

Abstract
As a member of the myotubularin family, myotubularin related protein 3 (MTMR3) has been demonstrated to participate in tumor development, including oral and colon cancer. However, little is known about its functional roles in breast cancer. In the present study, the expression of MTMR3 in breast cancer was evaluated by immunohistochemical staining of tumor tissues from 172 patients. Online data was then used for survival analysis from the PROGgeneV2 database. In vitro, MTMR3 expression was silenced in MDA-MB-231 cells via lentiviral shRNA transduction. MTT, colony formation and flow cytometry assays were performed in the control and MTMR3-silenced cells to evaluate the cell growth, proliferation and cell cycle phase distribution, respectively. Western blotting was used to evaluate the protein expression levels of autophagy-related markers. The results demonstrated that the expression of MTMR3 in breast cancer tissues was significantly increased compared with adjacent normal tissues. MTMR3 was highly expressed in triple-negative breast cancer and was associated with disease recurrence. MTMR3 knockdown in MDA-MB-231 cells inhibited cell proliferation and induced cell cycle arrest and autophagy. The present results indicated that MTMR3 may have an important role in promoting the progression of breast cancer, and its inhibition may serve as a promising therapeutic target for breast cancer treatment.
Keywords: breast cancer, myotubularin related protein 3, proliferation, cell cycle, autophagy
Introduction
Breast cancer is the second leading cause of cancer-related deaths in women, and the incidence of breast is estimated to increase by ~0.5% annually (1,2). Despite enormous progress on breast cancer therapy, 20% of patients eventually die of their disease (3,4). Multiple studies have revealed that breast cancer is a heterogeneous and complex disease, whose pathophysiology cannot be explained by one or several mechanisms (5). A number of reliable biological markers have been found to predict the risk of recurrence and metastasis of breast cancer. Among these prognostic factors, estrogen receptor (ER), progesterone receptor (PR) and erb-b2 receptor tyrosine kinase 2 (HER2) status are significantly associated with overall clinical outcome (6). In addition, triple-negative breast cancer (TNBC), which is characterized by absent or minimal expression of ER, PR and HER2, is more aggressive and has a worse prognosis compared with the other subtypes of breast cancer (7). Recently, the expression profile of TNBC was described in a homogeneous population from northeastern Mexico and a novel gene signature related to metabolism was proposed (8). Such expression profiles are valuable for elucidating the mechanisms involved in tumorigenesis, diagnosis, prognosis and potential therapies.
Myotubularin related protein 3 (MTMR3) was first identified as an inositol lipid 3-phosphatase and belongs to the myotubularin (MTM) family (9). It confers a unique substrate specificity to phosphatidylinositol(3)-phosphate (PtdIns3P) and phosphatidylinositol(3,5)-biphosphate [PtdIns(3,5)P2], and the hydrolysis products, PtdIns and PtdIns5P, regulate cell activity and tumor progression (9,10). MTMR3 contains a N-terminal phosphatidylinositol lipids binding domain, a pleckstrin homology-GRAM (PHG) domain, and a C-terminal modulatory domain, that is responsible for endosomal localization and for binding to Ptdins3P (10). Inositol lipids are thought to be involved in various cellular functions, including proliferation, invasion, survival, cell cycle progression, migration and membrane trafficking (11). Several investigations into MTMR3 have underlined the key role of this gene in cancer development (12–14). Oppelt et al (14) reported that MTMR3 is widely expressed in most cancer cell lines and MTMR3-deficiency leads to reduced motility in rhabdomyosarcoma Rh30 cells and osteosarcoma U2OS cells.
Previously, a key role for MTMR3 was revealed in oral cancer. Kuo et al (15) demonstrated that miR-99a exerts antimetastatic effects through decreasing MTMR3 levels, suggesting that MTMR3 may serve as a potential therapeutic target for oral cancer. In breast cancer, although MTMR3 has been shown to be associated with cell cycle regulation and apoptosis in the SK-BR-3 cell line (16), the clinical impacts and functional role of MTMR3 remain unclear. Autophagy is a critical intracellular pathway that is associated with the bulk degradation of cytoplasmic components (17). In addition to acting as a tumor inhibitor, autophagy can also enhance cell survival to drive tumor growth and metastasis (18). Notably, depletion of MTMR3 was demonstrated to trigger autophagosome formation, but overexpression of MTMR3 resulted in smaller nascent autophagosomes, subsequently blocking autophagy (19). In breast cancer, MTMR3 has been reported to be regulated by miR-100, which could mediate apoptosis of breast cancer (16). However, the functions of MTMR3 in breast cancer have not been elucidated to date.
The present study explored the prognostic role of MTMR3 in breast cancer, and the effects of MTMR3 silencing in MDA-MB-231 cells. The aim of the present study was to investigate the clinical implication of MTMR3 and its potential biological or functional mechanisms.
Materials and methods
Tissue specimen collection and follow-up
A total of 172 patients were enrolled in the present study. All samples were collected at Xiangya Hospital (Changsha, China) between January 2013 and December 2013. For 52 of them, paired primary tumor tissues and adjacent normal tissues (>5 cm away from tumor area) were obtained. Formalin-fixed paraffin-embedded (FFPE) tumor tissues from 120 patients that underwent surgical removal were used to analyze MTMR3 protein expression levels. These patients were divided into two groups: Relapse group and non-relapse group. Relapse was defined as metastases or local recurrence occurring within 5 years; the terminal date for follow-up was January 2018. The clinicopathological information was obtained from the patients' records: age, pathology subtypes, status of ER, PR and HER2 expression levels, and clinical stage. All cases met the following inclusion criteria: i) Histologically confirmed primary breast cancer; ii) patients underwent surgery, following which there was adequate specimen of tumor tissue; iii) no metastasis before operation; iv) patient underwent full follow-up at the hospital after treatment; and v) patients did not receive preoperative chemotherapy, immunotherapy or radiotherapy. All samples were evaluated and subjected to histological diagnosis by pathologists. This study was approved by the Ethics Committee of the Xiangya Hospital of Central South University and all patients provided written informed consent.
Immunohistochemistry (IHC)
Staining of all the FFPE tissue sections (4-µm thick) was performed as described previously (20). Briefly, following 4% paraformaldehyde fixation for 24 h at room temperature, samples were embedded in paraffin and sectioned at 4 µm. Sections of tumors were dewaxed with xylenes and dehydrated in gradient ethanol, followed by antigen retrieval in citrate antigen retrieval solution (cat. no. P0081; Beyotime Institute of Biotechnology). Endogenous peroxidase blocking buffer (100 µl; cat. no P0100A; Beyotime Institute of Biotechnology) was added for 10 min to block the endogenous peroxidase activity. Then, the sections were treated with 100 µl blocking solution (cat. no. B10710; Invitrogen; Thermo Fisher Scientific, Inc.) and covered with parafilm. Subsequently, the sections were incubated with primary anti-MTMR3 antibody (1:100; cat. no. 12443; Cell Signaling Technology, Inc.) overnight at 4°C. After incubation with horseradish peroxidase (HRP)-conjugated secondary antibody (1:5,000; cat. no. ab205718; Abcam) for 1 h at room temperature, the slides were stained with diaminobenzidine (cat. no. D3939; Sigma-Aldrich; Merck KGaA) for 60 min at room temperature, followed by counterstaining with hematoxylin (cat. no. C0107; Beyotime Institute of Biotechnology). The staining was visualized using a light microscope (CKX41; Olympus Corporation) at ×100 and ×400 magnification. Positivity and intensity were assessed by two independent pathologists in a blinded manner, according to a previous report (21). The staining intensity and proportion of immune reactive cells were scored, as previously described (22).
Online overall survival analysis
Survival analysis for the MTMR3 gene in breast cancer was performed using the PROGgeneV2 prognostic database (http://genomics.jefferson.edu/proggene/) (18). This database includes the prognostic data from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases, while providing an easy operator interface. The following parameters were selected in the first interface: ‘MTMR3’ in Single/multiple user input genes; ‘breast cancer’ in cancer type; ‘death’ in survival measure; and ‘median’ in bifurcate gene expression. Then, in the second interface, all the filters were selected, and the plot was created. According to the median expression level of MTMR3 in breast cancer tissues, the patient samples were classified into two groups, higher and lower expression levels. The survival patient information, including 3 and 5 year-survival rates, was compared using Kaplan-Meier survival curves.
Cell lines and culture
Human breast cancer cell lines MCF-7, T47D, BT474, MDA-MB-231 and ZR-75-30, as well as human embryonic kidney 293T cells, were purchased from The Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The MCF-7, BT474 and ZR-75-30 cell lines were maintained in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Inc.). The T47D, MDA-MB-231 and 293T cells were grown in DMEM (Sigma-Aldrich; Merck KGaA) with 10% FBS. All cells were cultured in a humidified atmosphere with 5% CO2 at 37°C.
Construction of recombinant lentivirus vector and cell infection
The short hairpin (sh) RNA targeting MTMR3 (shMTMR3, 5′-CCAGTCGAGTATGCAAGTCTTGGTACCAAGACTTGCATACTCGACTGG-3′) and a scrambled negative control shRNA (shCon, 5′-TTCTCCGAACGTGTCACGTCTCGAGACGTGACACGTTCGGAGAA-3′) were designed using the sequence for the human MTMR3 mRNA under accession no. NM_021090.3. The lentiviral vectors (transfer, pFH-L-GFP; packaging, pVSVG-I and pCMVΔR8.92) were purchased from Shanghai Holly Biotechnology Co., Ltd. The chemically synthesized oligonucleotides were cloned into the lentivirus transfer vector, and then co-transfected into 293T cells with the packing vectors, using Lipofectamine 2000 (Thermo Fisher Scientific, Inc.), following the manufacturer's protocol. At 48 h post-transfection, the culture media were collected, filtered through a 45 µm filter (EMD Millipore), and ultra-centrifuged at 100,000 × g at 4°C for 30 min. Finally, 200 µl concentrated lentiviral particles were obtained. MDA-MB-231 cells, as the target cells, were seeded at 5×104 cells/well in 6-well plates prior to transduction with shMTMR3 or control lentiviral particles. Cells without transduction served as the control (Con) group. The transfection efficiency was monitored via green fluorescent protein (GFP) expression.
RNA isolation, reverse transcription and quantitative PCR (RT-qPCR)
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, Inc.) at 4 days post-infection. Total RNA was then treated with DNase (Ambion; Thermo Fisher Scientific, Inc.), tested by agarose gel electrophoresis for its integrity, and converted to cDNA using a RevertAid H Minus First Strand cDNA Synthesis kit (Thermo Fisher Scientific, Inc.). qPCR was performed on a Bio-Rad CFX-96 Real-Time PCR platform using SYBR Green Supermix reagents (Bio-Rad Laboratories, Inc.). The primers were as follows: MTMR3, 5′-AGCAGAGTGGGCTCAGTGTT-3′ (forward) and 5′-ACTGTCCACGTTTGGTCCTC-3′ (reverse); β-actin, 5′-GTGGACATCCGCAAAGAC-3′ (forward) and 5′-AAAGGGTGTAACGCAACTA-3′ (reverse). The thermocycling conditions were as follows: 95°C for 1 min for denaturation, followed by 40 cycles of 95°C for 5 sec and 60°C for 20 sec. Relative fold changes in mRNA expression were calculated using the formula 2−ΔΔCq (23). β-actin was used as the internal reference for normalization.
Western blotting
Protein sample preparation and western blot assay were performed as previously described (24). The primary antibodies used were: anti-MTMR3 (1:500; cat. no. 12443; Cell Signaling Technology, Inc.), anti-Cyclin D1 (1:1,000; cat. no. 60186-1-1g; ProteinTech Group, Inc.), anti-cyclin-dependent kinase 2 (1:1,000; CDK2; cat. no. 11026-2-AP; ProteinTech Group, Inc.), anti-p62 (1:1,000; cat. no. 19117-1-AP; ProteinTech Group, Inc.), anti-p21 (1:1,000; cat. no. 2947; Cell Signaling Technology, Inc.), anti-Cyclin E (1:1,000; cat. no. sc-247; Santa Cruz Biotechnology, Inc.), anti-Cyclin A (1:1,000; cat. no. sc-751; Santa Cruz Biotechnology, Inc.), anti-cell division cycle 25 A (1:1,000; cdc25A; sc-7157; Santa Cruz Biotechnology, Inc.), anti-microtubule associated protein 1 light chain 3 (LC3) A (1:500; cat. no. 4599; Cell Signaling Technology, Inc.), anti-LC3B (1:500; cat. no. 3868; Cell Signaling Technology, Inc.) and anti-GAPDH (1:20,000; cat. no. 10494-1-AP; ProteinTech Group, Inc.). Bound HRP-labeled secondary antibody (1:5,000; cat. no. SC-2005 or SC-2054; Santa Cruz Biotechnology, Inc.) was assayed by super ECL detection reagent (Pierce; Thermo Fisher Scientific, Inc.). Protein density of western blots was analyzed using ImageJ 1.51k software (National Institutes of Health).
MTT assay
MTT assay was performed to detect the effect of MTMR3 knockdown on MDA-MB-231 cell viability. Briefly, Con, shCon and shMTMR3 cells were seeded in 96-well plates at a density of 2.5×103 cells/well and incubated for 5 days. At day 1, 2, 3, 4, and 5, 20 µl MTT solution (5 mg/ml; Sigma-Aldrich; Merck KGaA) was added in each well and the cells were incubated for 4 h. Then, 100 µl acidic isopropanol (10% SDS, 5% isopropanol and 0.01 M HCl) was added to dissolve the purple formazan crystals. Finally, the absorbance at 595 nm was measured with an ELISA reader (Bio-Rad Laboratories, Inc.).
Colony formation assay
For colony formation assay, 400 cells per well were seeded in 6-well plates and allowed to grow for 9 days. Wells containing colonies were washed with PBS, fixed with paraformaldehyde (3.7%) for 15 min, rinsed once more with PBS, and then stained with crystal violet for 10 min. The excess stain was removed by washing three times with ddH2O, and then the colonies were photographed with a digital camera. The number of cell colonies (>50 cells/colony) was manually counted using ImageJ software.
Cell cycle phase distribution analysis
For cell cycle distribution assay, transduced cells were incubated into 6-cm dishes at a density of 1×105 cells/dish. After 20 h of incubation, cells were digested with trypsin, centrifuged, washed twice with cold PBS, and fixed in pre-chilled 70% ethanol at −20°C overnight. Then, cells were washed with PBS once again and stained with propidium iodide. Finally, the cells were analyzed using a Cell Lab Quanta™ flow cytometer (Beckman Coulter, Inc.) and the ModFit LT software (version 3.1.0.0; Verity Software House, Inc.).
Statistical analysis
All quantitative data were expressed as the mean ± standard deviation from three independent experiments. For the IHC results, P-values were calculated using the χ2 test and Wilcoxon matched-pairs signed rank test. For the MTT and cell cycle analysis experiments, two-way ANOVA with Dunnett's multiple comparisons test was used. For the colony formation experiments, one-way ANOVA with Dunnett's multiple comparisons test was used. Statistical analyses were performed in GraphPad Prism (version 7.0, GraphPad Software) or SPSS 13.0 software (SPSS, Inc.). P<0.05 was considered to indicate a statistically significant difference.
Results
Correlation between MTMR3 expression and survival in patients with BC
To explore the role of MTMR3 in breast cancer, first, its expression in breast cancer tissues was assessed using IHC. Fig. 1A presents representative photomicrographs of four degrees of MTMR3 expression intensity. Further analysis demonstrated that MTMR3 expression was significantly higher in breast cancer tissues compared with adjacent paracarcinoma tissues (Fig. 1B and C; P<0.001). Furthermore, the association between MTMR3 mRNA expression levels and overall survival (OS) in breast cancer patients was analyzed using the publicly-available PROGgeneV2 prognostic database. The results revealed that high expression of MTMR3 mRNA in breast cancer may be correlated with poor overall survival (Fig. 2A; P=0.003), and associated with poor relapse-free survival (Fig. 2B; P<0.02).
Figure 1.
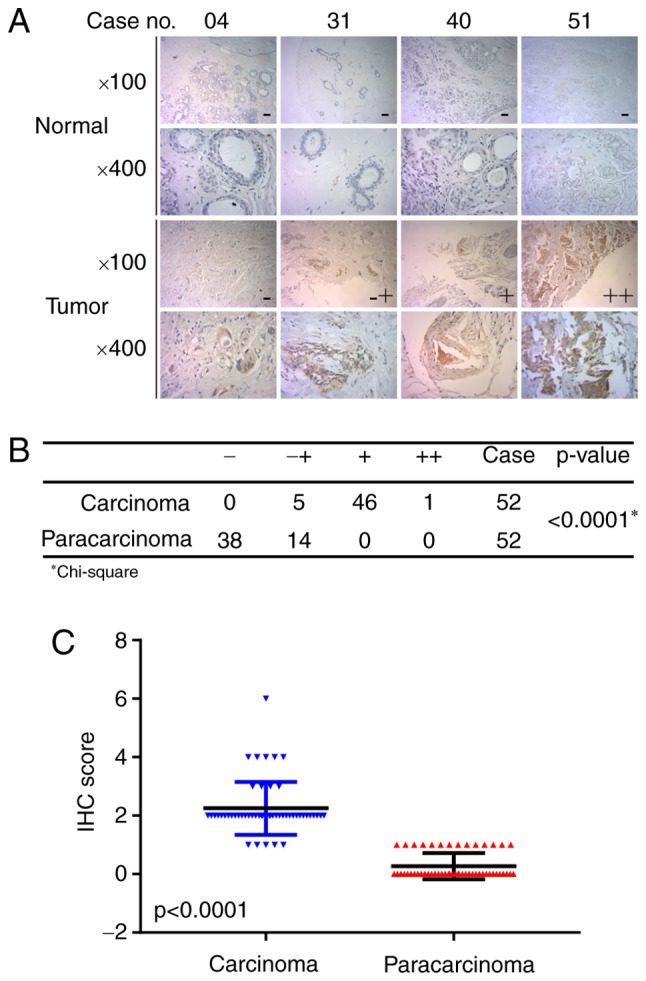
MTMR3 protein expression in breast cancer tissues and adjacent paracarcinoma tissues. (A) Representative images of negative (−), weak positive (−+), positive (+) and strongly positive (++) MTMR3 staining by IHC analysis. (B) Statistical analysis of different degrees of MTMR3 staining in breast cancer tissues. (C) Expression levels (IHC score) of MTMR3 in breast cancer tissues and paired adjacent paracarcinoma tissues. MTMR3, myotubularin related protein 3; IHC, immunohistochemistry.
Figure 2.

Survival analysis for MTMR3. Kaplan-Meier plots of the 3-year and 5-year (A) overall survival and (B) relapse-free survival according to MTMR3 expression in the indicated patient databases. MTMR3, myotubularin related protein 3.
MTMR3 in predicting metastasis and local recurrence of breast cancer
The clinical characteristics of 120 cases were reviewed. The median follow-up time was 52 months. The expression score of 2 (≤2 vs. >2), as optimal cut-off value for MTMR3 evaluation, was determined by receiver operating characteristic (ROC) curve estimation, giving the score closest to the point of maximum Youden's index. Based on the outcome of patients, all samples were divided into two groups. To elucidate whether MTMR3 expression was associated to relapse, χ2 univariate analysis was applied (Table I). The results demonstrated that there were no significant differences in 5-years relapse between PR (P=0.102), HER2 (P=0.115) status and age (P=0.633). By contrast, MTMR3 expression (P=0.038), ER status (P=0.031), and clinical stage (P=0.004) were strongly associated with disease recurrence. Thus, these three variables were included in multivariate logistic regression model. As described in Table II, although there was no significant association between MTMR3 expression and recurrence of breast cancer, lower expression conferred a moderate reduction in relapse during a 5-year period (OR=0.412, 95% CI=0.158–1.075, P=0.070). In addition, because of the observation that ER expression and clinical stage were strongly associated with clinical outcome, it was speculated that MTMR3 may be a predictor for recurrence of breast cancer due to other prognostic factors. To examine this possibility, Spearman rank correlation analysis was performed. Notably, MTMR3 expression was demonstrated to have significant positive correlation with the TNBC subtype (r=0.209, P=0.022; Table III). However, there was no correlation between MTMR3 staining and ER, PR and HER2 expression or clinical stage (Table III).
Table I.
Univariate analysis for recurrence within 5 years in relation to predictive factors.
| Variable | Relapse | Non-relapse | χ2 | P-value |
|---|---|---|---|---|
| Age (years) | ||||
| <50 | 30 | 45 | 0.229 | 0.633 |
| ≥50 | 20 | 25 | ||
| Clinical stage | ||||
| I–II | 25 | 53 | 8.477 | 0.004 |
| III–IV | 25 | 17 | ||
| ER status | ||||
| Negative | 30 | 28 | 4.672 | 0.031 |
| Positive | 20 | 42 | ||
| PR status | ||||
| Negative | 29 | 30 | 2.676 | 0.102 |
| Positive | 21 | 40 | ||
| HER2 status | ||||
| Negative | 28 | 29 | 2.483 | 0.115 |
| Positive | 22 | 41 | ||
| MTMR3 expression | ||||
| Low | 8 | 23 | 4.326 | 0.038 |
| High | 42 | 47 |
Significant P-values are denoted in bold font. ER, estrogen receptor; PR, progesterone receptor; HER2, erb-b2 receptor tyrosine kinase 2; MTMR3, myotubularin related protein 3.
Table II.
Results from multivariate logistic regression analyses comparing selected variables.
| Variable | Odds ratio | 95% confidence interval | P-value |
|---|---|---|---|
| Clinical stage (I–II vs. III) | 0.295 | 0.129–0.672 | 0.004 |
| ER status (negative vs. positive) | 2.643 | 1.192–5.864 | 0.017 |
| MTMR3 expression (low vs. high) | 0.412 | 0.158–1.075 | 0.070 |
Significant P-values are denoted in bold font. ER, estrogen receptor; MTMR3, myotubularin related protein 3.
Table III.
Spearman rank correlation analysis of MTMR3 expression and clinical features.
| Clinical features | ER | PR | HER2 | TNBC | Clinical stage |
|---|---|---|---|---|---|
| Correlation coefficient (r) | −0.66 | −0.162 | −0.37 | 0.209 | 0.074 |
| P-value | 0.475 | 0.078 | 0.685 | 0.022 | 0.423 |
Significant P-values are denoted in bold font. ER, estrogen receptor; PR, progesterone receptor; HER2, erb-b2 receptor tyrosine kinase 2; TNBC, triple-negative breast cancer; MTMR3, myotubularin related protein 3.
MTMR3 knockdown suppresses cell proliferation and colony formation
As presented in Fig. 3A, all five breast cancer cell lines tested in the present study expressed the MTMR3 protein. The MDA-MB-231 cell line had the highest level of MTMR3 expression, and it belongs in the TNBC subtype, thus, it was selected in the present study for subsequent loss-of-function experiments. MTMR3 was silenced in MDA-MB-231 cells via lentiviral shRNA transduction. The efficacy of MTMR3 knockdown was determined by RT-qPCR analysis (Fig. 3B); the mRNA expression levels of MTMR3 was significantly decreased in the shMTMR3 group compared with the shCon group (P<0.001). Western blotting further confirmed the successful knockdown of MTMR3 in MDA-MB-231 cells (Fig. 3C).
Figure 3.

Knockdown of MTMR3 suppresses the proliferative and colony formation capacities of MDA-MB-231 cells. (A) Western blot analysis of MTMR3 protein expression in five different breast cancer cell lines. (B) MTMR3 mRNA expression levels detected in shMTMR3, shCon, and Con MDA-MB-231 cells. (C) Western blot analysis of MTMR3 knockdown efficacy in MDA-MB-231 cells. (D) Comparative growth curves of tumor cells in the Con, shCon and shMTMR3 groups, as measured by MTT assay. (E) Representative images of formed colonies and statistical analysis of colony numbers in the Con, shCon, and shMTMR3 groups. Representative images of individual colonies in each group were captured at magnification ×40. Three independent experiments were performed. ***P<0.001 vs. shCon. MTMR3, myotubularin related protein 3; sh, short hairpin; Con, control; OD, optical density.
Next, the functional role of MTMR3 was explored. The proliferative capability was determined by MTT assay. MTMR3 knockdown resulted in a significant decrease in cell numbers in a time-dependent manner (Fig. 3D; P<0.001). However, no change in cell numbers was observed in cells infected with a control lentivirus compared with untreated cells. In addition, the clone-forming capacity of MDA-MB-231 cells was evaluated in the three experimental groups (shMTMR3, shCon and Con). This assay represents the ability to sustain cell growth without contact attenuation (25). As presented in Fig. 3E, the single colony size and the numbers of total colonies formed were reduced in MTMR3-silenced cells compared with control cells (P<0.001). These results indicated that MTMR3 may have important roles in the tumorigenesis of breast cancer cells.
MTMR3 regulates cell cycle progression and autophagy-related protein expression
To explore the potential molecular mechanism of cell growth inhibition induced by MTMR3 knockdown, flow cytometry was conducted to determine the cell cycle distribution. Compared with shCon or Con groups, the percentage of shMTMR3-cells in the G0/G1 phase and G2/M phase were significantly increased, while they were significantly decreased in the S phase (Fig. 4A and B). These results suggested that MTMR3 knockdown resulted in disturbed cell cycle progression, thereby contributing to the decline in cell proliferation and colony formation. Furthermore, the protein expression levels of several cell cycle regulators were detected in MDA-MB-231 cells using western blotting. As presented in Fig. 4C, MTMR3 knockdown markedly elevated the expression of p21 and Cyclin A, but reduced the expression of cdc25A and Cyclin E. The protein expression levels of CDK2 were slightly increased in the shMTMR3 group compared with the shCon group (Fig. 4C).
Figure 4.


Knockdown of MTMR3 alters cell cycle progression and related protein expression in MDA-MB-231 cells. (A and B) Cell cycle distribution was analyzed by flow cytometry. Representative plots and quantification of percentage of cells in each cell cycle phase is shown. (C) Western blot analysis of proteins associated with cell cycle regulation in MDA-MB-231 cells. (D) Western blot analysis of proteins associated with autophagy regulation in MDA-MB-231 cells. Three independent experiments were performed. **P<0.01, ***P<0.001 vs. shCon. MTMR3, myotubularin related protein 3; sh, short hairpin; Con, control; cdc25A, cell division cycle 25 A; CDK2, cyclin-dependent kinase 2; LC3, microtubule associated protein 1 light chain 3.
In addition to cell cycle progression, autophagy is closely associated with the regulation of cell survival control (26). Therefore, it was speculated that MTMR3 knockdown may affect cell autophagy in MDA-MB-231 cells. As presented in Fig. 4D, MTMR3 knockdown induced a significant increase in the LC3-II/LC3-I ratio and a reduction in p62, which suggested that MTMR3 knockdown induced autophagy in MDA-MB-231 cells.
Discussion
Breast cancer is the most common malignancy among women. Large numbers of research publications have indicated the essential roles of numerous genes in growth and viability of breast cancer cells (27,28). The present study indicated that high expression levels of MTMR3 may have negative effects on overall survival and relapse-free survival in patients with breast cancer. Next, the MTMR3 protein expression levels were detected in two different groups of patients by IHC. The results of univariate analysis revealed a significant association of MTMR3 protein expression with cancer relapse within 5 years; however, multivariate analysis revealed that MTMR3 expression was not a significant independent prognostic factor. This negative result may be due to low sample size and follow-up time. Of note, MTMR3 expression was demonstrated to be positively correlated with TNBC, which is known to be enriched for genes involved in metastasis or recurrence-associated signaling pathways.
Notably, MTMR3 acts as a dual function regulatory molecule to attenuate or enhance tumor growth in different types of cancer. Briefly, lack of MTMR3 significantly repressed the proliferative, migratory and invasive potential of oral cancer cells (15), while MTMR3 exogenous expression inhibited the clonal growth of transfected lung carcinoma cells (29). In order to reveal the phenotypes of MTMR3 in TNBC cells, MTMR3 was silenced in the MDA-MB-231 cell line, which had high expression of MTMR3, via shRNA. The results revealed that MTMR3 knockdown resulted in suppressed proliferation and colony formation, suggesting that MTMR3 may exert protumor activity in TNBC cells.
Dysregulation of cell cycle progression is a key intrinsic factor in driving tumorigenesis (30). In the present study, MTMR3 knockdown resulted in an increase of cells in the G0/G1 and G2/M phases and a decrease of cells in the S phase, suggesting that MTMR3 has a role in cell cycle. Based on the present findings, a mechanism of proliferation and colony formation inhibition induced by knockdown of MTMR3 may be mediated through G0/G1 cell cycle arrest. In addition, the molecular mechanism of this phenotype was explored via western blot analysis, and the results revealed increased expression of p21, Cyclin A and CDK2, and suppressed expression of cdc25A and Cyclin E, following MTMR3 silencing. The decision for a cell to exit from G1 and enter into the S phase is closely controlled by these genes (31,32). Adenovirus-mediated overexpression of p21 can prevent proliferation of rat vascular smooth muscle cells (33). Lim et al (34) demonstrated that knockdown of metallothionein-2A abrogated cell growth via prevention of G1 to S phase transition though upregulation of ataxia telangiectasia mutated and downregulation of cdc25A. It was reported that E2F-induced cell growth can be inhibited by miR-15 and miR-16, due to their role in downregulating Cyclin E (35). Suppression of CDK2 and CDK4 is responsible for attenuation of cell proliferation mediated by C/EBPα (36). By contrast, the activation of CyclinA/CDK2 complex is considered a key factor for primary lung cancer cell proliferation (37). The present results suggested that knockdown of MTMR3 repressed the proliferation and colony formation of MDA-MB-231 cells via blocking G1-to-S transition through effects on regulators of S-phase entry, p21, cdc25A, Cyclin E, Cyclin A and CDK2.
Autophagy is a cellular degradation process that has an important role in tumor development, progression and therapy response (38–40). Activation of autophagy may prolong survival of tumor cells, and paradoxically, a defect in autophagy may also have a role in carcinogenesis and tumor progression (41). A previous study has reported that MTMR3 is an autophagy-related molecule that participates in initiation of autophagy (42). Taguchi-Atarashi et al (19) demonstrated that specific knockdown of MTMR3 enhanced autophagosome formation, which was modulated by local levels of Ptdlns3P. Conversely, MTMR3 caused a reduced autophagic activity and smaller nascent autophagosomes when overexpressed (19). LC3A and LC3B are widely used as autophagy biomarkers, and the type I (16 kDa) is converted to type II (14 kDa) (43). A significant upregulation in the LC3-II/LC3-I ratio was observed in the present study following MTMR3 knockdown, implying an activation of autophagy. A previous study has reported that autophagy suppresses carcinogenesis via elimination of p62, a multidomain protein (44). In the present study, downregulation of p62 was observed in the MTMR3-knockdown group. Based on the present results, MTMR3 knockdown inducing activation of autophagy may be related to the decreased proliferation and colony formation in MDA-MB-231 cells. One of the limitations of the present study is that experiments were performed only in vitro and only in one cell line; therefore, further studies are needed with additional cell lines and in vivo models to confirm these results.
In conclusion, the present study demonstrated that high levels of MTMR3 were associated with relapse-free survival and the TNBC subtype, as well as with a protumorigenic function in human breast cancer cells. Knockdown of MTMR3 suppressed proliferation and colony formation through cell cycle arrest and autophagy in MDA-MB-231 cells. These results provided novel insights into the pathological mechanism of breast cancer and may facilitate the exploration of novel therapeutic targets.
Acknowledgments
Not applicable.
Funding
This study was supported by grants from the National Natural Science Foundation of China (grant nos. 81572612, 81372842 and 81803640), the Hunan Provincial Natural Science Foundation (grant no. 2015JJ2183), the Youth Science Foundation of Xiangya Hospital, Central South University (grant no. 2017Q02) and the Research Innovation Program for Graduate Students of Central South University (grant no. 2018zzts912). This study was also supported by the National Key Clinical Specialist Construction Programs of China (grant no. 2014kll).
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
ZW, RS, WBZ and LQS conceived and designed the experiments. ZW and RS performed the experiments. ZW and YJW analyzed the data. MZ, JC and JH collected the clinical samples and clinical data. ZW wrote the manuscript. ZW, JC, LQS and WBZ reviewed/edited the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was approved by the Ethics Committee of the Xiangya Hospital of Central South University. All of the patients provided written informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin. 2014;64:52–62. doi: 10.3322/caac.21203. [DOI] [PubMed] [Google Scholar]
- 2.Alkabban FM, Ferguson T. StatPearls. Treasure; Island (FL): 2019. Cancer, breast. [Google Scholar]
- 3.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 4.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 5.Guan X, Liu Z, Zhao Z, Zhang X, Tao S, Yuan B, Zhang J, Wang D, Liu Q, Ding Y. Emerging roles of low-density lipoprotein in the development and treatment of breast cancer. Lipids Health Dis. 2019;18:137. doi: 10.1186/s12944-019-1075-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parise CA, Caggiano V. Risk of mortality of node-negative, ER/PR/HER2 breast cancer subtypes in T1, T2, and T3 tumors. Breast Cancer Res Treat. 2017;165:743–750. doi: 10.1007/s10549-017-4383-5. [DOI] [PubMed] [Google Scholar]
- 7.Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin Cancer Res. 2007;13:4429–4434. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- 8.Santuario-Facio SK, Cardona-Huerta S, Perez-Paramo YX, Trevino V, Hernandez-Cabrera F, Rojas-Martinez A, Uscanga-Perales G, Martinez-Rodriguez JL, Martinez-Jacobo L, Padilla-Rivas G, et al. A new gene expression signature for triple negative Breast cancer using frozen fresh tissue before neoadjuvant chemotherapy. Mol Med. 2017;23:101–111. doi: 10.2119/molmed.2016.00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clague MJ, Lorenzo O. The myotubularin family of lipid phosphatases. Traffic. 2005;6:1063–1069. doi: 10.1111/j.1600-0854.2005.00338.x. [DOI] [PubMed] [Google Scholar]
- 10.Lorenzo O, Urbé S, Clague MJ. Analysis of phosphoinositide binding domain properties within the myotubularin-related protein MTMR3. J Cell Sci. 2005;118:2005–2012. doi: 10.1242/jcs.02325. [DOI] [PubMed] [Google Scholar]
- 11.Clarke JH, Letcher AJ, D'Santos C S, Halstead JR, Irvine RF, Divecha N. Inositol lipids are regulated during cell cycle progression in the nuclei of murine erythroleukaemia cells. Biochem J. 2001;357:905–910. doi: 10.1042/bj3570905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin Y, Zhao J, Wang H, Cao J, Nie Y. miR-181a modulates proliferation, migration and autophagy in AGS gastric cancer cells and downregulates MTMR3. Mol Med Rep. 2017;15:2451–2456. doi: 10.3892/mmr.2017.6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng B, Yu X, Chai R. Myotubularin-related phosphatase 3 promotes growth of colorectal cancer cells. ScientificWorldJournal. 2014;2014:703804. doi: 10.1155/2014/703804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oppelt A, Haugsten EM, Zech T, Danielsen HE, Sveen A, Lobert VH, Skotheim RI, Wesche J. PIKfyve, MTMR3 and their product PtdIns5P regulate cancer cell migration and invasion through activation of Rac1. Biochem J. 2014;461:383–390. doi: 10.1042/BJ20140132. [DOI] [PubMed] [Google Scholar]
- 15.Kuo YZ, Tai YH, Lo HI, Chen YL, Cheng HC, Fang WY, Lin SH, Yang CL, Tsai ST, Wu LW. MiR-99a exerts anti-metastasis through inhibiting myotubularin-related protein 3 expression in oral cancer. Oral Dis. 2014;20:e65–e75. doi: 10.1111/odi.12133. [DOI] [PubMed] [Google Scholar]
- 16.Gong Y, He T, Yang L, Yang G, Chen Y, Zhang X. The role of miR-100 in regulating apoptosis of breast cancer cells. Sci Rep. 2015;5:11650. doi: 10.1038/srep11650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: Caution in the interpretation of LC3 localization. Autophagy. 2007;3:323–328. doi: 10.4161/auto.4012. [DOI] [PubMed] [Google Scholar]
- 18.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: Therapeutic implications. Mol Cancer Ther. 2011;10:1533–1541. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taguchi-Atarashi N, Hamasaki M, Matsunaga K, Omori H, Ktistakis NT, Yoshimori T, Noda T. Modulation of local PtdIns3P levels by the PI phosphatase MTMR3 regulates constitutive autophagy. Traffic. 2010;11:468–478. doi: 10.1111/j.1600-0854.2010.01034.x. [DOI] [PubMed] [Google Scholar]
- 20.Zhou W, Guan X, Wang L, Liao Y, Huang J. p12(CDK2-AP1) inhibits breast cancer cell proliferation and in vivo tumor growth. J Cancer Res Clin Oncol. 2012;138:2085–2093. doi: 10.1007/s00432-012-1286-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshikawa D, Ojima H, Iwasaki M, Hiraoka N, Kosuge T, Kasai S, Hirohashi S, Shibata T. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br J Cancer. 2008;98:418–425. doi: 10.1038/sj.bjc.6604129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z, Chen J, Zhong MZ, Huang J, Hu YP, Feng DY, Zhou ZJ, Luo X, Liu ZQ, Jiang WZ, Zhou W. Overexpression of ANLN contributed to poor prognosis of anthracycline-based chemotherapy in breast cancer patients. Cancer Chemother Pharmacol. 2017;79:535–543. doi: 10.1007/s00280-017-3248-2. [DOI] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Zhou W, Wang Z, Shen N, Pi W, Jiang W, Huang J, Hu Y, Li X, Sun L. Knockdown of ANLN by lentivirus inhibits cell growth and migration in human breast cancer. Mol Cell Biochem. 2015;398:11–19. doi: 10.1007/s11010-014-2200-6. [DOI] [PubMed] [Google Scholar]
- 25.Ma D, Fang Q, Li Y, Wang J, Sun J, Zhang Y, Hu X, Wang P, Zhou S. Crucial role of heme oxygenase-1 in the sensitivity of acute myeloid leukemia cell line Kasumi-1 to ursolic acid. Anticancer Drugs. 2014;25:406–414. doi: 10.1097/CAD.0000000000000068. [DOI] [PubMed] [Google Scholar]
- 26.Swart C, Du TA, Loos B. Autophagy and the invisible line between life and death. Eur J Cell Biol. 2016;95:598–610. doi: 10.1016/j.ejcb.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 27.Odle TG. Precision medicine in breast cancer. Radiol Technol. 2017;88:M401–M421. [PubMed] [Google Scholar]
- 28.Weaver O, Leung JWT. Biomarkers and imaging of breast cancer. AJR Am J Roentgenol. 2018;210:271–278. doi: 10.2214/AJR.17.18708. [DOI] [PubMed] [Google Scholar]
- 29.Yoo YD, Cho SM, Kim JS, Chang YS, Ahn CM, Kim HJ. The human myotubularin-related protein suppresses the growth of lung carcinoma cells. Oncol Rep. 2004;12:667–671. [PubMed] [Google Scholar]
- 30.Wang YC, Yang X, Xing LH, Kong WZ. Effects of SAHA on proliferation and apoptosis of hepatocellular carcinoma cells and hepatitis B virus replication. World J Gastroenterol. 2013;19:5159–5164. doi: 10.3748/wjg.v19.i31.5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gartel AL, Serfas MS, Tyner AL. p21-negative regulator of the cell cycle. Proc Soc Exp Biol Med. 1996;213:138–149. doi: 10.3181/00379727-213-44046. [DOI] [PubMed] [Google Scholar]
- 32.Tan Y, Sun D, Jiang W, Klotz-Noack K, Vashisht AA, Wohlschlegel J, Widschwendter M, Spruck C. PP2A-B55β antagonizes cyclin E1 proteolysis and promotes its dysregulation in cancer. Cancer Res. 2014;74:2006–2014. doi: 10.1158/0008-5472.CAN-13-1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang MW, Barr E, Lu MM, Barton K, Leiden JM. Adenovirus-mediated over-expression of the cyclin/cyclin-dependent kinase inhibitor, p21 inhibits vascular smooth muscle cell proliferation and neointima formation in the rat carotid artery model of balloon angioplasty. J Clin Invest. 1995;96:2260–2268. doi: 10.1172/JCI118281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lim D, Jocelyn KM, Yip GW, Bay BH. Silencing the Metallothionein-2A gene inhibits cell cycle progression from G1- to S-phase involving ATM and cdc25A signaling in breast cancer cells. Cancer Lett. 2009;276:109–117. doi: 10.1016/j.canlet.2008.10.038. [DOI] [PubMed] [Google Scholar]
- 35.Ofir M, Hacohen D, Ginsberg D. MiR-15 and miR-16 are direct transcriptional targets of E2F1 that limit E2F-induced proliferation by targeting cyclin E. Mol Cancer Res. 2011;9:440–447. doi: 10.1158/1541-7786.MCR-10-0344. [DOI] [PubMed] [Google Scholar]
- 36.Wang H, Iakova P, Wilde M, Welm A, Goode T, Roesler WJ, Timchenko NA. C/EBPalpha arrests cell proliferation through direct inhibition of Cdk2 and Cdk4. Mol Cell. 2001;8:817–828. doi: 10.1016/S1097-2765(01)00366-5. [DOI] [PubMed] [Google Scholar]
- 37.Dobashi Y, Shoji M, Jiang SX, Kobayashi M, Kawakubo Y, Kameya T. Active cyclin A-CDK2 complex, a possible critical factor for cell proliferation in human primary lung carcinomas. Am J Pathol. 1998;153:963–972. doi: 10.1016/S0002-9440(10)65638-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grander D, Panaretakis T. Autophagy: Cancer therapy's friend or foe? Future Med Chem. 2010;2:285–297. doi: 10.4155/fmc.09.155. [DOI] [PubMed] [Google Scholar]
- 39.Duan X, Chen B, Cui Y, Zhou L, Wu C, Yang Z, Wen Y, Miao X, Li Q, Xiong L, He J. Ready player one? Autophagy shapes resistance to photodynamic therapy in cancers. Apoptosis. 2018;23:587–606. doi: 10.1007/s10495-018-1489-0. [DOI] [PubMed] [Google Scholar]
- 40.Xu Z, Yan Y, Zeng S, Qian L, Dai S, Xiao L, Wang L, Yang X, Xiao Y, Gong Z. Reducing autophagy and inducing G1 phase arrest by aloperine enhances radio-sensitivity in lung cancer cells. Oncol Rep. 2017 Jun 19; doi: 10.3892/or.2017.5732. (Epub ahead of print) doi: 10.3892/or.2017.5732. [DOI] [PubMed] [Google Scholar]
- 41.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nature reviews Cancer. 2007;7:961–967. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roberts R, Ktistakis NT. Omegasomes: PI3P platforms that manufacture autophagosomes. Essays Biochem. 2013;55:17–27. doi: 10.1042/bse0550017. [DOI] [PubMed] [Google Scholar]
- 43.Fernández-Araujo A, Sánchez JA, Alfonso A, Vieytes MR, Botana LM. Different toxic effects of YTX in tumor K-562 and lymphoblastoid cell lines. Front Pharmacol. 2015;6:124. doi: 10.3389/fphar.2015.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lamark T, Svenning S, Johansen T. Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays Biochem. 2017;61:609–624. doi: 10.1042/EBC20170035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.