

Abstract
Diversity indices have been routinely computed in the study of human microbiome-associated diseases (MADs). However, it is still unclear whether there is a consistent diversity-disease relationship (DDR) for the human MADs, and whether there are consistent differences in the taxonomic composition of microbiomes sampled from healthy versus diseased individuals. Here we reanalyzed raw data and used a meta-analysis to compare the microbiome diversity and composition of healthy versus diseased individuals in 41 comparisons extracted from 27 previously published studies of human MADs. In the DDR analysis, the average effect size across studies did not differ from zero for a comparison of healthy versus diseased individuals. In 30 of 41 comparisons (73%) there was no significant difference in microbiome diversity of healthy versus diseased individuals, or of different disease classes. For the species composition analysis (shared species analysis), the effect sizes were significantly different from zero. In 33 of 41 comparisons (80%), there were fewer OTUs (operational taxonomic units) shared between healthy and diseased individuals than expected by chance, but with 49% (20 of 41 comparisons) statistically significant. These results imply that the taxonomic composition of disease-associated microbiomes is often distinct from that of healthy individuals. Because species composition changes with disease state, some microbiome OTUs may serve as potential diagnostic indicators of disease. However, the overall species diversity of human microbiomes is not a reliable indicator of disease.
Subject terms: Microbial ecology, Microbial communities
Introduction
The question of whether or not microbiome species diversity plays an important role in human diseases was first raised in the 1980s (e.g., [1–5]). Recent advances in metagenomics technology and the development of human microbiome project (HMP) have revolutionized the exploration of this relationship (e.g., [1, 6–9]). The human microbiome includes bacteria, virus, bacteriophages, and plasmids, but at this stage, diversity analysis has been applied exclusively to bacteria. Traditional ecological diversity indices such as species richness and the Shannon index have been routinely reported in studies that compare the microbiomes of diseased and healthy individuals (see Table S1). So far, published studies have generated inconsistent results: the microbiome diversity of diseased individuals may be higher, lower, or no different than the microbiome diversity of healthy individuals. However, most of the studies did not employ formal statistical tests or use consistent diversity metrics, which makes a simple meta-analysis problematic (see Table S1). Moreover, it is still unclear whether diversity change is a cause of microbiome-associated disease or a consequence, and this question is rarely addressed explicitly. For this reason, we use the term microbiome associated diseases (MAD) to refer to those diseases that are associated with the changes occurring in the human microbiome, but do not specify the direction of cause and effect.
Even in the absence of disease, microbiome diversity can vary widely among human populations, among individuals within a population, and among different microbiome habitats within the same individual [6, 7, 9–11]. Although different researchers may use different thresholds for clustering and distinguishing OTUs (operational taxonomic units) [12–18], it is generally accepted that in healthy individuals, microbiomes of gut, oral and skin habitats are relatively species-rich, whereas the microbiomes of vaginal and lung habitats are relatively species-poor [6–9, 11, 19–21]. In other words, different microbiome habitats may have different core microbiota and different baseline diversities, which, makes it rather challenging to discover a general DDR pattern.
An additional challenge in comparing microbiome diversity is that most species diversity indices are sensitive to the sample size. In the existing literature, the term “sample size” is often interchangeably used with several other terms including sampling effort, sampling intensity, sequencing coverage or sequencing depth. The first three terms are often used in ecology in the discussion of rarefaction, and they are the counterparts of sequencing coverage (depth) in microbial metagenomics. As the sample size increases, the number of OTUs (operational taxonomic units) recorded inevitably increases. This problem is even more acute for studies of hyper-diverse microbiomes [22] than it is for traditional studies of plant and animal communities. Thus, some of the heterogeneity among and within studies of human microbiomes may reflect this source of statistical variation. Ecologists have recognized this sampling problem for many decades [23] and used rarefaction (a form of interpolation) and asymptotic species richness estimators (a form of extrapolation) to standardize biodiversity comparisons [24–26].
Although existing studies on human MADs have routinely computed diversity indices, basic patterns of DDR in human microbiomes are still not well established. Here we re-analyze raw data from published studies in which sequence counts or OTU tables were provided, allowing for a rigorous statistical analysis of the patterns. We ask two questions: (i) Is there evidence for a distinctive microbiome composition in diseased versus healthy individuals, or could the differences in taxonomic composition (which inevitably include many rare OTUs) be explained by simple sampling effects? (ii) Are there consistent differences in the taxonomic diversity of diseased and healthy individuals for different microbiome-associated diseases?
To address the first question of microbiome species composition, we used a randomization test for performing shared species analysis (SSA). This test does not simply compare the OTU richness or diversity of healthy and diseased individuals, but instead quantifies the difference in species composition (OTU identity) between the two groups, which is a measure of beta diversity in terms of [27]. To strengthen the rigor of the SSA, we designed two algorithms (A1 & A2): A1 randomizes the assignments of the individual reads (bacterial individuals) to the healthy or diseased groups, and A2 randomizes the assignments of the entire sample from a single subject (and its associated reads) to the healthy or diseased groups. The difference between both the algorithms is that A1 treats the individual reads as independent elements, whereas the more conservative A2 treats the entire sample of reads from a single subject as the independent sampling element.
To answer the second question of microbiome species diversity, we adopted Hill numbers as a unified measure of community diversity ([25, 28, 29]). Hill numbers present a series of values of Renyi’s entropy corresponding to the so-termed diversity orders (q = 0, 1, 2,…) and the series (also known as diversity profile) allows for more comprehensive measuring of diversity than any single diversity index such as species richness, the Shannon index or the Simpson index. Furthermore, when q = 0, the Hill number is species richness; when q = 1, the Hill number is the exponential of the Shannon index; when q = 2, the Hill number is the inverse of the Simpson index. Therefore, the diversity results we computed from adopting Hill numbers can still be compared qualitatively with the studies in existing literature that use traditional diversity measures. One additional advantage from using Hill numbers is that the familiar rarefaction approach for interpolating species richness in small subsamples as well as extrapolating species richness to asymptotic values has been extended to them [30]. In microbiome metagenomic research, these methods for interpolating or extrapolating Hill numbers can help to standardize comparisons that are based on unequal sample sizes.
Materials and methods
Datasets of human microbiome associated diseases
The systematic investigation of the MADs started approximately a decade ago with the landmark US-NIH HMP and EU MetaHIT projects [6, 31]. Most data samples from the HMP/MetaHIT are from healthy human cohorts because the mission of HMP/MetaHIT was to establish a ‘baseline’ of human microbiomes. Samples of MADs from healthy versus diseased individuals were mostly collected by individual research teams and consequently the datasets are rather scattered. The EMP (Earth Microbiome Project) appears to have hosted the single largest database of the human MADs, and we obtained approximately 2/3 of the MAD case studies from EMP data depository. Indeed, the datasets from EMP source include majority of the high-profile MADs, including neuronal degenerative diseases, IBD (inflammatory bowel disease), obesity, and diabetes. One important advantage of EMP datasets is that they are based on standardized sequencing operations and bioinformatics analysis, which facilitated our meta-analyses of the human DDR. We selected the remaining 1/3 of the case studies from a variety of published sources with the goals of (i) covering all five major microbiome habitats (gut, oral, skin, lung and vaginal) as well as two important fluid habitats (milk and semen) and (ii) representing the most widely investigated MADs from individual research publications. Although our selected datasets are not exhaustive, they are representative of state-of-the-art research in the human MADs. A brief description of the MAD case studies is provided in Table S1 of the online supplementary information (OSI).
Quantifying microbiome diversity with hill numbers
There are two common challenges in all biodiversity studies that are based on counts of individuals classified into species (or sequences classified into OTUs, as in metagenome studies). The first challenge is that traditional analyses of species richness do not incorporate data on the evenness or relative abundance of different taxa [28]. The second challenge is that biodiversity indices are sensitive to the sample size: indices with more weight given to rare species (such as species richness) are more sensitive to sampling biases [23].
To address the first challenge, we quantified diversity using the [28] numbers, a family of diversity indices that use a standardized scale of the equivalent number of “equally abundant species” [25, 29]. For a given diversity order q, the alpha diversity is:
| 1 |
where S is the number of species, pi is the relative abundance of species i, and q is the order number of diversity. Different values of q correspond to different ecological diversity indices. q = 0 corresponds to species richness, which places maximum weight on rare species (abundances are ignored). q = 1, and q = 2 correspond respectively to algebraic transformations of the Shannon index and Simpson’s index of diversity. As q increases, the Hill number index is increasingly weighted by the relative abundances of the common species and is less affected by the number of rare species.
Note that when q = 1, the Hill number is undefined, but its limit as q approaches 1 exists in the following form:
| 2 |
Standardizing biodiversity comparisons of healthy and diseased subjects with asymptotic diversity measures
Although the Hill numbers provide for an important standardization of biodiversity in common units of “equivalent numbers of equally abundant species”, these indices are still sensitive to sampling effects, particularly for low-order values of q [25]. To address the second challenge (sampling intensity), ecologists have traditionally used rarefaction to interpolate sampling curves to a standardized sampling level for comparing species richness and other biodiversity indices [23]. The weakness of rarefaction is that samples are inevitably standardized to the lowest abundance sample, and so much data is discarded to make the comparison. This problem is especially severe for hyper-diverse microbial assemblages [32].
As an alternative strategy to rarefaction, diversity can be standardized by using asymptotic species richness estimators [33]. These statistics estimate the number of species expected when sampling is presumably maximal and no further species would be encountered with additional sampling [34]. The only disadvantage of this approach is that the extrapolation of species richness for hyper-diverse assemblages may be uncertain and have large associated variances [30, 35]. However, the higher-order Hill numbers (q = 1, q = 2) are much less prone to uncertainty when they are extrapolated out to the asymptote. Using the asymptotic estimators allows for standardization of all samples but does not discard any of the data. For each healthy or diseased individual sampled in the different studies, we used the iNEXT R library [30, 36] to compute the analytical solutions of asymptotic richness for Hill numbers q = 0, 1, and 2.
Statistical tests of effect sizes
We used [37] d-statistic to measure the effect size for each study as the difference in the average diversity metric between the healthy (H) and diseased (D) microbiome treatments. Before performing the effect size test, we applied a square-root transformation to the Hill numbers to address non-normality and because the Hill numbers are measured in units of equally abundant species (counts). We used the Compute.es R-Package (https://CRAN.R-project.org/package=compute.es), to compute Cohen’s [37] d-statistic from standard t-test values. If d > 0, the healthy group has higher (standardized) diversity than the diseased group, and vice versa if d < 0. Finally, we repeated the entire meta-analysis using the unstandardized OTU counts from each study to compare results with the meta-analysis of the asymptotic estimators [38].
The d-statistic is calculated as:
| 3 |
where t is the t-value from standard t test, n1 and n2 are the sample sizes of two treatments. Obviously, the d-statistic is not influenced by the possible difference in the sample sizes.
Statistical tests of shared species (OTUs)
The number of shared OTUs between healthy and diseased individuals varied widely among studies, and depends in part on the number of individuals per group and the number of reads per individual sample. If there are distinctive OTUs associated with the diseased and healthy state, then there should be relatively few shared OTUs between these two groups. Alternatively, if the same microbiome is associated with healthy and diseased individuals, the distinctive OTUs in each group would represent random sampling effects (which are especially strong for rare or under-sampled taxa), and the number of shared OTUs would be no different than expected by chance (H0). This analysis compares the composition, or beta diversity [27], of the treatments, whereas the previous Hill number analyses above compare the alpha diversity, or taxon richness, between treatments.
We used two algorithms to estimate the number of shared OTUs expected under H0. In the first algorithm (A1), the expected number of shared OTUs was generated by pooling all the reads (bacterial individuals) within each study (including the healthy and diseased treatments) together and then randomly assigning each read to the healthy or diseased category. A1 maintains the total number of reads in each of the two original groups. In the second algorithm (A2), we randomly assigned each microbiome sample in the study to the diseased or healthy group, and then pooled the reads within each of the randomized pseudo-groups. A2 maintains the numbers of microbiome samples in each of the two original groups.
After randomization with A1 or A2, we then pooled the reads within each pseudo-group and calculated the number of shared OTUs between the two pseudo-groups. The randomization was repeated 1000 times to generate a distribution of the expected number of shared OTUs under the null hypothesis of random sampling (H0). We then compared to the observed number of OTUs to the simulated distribution to estimate the tail probability of obtaining the observed results with random sampling p(# Shared OTUs|H0) We converted these null model results into a standardized effect size:
| 4 |
where SOTUobs = the observed number of shared OTUs, mean(SOTUsim) = the average number of shared OTUs in the 1000 simulated assemblages, and sd(SOTUsim) is the sample standard deviation of the 1000 simulated assemblages. A detailed description on both A1 and A2 algorithms is presented on the online supplementary information (OSI).
Results
Differences in microbiome diversity between healthy and diseased individuals
Overall, the effect sizes in the 41 comparisons of microbiome diversity from healthy versus diseased individuals did not differ statistically from an average effect size of 0 (one-sample t test = −0.742, p = 0.463, q = 0). Table S2 summarizes all 41 comparisons of microbiome diversity in diseased and healthy individuals from 27 published studies. For the DDR analysis, in 30 of 41 comparisons (73%) there was no significant difference in microbiome diversity of healthy (H) versus diseased individuals (D), or of microbiome diversity of individuals classified into different disease or treatment groups. In 5 cases (12%) the microbiome diversity of healthy individuals significantly exceeded that of diseased (H > D), and in 6 cases (15%) the pattern was reversed (H < D). To avoid non-independence of multiple comparisons (including different disease states) within each study, Fig. 1 summarizes the patterns for single comparisons of effect size within each of the 27 studies, calculated for 3 different orders of Hill number. For these asymptotic estimators of species diversity, results were consistent with the full analysis of all 41 cases: In the majority of cases (67%), there was no significant difference between healthy and diseased individuals (H = D, 18 cases). In 4 cases H > D (15%), and in 5 cases H < D (18%). Within each case study, effect sizes were qualitatively similar for the different Hill numbers (Fig. 1).
Fig. 1.

Effect sizes for a comparison of microbiome diversity of healthy versus diseased individuals. Bar color indicates Hill number exponent (black: q = 0; dark grey: q = 1, light grey: q = 2). If the effect size is positive, the microbiome diversity of healthy individuals was higher than that of diseased individuals (d = H−D > 0, indicated by a right bar). If the effect is negative, the microbiome diversity of healthy individuals was lower than that of diseased individuals (d = H−D < 0, indicated by a left bar). Asterisks indicate p < 0.05 for test of Cohen’s (1988) [37] d statistic. T1D type-1 diabetes, PD Parkinson’s disease, MHE minimal hepatic encephalopathy, CRC colorectal cancer, IBD irritated bowel disease, HIV human immunodeficiency virus, CF cystic fibrosis, BV bacterial vaginosis. To simplify the graphs, only one comparison for each case study is displayed here. Occasionally, some case studies may have more than one comparison (i.e., more than two groups to compare, e.g., IBD has healthy, CD and UC groups). The results for all 41 possible comparisons are exhibited in OSI Table S2
Raw OTU counts were significantly correlated with asymptotic richness estimators (Fig. 2), although the slope of the relationship was significantly greater than 1.0, indicating more missing taxa for assemblages sampled with deeper coverage (larger sample size). However, effect sizes calculated for raw OTUs and asymptotic diversity were extremely similar (Fig. 3), so the results would not have changed if the data had not been standardized with asymptotic estimators of Hill numbers.
Fig. 2.
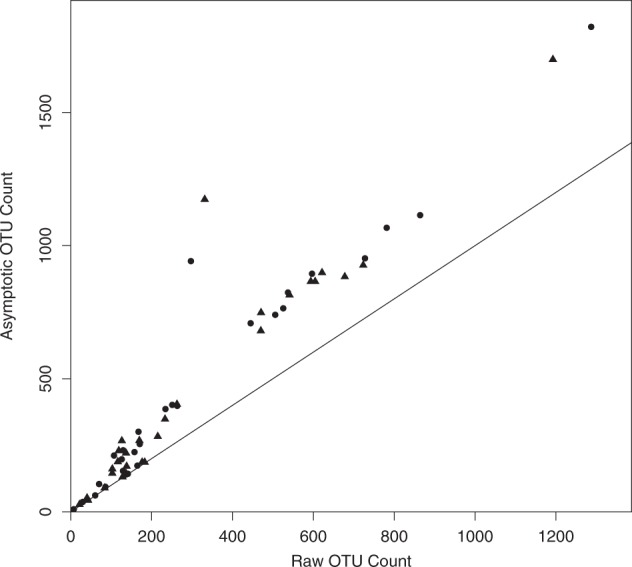
Comparison of observed and asymptotic OTU counts (species richness; q = 0) for healthy individuals (circles) and diseased individuals (triangles). The solid line has intercept 0 and slope 1, which would be found if the observed OTU count and the asymptotic estimator were the same. The asymptotic estimator is the observed OTU count plus the estimated number of undetected OTUs in the group. The asymptotic OTU count has a linear correlation with the raw OTU count (r = 0.98, p < 0.001)
Fig. 3.

Meta-analysis of effect sizes using untransformed data (x-axis) versus estimates of asymptotic diversity (y-axis). Results are shown for q = 0 (species richness), but results are similar for other Hill numbers. Each point represents a different microbiome study in which the diversity of diseased versus healthy individuals was compared (see Fig. 1)
Differences in shared OTUs between healthy and diseased individuals
Table S3A (with A1 algorithm) and Table S3B (with A2 algorithm) listed the results of shared species analysis between the healthy and diseased treatments. With the A1 algorithm (reshuffling reads), the observed number of shared OTUs between healthy and diseased individuals was significantly smaller than expected by chance in 40 of 41 comparisons. Only in the bacterial vaginosis (BV) study, the observed number of shared OTUs was similar to the number expected by chance. With the more conservative A2 algorithm (reshuffling individuals) the observed number of shared OTUs between healthy and diseased individuals was significantly smaller than expected by chance. in 20 of 41 comparisons, and was smaller, but not statistically significant in an additional 13 comparisons. Across all comparisons, the SES for the number of shared OTUs was statistically smaller than expected for both null model algorithms [A1: mean (SES) = −71.956, one-sample t-test = −3.076, p = 0.004; A2: mean (SES) = −2.24, one-sample t-test = −5.027, p < 0.001] (Fig. 4).
Fig. 4.

The shared species analysis (SSA) with two algorithms: A1 (read-level reshuffling) & A2 (sample-level reshuffling; see the Online Supplementary Information for the details). According to more conservative A2, in 33 of 41 comparisons (80%), there were fewer OTUs (operational taxonomic units) shared between the healthy and diseased individual subjects than expected by chance, but with 49% (20 of 41 comparisons, marked with “*”) being statistically significant at p-value < 0.05. Since A2 is more conservative (reliable), we only marked the cases with statistically significant differences according to the A2
Discussion
Until the recent decade, mainstream biomedicine has largely ignored community ecology theory, but epidemiologists, entomologists, and plant pathologists have been investigating disease ecology for decades (e.g. [39–41]). In the disease ecology of zoonoses (infectious diseases of animals that can be transmitted to humans) [39–42], the idea that the diversity of an ecological community may influence the transmission and dynamics of pathogens can be traced back to [43]. A fundamental premise was that persistence of a pathogen often requires a minimum threshold of host diversity for infections to occur. Recent studies have been conducted to evaluate how the diversity of free-living species (disease vectors such as mosquitoes) may influence the transmission of established pathogens among suitable hosts, in particular the transmission from wildlife to humans and to husbandry animals [40, 41]. But a typical transmission system of zoonoses can implicate three types of communities: hosts, vectors and pathogens (parasites).
Two prevalent hypotheses to explain complex DDR relationships among these communities (which potentially involve the diversities of three categories of interacting communities) in zoonoses are the dilution effect and the amplification effect. Dilution effects are anticipated to occur when ecological communities of pathogens (parasites) are nested in their occurrence in hosts, and interactions between the pathogen and the most suitable hosts persist or increase when biodiversity declines [39–42]. Amplification effects refer to the opposite trend in which the rising host diversity actually “amplifies” the pathogen (parasite) infections [41]. concluded that there is now clear empirical evidence suggesting that biodiversity loss is associated with rising transmission or disease severity for a wide range of important pathogens of plants, wildlife and humans.
DDR relationships have also been investigated in plant pathology and economic entomology (e.g., [44, 45]). Increasing biodiversity in crop and forest ecosystems has been well recognized as one of the more effective ways to control agricultural and forest pests, suggesting that “dilution effects” may be prevalent in these systems.
Our study of human MADs suggests yielded little evidence for a consistent DDR: in most studies, there was no statistically significant difference in the diversity of healthy versus diseased individuals. Regardless of statistical significance, in 14 of 27 comparisons (52%) healthy individuals had higher microbiome diversity than diseased individuals. Moreover, results were not consistent for the similar microbiome sites used in different studies. Overall, the effect sizes in the 41 comparisons did not differ statistically from an average effect size of 0 (one-sample t test = −0.742, p = 0.463, q = 0). In contrast, Johnson et al. [41]. detected a disease effect in 87% of studies (we calculated the percentage from their compilation, see Table S4), although no standardization of data and no statistical tests were applied.
However, this difference between human MAD data and Johnson et al. [41]. compilation (see Table S4) in the percentages of disease effects cannot be entirely attributed to differences in statistical methods, because we obtained virtually identical results for comparisons of untransformed OTU data (Fig. 3). The consistency of our results with standardized and unstandardized data probably represents the fact that, within a study, the same sampling methods and comparable sampling intensities (DNA sequencing coverage) were used for diseased and healthy individuals. Moreover, there was replicated, independent sampling of individuals within groups. The concordance of the raw and standardized results (Fig. 3) strengthens the use of meta-analyses with standardized effect size measurements. Moreover, the results were qualitatively consistent for different diversity indices that weight the contributions of rare and common species differently (Table S2).
Our results do not imply there is no effect of disease on diversity (or vice versa). Across most comparisons (40/41 for A1, 33/41 for A2), there were fewer shared OTUs than expected by chance, suggesting that at least some OTUs were consistently associated with the diseased versus the healthy state. Although we failed to detect a consistent pattern of changes in overall microbiome diversity, there were reliable changes in the species composition of OTUs associated with diseased and healthy individuals. Indeed, the change of shared species should offer promising diagnosis indicators for human MADs. Further research, including experimental studies with animal models, is needed to decide whether the DDR patterns in humans is atypical, or different from DDR patterns in zoonoses or crop and forest diseases (pests). Our opinion is that human MAD systems are rather different because, in many cases, the human microbiome may not be a pathogen or etiological cause at all. Mechanistic (etiological) understanding of human MADs will take additional research, and we believe establishing a formal theory of the DDR patterns for human MADs at this time is still premature.
Supplementary information
Acknowledgements
This study received funding from the following sources: National Science Foundation of China (Grant No. 71473243), Cloud-Ridge Industry Technology Leader Grant, A China-US International Cooperation Project on Genomics/Metagenomics Big Data.
Author contributions
ZSM and NJG defined the research objective. ZSM and LWL conducted data analysis and interpreted the results. ZSM wrote the manuscript and NJG revised the manuscript. All authors read and approved the final manuscript.
Data availability
All datasets analyzed in this study are available in public domain and see Table S1 for the detailed access information for each of the 27 datasets.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Zhanshan (Sam) Ma, Email: ma@vandals.uidaho.edu.
Nicholas J. Gotelli, Email: Nicholas.Gotelli@uvm.edu
Supplementary information
The online version of this article (10.1038/s41396-019-0395-y) contains supplementary material, which is available to authorized users.
References
- 1.Relman DA. The human microbiome: ecosystem resilience and health. Nutr Rev. 2012;70(Suppl. 1):S2–S9. doi: 10.1111/j.1753-4887.2012.00489.x.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Theilade E. The non-specific theory in microbial etiology of inflammatory periodontal diseases. J Clin Periodontol. 1986;13:905–11. doi: 10.1111/j.1600-051X.1986.tb01425.x. [DOI] [PubMed] [Google Scholar]
- 3.Sobel JD. Is there a protective role for vaginal flora? Curr Infect Dis Rep. 1999;1:379–83. doi: 10.1007/s11908-999-0045-z. [DOI] [PubMed] [Google Scholar]
- 4.Marsh PD. Microbial ecology of dental plaque and its significance in health and disease. Adv Dent Res. 1994;8:263. doi: 10.1177/08959374940080022001. [DOI] [PubMed] [Google Scholar]
- 5.Grenier D, Mayrand D. Adult periodontitis: an ecological perspective of mixed infections. Trends Microbiol. 1995;3:148. doi: 10.1016/S0966-842X(00)88904-0. [DOI] [PubMed] [Google Scholar]
- 6.HMP Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–14. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li K, Bihan M, Yooseph S, Methe BA. Analyses of the microbial diversity across the human microbiome. PLoS One. 2012;7:e32118. doi: 10.1371/journal.pone.0032118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma B, Forney LJ, Ravel J. The vaginal microbiome: rethinking health and disease. Annu Rev Microbiol. 2012;66:371–89. doi: 10.1146/annurev-micro-092611-150157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lozupone CA, Stombaugh JI, Gordon J, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–30. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lloyd-Price J, Abu-Ali G, Huttenhower C. The healthy human microbiome. Genome Med. 2016;8:51. doi: 10.1186/s13073-016-0307-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knight R, Callewaert C, Marotz C, Hyde ER, Debelius JW, McDonald D, et al. The The microbiome and human biology. Annu Rev Genom Hum Genet. 2017;18:65–86. doi: 10.1146/annurev-genom-083115-022438. [DOI] [PubMed] [Google Scholar]
- 12.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caporaso J, Kuczynski J, Stombaugh J. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Logares R, Audic S, Bass D, Bittner L, Boutte C, Christen R, et al. Patterns of rare and abundant marine microbial eukaryotes. Curr Biol. 2014;24:813–21. doi: 10.1016/j.cub.2014.02.050. [DOI] [PubMed] [Google Scholar]
- 15.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 16.Lundberg DS, Yourstone S, Mieczkowski P, Jones CD, Dangl JL. Practical innovations for high-throughput amplicon sequencing. Nat Methods. 2013;10:999. doi: 10.1038/nmeth.2634. [DOI] [PubMed] [Google Scholar]
- 17.Sczyrba A, Hofmann P, Belmann P, Koslicki D, Janssen S, Dröge J, et al. Critical assessment of metagenome interpretation-a benchmark of metagenomics software. Nat Methods. 2017;14:1063–71. doi: 10.1038/nmeth.4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.QIIME-2. 2018. https://qiime2.org/. Accessed 12 Sept 2018.
- 19.Dickson RP, Huffnagle GB. The Lung microbiome: new principles for respiratory bacteriology in health and disease. PLoS Pathog. 2015;11:e1004923. doi: 10.1371/journal.ppat.1004923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat Rev Microbiol. 2018;16:143–55. doi: 10.1038/nrmicro.2017.157. [DOI] [PubMed] [Google Scholar]
- 21.Verma D, Garg PK, Dubey AK. Insights into the oral microbiome. Arch Microbiol. 2018;200:525–40. doi: 10.1007/s00203-018-1505-3. [DOI] [PubMed] [Google Scholar]
- 22.Hughes JB, Hellmann JJ, Ricketts TH, Bohannan BJM. Counting the uncountable: Statistical approaches to estimating microbial diversity. Appl Environ Microbiol. 2001;67:4399–406. doi: 10.1128/AEM.67.10.4399-4406.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gotelli NJ, Colwell RK. Quantifying biodiversity: procedures and pitfalls in the measurement and comparison of species richness. Ecol Lett. 2001;4:379–91. doi: 10.1046/j.1461-0248.2001.00230.x. [DOI] [Google Scholar]
- 24.Colwell RK, Chao A, Gotelli NJ, Lin SY, Mao CX, Chazdon RL, et al. Models and estimators linking individual-based and sample-based rarefaction, extrapolation, and comparison of assemblages. J Plant Ecol. 2012;5:3–21. doi: 10.1093/jpe/rtr044. [DOI] [Google Scholar]
- 25.Chao A, Chiu CH, Jost L. Unifying species diversity, phylogenetic diversity, functional diversity and related similarity and differentiation measures through Hill numbers. Annu Rev Ecol, Evol, Syst. 2014;45:297–324. doi: 10.1146/annurev-ecolsys-120213-091540. [DOI] [Google Scholar]
- 26.Gotelli NJ, Shimadzu H, Dornelas M, McGill B, Moyes F, Magurran AE. Community-level regulation of temporal trends in biodiversity. Sci Adv. 2017;3:e170031. doi: 10.1126/sciadv.1700315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson MJ, Crist TO, Chase JM, Vellend M, Inouye BD, Freestone AL, et al. Navigating the multiple meanings of beta-diversity: a roadmap for the practicing ecologist. Ecol Lett. 2011;14:19–28. doi: 10.1111/j.1461-0248.2010.01552.x. [DOI] [PubMed] [Google Scholar]
- 28.Hill MO. Diversity and evenness: a unifying notation and its consequences. Ecology. 1973;54:427–342. doi: 10.2307/1934352. [DOI] [Google Scholar]
- 29.Chao A, Chiu CH, Hsieh TC. Proposing a resolution to debates on diversity partitioning. Ecology. 2012;93:2037–51. doi: 10.1890/11-1817.1. [DOI] [PubMed] [Google Scholar]
- 30.Chao A, Gotelli NG, Hsieh TC, Sander EL, Ma KH, Colwell RK, et al. Rarefaction and extrapolation with Hill numbers: a framework for sampling and estimation in species biodiversity studies. Ecol Monogr. 2014;84:45–67. doi: 10.1890/13-0133.1. [DOI] [Google Scholar]
- 31.Ehrlich SD, Consortium TM. MetaHIT: The European Union Project on Metagenomics of the Human Intestinal Tract. Metagenomics Human Body. 2011;25:968. [Google Scholar]
- 32.McMurdie PJ, Holmes S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol. 2014;10:e1003531. doi: 10.1371/journal.pcbi.1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colwell RK, Coddington JA. Estimating terrestrial biodiversity through extrapolation. Philos Trans R Soc Lond. 1994;345:101–18. doi: 10.1098/rstb.1994.0091. [DOI] [PubMed] [Google Scholar]
- 34.Chao A. Nonparametric estimation of the number of classes in a population. Scand J Stat. 1984;11:265–70. [Google Scholar]
- 35.Chao A, Colwell RK, Lin CW, Gotelli NJ. Sufficient sampling for asymptotic minimum species richness estimators. Ecology. 2009;90:1125–33. doi: 10.1890/07-2147.1. [DOI] [PubMed] [Google Scholar]
- 36.Hsieh T. C., Ma K. H., Chao Anne. iNEXT: an R package for rarefaction and extrapolation of species diversity (Hill numbers) Methods in Ecology and Evolution. 2016;7(12):1451–1456. doi: 10.1111/2041-210X.12613. [DOI] [Google Scholar]
- 37.Cohen J. Statistical Power Analysis for the Behavioral Sciences (2nd). Hillsdale, NJ: Lawrence Erlbaum Associates; 1988.
- 38.Cooper H, Hedges LV, Valentine JC. The handbook of research synthesis and meta-analysis. 2nd ed. New York, NY, USA: Russell Sage Foundation; 1997. p. 592.
- 39.Ostfeld RS, Keesing F, Eviner VT (eds.). Infectious disease ecology: effects of ecosystems on disease and of disease on ecosystems. Princeton, NJ, USA: Princeton University Press; 2008.
- 40.Johnson PTJ, Preston DL, Hoverman JT, Richgels KLD. Biodiversity decreases disease through predictable changes in host community competence. Nature. 2013;494:230–4. doi: 10.1038/nature11883. [DOI] [PubMed] [Google Scholar]
- 41.Johnson PTJ, Ostfeld RS, Keesing F. Frontiers in research on biodiversity and disease. Ecol Lett. 2015;18:1119–33. doi: 10.1371/journal.pone.0041606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Myers SS, Gaffikin L, Golden CD, Ostfeld RS, Redford KH, Ricketts TH, et al. Human health impacts of ecosystem alteration. Proc Natl Acad Sci USA. 2013;110:18753–60. doi: 10.1073/pnas.1218656110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elton Charles S. The Ecology of Invasions by Animals and Plants. Boston, MA: Springer US; 1958. [Google Scholar]
- 44.Zadoks JC. Reflections on space, time, and diversity. Annu Rev Phytopathol. 1999;37:1–17. doi: 10.1146/annurev.phyto.37.1.1. [DOI] [PubMed] [Google Scholar]
- 45.Wan FH, Yang NW. Invasion and management of agricultural alien insects in China. Annu Rev Entomol. 2016;61:77–98. doi: 10.1146/annurev-ento-010715-023916. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets analyzed in this study are available in public domain and see Table S1 for the detailed access information for each of the 27 datasets.