

Abstract
This manuscript describes an enantioselective synthesis of the naturally occurring alkaloid Citrinadin B. The synthetic effort revealed an anomaly in the original structural assignment that has led to the proposal of a stereochemical revision. This revision is consistent with the structures previously reported for a closely related family of alkaloids, PF1270A-C. The synthesis is convergent and employs a stereoselective intermolecular nitrone cyloaddition reaction as a key step.
Graphical Abstract
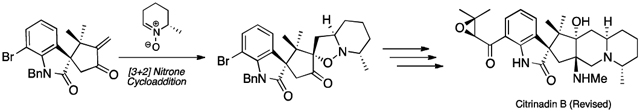
Citrinadin B (1) is the structurally least complex member in a family of related alkaloids that includes citrinadin A (2) and PF1270 A–C (3–5, respectively, Figure 1). Isolated from Penicillium citrinum N059 by Kobayashi, the citrinadins display modest cytotoxicity against murine leukemia L1210 cells and were assigned the structures illustrated below, including relative and absolute stereochemistry, via a combination of spectroscopic and chemical correlation studies.1 In contrast, the related PF1270 compounds (3–5), isolated from Penicillium waksmanii by Kushida, were found to display high affinity for the histamine H3 receptor.2 In this latter series, the assignment of structural and relative stereochemical features was based upon an X-ray crystallographic analysis of 3. Although the absolute stereochemistry of PF1270 A was not formally assigned by Kushida, the structures of alkaloids 3–5 as illustrated in Scheme 1 reflects what was proposed in the isolation paper. Notably, comparison of the published structures of the citrinadins (1 and 2) and the PF1270 compounds (3–5) reveals a critical difference in the proposed relative stereochemistry between the spirooxindole core of the molecule and the epoxyketone sidechain.3 Intrigued by the biological activity, structural complexity, and interesting stereochemical discrepancies found within this group of alkaloids, we endeavored to complete their total synthesis.4 Our initial efforts focused upon the preparation of citrinadin B (1), and our strategy was designed with the synthesis of alkaloids 2–5 in mind. Herein we report the initial phase of our efforts.
Figure 1.

Reported Structures for Citrinadins A, B and PF1270 A-C
Scheme 1.

Retrosynthetic Analysis of ent-Citrinadin B.
Mindful of the versatility that would be required of a synthesis designed to deliver the citrinadin and the PF1270 compounds, we developed a convergent approach toward citrinadin B (1) wherein the epoxyketone, spirooxindole, and methylpiperidine moieties would be introduced as independent components (Scheme 1). This strategy enables the preparation of various structurally and stereochemically diverse products.5 As illustrated, the aryl bromide 6 was envisioned to engage in a late stage coupling, which would allow for incorporation of either antipode of an intact and potentially labile epoxyketone. Likewise, to allow the construction of either spirooxindole antipode we planned to employ enone 8 which was envisioned as serving as the dipolarophile in an intermolecular [3+2] cycloaddition that would deliver the pertinent stereochemical features of the polycyclic core (8 + 9 → 7). A particularly intriguing aspect of this cycloaddition approach would be determining the effectiveness of asymmetric induction from the stereochemistry resident in nitrone 9. Given that either antipode of 9 is readily available from either D- or L- alanine (10) and both stereochemistries were potentially present in the natural products (vide supra) we opted to initially prepare the spirooxindole component (8) as a racemate and explore the ability of 9 to act as a resolving agent.6 We anticipated that enone (±)-8 would be available from dibromoaniline 11.
In the forward sense, exposure of dibromoaniline 11 to trimethylaluminum followed by lactone 127 furnished an intermediate alcohol that was protected as the corresponding silyl ether 13 (Scheme 2).8 Cyclization of amide 13 to oxindole (±)-14 under Heck conditions9 was followed by benzyl protection, silyl group cleavage, and alcohol oxidation to furnish aldehyde (±)-15. At this point we began setting the stage for an eventual reductive ene-yne cyclization by treating oxindole (±)-15 with ethynyl Grignard. Addition of the Grignard was immediately followed by protection of the resulting diastereomeric alcohols as their corresponding silyl ethers (±)-16. Cyclization of (±)-16 was thereafter accomplished under conditions developed by Trost,10 which was then followed by TBAF mediated deprotection to afford a diastereomeric mixture of alcohols ((±)-17). Oxidation of the latter under Swern conditions provided the dipolar cycloaddition substrate, enone (±)-8.
Scheme 2.

Synthesis of Enone (±)-8
With both enone (±)-8 and nitrone (−)-911 in hand we began exploring the critical [3+2] cycloaddition (Scheme 3). Given the potential for formation of numerous diastero- and regioisomeric products, we were delighted to find that under a variety of conditions only two diastereomeric cycloaddition products, (+)-7 and (−)-18, are observed. After considerable experimentation, we found that addition of L-proline to this reaction had a beneficial affect on both rate and the observed dr. Extensive studies into the resultant stereochemistry revealed that cycloadducts (+)-7 and (−)-18 differed only by their configuration at the spirooxindole center (C(3)).12 Moreover, the relative stereochemistry resident in the minor product ((+)-7) was as needed for the conversion to the natural product.
Scheme 3.

Cycloaddition of Enone (±)-8 and Nitrone (−)-9
Although establishing the viability of the critical [3+2] cycloaddition was an important milestone, many challenges remained in advancing cycloadduct (+)-7 to citrinadin B (1). In particular, completion of the natural product would require the addition of a carbon atom, as well as closure of the central fused D ring of the citrinadin core. In order to address both of these concerns, we reasoned that a Corey-Chaykovsky epoxidation would allow for introduction of a single carbon and set the stage for subsequent ring closure via intramolecular nucleophilic attack of the proximal nitrogen onto the derived epoxide. In the event, exposure of oxindole (+)-7 to dimethylsulfonium methylide furnished the corresponding spiroepoxide (+)-19 as a single diastereomer.13 While initial attempts to promote intramolecular opening of the epoxide were unsuccessful, we eventually found that exposure of epoxide (+)-19 to in situ generated TMSI furnished ammonium salt (+)-21 in good yield, presumably via the intermediacy of iodide 20.14 Moreover, ammonium salt (+)-21 proved to be an excellent substrate for a Zn mediated N-O bond cleavage reaction that delivered diol (+)-22. Importantly, diol (+)-22 furnished crystals suitable for an X-ray analysis that confirmed our previous structural assignment, as well as the stereochemical fidelity in the steps leading to this key polycyclic intermediate.
With the carbocyclic core completed, our attention next turned to installation of the methylamine group and oxindole deprotection. Unfortunately, we quickly found the benzyl protecting group to be intransigent to removal. After an exhaustive screening of conditions the most reliable combination of reagents was found to be t-BuLi and O2.15 Unfortunately these harsh conditions were not compatible with the aryl bromide moiety of diol (+)-22. (Scheme 1).
In light of this fact, we planned to make use of the aryl bromide to install a side chain surrogate prior to benzyl deprotection. To this end, diol (+)-22 was converted to the corresponding epoxide (+)-6, which was subsequently coupled with 3-methyl-1-butyne under Sonogashira conditions to give alkyne (+)-24 (Scheme 5).16Importantly the alkyne moiety in the derived product ((+)-24) proved very resilient to the harsh benzyl deprotection conditions, and tolerated a subsequent epoxide opening reaction with MgCl2/NaN3 to furnish azide (+)-25. Thereafter, exposure of azide (+)-25 to gold mediated oxidation conditions reported by Zhang allowed conversion of the alkyne moiety to an enone, thus delivering azido alcohol (+)-26.17
Scheme 5.

Epoxyketone Installation and Elaboration
Given the necessity of incorporating an epoxyketone precursor, a diastereoselective enone epoxidation was now required to complete citrinadin B. Although many different epoxidation conditions were explored, a protocol developed by Enders proved most effective.18 At the outset of our epoxidation studies we explored the intrinsic stereochemical bias of (+)-26 under the Enders conditions, and thus ran the reaction in the absence of any chiral additive. As illustrated in Scheme 5, this resulted in a mixture (1:1) of the corresponding epoxides, which proved to be inseparable until converted to the corresponding Boc-protected variants (+)-27 and (−)-28. In further experiments with the Enders system we discovered that the ratio of epoxide diastereomers could be increased in favor of (−)-28 when (S,S)-ephedrine was used as the additive; thus, in accord with the predictive models put forth by Enders, we assigned (−)-28 as possessing the (S)-configuration at the epoxide stereocenter.19 The Enders model, coupled with the fact that our synthesis relied upon L-alanine, meant that advancement of the (R)-configured epoxide ((+)-27) would be expected to deliver enantiomeric citrinadin B, in accord with Kobayashi’s structural assignment.
At this point, the final transformation required was the conversion of the azide moiety to the corresponding methylamine. Given that we had access to (+)-27 and (−)-28, we decided to subject both to a final three-step sequence that involved azide reduction, monomethylation, and deprotection. To our delight, both (+)-27 and (−)-28 were readily advanced through this sequence and furnished what we expected to be 1 and (+)-29, respectively. To our surprise,1H and13C NMR data obtained for 1, the material deriving from (+)-27 and anticipated to be enantiomeric to citrinadin B, did not match that reported for the natural product.20 However, we found that the spectral data obtained for (+)-29 was a much closer match to Kobayashi’s reported data. Moreover, scrutinizing the circular dichroism spectra of both 1 and (+)-29 yielded an unanticipated result. While the CD spectra of 1 appeared mismatched to that of citrinadin B, the CD spectra of (+)-29 was an excellent match. In light of the chiroptical data obtained, as well as the use of L-alainine as our nitrone starting material, we conclude that a structural reassignment of citrinadin B is necessary. We propose that the absolute stereochemistry of the spirooxindole and stereogenic centers residing in the fused tricyclic region are opposite to that originally assigned by Kobayashi, and thus the structure of citrinadin B is in fact (+)-29. In addition to being consistent with the data we have obtained to date, this reassignment brings the structure of citrinadin B in line with that determined crystallographically for PF 1270A (vide supra). Similar conclusions were reached by Martin in the course of his elegant synthesis of citrinadin A.21
In conclusion, we have developed a convergent synthetic strategy that can be used to provide stereocontrolled access to citrinadin B ((+)-29). Completion of this stereoselective synthesis resulted in a stereochemical revision that more closely aligns the stereostucture of citrinadin B to the PF1270A-C alkaloids (3–5). Based on these studies, efforts in our laboratories are focused on adapting this synthetic strategy to the preparation of Citrinadin A (2), as well as PF1270A-C (3–5).
Supplementary Material
Scheme 6.

Completion and Structural Revision of (+)-Citrinadin B
Acknowledgment.
Financial support was provided by Amgen, Bristol-Myers Squibb, and the NSF (CHE-1058292). J.E. thanks the NIH (GM095076) a postdoctoral fellowship. T.M. thanks the Uehara Foundation, Professor Toshiaki Sunazuka, Professor Satoshi Ōmura, and the Kitasato Institute, for postdoctoral support. In addition, Dr. Chris Rithner, Don Heyse, and Don Dick are acknowledged for their assistance in obtaining crystallographic and spectroscopic data. Finally, we thank Professor Stephen Martin for the collegial exchange of information regarding his work on Citrinadin A.
Footnotes
Supporting Information Available: Experimental and characterization details (PDF, CIF). This material is available free of charge via the Internet at http://pubs.acs.org
References:
- (1).(a) Tsuda M; Kasai Y; Komatsu K; Sone T; Tanaka M; Mikami Y; Kobayashi J Org. Lett 2004, 6, 3087–3089; [DOI] [PubMed] [Google Scholar]; (b) Mugishima T; Tsuda M; Kasai Y; Ishiyama H; Fukushi E; Kawabata J; Watanabe M; Akao K; Kobayashi JJ Org. Chem 2005, 70, 9430–9435. [DOI] [PubMed] [Google Scholar]
- (2).Kushida N; Watanabe N; Okuda T; Yokoyama F; Gyobu Y; Yaguchi TJ Antibiotics 2007, 60, 667–673. [DOI] [PubMed] [Google Scholar]
- (3).If Kushida had illustrated the antipode, one might have attributed the difference to the epoxide stereochemistry. Based on the current study (vide infra) we believe Kushida’s depiction of absolute stereochemistry to be correct.
- (4).For previously reported synthetic approaches to the citrinadins, see:; (a) Pettersson M; Knueppel D; Martin SF Org. Lett 2007, 9, 4623–4626. [DOI] [PubMed] [Google Scholar]; (b) Guerrero CA; Sorensen EJ; Org. Lett 2011, 13, 5164–5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).It is important to note that the retrosynthetic analysis illustrated in Scheme 2 reflects the fact that our synthetic studies to date have been performed using L-alanine, the natural and least expensive antipode. Based on Kobayashi’s stereochemical assignment we expected our synthesis to furnish enantiomeric citrinadin B.
- (6).For the enantioselective preparation of 9, see:; Chackalamannil S; Wang Y Tetrahedron 1997, 53, 11203–11210. [Google Scholar]
- (7).Murphy JA; Rasheed F; Roome SJ; Scott KA; Lewis NJ Chem. Soc., Perkin Trans 1 1998, 2331–2339. [Google Scholar]
- (8).Lipton MF; Basha A; Weinreb SM Org. Synth 1979, 59, 49–52. [Google Scholar]
- (9).(a) Ableman MM; Oh T; Overman LE J. Org. Chem 1987, 52, 4130–4133; [Google Scholar]; (b) Artman III GD; Weinreb SM Org. Lett 2003, 5, 1523– 1526. [DOI] [PubMed] [Google Scholar]
- (10).Trost BM; Rise FJ Am. Chem. Soc 1987, 109, 3161–3163. [Google Scholar]
- (11).This material was prepared from L-alanine in accord with the procedure reported by Chakalamanil (see ref. 5). However, in our hands the ee of 9 varied between 85–95%. We illustrate here the worst-case scenario.
- (12).The stereochemistry present at C(3) of spirooxindoles 7 and 18 was assigned via NOESY studies, as well as correlation to X-ray structures of analogues lacking the bromo group. The relative and absolute stereochemical assignment of (+)-7 was verified via X-ray crystallography of a later intermediate (See (+)-22, Scheme 4).
- (13).(a) Corey EJ; Chaykovsky MJ Am. Chem. Soc 1965, 87, 1353– 1364; [Google Scholar]; (b) Aggarwal VK; Winn CL Acc. Chem. Res 2004, 37, 611–620. [DOI] [PubMed] [Google Scholar]
- (14).Caputo R; Mangoni Li; Nerl O; Palumbo G Tetrahedron Lett. 1981, 22, 3551–3552. [Google Scholar]
- (15).Williams RM; Kwast E Tetrahedron Lett. 1989, 30, 451–454. [Google Scholar]
- (16).Sonogashira K; Tohda Y; Hagihara N Tetrahedron Lett. 1975, 16, 4467–4470. [Google Scholar]
- (17).Lu B; Li C; Zhang LJ Am. Chem. Soc 2010, 132, 14070–14072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Enders D; Zhu J; Raabe G Angew. Chem., Int. Ed. Engl 1996, 35, 1725–1728. [Google Scholar]
- (19).This stereochemical assignment was also consistent with a comparison of chiroptical properties akin to that employed by Kobayashi in his original isolation paper.
- (20).The original isolation paper reports NMR data as being obtained on a free base. However, in our hands the greatest spectroscopic similarities were seen upon conversion to the corresponding TFA salt form. Identical NMR spectra were not obtainable, which we attribute to the presence of an unknown salt form in the natural sample. Unfortunately, authentic samples, original NMR data, and even the producing fungal strain were not available. Efforts to reisolate the citrinadins from a related penicillium are currently underway and will be reported in due course.
- (21).See the accompanying manuscript in this issue.
Scheme 4.

Completion of the Citrinadin Carbocyclic Core
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.