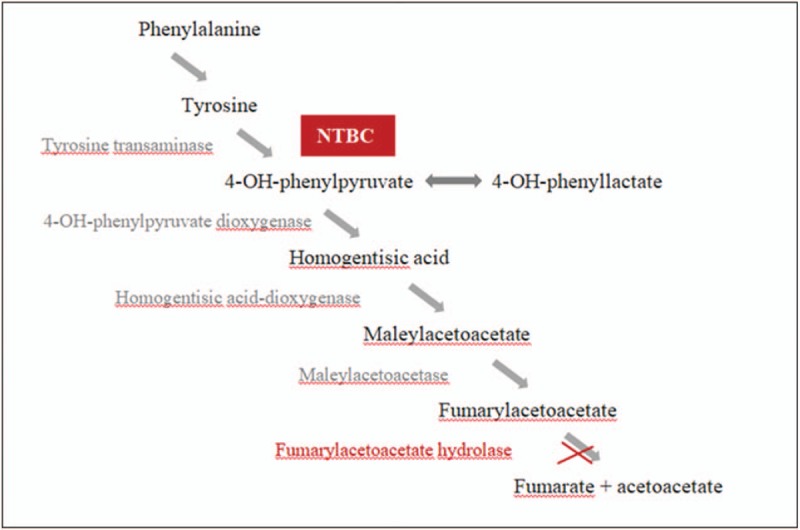
Metabolic pathway of tyrosine. Fumarylacetoacetate hydrolase is the deficient enzyme in tyrosinemia type 1. NTBC inhibits the upstream enzyme 4-hydroxyphenylpyruvate dioxygenase preventing the formation of the toxic metabolites fumarylacetoacetate and succinylacetoacetate. NTBC = nitisinone.
Few studies until now had assessed in wide series the evolution of HT1 patients treated with NTBC.[6,11] The aim of this multicentric study is to evaluate Spanish patients with HT1 in treatment with nitisinone, including novels aspects, not previously studied, as nutritional assessment and anthropometric parameters.
2. Methods
2.1. Study design
This is a retrospective observational multicentric study developed in Spanish HT1 patients treated with NTBC. The study protocol was approved by the local research ethics committee of each Center. All HT1 patients who received treatment with NTBC were considered eligible and were recruited after informed consent had been obtained.
2.2. Population
Patients were recollected through a noninterventional post-authorization safety study to evaluate long-term safety of NTBC (Orfadin) treatment in standard clinical care. Data were covered by the physicians responsible for these patients from 14 regions in Spain. The contact was made through the Scientific Society of Inborn Errors of Metabolism of Spain.
At diagnosis, the following parameters were evaluated: age and year of diagnosis, sex, type of diagnosis (by clinical symptoms or by newborn screening [NBS]), mutational analysis of the FAH gene, phenotype (acute, subacute, or chronic attending to the onset of symptoms: <6 months; 6–24 months or >24 months, respectively), presence of clinical symptoms and/or biochemical markers and/or imaging studies suggesting diagnosis (liver, renal, haematological, cognitive, ophthalmologic status, and others). Treatment was held according to Spanish Guidelines,[17] during follow-up patients received not only NTBC treatment but also a dietary restricted in Tyr and Phe, according to age and tolerance. Clinical course was subsequently monitored. Follow-up included start date of NTBC treatment and follow-up time; dose of NTBC per kg administered; adherence to diet and pharmacological treatment (good, regular, poor, and very poor); measurement of SA, Phe, Tyr, alpha-fetoprotein (AFP) and NTBC blood concentrations, SA in urine; assessment of anthropometric parameters (weight, height, body mass index [BMI]); evolution of clinical symptoms (liver, renal, haematological, cognitive, ophthalmologic status, and others); development or not of HCC; behavioral assessment (deficit or not of attention and hyperactivity syndrome); schooling (normal or with support); intellectual quotient (IQ) testing.
2.3. Methods
2.3.1. Anthropometric methods
Recumbent length was measured with a measuring board and weight with a manual baby scale until the age of 24 months. Thereafter, standing height was measured with a wall-mounted stadiometer and body weight, to the nearest 100 g, with digital scales. Patients were weighted barefoot and after overnight fasting. Weight and height percentiles and z-scores were calculated using the online nutritional assessment tool of the Spanish Society of Gastroenterology, Hepatology, and Nutrition (www.gastroinf.es).
The nutritional status was performed by calculating the BMI as BMI = weight (kg)/height2 (m2). Subjects were classified according to BMI by using the WHO Child Growth Standards (underweight: BMI percentile <15; normal: BMI percentile 15–85, overweight: BMI percentile 85–95, obese: BMI percentile >95).[18,19]
2.3.2. Analytical methods
NBS for HT1 by levels of Tyr (cut-off level: 200 μmol/L) and SA as the second marker or only SA on filter paper blood spots using tandem mass spectrometry was done in the regions of Spain where expanded NBS was first performed (Galicia, Cataluña, Madrid, and Murcia).
Quantitative analysis of amino acids (Phe, Tyr) in plasma samples was carried out by ion-exchange chromatography (Biochrom 30 autoanalyzer) after deproteinization of the sample (plasma or urine) with 5-sulfosalicylic acid. SA was detected in dried blood spot by a kit of Chromsystems (Teknokroma, Barcelona, Spain) by tandem mass spectrometry. AFP and some other routine biochemical parameters were analyzed in the respective laboratories associated with each center, but because the reference ranges of these parameters are standardized, we assumed that the slight variability among them is of no clinical significance. NTBC concentrations were evaluated by using tandem-mass spectrometry.[20]
Reference range values for biochemical parameters were: SA (<0.1 μmol/L in blood and <1 mmol/mol creatinine in urine), Phe (35–120 μmol/L), Tyr (<400 μmol/L), AFP (<10 μg/L, in the first year of life less than 77 μg/L in girls and 22 μg/L in boys), NTBC (30–60 μmol/L).
Abdominal ultrasound and/or liver magnetic resonance imaging, and ophthalmologic examination were also done in each reference center according to the chronology indicated in the Spanish follow-up protocol.
Molecular testing: DNA was isolated and sequenced by standard procedures for blood samples of all patients and their parents. Molecular analysis of FAH was performed using standard procedures. Primers were designed to overlap the coding sequences and their flanking regions (sequences available on request). Polymerase chain reaction products were purified by ExoSap (usb) enzyme and sequenced using a Big Dye Terminator Cycler Sequencing Ready reaction kit and the manufacturer's protocol (Applied Biosystems, Waltham, Massachusetts). The sequencing reactions were performed in an ABI 3130XL Genetic Analyser.
2.3.3. Cognitive function
Cognitive function was assessed by psychomotor development index (PDI) or IQ using the Wechsler intelligence scale R for school-age children, the McCarthy Scales of Children's Abilities for preschool-age children, and the Brunet–Lèzine scale in infants. The overall index score of PDI or IQ is considered in the normal range when it is above 85.
2.4. Statistical analysis
Data were analyzed using the R statistical package v3.4.0 [R Core Team (2017). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.r-project.org/, last accessed: 12/07/2018]. Sample normality was assessed using both the Kolmogorov–Smirnov and the Shapiro–Wilk tests. In cases where one of the variables was quantitative and the other qualitative, we applied the Student t test if the quantitative variable was normally distributed, and the Wilcoxon signed-rank test otherwise. Fisher exact test or Chi-squared tests were used when both variables were qualitative. To assess correlations between quantitative variables, we used the Pearson correlation for normally distributed data and the Spearman rho otherwise. The resulting P-values were adjusted using the Bonferroni correction and only values lower than .05 were considered significant.
3. Results
3.1. Diagnosis
We evaluated 52 Spanish patients, 7 adults and 45 children (57.7% male; mean age: 11 years and 8 months, range: 2 months–24 years) with HT1. The characteristics of the study population are detailed in Table 1 . All patients receive treatment with NTBC from diagnosis. Eight of them (p1–p8) were early detected by NBS, all being asymptomatic except for 1 that already had liver failure data at diagnosis. In 5 patients NBS was performed using Tyr in blood spot as a primary marker and SA as a secondary marker, and in the remaining 3 patients SA was the primary marker. The average age for diagnosis was 12.7 days (range 7–21).
Table 1.
Characteristics of tyrosinemia 1 patients at diagnosis.
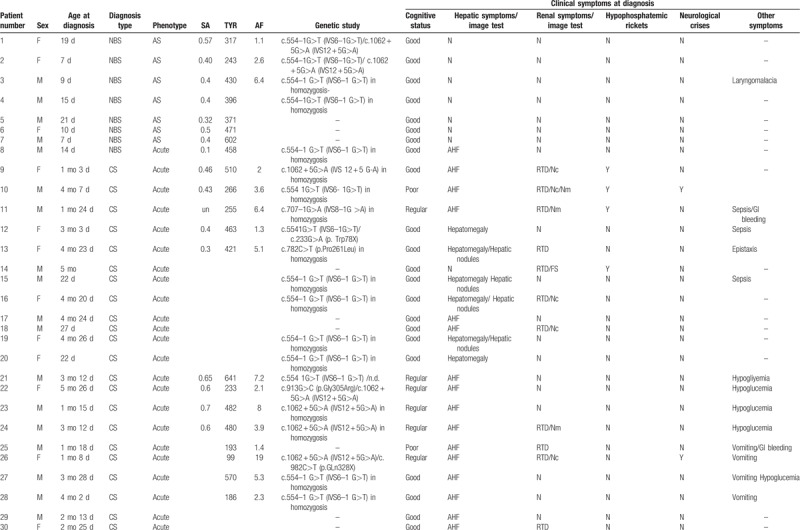
A total of 44 (84.6%) of our patients were diagnosed at a later stage due to by the appearance of clinical symptoms (no-NBS group) (median age at diagnosis: 241 days, range: 22–1428) with noteworthy delay in diagnosis respect with NBS cases (P < .001). Acute presentation was the most frequent form (24/44; 54.5%), followed by subacute presentation in 31.8% (14/44) and chronic presentation in 13.6% (6/44). Liver affectation was markedly more frequent at diagnosis in cases with late diagnosis (43/44) compared to cases detected by NBS (1/8) (P < .001). In acute forms, acute liver failure stands out as the most frequent hepatic manifestation (75% – 18/24) followed by hepatomegaly (25% – 6/24) and hepatic nodules (20.8% – 5/24). In those patients with subacute/chronic form the incidence of hepatomegaly, in 3 patients together with splenomegaly, and hepatic nodules is greater (90% and 35%, respectively). In a patient with a chronic form the debut was due to a hepatoblastoma.
In no-NBS group kidney involvement at diagnosis was present in 20/44 (45.4%), also more frequent than in NBS-group (P = .03), including renal tubular dysfunction (17/44), nephromegaly (7/44), nephrocalcinosis (7/44), renal dysfunction (1/44), and Fanconi syndrome (1/44, patient 14), with higher frequency in acute forms of tubulopathy and nephrocalcinosis and in subacute/chronic forms of nefromegaly. Hypophosphatemic rickets was observed in 4 acute and 1 subacute patients, and 3 patients had neurological crises at diagnosis. Other manifestations at diagnosis in no-NBS group were vomiting (18%), hypoglycemia (15.9%), sepsis (9%), and growth failure (6.8%).
As reflected in Table 2, Tyr, AFP, and SA levels at diagnosis were slightly lower in NBS-group than in those with late diagnosis (Tyr: 416.7 ± 153.5 vs 469.95 ± 231 μmol/L, P = .79; AFP: 4.3 vs 5.9 ng/mL, P = .84; and SA: 0.42 vs 0.5 μmol/L, P = .53). Both AFP and Tyr levels show a lineal correlation with the time of evolution of the disease at diagnosis with higher levels in chronic forms of tyrosinemia (P = 0.048 for AFP). No relevant differences were observed in Phe levels between the 2 groups.
Table 1 (Continued).
Characteristics of tyrosinemia 1 patients at diagnosis.
3.2. Follow-up
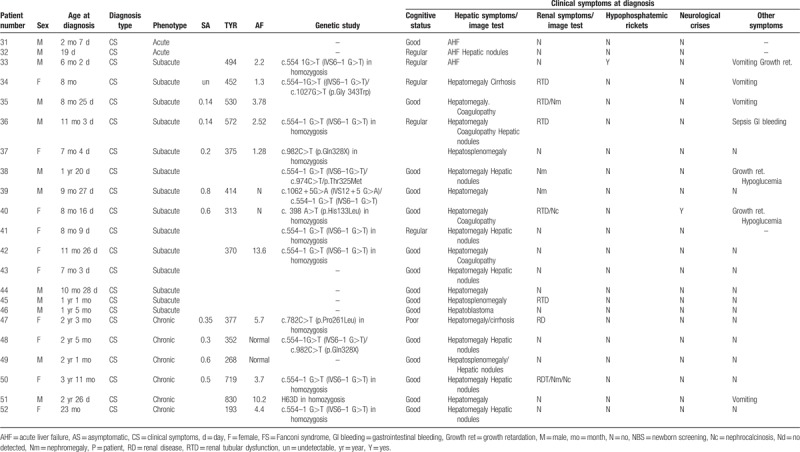
The average follow-up period was 6.1 ± 4.9 years in patients with early diagnosis and 10.6 ± 5.4 years in those with late diagnosis. All patients received NTBC from diagnosis with an evolving average dose of 0.82 mg/kg/d (range: 0.44–1.34) according to NTBC levels. A good diet adherence was observed in 87% of NBS-patients and 65% of patients with late diagnosis, while only 1 patient showed poor adherence to pharmacological treatment. General clinical outcome was positive in most patients; only 6 patients, all of them not diagnosed by NBS, developed hepatic manifestations, in addition to the patient with hepatoblastoma at diagnosis: hepatic nodules (3; cases number 32, 39, and 50), hepatic hyperechogenicity (case number 40), hepatic cirrhosis (case 50), and hepatic steatosis (case 45). Only 1 case of new hepatic malignancy was observed in our series: a female patient with a subacute form (case number 42), good dietetic and therapeutic adherence, and Tyr and NTBC levels within range, who developed a hepatocarcinoma at the age of 13 years and needed liver transplantation. All NBS patients continued free of liver and renal disease.
In no-NBS group renal evolution was satisfactory after treatment in 41/44 patients, 1 patient developed renal dysfunction secondary to chemotherapy treatment of hepatoblastoma and 2 maintained nephrocalcinosis. Hematological and ophthalmological evolution was good except in 1 patient, with very poor diet and treatment adherence, who developed ocular manifestations. Six patients, 5 of them with acute form and 1 with subacute diagnosis, presented seizures: photosensitive epilepsy (1/6), myoclonic epilepsy (2/6), absence crisis (1/6), and 2 with an isolated seizure.
It highlights that anthropometric evolution is our series is satisfactory as almost half of the HT1 patients maintained a BMI adequate to their age and sex (47.06%). Only 3 patients, all of them of late diagnosis, showed underweight in their follow-up, nevertheless the percentage of obesity and overweight in our study population is high, and interestingly, no significant differences were observed in this percentage between NBS and no-NBS groups. No patient has shown development of diabetes or hyperglycemia so far.
We evaluate the correlation between BMI with several variables: Tyr levels, duration of pharmacologic or dietetic treatment, and adherence to both treatments, and we found a significant correlation between BMI and NTBC treatment (P-value: 9.62e−05; rho = 0.5987515). BMI was both lower in patients with good dietary adherence (20.40 ± 4.43 vs 24.30 ± 6.10; P = .08) and in those with good adherence to NTBC treatment (21.19 ± 4.68 vs 28.58 ± 213.79).
Learning difficulties were not observed in NBS-patients but in late-diagnosis group pedagogical support or curricular adaptation was necessary in 15% of patients, and 6.8% (3/44) showed school failure, 1 case with concomitant diagnosis of Angelman syndrome and 2 with poor diet and pharmacological adherence. Thirty-eight patients had cognitive evaluation. IQ was ≥85 in all NBS- and 68.75% of late diagnosis cases evaluated; in this second group, 3 patients with acute-onset and 1 with chronic presentation, showed IQ ≤70. A negative correlation between the duration of NTBC treatment and IQ was not verified in our series by a logistic regression model. Seventeen percent of HT1 patients showed attention hyperactivity and attention deficit and no neurological crises were observed in the study cohort during the follow-up period.
After treatment with NTBC a reduction in Tyr and AFP levels was observed in all the study groups, significant for AFP in the group with clinical diagnosis (P = .03) and specially in the subacute/chronic forms (P = .018) (Fig. 2).
Figure 2.
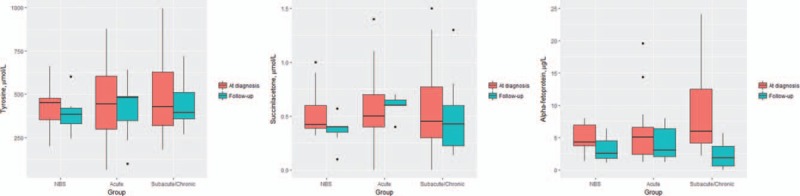
Comparison between tyrosine, succinylacetone and alpha-fetoprotein levels in the NBS, acute and subacute/chronic patients at diagnosis and in the follow-up, after treatment with NTBC. NBS = neonatal screening, NTBC = nitisinone.
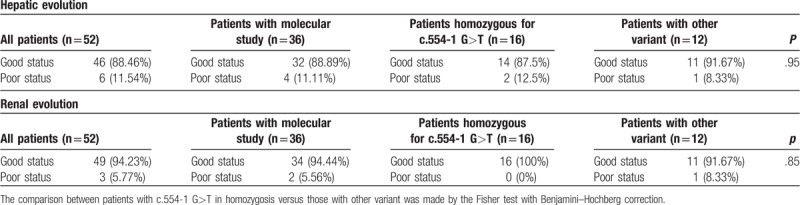
Genetic study was carried out in 36 patients and 12 different variants in FAH gene were detected. All variants were previously described,[26–31] except a novel variant (H63D) identified in homozygosis in a male patient with a chronic form of HT1. The most prevalent Spanish mutation is c.554–1 G>T (IVS6–1 G>T) (63.89% – 23/36), 16 patients showed this mutation in homozygosis (16/23 – 69.57%) and 7 in heterozygosis (7/23 – 30.43%) (Table 3). In the group of patients homozygous for c.554–1 G>T, 93.8% had hepatic and 50% renal involvement at diagnosis. The hepatic evolution of HT1 patients with the aforementioned variant, considered together in both homo and heterozygosis, is slightly worse when compared to patients with other mutations (poor hepatic status in the follow-up: 12.5% vs 8.33%, respectively, P = .95). Moreover, the patient who developed hepatocarcinoma is homozygous for this mutation. Thus, the mutation c.554–1 G>T, despite the absence of statistical significance with patients harboring other mutations, probably due to the low sample size in mutational comparison groups, seems to condition a worse hepatic prognosis even in spite of the treatment. The comparison between the follow-up according to the genotype is shown in Table 4.
Table 2.
Clinical, analytical, and anthropometric evolution of tyrosinemia type 1 Spanish patients.
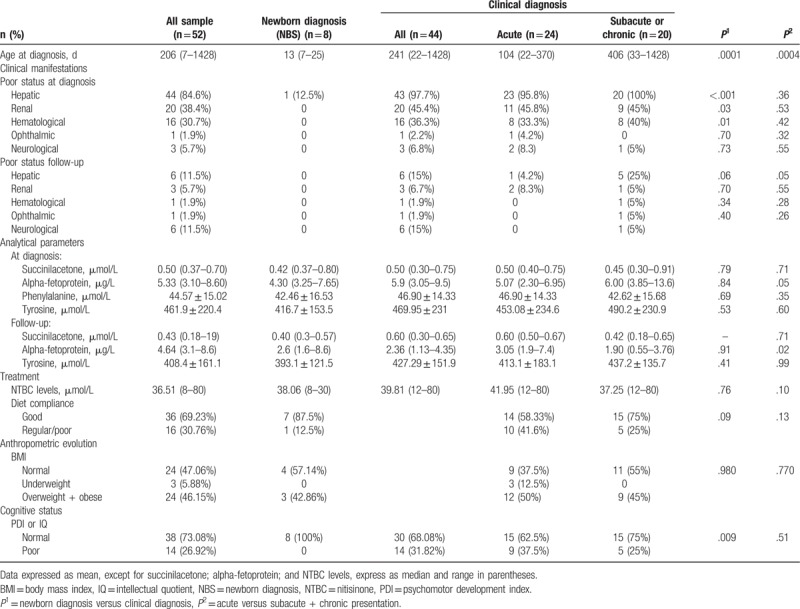
Table 3.
Molecular study of the Spanish HT1 patients.
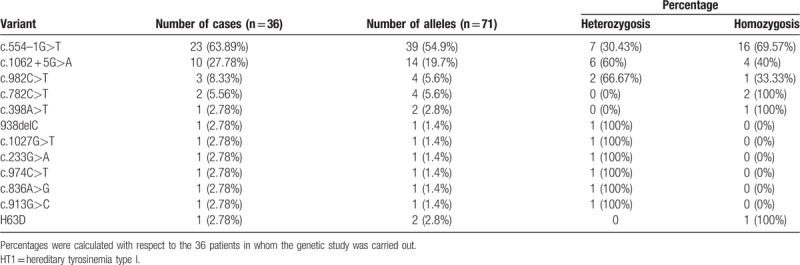
Table 4.
Comparison between hepatic and renal evolution according to the genotype.
4. Discussion
NTBC has brought about a drastic improvement in the treatment and prognosis of HT1 as all the clinical multiorganic complications associated to toxic metabolites accumulation in this pathology have been shown to be preventable with NTBC therapy.[6,11,21] Evidence supported that earlier treatment with NTBC and dietary protein restriction within the first 2 months of life showed a lower rate of complications (hepatic and renal dysfunction)[12]; shorter hospital admissions and a reduced need for liver transplantation.[22] In accordance with previous series of HT1,[6,11,21,23] our revision confirms a clear clinical benefit with the combination of NTBC and dietary treatment. From the 43 patients with hepatic affectation at diagnosis (1 NBS-patient, 23 with acute onset and 19 with subacute-chronic presentation) only 6 maintain hepatic damage in the follow-up. In The Québec NTBC study no patient with early treatment (before 1 month of age) had developed to date HCC; in our series, with a long-follow up period (up to 15 years in some patients), but shorter than the surveillance of the referred study (up to 20 years), a new hepatic malignancy was observed in the follow-up in a patient diagnosed at about 1 year of age who maintained a good metabolic control.
As signaled by Geppert et al systematic review,[22] any of our patients developed renal dysfunction after NTBC treatment and, moreover, renal outcome of those HT1 patients with renal pretreatment involvement was meanly satisfactory.
A good metabolic control, judged by normalization of plasma SA and Tyr levels, was achieved quickly in treated patients. In our series, the greatest effect in reducing AFP levels was observed in no-NBS group, especially in those patients with subacute/chronic forms (Fig. 1).
Actually, despite this good metabolic response, a main concern in HT1 treatment is the high frequency of neurocognitive impairment of HT1 patients showing a lower IQ, impaired executive functioning, motor abilities, and social cognition and schooling problems.[2,15,16] The exact pathophysiological mechanisms underlying these impairments were not well elucidated.[24] Despite that no significant correlation between Tyr or Phe plasma levels and IQ at school age was found in a recent study,[25] a relation with chronic hypertyrosinemia has been suggested and some clinical reports seemed to support this hypothesis.[23,26]
Our series showed a clearly better neurocognitive outcome in NBS-patients with early treatment. Learning difficulties were only observed in 23% of late-diagnosis patients The finding of frequent attention hyperactivity and attention deficit (17%) is consistent with the results reported in other studies[27] and less of 10% of HT1 patients presented seizures, percentage similar to the observed in an European cross-sectional study.[6]
It is remarkable in our series that QI in the follow-up remains within a normal range in all the NBS-patients evaluated and in 73% of those with clinical diagnosis and only 4 patients from the 38 with QI assessment showed QI <70. This results contrast with those reported previously with a high number of patients with intellectual and psychomotor delay.[15,21,25]
Although the observation of a negative correlation between the duration of treatment with NTBC and IQ,[2] a recent animal model showed that this slower learning and cognitive impairment is caused by HT1 and not by NTBC treatment itself.[28] Our results endorse it, as no significant relation was demonstrated by a logistic regression model between the duration of NTBC treatment and IQ. Neither correlation was objectified between IQ and Phe/Tyr quotient (Spearman rho: 0.11; P-value: .57).
Although the low number of patients with poor dietetic-therapeutic adherence prevents an adequate statistical comparison, the mean BMI is clearly lower in those patients with good treatment compliance, which does not suggest that overweight/obesity is related to the use of special formulas free of Tyr and phenylalanine, which contrasts with studies carried out on other inborn errors of amino acid metabolism as phenylketonuria.[30]
The most prevalent mutation in our cohort, c.554–1 G>T (IVS6–1 G>T), is a splice mutation with higher prevalence in Southern Europe and in Mediterranean region, previously reported in other studies of Spanish HT1 patients,[10,29,31] and ranks second in prevalence in European HT1 patients.[4] The second mutation in frequency was c.1062 + 5 G>A [IVS 12 + 5 (G->A)], also a splice mutation previously described in our country.[10,31,32] Both mutations together accounts for 74.6% of the FAH alleles investigated (c.554–1 G>T: 39 alleles and c.1062 + 5 G>A: 14 alleles). The rest of mutations are rare in our population: c.982C>T,[30] c.836A>G,[31] c.233G>A,[31] c.974C>T[10] and 1 of them (c.782C>T) was identified for the first time in Spanish HT1 patients. This mutation, c.782C>T, was found in 2 Caucasians patients from Madrid is a missense mutation found in 100% of Ashkenazi-Jewish patients examined in Israel[33] and also described in Saudi Arabia and Egypt.[34]
The main limitations of our study are the small number of NBS patients included, reflecting that HT1 screening is not included in the neonatal screening of most regions of our country which results in a limitation of the statistical power of some comparisons as well as the fact that the mutational study was not performed in the entire population studied.
This series confirms that NTBC treatment had clearly improved the vital prognosis and quality of life of HT1 patients, but it also reveals cognitive manifestations and learning difficulties a medium term in a considerable number of patients, as well as, in a novel way, a high percentage of overweight/obesity in HT1 patients.
Author contributions
Conceptualization: María-Luz Couce, Luis Aldámiz-Echevarría, Isidro Vitoria, Luis Peña-Quintana.
Data curation: Victor Navas, Elena Martín-Hernández, Camila García-Volpe, Guillem Pintos, Luis Peña-Quintana, Tomás Hernández, David Gil-Ortega, Félix Sánchez-Valverde, María Bueno, Encarna López-Ruzafa.
Formal analysis: María-Luz Couce, Elena Martín-Hernández, Luis Peña-Quintana, Iria Roca.
Funding acquisition: Carmen Diaz.
Methodology: María-Luz Couce, Isidro Vitoria, Victor Navas, Elena Martín-Hernández, Camila García-Volpe, Guillem Pintos, Tomás Hernández, David Gil-Ortega, Félix Sánchez-Valverde, María Bueno, Encarna López-Ruzafa.
Software: Iria Roca.
Supervision: María-Luz Couce, Luis Aldámiz-Echevarría, Isidro Vitoria, Iria Roca, Carmen Diaz.
Validation: María-Luz Couce, María Bueno.
Writing – original draft: María-Luz Couce, Paula Sánchez-Pintos.
Footnotes
Abbreviations: AFP = alpha-fetoprotein, BMI = body mass index, FAH = fumarylacetoacetate hydrolase, HCC = hepatocellular carcinoma, HT1 = hereditary tyrosinemia type 1, IQ = intellectual quotient, NBS = newborn screening, NTBC = nitisinone, SA = succinylacetone, Tyr = tyrosine.
How to cite this article: Couce ML, Sánchez-Pintos P, Aldámiz-Echevarría L, Vitoria I, Navas V, Martín-Hernández E, García-Volpe C, Pintos G, Peña-Quintana L, Hernández T, Gil D, Sánchez-Valverde F, Bueno M, Roca I, López-Ruzafa E, Díaz-Fernández C. Evolution of tyrosinemia type 1 disease in patients treated with nitisinone in Spain. Medicine. 2019;98:39(e17303).
Article-processing charge was covered by the Fundación IDIS-C012.
The authors have no conflicts of interest to disclose.
References
- [1].De Laet C, Dionisi-Vici C, Leonard JV, et al. Recommendations for the management of tyrosinaemia type 1. Orphanet J Rare Dis 2013;8:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].van Ginkel WG, Jahja R, Huijbregts SCJ, et al. Neurocognitive outcome in tyrosinemia type 1 patients compared to healthy controls. Orphanet J Rare Dis 2016;11:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].De Laet C, Munoz VT, Jaeken J, et al. Neuropsychological outcome of NTBC-treated patients with tyrosinaemia type 1. Dev Med Child Neurol 2011;53:962–4. [DOI] [PubMed] [Google Scholar]
- [4].Angileri F, Bergeron A, Morrow G, et al. Geographical and ethnic distribution of mutations of the fumarylacetoacetate hydrolase gene in hereditary tyrosinemia type 1. JIMD Rep 2015;19:43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sander J, Janzen N, Peter M, et al. Newborn screening or hepatorenal tyrosinemia: tandem mass spectrometric quantification of succinylacetone. Clin Chem 2006;52:482–7. [DOI] [PubMed] [Google Scholar]
- [6].Mayorandan S, Meyer U, Gokcay G, et al. Cross sectional study of 168 patients with hepatorenal tyrosinaemia and implications for clinical practice. Orphanet J Rare Dis 2014;9:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lindstedt S, Holme E, Lock EA, et al. Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet 1992;340:813–7. [DOI] [PubMed] [Google Scholar]
- [8].Holme E, Lindstedt S. Tyrosinaemia type I and NTBC (2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione). J Inherit Metab Dis 1998;21:507–17. [DOI] [PubMed] [Google Scholar]
- [9].Russo PA, Mitchell GA, Tanguay RM. Tyrosinemia: a review. Pediatr Dev Pathol 2001;4:212–21. [DOI] [PubMed] [Google Scholar]
- [10].Couce ML, Dalmau J, del Toro M, et al. Spanish Working Group on Tyrosinemia Type 1. Tyrosinemia type 1 in Spain: mutational analysis, treatment and long-term outcome. Pediatr Int 2011;53:985–9. [DOI] [PubMed] [Google Scholar]
- [11].Larochelle J, Alvarez F, Bussières JF, et al. Effect of nitisinone (NTBC) treatment on the clinical course of hepatorenal tyrosinemia in Québec. Mol Genet Metab 2012;107:49–54. [DOI] [PubMed] [Google Scholar]
- [12].Das AM. Clinical utility of nitisinone for the treatment of hereditary tyrosinemia type-1 (HT-1). Appl Clin Genet 2017;10:43–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nobili V, Jenkner A, Francalanci P, et al. Tyrosinemia type 1: metastatic hepatoblastoma with a favorable outcome. Pediatrics 2010;126:e235–8. [DOI] [PubMed] [Google Scholar]
- [14].Holme E, Lindstedt S. Nontransplant treatment of tyrosinemia. Clin Liver Dis 2000;4:805–14. [DOI] [PubMed] [Google Scholar]
- [15].Thimm E, Richter-Werkle R, Kamp G, et al. Neurocognitive outcome in patients with hypertyrosinemia type I after a long-term treatment with NTBC. J Inherit Metab Dis 2012;35:263–8. [DOI] [PubMed] [Google Scholar]
- [16].Bendadi F, de Koning TJ, Visser G, et al. Impaired cognitive functioning in patients with tyrosinemia type I receiving nitisinone. J Pediatr 2014;164:398–401. [DOI] [PubMed] [Google Scholar]
- [17].Couce ML, Aldámiz-Echevarría L, Baldellou A, et al. Recommendations and management of type I hereditary or hepatorenal tyrosinemia. An Pediatr (Barc) 2010;73:279.e1–4. [DOI] [PubMed] [Google Scholar]
- [18].WHO. Physical status: the use, interpretation of, anthropometry. Report of a WHO expert committee. World Health Organ Tech Rep Ser 1995;854:1–452. [PubMed] [Google Scholar]
- [19].WHO, Multicentre Growth Reference Study Group. WHO child growth standards based on length/height, weight and age. Acta Paediatr 2006;450:S76–85. [Google Scholar]
- [20].Herebian D, Spiekerkötter U, Lamshöft M, et al. Liquid chromatography tandem mass spectrometry method for the quantitation of NTBC (2-(nitro-4-trifluoromethylbenzoyl)1,3-cyclohexanedione) in plasma of tyrosinemia type 1 patients. J Chromatogr B Analyt Technol Biomed Life Sci 2009;877:1453–9. [DOI] [PubMed] [Google Scholar]
- [21].Masurel-Paulet A, Poggi-Bach J, Rolland MO, et al. NTBC treatment in tyrosinaemia type I: long-term outcome in French patients. J Inherit Metab Dis 2008;31:81–7. [DOI] [PubMed] [Google Scholar]
- [22].Geppert J, Stinton C, Freeman K, et al. Evaluation of pre-symptomatic nitisinone treatment on long-term outcomes in tyrosinemia type 1 patients: a systematic review. Orphanet J Rare Dis 2017;12:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Raimann E, Cornejo V, Arias C, et al. Clinical follow up of Chilean patients with tyrosinemia type 1 treated with 2-(2-nitro-4-trifl uoromethylbenzoyl)-1,3-ciclohexanedione (NTBC). Rev Med Chile 2012;140:169–75. [DOI] [PubMed] [Google Scholar]
- [24].van Ginkel WG, Jahia R, Huijbregts SCJ, et al. Neurological and neuropsychological problems in tyrosinemia type 1 patients. Adv Exp Med Biol 2017;959:111–22. [DOI] [PubMed] [Google Scholar]
- [25].García MI, de la Parra A, Arias C, et al. Long-term cognitive functioning in individuals with tyrosinemia type 1 treated with nitisinone and protein-restricted diet. Mol Genet Metab Rep 2017;11:12–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Walker H, Pitkanen M, Barrington SF. Three cases of hereditary tyrosinaemia type 1: neuropsychiatric outcomes and brain imaging following treatment with NTBC. JIMD Rep 2018;40:97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pohorecka M, Biernacka M, Jakubowska-Winecka A, et al. Behavioral and intellectual functioning in patients with tyrosinemia type 1. Pediatr Endocrinol Diabetes Metab 2012;18:96–100. [PubMed] [Google Scholar]
- [28].Hillgartner MA, Coker SB, Koening AE, et al. Tyrosinemia type I and not treatment with NTBC causes slower learning and altered behavior in mice. J Inherit Metab Dis 2016;39:673–82. [DOI] [PubMed] [Google Scholar]
- [29].Bergman AJ, van den Berg IE, Brink W, et al. Spectrum of mutations in the fumarylacetoacetate hydrolase gene of tyrosinemia type 1 patients in northwestern Europe and Mediterranean countries. Hum Mutat 1998;12:19–26. [DOI] [PubMed] [Google Scholar]
- [30].Gokmen Ozel H, Ahring K, Bélanger-Quintana A, et al. Overweight and obesity in PKU: the results from 8 centres in Europe and Turkey. Mol Genet Metab Rep 2014;1:483–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Arranz JA, Pinol F, Kozak L, et al. Splicing mutations, mainly IVS6-1(G>T), account for 70% of fumarylacetoacetate hydrolase (FAH) gene alterations, including 7 novel mutations, in a survey of 29 tyrosinemia type I patients. Hum Mutat 2002;20:180–8. [DOI] [PubMed] [Google Scholar]
- [32].Pérez-Carro R, Sánchez-Alcudia R, Pérez B, et al. Functional analysis and in vitro correction of splicing FAH mutations causing tyrosinemia type I. Clin Genet 2014;86:167–71. [DOI] [PubMed] [Google Scholar]
- [33].Elpeleg ON, Shaag A, Holme E, et al. Mutation analysis of the FAH gene in Israeli patients with tyrosinemia type I. Hum Mutat 2002;19:80–1. [DOI] [PubMed] [Google Scholar]
- [34].Imtiaz F, Rashed MS, Al-Mubarak B, et al. Identification of mutations causing hereditary tyrosinemia type I in patients of Middle Eastern origin. Mol Genet Metab 2011;104:688–90. [DOI] [PubMed] [Google Scholar]