

Abstract
The gut microbiota has an important role in animal health and performance, but its contribution is difficult to determine, in particular given the effects of host genetic factors. Here, whole-genome sequencing of the hosts and 16S rRNA gene sequencing of the microbiota were performed to separate the effects between host genetics and the microbiota in the duodenum, jejunum, ileum, caecum and faeces on fat deposition in 206 yellow broilers reared under identical conditions. Despite the notable spatial variation in the diversity, composition and potential function of the gut microbiota, host genetics exerted limited effects on the gut microbial community. The duodenal and caecal microbiota made greater contributions to fat deposition and could separately account for 24% and 21% of the variance in the abdominal fat mass after correcting for host genetic effects. We further identified two caecal microbial taxa, Methanobrevibacter and Mucispirillum schaedleri, which were significantly correlated with fat deposition. Chickens with a lower Methanobrevibacter abundance had significantly lower abdominal fat content than those with a higher abundance of Methanobrevibacter (35.51 vs. 55.59 g), and the body weights of these chickens did not notably differ. Chickens with a higher M. schaedleri abundance exhibited lower abdominal fat accumulation (39.88 vs. 55.06 g) and body weight (2.23 vs. 2.41 kg) than those with a lower abundance of this species. These findings may aid the development of strategies for altering the gut microbiota to control fat deposition during broiler production.
Subject terms: Microbial communities, Genomics, Obesity
Introduction
With the increasing world population, ensuring an adequate supply of safe and high-quality food has rapidly become a major global concern [1]. Broiler chickens, as an indispensable source of animal protein for human growth and development without any religious taboos, are the most commonly farmed animals in the world [2] and serve as powerful experimental model for basic and applied research. However, the rapid growth of modern broilers has led to a series of developmental and metabolic disorders [3, 4]. In particular, excessive abdominal fat deposition is a widespread problem in poultry production, because it not only reduces feed efficiency and the yield of edible carcasses [3] but also results in consumer dissatisfaction [5]. More than 65 billion chickens are produced annually (FAO 2016) and ~2% of the live body weight (BW) of broilers is abdominal fat [5]. Based on these values, ~3 million tons of abdominal fat are produced by broilers and discarded during processing each year worldwide. Thus, the economic losses associated with this problem are estimated to be over $2.7 billion.
Despite substantial heritability (h2, 0.40 ~ 0.53) [6, 7], genetic progress in abdominal fat deposition has been slow due to the difficulty and cost of measuring this phenotype in a non-invasive manner [8]. In addition, the genetic selection against fatness is hindered by the high positive correlation between BW and abdominal fat content [9]. Therefore, another effective approach for modulating fat deposition in broiler chickens is urgently needed. In addition to breeding and selection, fat accumulation may also be decreased through the regulation of the gut microbiota, which has an important role in detoxification, immune function, food digestion, nutrient and energy acquisition [10–12]. In particular, microbial fermentation is most active in the chicken caecum and can produce energy-rich short-chain fatty acids (SCFAs) [13].
The composition of the gut microbiota is influenced by multiple environmental factors, such as diet [14, 15] and medication [16], thereby offering opportunities to improve host physiological status by modulating gut microbial communities. Accumulating and emerging lines of evidence from humans [17–20], mice [15, 21–23] and farm animals [24–26] have revealed a strong association between the gut microbiota and adiposity. Furthermore, the gut microbiota can modulate obesity, as demonstrated by the transplantation of two specific species (Bacteroides thetaiotaomicron and Methanobrevibacter smithii) [21], the caecal contents of mice [22, 23] or the faecal microbiota from obese and lean humans [19, 27] into germ-free mice. Notably, the colonization of germ-free mice with ‘obese microbiota’ resulted in a significantly greater increase in total body fat than the colonization with ‘lean microbiota’ [22, 27]. However, to the best of our knowledge, the vast majority of the published studies in this field were based on rodent or human models, which may not be suitable for poultry, owing to the unique digestive system and gut microbiota composition of birds. Lacking teeth and jaw muscles, birds grind and mash feed with their gizzard and primarily digest and absorb nutrients in their small intestine (SI, including the duodenum, jejunum and ileum) [28]. The shared core microbiota of the SI is dominated by Firmicutes and Proteobacteria, with lower abundances of Bacteroidetes and Actinobacteria [29]. The pair of caeca is a unique feature of the chicken gastrointestinal tract (GIT) and constitutes an important site for the fermentation of undigested nutrients (such as cellulose and starch) [13], with Bacteroidetes and Firmicutes being the dominant phyla in the caecum [2]. As the avian colon is very short and located in such proximity to the cloaca, few nutrients are absorbed there.
Although there is clear evidence demonstrating the effect of the gut microbiota on host phenotypes, the degree to which host genetics can modulate the microbial composition remains an open question. Although several previous studies for candidate genes found that the gut microbiota is associated with host genetic variations [30–33], many recent studies have not observed strong evidence for this correlation [18, 34, 35].
Therefore, in this study, we performed whole-genome sequencing on 206 yellow broilers and 16S rRNA gene sequencing on 1026 samples from multiple gut segments to explore the extent to which host genetics shape the gut microbial composition. The single-nucleotide polymorphism (SNP) and microbiota data were then used to systematically evaluate the contribution of the gut microbial community to fat deposition using a linear mixed model (LMM). We further aimed to identify specific microorganisms whose abundances are significantly associated with fat accumulation. The findings of this study will provide insights into the microbial communities in different gut sections and their association with fatness.
Materials and methods
Animals, phenotypic data and sample collection
The yellow broiler from Wen’s Nanfang Poultry Breeding, Co., Ltd. (Xinxing County, Guangdong Province, China) was used in this study; this broiler is characterized by yellow feathers and excellent meat quality, and their mean age at slaughter is 78 days. All male broilers (n = 206) were hatched on the same day and reared in a poultry facility under standardized conditions of a 20:4 h light:dark cycle at 19–28 °C, with free access to water and corn-soybean-based diets (containing 2900 kcal/kg metabolizable energy and 190 g/kg crude protein). The birds included in our study were not administered any antibiotics or other veterinary drugs. The individual BW and feed intake were continuously recorded by the automatic feeder, during the fast-growing period from 56 to 76 days of age (see Supplementary Figure S1). The feed conversion ratio (FCR) was then calculated for each broiler.
At the age of 78 days, the BW was measured using an electronic scale (to the nearest 5 g). Whole blood was obtained from the wing vein and stored at −20 °C. Each bird was then euthanized by cervical dislocation followed by decapitation. After the abdomen was opened, the abdominal fat tissue (surrounding the gizzard, cloaca and adjacent abdominal muscles) was carefully dissected and weighed promptly using an electronic balance with a precision of 0.1 g. Moreover, as the gut microbiota, including that from the digesta and mucosa, may contribute to host interactions with respect to nutrient metabolism and immunity [36], the luminal content and mucosal surfaces of each gut segment (including the duodenum, jejunum, ileum and caecum) were collected and droppings were gathered from the cloaca (Fig. 1a). All the samples were snap-frozen in liquid nitrogen, transported to the laboratory and stored at −80 °C for subsequent studies. The abdominal fat percentage (AFP) was calculated based on the BW and abdominal fat weight (AFW).
Fig. 1.

Spatial variation in microbial diversity and community composition in diverse gut segments in chickens. a Spatial structure of the digestive tract of chickens and our five sampling sites. b ɑ-Diversity comparison based on the Shannon diversity index, using ANOVA to determine significant differences (**p < 0.01). The plots are a combination of violin plots and box plots, with the box plots showing the median and the 25% and 75% quantiles. The centre red point indicates the mean value in the corresponding group and the data are expressed as the means ± SD. c Principal coordinate analysis plot generated using OTU metrics based on the Bray–Curtis dissimilarities. Each point represents a sample. Differences were assessed by ANOSIM and significance was established at p < 0.05. An R-value close to ‘1’ suggests dissimilarity between groups, whereas an R-value close to ‘0’ suggests an even distribution of high and low ranks within and between groups. d Relative abundance of the dominant microbial phyla in different segments. e Relative abundance of microbiota in the five gut sites at the genus level. Only genera with an abundance of >1.0% in any of the sites are plotted
Extraction of host and gut microbial DNA
Host DNA was isolated from each blood sample using a Tiangen DNA Extraction Kit (Tiangen Biotech, Beijing, China) according to the manufacturer’s instructions. The gut (digesta and mucosa) and faecal samples were separately thawed on ice and homogenized, and ~200 mg of each sample was used for extraction of microbial genome DNA using the QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) in strict accordance to the manufacturer’s standard protocol. To ensure successful DNA isolation, the concentration and purity of the DNA extract was measured using a Nanodrop instrument (Thermo Fisher Scientific, Waltham, MA, USA) and the completeness of the DNA sample was then assessed by 1% agarose gel electrophoresis. Four faecal samples were excluded due to an insufficient stool amount.
Whole-genome sequencing and data processing
The host DNAs were sheared by sonication and the fragmented DNA was then end-repaired, dA-tailed, ligated to Illumina paired-end adaptors and amplified using PCR with 500 bp inserts for library construction. After PCR amplification and purification, resequencing was performed using the Illumina HiSeq 2500 Sequencer (Illumina, Inc., San Diego, CA, USA) to generate 150 bp paired-end reads. To minimize mapping errors, low-quality reads were removed using FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) with the parameters supplied by Yan et al. [37]. The clean reads from each bird were aligned to the chicken reference genome (galGal5, http://hgdownload.cse.ucsc.edu/goldenPath/galGal5/bigZips/) using the Burrows–Wheeler Alignment tool (BWA ver 0.7.15) [38], with the default parameters. We subsequently used the Picard toolkit (ver 1.119, http://broadinstitute.github.io/picard/) to sort the alignment results and remove potential PCR duplicate reads. The resulting alignments were indexed using SAMtools (ver 1.6) [39] and processed according to the best practices for the Genome Analysis Toolkit (GATK, ver 3.7) [40]. To obtain high-quality SNPs, we set a minimum quality score of 20 for both bases and mapped reads to call variants [41]. Finally, the SNPs of each individual bird were combined to obtain a common set of SNP data and the resulting dataset was subjected to filtering based on the following rigorous criteria using the GATK VariantFiltration module as follows: (a) quality by depth > 10.0; (b) mapping quality score > 40.0; (c) FS < 60.0; (d) MQRankSum > −12.5; and (e) ReadPosRankSum > −8.0. In addition, if more than three SNPs were clustered in a 10 bp window, they were all regarded as false positives and were removed [41].
Subsequently, only biallelic variants were used to achieve more robust quality control (QC) using PLINK (ver 1.9) [42] with the following parameters: sample call rate > 90%, SNP call rate > 95%, minor allele frequencies > 1% and Hardy–Weinberg equilibrium p-value < 10−5. The remaining SNPs and individuals were used for imputation in BEAGLE (ver 4.0) [43], and the PLINK analysis was re-performed using the same above-described criteria. After these steps, a total of 10,902,776 SNPs distributed across 34 chromosomes and 205 birds were obtained for subsequent analyses.
16S rRNA gene sequencing and analysis
The V4 region of the 16S rRNA gene was amplified using the primer pair 520F/802R (5′-AYTGGGYDTAAAGNG-3′ and 5′-TACNVGGGTATCTAATCC-3′) [44] and the amplicons were purified and quantified using Agencourt AMPure Beads and the PicoGreen dsDNA Assay Kit (Invitrogen, Carlsbad, CA, USA), respectively. After quantification, the barcoded V4 amplicons were pooled and subsequently sequenced using an Illumina MiSeq platform to generate 300 bp paired-end reads. The paired-end reads were assembled with FLASH [45]. The Quantitative Insights Into Microbial Ecology (v1.8.0) pipeline [46] was used for data QC and analyses. In brief, low-quality reads that met the following criteria were discarded: (a) an average Phred quality score below 20; (b) read lengths shorter than 150 bp; (c) sequence containing unknown bases or primer mismatches; and (d) mononucleotide repeats exceeding 8 bp. All remaining high-quality reads were aligned and clustered into operational taxonomic units (OTUs) at a similarity threshold of 97% using an open-reference OTU picking protocol [47, 48] (http://qiime.org/tutorials/open_reference_illumina_processing.html).
The SILVA 128 release [49] (https://www.arbsilva.de) was used as the reference database and the UCLUST algorithm [50] was used in the de novo clustering step. Afterward, chimaeras and singletons were filtered from the dataset, and OTUs with an average relative abundance < 10−6 were removed from the analysis [51]. The OTU abundance of each sample and the six-level taxonomic classification from phylum to species were then obtained. To determine the relationships among each microorganism in the five diverse gut segments, the Spearman’s correlation for microbial genera existing in at least 616 samples (60% of the total) was calculated using the psych package in R (https://www.r-project.org/).
Qualified OTU data were used to calculate ɑ-diversity metrics of the Shannon index using the vegan package [52]. Bray–Curtis dissimilarities were produced as β-diversity measures and then subjected to principal coordinate analysis (PCoA) with the ape package [53]. The different sites were statistically compared through analysis of similarity (ANOSIM) with 10,000 permutations and Tax4fun [54] was applied to predict the potential functional capabilities of the microbial communities detected in each of the five locations (duodenum, jejunum, ileum, caecum and faeces). The Wilcoxon rank-sum test was used to investigate the differences in KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways among the five sites. The p-values were adjusted by the false discovery rate (FDR) using the Benjamini–Hochberg (BH) method with the p.adjust function in R.
Construction of the microbial relationship matrix and host genetic relatedness matrix
All OTUs from each site that passed QC were normalized to have a zero mean and a unit variance, and then used to construct the microbial relationship matrix (MRM) [35, 55] with an R script based on the following equation:
where msij represents the estimated microbial relationship in site s between birds i and j; xsio and xsjo are the relative abundances of OTU o in site s in birds i and j, respectively; is the average relative abundance of the OTU o in site s in the population; is the variance of the abundance of OTU o in site s; and Ns is the total number of OTUs in site s used for the relatedness computation.
The eligible SNPs were further pruned to select independent markers using the indep-pairwise command in PLINK with the following parameters: window size of 50 kb, a step of 10 SNPs and a squared Pearson’s correlation coefficient (r2) value of 0.2. Afterwards, the independent dataset of 801,438 SNPs was used to estimate the principal components (PCs) and genetic relatedness matrix (GRM) [56] using GCTA (ver 1.91.1): [57]
in which gij represents the estimated genetic relationship between broilers i and j; xiv and xjv are the counts of the reference alleles in birds i and j, respectively; is the frequency of the reference allele in the population; and N is the number of variants.
Analysis of the association between the gut microbial community and host genetics
To explore the effects of the host genetics on the gut microbiota in diverse locations, we first evaluated the correlation between the GRM and Bray–Curtis distances within each gut section through both Spearman’s and Pearson’s correlation-based Mantel tests with 10,000 permutations. The relationship between the GRM and the MRM was also examined. In addition to investigating the correlation between the GRM and the community structures of the gut microbiota, we estimated the heritability at the genus level. Before this analysis, we normalized the relative abundances of the microorganisms that were detected in ≥ 60% of the samples from a specific segment through rank-based inverse normal transformations using the GenABEL package in R [58]. The microorganisms that were present in < 60% but ≥ 30% of the samples were dichotomized as present or absent [59] and the microorganisms that were detected in < 30% of the samples were excluded from this analysis.
Phenotype prediction based on host genetics and gut microbial communities
We then analysed how well fat deposition can be inferred from the gut microbial communities in comparison with the host genetics. As the chickens used in this experiment had no pedigree information, we estimate the SNP-based heritability of the phenotypes (AFW and AFP) instead, using the following model: [56]
where y is an observation vector (AFW or AFP); c is a vector of fixed covariates with the corresponding design matrix K; e is the residual effect; and g is a vector of aggregate effects of all SNPs with an ~N(0,G), where G and are the GRM and polygenetic variance (overall SNP effects), respectively. The first five host genetic PCs calculated as described above were considered covariates in the model to account for population stratification.
The phenotypic variance explained by the gut microbial variance was estimated as , where and are the phenotypic variance and gut microbial variance, respectively. This fraction has been termed ‘microbiability’ (m2) in animals [55, 60, 61] and ‘microbiome-association index’ in humans [35]. The LMM was calculated as follows:
where the model parameters are as described in model [A] except ms, which is the random effect of the gut microbiota in site s following the multinomial distribution ms ~ N(0,M) and M is the MRM. We estimated the m2 using GCTA. Instead of the GRM, which is usually used in GCTA, we used MRM, which was constructed as described above. To adjust for host genetic factors, all valid individuals and SNPs were used in a genome-wide association analysis (GWAS) with a univariate LMM, which was performed using GEMMA [62]. The likelihood ratio test p-value was selected as a criterion for examining the significance of the association between SNPs and host phenotypes. The genome-wide significant threshold was determined using a modified Bonferroni correction as previously described [63]. Using this approach, the genome-wide significance and suggestive significance were calculated as 9.17 × 10−8 (0.05/545,351) and 1.83 × 10−6 (1/545,351), respectively. We then extracted these SNPs with significant or suggestive significant effects on both the AFW and AFP, and calculated the PCs using PLINK. The first two PCs and the top five host genetic PCs were then considered covariates in model [B] to account for host genetics.
Identification of fatness-related microbiota
For the detection of microorganisms that are significantly associated with fat accumulation, we excluded the taxa that were presented in < 30% of samples from one gut section. The remaining microorganisms were regarded as quantitative traits. All the samples were then successively sorted by host phenotypes (AFW and AFP) and the abundance of each microorganism. The lowest 20% and highest 20% of the ranked birds were considered two distinct groups, and a statistical comparison of all the traits was then performed between the two groups using the Wilcoxon rank-sum test. We subsequently extracted the p-value from each test to construct the following matrix:
where element pi–j represents the p-value obtained from the Wilcoxon rank-sum test of trait j between the 20% lowest- and 20% highest-ranked birds based on trait i. The microorganism m was considered to have a potential relationship with fat deposition if pAFW–m, pm–AFW, pAFP–m and pm–AFP were all < 0.05, and the association was considered significant if FDR < 0.05.
We subsequently characterized the spatial distribution of the fatness-associated microbiota at multiple sites. Furthermore, we calculated the Spearman’s and Pearson’s correlations between the fatness-associated microbiota and other microbial genera (detected in at least 30% of the samples of any of the five gut sites) using the psych package in R and adjusted the p-value using the BH method.
Results
Characterization of host phenotypes and sequencing output
The descriptive statistics of the host phenotypes are summarized in Supplementary Table S1. All phenotypic observations in our study, with the exception of FCR, fit a normal distribution (Shapiro–Wilk test, p > 0.05). The correlations of FCR with AFW and AFP were negligible, whereas the correlation between AFW and BW was 0.53 (p < 0.001), and these two fat deposition-related traits (AFW and AFP) exhibited a high phenotypic correlation (r = 0.98, p < 0.001, Supplementary Figure S2).
To obtain sets of host genomic information, we generated 2.26 Tb of clean reads from 206 birds. The sequence data for each individual reached a 9.22-fold depth and 95.25% genome coverage, which allowed us to call variants with high confidence. After stringent QC, a final set of 10,902,776 SNPs (10.73 SNPs per kb) was obtained (Supplementary Table S2). With respect to the gut microbial community, the 16S rRNA gene sequencing analysis produced a total of 58,959,487 quality-filtered sequences from 1026 samples with an average of 57,465 reads (Supplementary Table S3), and 3,039 OTUs were then clustered with 97% sequence identity. We subsequently classified the OTUs into 536 species, 464 genera, 271 families, 151 orders, 83 classes and 33 phyla (Supplementary Figure S3).
Spatial changes in the diversity, composition and potential function of the gut microbiota
The ɑ-diversity of the microbiota in the caecum was significantly higher than the diversity in the other four sites (Fig. 1b). A PCoA was then conducted to visualize the differences in taxa composition among the diverse sites. The PCoA score plot showed an obvious separation among the SI, caecum and faeces (Fig. 1c), and the ANOSIM enhanced this separation or dissimilarity (R > 0.75, p < 10–4). In addition, an obvious discrepancy was also detected in the three sections of the SI (p < 10–4), even though the R-value was close to zero (R = 0.08).
At the phyla level, the three segments of the SI had similar dominant microorganism communities, which included Proteobacteria, Firmicutes, Actinobacteria and Bacteroidetes. However, visible differences were detected among the SI, caecum and faeces (Fig. 1d). For instance, Firmicutes and Bacteroidetes were two dominant phyla in the SI, with a total combined abundance of 37%, whereas the abundance of Firmicutes in the faeces and Bacteroidetes in the caecum accounted for ~60% and 48% of the total abundance, respectively.
At the genus level, Ochrobactrum and Rhodococcus were the two most abundant genera in the duodenum (7.48% and 6.40%) and ileum (7.70% and 6.34%), whereas unclassified Clostridiaceae exhibited the highest abundance (6.58%) in the jejunum (Fig. 1e). In the caecal samples, instead of the abovementioned genera, Bacteroides, unclassified Bacteroidales and unclassified Clostridiales constituted a notable fraction (51.38%), whereas unclassified Clostridiaceae, Lactobacillus and Gallibacterium represented the majority of the genera in the faecal microbiota, with relative abundances of 27.33%, 20.15% and 9.55%, respectively. Furthermore, for the majority of microorganisms, the relative abundance of a microorganism in one gut segment was not associated with that in other segments (Supplementary Figure S4). A limited number of genera belonging to the Firmicutes and Proteobacteria phyla showed significant and positive correlations among the three sections of SI, but the correlations of two dominant genera, Ochrobactrum and Rhodococcus, were not significant between any two of the three SI segments.
A functional capacity analysis of the gut microbial communities revealed that the top 50 pathways enriched in each gut segment were mostly the same (shared/total: 43/50, Fig. 2a) and primarily associated with metabolism. The abundances of those most common pathways showed distinct differences (FDR < 0.05) among the SI, caecum and faeces (Fig. 2b and Supplementary Table S4). The comparison of the predicted microbial functions between the faeces and gut samples revealed that the faecal microbiota was significantly enriched in genetic information processing pathways. The SI microbial community has important roles in the metabolism of small-molecule nutrients, whereas the caecal microbial community exhibits a stronger ability to degrade macromolecules. Specifically, fatty acid biosynthesis and metabolism of pyruvate and butanoate were overrepresented in the SI in comparison with the caecum and faeces, whereas carbohydrate and energy metabolism was overrepresented in the caecal samples compared with the SI and faeces. Of great significance, the abundance of methane metabolism was significantly higher in the caecum.
Fig. 2.

Comparison of the functional capacities of the gut microbial communities in diverse segments. a Overlap of the top 50 predictions among the five gut sections. b Heatmap showing the 43 shared predictions with different abundances among the diverse gut locations (see Supplementary Table S4). The heatmap is colour-coded based on row z-scores
Weak association between gut microbial communities and host genetics
To explore the effects of host genetics on the gut microbial community, we first examined the correlation between host genetic kinship and microbial β-diversity (based on Bray–Curtis dissimilarities) in diverse gut sites and found no correlation (r2 < 0.02, Supplementary Table S5). We subsequently tested the association between the GRM and MRM, and obtained similar results: both Spearman’s and Pearson’s correlation coefficients of the GRM with any of the five MRMs were close to zero (Supplementary Table S6). We then estimated the heritability of each microorganism at the genus level, including 505 quantitative traits (abundance as phenotype) and 334 binary traits (presence/absence) (Fig. 3a). The sum of the relative abundances of these microbial genera used for heritability estimation (with over 30% detection rate) in the duodenal, jejunal, ileal and faecal samples accounted for 93.42%, 95.30%, 92.28%, 99.85% and 98.54% of the total microbial composition, respectively (Fig. 3b). Of the 839 microbial genera, we found that five in the duodenum (accounting for 2.81% of the tested duodenal microbiota), 10 in the jejunum (5.41%), 17 in the ileum (9.44%), 14 in the caecum (15.05%) and 12 in the faeces (5.91%) exhibited significant (p < 0.05) SNP-based heritability (Supplementary Table S7). Most of these heritable bacteria belonged to the Firmicutes and Proteobacteria phyla (Fig. 3c). The cumulative abundance of these heritable bacteria was only 2.28%, 1.05%, 3.46%, 2.37% and 1.04% (p < 0.05) or, at most, 3.81%, 1.56%, 5.06%, 3.05% and 7.27% (p < 0.1) in the duodenum, jejunum, ileum, caecum and faeces, respectively (Fig. 3d). This result corroborated our aforementioned results that host genetics play a minor role in the shaping of the gut microbial composition.
Fig. 3.

Limited effects of host genetics on gut microbiota. a Number of identified microbial genera. Overall, 102, 99, 109, 79 and 116 microbial genera were detected in at least 60% of the duodenal, jejunal, ileal, caecal and faecal samples, respectively. Overall, 76, 86, 71, 14 and 87 microbial genera were detected in < 60% but ≥ 30% of the duodenal, jejunal, ileal, caecal and faecal samples, respectively. The former 505 microorganisms were regarded as quantitative traits and the latter 334 were dichotomized as present/absent for SNP heritability estimates. The microorganisms with a detection rate < 30% were excluded from further analysis. b The sum of the relative abundances of microbial genera with different detection rates in specific segment. c The number of significantly heritable microorganisms (p < 0.05) grouped by sampling site and phyla. d The cumulative relative abundance of heritable microbial genera in each gut segment (see Supplementary Table S7)
Heritability and microbiability of abdominal fat accumulation
As there was no firm evidence indicating an association between host genetics and the gut microbiota, we considered the abdominal fat mass as the target trait and dissected the contributions of host genetics and the microbiota in different gut segments to this target trait. The relatively high h2 for the AFW (0.33) and AFP (0.39) estimated in our study (Supplementary Table S8) revealed that host genetics have a substantial role in the determination of fat accumulation. Thus, we performed a GWAS and obtained 19 fat deposition-associated SNPs (Supplementary Figure S5 and Supplementary Table S9), and we then used these SNPs as additional covariates to correct for host genetic factors in the m2 estimation. The m2 of the AFW estimated for the duodenum (0.24) and caecum (0.21) was higher than that estimated for other anatomical sites (0.06 for the jejunum, 0.03 for the ileum and 0.02 for the faeces). Similar results were observed for the AFP (0.24 for the duodenum, 0.20 for the caecum, 0.03 for the faeces and 0.10 for the jejunum) and the m2 obtained for the ileum was close to zero (Supplementary Table S10 and Fig. 4).
Fig. 4.
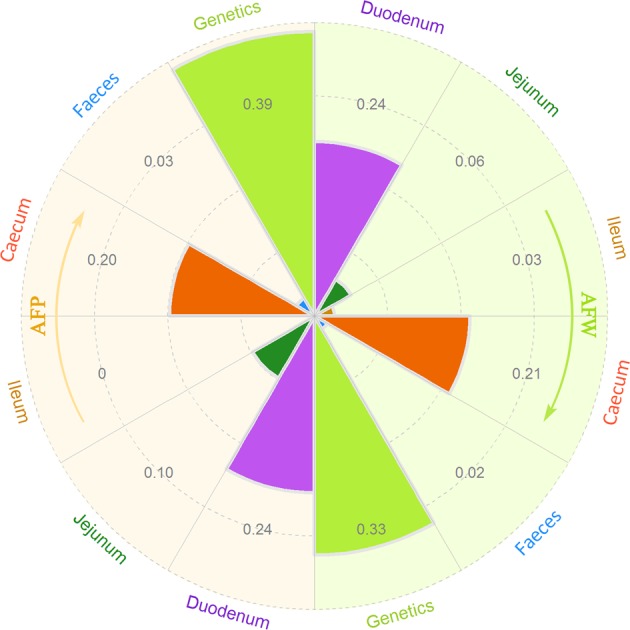
The contribution of the gut microbial community and host genetics to fat deposition. The results of abdominal fat weight (AFW) and abdominal fat percentage (AFP) are shown in the right and left, respectively. Dashed circles indicate the scale of microbiability or heritability from 0.1 to 0.4 (see Supplementary Tables S8 and S10)
The genus Methanobrevibacter and Mucispirillum are significantly associated with fat deposition
As noted above, the gut microbiota is involved in abdominal fat deposition. We further screened which microorganisms are indeed associated with fat accumulation. Given the high correlation between the AFW and the AFP (0.98, see Fig. 5a and Supplementary Figure S2), only microorganisms that exhibited a significant association with both AFW and AFP were considered causal. A double-divergent Wilcoxon rank-sum test with both AFW and AFP identified 21 related microbial genera (Fig. 5b, c). Among these, 16 and 17 genera were significantly associated with the AFW and AFP, and 12 genera were associated with both traits (Supplementary Table 11–13). Of the 12 shared genera, 3, 2, 1 and 6 were located in the caecum, faeces, ileum and jejunum, respectively. To visualize the relationship between fat deposition and the gut microbiota, Pearson’s and Spearman’s correlation coefficients were calculated for these two traits with the relative abundance of the 12 shared microbial genera (Fig. 5d). Most microorganisms were negatively correlated with these two fat-related traits (negative/positive: 10/2) and the correlations among these microorganisms that were present in different gut segments were not significant or weak. The microorganisms in the jejunum were positively and strongly correlated with each other, which implied a strong symbiotic relationship.
Fig. 5.

Adiposity-associated microorganisms. a Overlap analysis of the shared individuals in both the abdominal fat weight (AFW) and abdominal fat percentage (AFP) ranked groups of the highest 20% (N = 40) and lowest 20% (N = 40), respectively. b Significant p-values for each Wilcoxon rank-sum test (see methods). Displayed from the outer to the inner circle are the gut segment, the significance test in each microbial abundance between the highest- and lowest-AFP birds (pAFP–m), the significance test in each microbial abundance between the highest- and lowest-AFW birds (pAFW–m), the significance test in AFP between the two groups with the highest and lowest microbial abundance (pm–AFP), and the significance test in AFW between the two groups with the highest and lowest microbial abundance (pm–AFW), where p-values are plotted as −log2 (p-value); the blue dashed line shows the significance threshold (p < 0.05). Each point represents a microorganism and the red point indicates the p-value passed the significance threshold. The grey dashed line indicates that the pAFW–m, pm–AFW, pAFP–m and pm–AFP values for one microorganism are all < 0.05. c The number of microbial genera associated with AFP and AFW at p < 0.05 and their overlaps with each other. The 12 shared microbial genera are considered potentially related to fat deposition. d Pearson’s (upper diagonal) and Spearman’s (lower diagonal) correlations among the AFW, AFP and the shared 12 microbial genera. Red and blue tiles indicate positive and negative correlations, respectively; significant R-values are filled in numerically (p < 0.05). The diagonal shows the fatness traits or gut segments (C caecum, F faeces, I ileum, J jejunum)
After adjusting the p-value with FDR < 0.05, two caecal genera—Methanobrevibacter and Mucispirillum—showed significant association with the AFW and AFP. Moreover, we found that these two microorganisms were significantly correlated with fat disposition at six levels, ranging from species to phylum (Supplementary Table S14). Fortunately, one was identified as Mucispirillum schaedleri. Fat deposition was significantly lower in the 20% of birds with the lowest Methanobrevibacter abundance compared with the 20% of birds with the highest (the AFW was 35.51 vs. 55.59 g, FDR < 0.01; the AFP was 1.53 vs. 2.36%, FDR < 0.01; Fig. 6a, b) and no difference in BW was found between these two groups (2.28 vs. 2.33 kg, FDR > 0.5; Fig. 6c). The 20% of birds with the highest Mucispirillum abundance exhibited a significantly lower fat mass than the 20% of birds with the lowest abundance of this microorganism (the AFW was 39.88 vs. 55.06 g, FDR < 0.05; the AFP was 1.75 vs. 2.28%, FDR < 0.05; Fig. 6d, e), whereas the group with the highest abundance showed a significantly lower BW than the group with the lowest abundance (2.23 vs. 2.41 kg, FDR < 0.05; Fig. 6f).
Fig. 6.

Effects of fatness-associated microbial genera on host phenotypes. Spatial distribution of these related microbiota and its associations with other microbial genera. a–c Differences in the abdominal fat weight (AFW), abdominal fat percentage (AFP) and body weight (BW) between the two groups with the highest and lowest Methanobrevibacter abundance. d–f Differences in the AFW, AFP and BW between the two groups with the highest and lowest Mucispirillum abundance. For a–f, the plots are a combination of violin plots and box plots, with the box plots showing the median and the 25% and 75% quantiles. The centre red point indicates the mean value in the corresponding group, and data are expressed as the means ± SD. g Relative abundance and detection rate of Methanobrevibacter in the diverse gut segments. h Relative abundance and detection rate of Methanobrevibacter in the diverse gut segments. For g and h, the red curve denotes the average abundance, green bars indicate detection rate and blue bars indicate the percentage of undetected Methanobrevibacter. i Spearman’s correlation between Methanobrevibacter and other microbial genera (detected in at least 30% of samples in any of the five gut sites, see Supplementary Table S15). j Spearman’s correlation between Mucispirillum and other microbial genera (see Supplementary Table S16). For i and j, significance was established at an adjusted p < 0.05
Spatial distribution of the two adiposity-related microorganism and their correlations with other microorganisms and host genetics
We then characterized the spatial distribution of these two fatness-associated microorganisms at multiple sites. Methanobrevibacter and Mucispirillum were detected in almost all caecal samples (Fig. 6g, h) and accounted for 0.024% and 0.622% of the total abundance, respectively. However, the genus Methanobrevibacter was detected in < 4.37%, 2.93%, 12.33% and 49.01% of the duodenal, jejunal, ileal and faecal samples, respectively, and the genus Mucispirillum was not detected in 61.65%, 61.17%, 39.81% and 15.35% of the duodenal, jejunal, ileal and faecal samples, respectively. We also found that the abundances of Methanobrevibacter in the five gut sections showed no correlation with each other. The abundance of the genus Mucispirillum in the caecum was only slightly correlated with that in the ileum (0.26) and jejunum (0.15). These results implied that these two microorganisms were present in all gut segments but mainly thrived in the caecum. In addition, these two microorganisms in the caecum showed little to no association with the microorganisms in other gut locations.
We subsequently focused on the relationships between these two caecal microorganisms and other microorganisms, and the effects of host genetics on these two microorganisms. The majority of the Spearman’s correlations between the genus Methanobrevibacter and other gut microorganisms were negligible (Fig. 6i), and a similar result was found for the genus Mucispirillum (Fig. 6j). Only nine caecal microorganisms exhibited significant and positive relationships with Methanobrevibacter after adjustment (r = 0.22 ~ 0.40, both the Pearson’s and Spearman’s correlations; Supplementary Table S15), and five caecal microorganisms were significantly correlated with Mucispirillum (four positive values and one negative value, Supplementary Table S16). These results implied that the interactions between these two microorganisms and other gut microorganisms were limited. Moreover, according to the aforementioned heritability estimates, h2 was close to zero for both of these microorganisms, which indicated that these microorganisms were not influenced by host genetic regulation.
Discussion
The chicken GIT harbours a diverse microbial community that closely interacts with the host. The host provides a niche and nutrients for microbial growth or proliferation, and in return, the gut microbiota benefits the host in many ways. In addition to having a role in the gut-associated immune system, the microbiota aids in the breakdown and digestion of feed [12]. Although the lumen of the GIT is a contiguous space, the properties of the local microenvironment drive variations in both the identity and abundance of microbial taxa [64–66]. Oxygen gradients [67], pH levels [68] and nutrient availability [69] are the primary factors driving the spatial heterogeneity along the length of the GIT. In our study, substantial variations in Bacteroidetes, Firmicutes, Proteobacteria and Actinobacteria were observed among diverse gut sections. Oxygen availability significantly decreased from the SI to the caeca, because a fraction of the gut microbiota consumes oxygen [67]. In the SI, where transit occurs faster [70], the three sections have qualitatively similar but quantitatively different microbial communities. In fact, the communities of the SI are dominated by rapidly dividing aerobes, such as Ochrobactrum, which has very little fermentative activity, and is related to disease vulnerability [71], and Rhodococcus, which contains a broad range of enzymes that can degrade various toxic and xenobiotic compounds and transform or synthesize valuable molecules [72]. In contrast, the flow is slower in the caeca [73] and the metabolism favours the fermentation of cellulose, starch and resistant polysaccharides [13], which results in higher microbial diversity and dominance of the saccharolytic and anaerobic orders of Bacteroides and Clostridiales.
Although high-throughput sequencing can be applied to nearly any biological sample, the majority of studies on the gut microbiota use stool samples as a material owing to these samples being readily available and because multiple samples can be obtained from the same individual at different times. In the present study, we evaluated the differences in the microbiota of the SI, caecum and faecal matter of chickens. We observed that the faecal microbiota was very different from the microbiota of the other segments assayed. Our results support the notion that a single faecal sample is unable to capture the variations in the structure, diversity and function of the microbiota along the length of the GIT [12, 64, 66, 74]. Moreover, the abundances of the majority of microorganisms at different GIT segments did not affect their abundances in faeces. This result further corroborated the idea that focusing on measurements of microbial abundance in faeces cannot account for their spatial distributions.
As mentioned above, microbial communities are highly adapted to the environment of a particular gut location. However, our results are in accordance with those obtained in previous studies [18, 34] and suggest that host genetics have relatively small effects on the gut microbial composition. Specifically, we found that the host genetic relationship matrix is not significantly associated with any of the five different gut microbial relationship matrices or Bray–Curtis dissimilarities. In addition, the sum of the relative abundances of the heritable microbiota was relatively small, in the range of 1.04–3.46%. A recent large-scale study in humans further demonstrated that the gut microbiome is predominantly shaped by environmental factors [35]. Thus, the gut microbial community could be altered to improve production performance of hosts with diverse genetic backgrounds.
As our findings indicated that the spatial variations in the microbiota structure are largely independent of host genetics, we further evaluated the efficacy of predicting fat deposition from host genetics and diverse gut microbiota. Fat serves as the primary energy depot of the body [75]. The most common problem encountered by the broiler industry is that intensive selection for growth has resulted in the over-deposition of fat in chickens. Abdominal fat, as a type of adipose tissue, is recognized as the most important type of fat associated with excessive fat accumulation [5]. Our estimation of SNP-based h2 for the abdominal fat content was moderate (0.33) and the value was slightly lower but comparable to that obtained in previous estimates, which reported pedigree-based h2 values ranging from 0.40 [6] to 0.53 [7]. Given the moderate correlation (0.53) between the AFW and BW, we further assessed the heritability of the AFP, which was adjusted by BW. The h2 of the AFP remained moderate (0.39), indicating that the host genetics have considerable effects on abdominal fat deposition. Several previous studies have identified candidate genes for fat deposition traits in chicken [76, 77]. Thus, we conducted a GWAS to obtain adipose-associated SNPs and introduced these SNPs as additional covariates in the model [B] to adjust the effects of host genetics. It should be noted that the accuracy of predicting abdominal fat deposition by gut microbiota may be reduced if the host genetic factors are not taken into consideration [35].
‘Microbiability’ estimates can be used as a tool to quantify the effect of gut microbial abundance on host phenotypes and are defined as the fraction of phenotypic variance that can be inferred from the gut microbiota. This concept was originally proposed by Difford et al. [60, 61]. for dairy cattle and has been used to predict several complex traits in pigs [55]. Larger m2 values indicate that the gut microbial community is more informative of the investigated phenotype. A recent study in humans referred to this fraction as the microbiome-association index and demonstrated that it could be reliably estimated using sequence data from only hundreds of individuals [35]. Based on our m2 estimates, the duodenal and caecal microbial communities are more important predictors of fat deposition than the other three gut sections, and these two sections made contributions of ~24% and 21% to abdominal fat deposition in broilers after correcting for host genetic effects, respectively.
As the function of the duodenum is nutrient digestion and absorption, it is likely to be that the microbiota in this section may influence the health and nutrient utilization efficiency of the gut and thus chicken performance. More than 95% of fat is digested in the duodenum [78], and the released products are largely absorbed by the distal duodenum and jejunum [78, 79] and directly deposited in adipose tissues. A recent study in mice demonstrated that the proximal SI microbiota and their mediators play an important role in the regulation of host dietary fat metabolism and absorption [11]. Although much of the actual function of these microbial communities remains unclear, several previous observations in chickens revealed that the duodenal microbiota is significantly associated with energy utilization [10, 80]. The caecum consists of paired long anoxic blind pouches and the metabolic pathways in the microbiota harboured in these pouches are dominated by carbohydrate metabolism, with a lower occurrence of respiration-related genes [28]. The findings of the present study revealed that carbohydrate metabolism is more abundant in the caecum microbiota. Most indigestible residues enter the caeca and are fermented to produce SCFAs (principally acetate and butyrate) by the abundant microorganisms present [13]. These SCFAs are absorbed transepithelially and serve as a source of energy for the host [65]. Although their exact contribution in chickens has yet to be determined, butyrate has been shown to drive gut homoeostasis and improve growth performance in chickens [81].
Our results showed that the gut microbiota was significantly associated with fat accumulation, leading us to investigate which microorganisms play crucial roles in this association. We observed that the highly abundant species M. schaedleri was significantly associated with a lower abdominal fat mass and a lower BW of chickens. A recent genome prediction showed that M. schaedleri uses monosaccharides, oligopeptides, amino acids and SCFAs as substrates for its energy metabolism, whereas it has an extremely limited capacity to degrade polysaccharides [82]. Thus, it is thus likely to be that this microorganism serves as a consumer of breakdown products produced by hydrolytic or fermentative microbiota. Moreover, the presence of M. schaedleri could alter the expression of host immune-related genes [82], suggesting that it may be a pathobiont for certain diseases, such as murine colitis [83, 84].
Another fatness-related taxon identified in the present study was the genus Methanobrevibacter. Significantly lower fat deposition was observed in birds with lower Methanobrevibacter abundance than in birds with high Methanobrevibacter abundance, and the BW of these birds was not obviously different, consistent with the results of previous studies [21, 24]. Methanobrevibacter is a common and important methanogenic archaeon that primarily inhabits the caecum of chickens [85]. Methane-producing microorganisms can improve fermentation efficiency by consuming any excess hydrogen and formate in the bowel, which subsequently improves acetate production and allows the body to absorb more nutrients and calories [86–88]. Gordon et al. [21] revealed that the co-inoculation of gnotobiotic mice with M. smithii and the polysaccharide-fermenting bacterium B. thetaiotaomicron significantly increased the amounts of caecal fermentation products (such as acetate, propionate and butyrate), total liver triglycerides and host fat stores without affecting the total BW. Several previous studies showed that antimethanogenic compounds (such as bromochloromethane) could effectively decrease the abundance of methanogen populations and have no effect on BW [89, 90]. In addition, our results demonstrated that Methanobrevibacter had limited association with other gut microbiota and was not influenced by host genetic regulation. Thus, in modern broiler production, we may develop approaches to reduce fat deposition by inhibiting the caeca-associated genus Methanobrevibacter without affecting the proportion of carcass meat and the gut microbiota community. Although we noticed that methane-producing microorganisms are correlated with fermentation efficiency, we expect to further investigate the role of this microorganism in chickens in the future experiments.
The microbial community in the duodenum was able to explain 24% of fat deposition, but we did not identify any duodenal microorganisms that were significantly associated with AFP, primarily due to the stringent multiple testing criteria used and the relatively modest sample size. The gut microbial communities identified in our study were profiled by 16S rRNA gene sequencing, which can reliably detect microbial taxonomies and composition but exhibits limitations with respect to the identification of specific species and strains. With reductions in the cost of metagenome sequencing, we expect to obtain further information regarding the expression levels of genes of the chicken gut microbiota in the future.
In summary, our results demonstrated that the gut microbial community of chickens was largely independent of host genetics, but the microbiota was highly adapted to the environment of a particular gut location. A certain proportion of the variability in fat deposition was associated with the duodenal and caecal microbiota in chickens. In particular, the genus Methanobrevibacter and the species M. schaedleri were strongly associated with fat deposition, despite their low abundance. These findings provide insights into the roles of the gut microbiota in complex traits and contributes to the development of effective therapies for reducing fat accumulation in broiler production.
Supplementary information
Acknowledgements
This work was supported by Programs for Changjiang Scholars and Innovative Research in Universities (IRT_15R62) and Chinese Universities Scientific Fund (2018TC026 and 2018QC030). We thank Dr Bingkun Zhang from the Department Animal Nutrition and Feed Sciences of China Agricultural University for helpful discussion on chicken nutrition and gastrointestinal physiology.
Author contributions:
NY and JZ conceived and designed this study. CW, WY, JZ, CS, CJ, QZ and DZ performed the phenotype and sample collection. CW analyzed the data and wrote the manuscript. WY assisted in the 16S rRNA gene sequencing data analysis. CS assisted in the whole-genome sequencing data analysis. NY, CS and JZ contributed to the revisions. All authors reviewed and approved the final manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Chaoliang Wen, Wei Yan, Congjiao Sun
Contributor Information
Jiangxia Zheng, Phone: +86-10-6273 2741, Email: jxzheng@cau.edu.cn.
Ning Yang, Phone: +86-10-6273 1351, Email: nyang@cau.edu.cn.
Supplementary information
The online version of this article (10.1038/s41396-019-0367-2) contains supplementary material, which is available to authorized users.
References
- 1.Kearney J. Food consumption trends and drivers. Philos Trans R Soc B. 2010;365:2793–807. doi: 10.1098/rstb.2010.0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pandit RJ, Hinsu AT, Patel NV, Koringa PG, Jakhesara SJ, Thakkar JR, et al. Microbial diversity and community composition of caecal microbiota in commercial and indigenous Indian chickens determined using 16s rDNA amplicon sequencing. Microbiome. 2018;6:115. doi: 10.1186/s40168-018-0501-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel PB. Evolution of the modern broiler and feed efficiency. Annu Rev Anim Biosci. 2014;2:375–85. doi: 10.1146/annurev-animal-022513-114132. [DOI] [PubMed] [Google Scholar]
- 4.Tallentire CW, Leinonen I, Kyriazakis I. Breeding for efficiency in the broiler chicken: a review. Agron Sustain Dev. 2016;36:66. [Google Scholar]
- 5.Wen C, Yan W, Zheng J, Ji C, Zhang D, Sun C, et al. Feed efficiency measures and their relationships with production and meat quality traits in slower growing broilers. Poult Sci. 2018;97:2356–64. doi: 10.3382/ps/pey062. [DOI] [PubMed] [Google Scholar]
- 6.Leenstra FR, Pit R. Fat deposition in a broiler sire strain. 3. Heritability of and genetic correlations among body weight, abdominal fat, and feed conversion. Poult Sci. 1988;67:1–9. doi: 10.3382/ps.0670001. [DOI] [PubMed] [Google Scholar]
- 7.Gaya LG, Ferraz JB, Rezende FM, Mourao GB, Mattos EC, Eler JP, et al. Heritability and genetic correlation estimates for performance and carcass and body composition traits in a male broiler line. Poult Sci. 2006;85:837–43. doi: 10.1093/ps/85.5.837. [DOI] [PubMed] [Google Scholar]
- 8.Dong JQ, Zhang XY, Wang SZ, Jiang XF, Zhang K, Ma GW, et al. Construction of multiple linear regression models using blood biomarkers for selecting against abdominal fat traits in broilers. Poult Sci. 2018;97:17–23. doi: 10.3382/ps/pex319. [DOI] [PubMed] [Google Scholar]
- 9.Leclercq B, Blum JC, Boyer JP. Selecting broilers for low or high abdominal fat: initial observation. Br Poul Sci. 1980;21:107–13. [Google Scholar]
- 10.Yan W, Sun C, Yuan J, Yang N. Gut metagenomic analysis reveals prominent roles of Lactobacillus and cecal microbiota in chicken feed efficiency. Sci Rep. 2017;7:45308. doi: 10.1038/srep45308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martinez-Guryn K, Hubert N, Frazier K, Urlass S, Musch MW, Ojeda P, et al. Small Intestine microbiota regulate host digestive and absorptive adaptive responses to dietarylipids. Cell Host Microbe. 2018;23:458–69. doi: 10.1016/j.chom.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stanley D, Hughes RJ, Moore RJ. Microbiota of the chicken gastrointestinal tract: influence on health, productivity and disease. Appl Microbiol Biot. 2014;98:4301–10. doi: 10.1007/s00253-014-5646-2. [DOI] [PubMed] [Google Scholar]
- 13.Svihus B, Choct M, Classen HL. Function and nutritional roles of the avian caeca: a review. World Poult Sci J. 2013;69:249–64. [Google Scholar]
- 14.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–63. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Everard A, Lazarevic V, Gaia N, Johansson M, Stahlman M, Backhed F, et al. Microbiome of prebiotic-treated mice reveals novel targets involved in host response during obesity. ISME J. 2014;8:2116–30. doi: 10.1038/ismej.2014.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vangay P, Ward T, Gerber JS, Knights D. Antibiotics, pediatric dysbiosis, and disease. Cell Host Microbe. 2015;17:553–64. doi: 10.1016/j.chom.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serena C, Ceperuelo-Mallafre V, Keiran N, Queipo-Ortuno MI, Bernal R, Gomez-Huelgas R, et al. Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. ISME J. 2018;12:1642–57. doi: 10.1038/s41396-018-0068-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beaumont M, Goodrich JK, Jackson MA, Yet I, Davenport ER, Vieira-Silva S, et al. Heritable components of the human fecal microbiome are associated with visceral fat. Genome Biol. 2016;17:189. doi: 10.1186/s13059-016-1052-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fei N, Zhao L. An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J. 2013;7:880–4. doi: 10.1038/ismej.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2008;457:480–4. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Samuel BS, Gordon JI. A humanized gnotobiotic mouse model of host-archaeal-bacterial mutualism. Proc Natl Acad Sci USA. 2006;103:10011–6. doi: 10.1073/pnas.0602187103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–131. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 23.Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. 2004;101:15718–23. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hou Q, Kwok L, Zheng Y, Wang L, Guo Z, Zhang J, et al. Differential fecal microbiota are retained in broiler chicken lines divergently selected for fatness traits. Sci Rep. 2016;6:37376. doi: 10.1038/srep37376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang H, Huang X, Fang S, Xin W, Huang L, Chen C. Uncovering the composition of microbial community structure and metagenomics among three gut locations in pigs with distinct fatness. Sci Rep. 2016;6:27427. doi: 10.1038/srep27427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding J, Zhao L, Wang L, Zhao W, Zhai Z, Leng L, et al. Divergent selection-induced obesity alters the composition and functional pathways of chicken gut microbiota. Genet Sel Evol. 2016;48:93. doi: 10.1186/s12711-016-0270-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341:1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Svihus B. Function of the digestive system. J Appl Poult Res. 2014;23:306–14. [Google Scholar]
- 29.Choi JH, Kim GB, Cha CJ. Spatial heterogeneity and stability of bacterial community in the gastrointestinal tracts of broiler chickens. Poult Sci. 2014;93:1942–50. doi: 10.3382/ps.2014-03974. [DOI] [PubMed] [Google Scholar]
- 30.Blekhman R, Goodrich JK, Huang K, Sun Q, Bukowski R, Bell JT, et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 2015;16:191. doi: 10.1186/s13059-015-0759-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goodrich JK, Davenport ER, Beaumont M, Jackson MA, Knight R, Ober C, et al. Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe. 2016;19:731–43. doi: 10.1016/j.chom.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonder MJ, Kurilshikov A, Tigchelaar EF, Mujagic Z, Imhann F, Vila AV, et al. The effect of host genetics on the gut microbiome. Nat Genet. 2016;48:1407–12. doi: 10.1038/ng.3663. [DOI] [PubMed] [Google Scholar]
- 33.Kolde R, Franzosa EA, Rahnavard G, Hall AB, Vlamakis H, Stevens C, et al. Host genetic variation and its microbiome interactions within the Human Microbiome Project. Genome Med. 2018;10:6. doi: 10.1186/s13073-018-0515-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J, Thingholm LB, Skiecevičienė J, Rausch P, Kummen M, Hov JR, et al. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat Genet. 2016;48:1396–406. doi: 10.1038/ng.3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. 2018;555:210–5. doi: 10.1038/nature25973. [DOI] [PubMed] [Google Scholar]
- 36.Smith CC, Snowberg LK, Gregory CJ, Knight R, Bolnick DI. Dietary input of microbes and host genetic variation shape among-population differences in stickleback gut microbiota. ISME J. 2015;9:2515–26. doi: 10.1038/ismej.2015.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan L, Yang M, Guo H, Yang L, Wu J, Li R, et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat Struct Mol Biol. 2013;20:1131–9. doi: 10.1038/nsmb.2660. [DOI] [PubMed] [Google Scholar]
- 38.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a mapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–8. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81:1084–97. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Claesson MJ, O'Sullivan O, Wang Q, Nikkilae J, Marchesi JR, Smidt H, et al. Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS ONE. 2009;4:e0006669. doi: 10.1371/journal.pone.0006669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–63. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rideout JR, He Y, Navas-Molina JA, Walters WA, Ursell LK, Gibbons SM, et al. Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ. 2014;2:e545. doi: 10.7717/peerj.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jervis-Bardy J, Leong LEX, Marri S, Smith RJ, Choo JM, Smith-Vaughan HC, et al. Deriving accurate microbiota profiles from human samples with low bacterial content through post-sequencing processing of Illumina MiSeq data. Microbiome. 2015;3:19. doi: 10.1186/s40168-015-0083-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2012;41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 51.Benjamino J, Lincoln S, Srivastava R, Graf J. Low-abundant bacteria drive compositional changes in the gut microbiota after dietary alteration. Microbiome. 2018;6:86. doi: 10.1186/s40168-018-0469-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dixon P. VEGAN, a package of R functions for community ecology. J Veg Sci. 2003;14:927–30. [Google Scholar]
- 53.Paradis E, Claude J, Strimmer K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–90. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 54.Aßhauer KP, Wemheuer B, Daniel R, Meinicke P. Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics. 2015;31:2882–4. doi: 10.1093/bioinformatics/btv287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Camarinha-Silva A, Maushammer M, Wellmann R, Vital M, Preuss S, Bennewitz J. Host genome influence on gut microbial composition and microbial prediction of complex traits in pigs. Genetics. 2017;206:1637–44. doi: 10.1534/genetics.117.200782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang J, Bakshi A, Zhu Z, Hemani G, Vinkhuyzen AA, Lee SH, et al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat Genet. 2015;47:1114–20. doi: 10.1038/ng.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:1294–6. doi: 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- 59.Zierer J, Jackson MA, Kastenmüller G, Mangino M, Long T, Telenti A, et al. The fecal metabolome as a functional readout of the gut microbiome. Nat Genet. 2018;50:790–5. doi: 10.1038/s41588-018-0135-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Difford GF, Lassen J, Løvendahl P. Genes and microbes, the next step in dairy cattle breeding. In: Proceedings, EAAP–67th Annual Meeting, Belfast. Wageningen Academic Publishers: Netherlands, 2016, pp 285.
- 61.Difford GF, Plichta DR, Løvendahl P, Lassen J, Noel SJ, Højberg O, et al. Host genetics and the rumen microbiome jointly associate with methane emissions in dairy cows. PLoS Genet. 2018;14:e1007580. doi: 10.1371/journal.pgen.1007580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44:821–4. doi: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gao X. Multiple testing corrections for imputed SNPs. Genet Epidemiol. 2011;35:154–8. doi: 10.1002/gepi.20563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wielen PWJJ, Keuzenkamp DA, Lipman LJA, Knapen F, Biesterveld S. Spatial and temporal variation of the intestinal bacterial community in commercially raised broiler chickens during growth. Microb Ecol. 2002;44:286–93. doi: 10.1007/s00248-002-2015-y. [DOI] [PubMed] [Google Scholar]
- 65.Yeoman CJ, Chia N, Jeraldo P, Sipos M, Goldenfeld ND, White BA. The microbiome of the chicken gastrointestinal tract. Anim Health Res Rev. 2012;13:89–99. doi: 10.1017/S1466252312000138. [DOI] [PubMed] [Google Scholar]
- 66.Tropini C, Earle KA, Huang KC, Sonnenburg JL. The Gut Microbiome: connecting spatial organization to function. Cell Host Microbe. 2017;21:433–42. doi: 10.1016/j.chom.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Espey MG. Role of oxygen gradients in shaping redox relationships between the human intestine and its microbiota. Free Radic Bio Med. 2013;55:130–40. doi: 10.1016/j.freeradbiomed.2012.10.554. [DOI] [PubMed] [Google Scholar]
- 68.O'May GA, Reynolds N, Smith AR, Kennedy A, Macfarlane GT. Effect of pH and antibiotics on microbial overgrowth in the stomachs and duodena of patients undergoing percutaneous endoscopic gastrostomy feeding. J Clin Microbiol. 2005;43:3059–65. doi: 10.1128/JCM.43.7.3059-3065.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Berry D, Stecher B, Schintlmeister A, Reichert J, Brugiroux S, Wild B, et al. Host-compound foraging by intestinal microbiota revealed by single-cell stable isotope probing. Proc Natl Acad Sci USA. 2013;110:4720–5. doi: 10.1073/pnas.1219247110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weurding RE, Veldman A, Veen WA, van der Aar PJ, Verstegen MW. Starch digestion rate in the small intestine of broiler chickens differs among feedstuffs. J Nutr. 2001;131:2329–35. doi: 10.1093/jn/131.9.2329. [DOI] [PubMed] [Google Scholar]
- 71.Alonso CA, Kwabugge YA, Anyanwu MU, Torres C, Chah KF. Diversity of Ochrobactrum species in food animals, antibiotic resistance phenotypes and polymorphisms in the blaOCH gene. FEMS Microbiol Lett. 2017;364:fnx178. [DOI] [PubMed]
- 72.Bell KS, Philp JC, Aw DW, Christofi N. The genus Rhodococcus. J Appl Microbiol. 1998;85:195–210. doi: 10.1046/j.1365-2672.1998.00525.x. [DOI] [PubMed] [Google Scholar]
- 73.Hinton AJ, Buhr RJ, Ingram KD. Physical, chemical, and microbiological changes in the ceca of broiler chickens subjected to incremental feed withdrawal. Poult Sci. 2000;79:483–8. doi: 10.1093/ps/79.4.483. [DOI] [PubMed] [Google Scholar]
- 74.Oakley BB, Kogut MH. Spatial and temporal changes in the broiler chicken cecal and fecal microbiomes and correlations of bacterial taxa with cytokine gene expression. Front Vet Sci. 2016;3:11. doi: 10.3389/fvets.2016.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maruvada P, Leone V, Kaplan LM, Chang EB. The human microbiome and obesity: moving beyond associations. Cell Host Microbe. 2017;22:589–99. doi: 10.1016/j.chom.2017.10.005. [DOI] [PubMed] [Google Scholar]
- 76.Resnyk CW, Carré W, Wang X, Porter TE, Simon J, Le Bihan-Duval E, et al. Transcriptional analysis of abdominal fat in chickens divergently selected on bodyweight at two ages reveals novel mechanisms controlling adiposity: validating visceral adipose tissue as a dynamic endocrine and metabolic organ. BMC Genom. 2017;18:626. doi: 10.1186/s12864-017-4035-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Abdalla BA, Chen J, Nie Q, Zhang X. Genomic insights into the multiple factors controlling abdominal fat deposition in a chicken model. Front Genet. 2018;9:262. doi: 10.3389/fgene.2018.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sklan D, Hurwitz S, Budowski P, Ascarelli I. Fat digestion and absorption in chicks fed raw or heated soybean meal. J Nutr. 1975;105:57–63. [Google Scholar]
- 79.Noy Y, Sklan D. Digestion and absorption in the young chick. Poult Sci. 1995;74:366–73. doi: 10.3382/ps.0740366. [DOI] [PubMed] [Google Scholar]
- 80.Konsak BM, Stanley D, Haring VR, Geier MS, Hughes RJ, Howarth GS, et al. Identification of differential duodenal gene expression levels and microbiota abundance correlated with differences in energy utilisation in chickens. Anim Prod Sci. 2013;53:1269–75. [Google Scholar]
- 81.Panda A. K., Rao S. V. Rama, Raju M. V. L. N., Sunder G. Shyam. Effect of Butyric Acid on Performance, Gastrointestinal Tract Health and Carcass Characteristics in Broiler Chickens. Asian-Australasian Journal of Animal Sciences. 2009;22(7):1026–1031. [Google Scholar]
- 82.Loy A, Pfann C, Steinberger M, Hanson B, Herp S, Brugiroux S, et al. Lifestyle and horizontal gene transfer-mediated evolution of Mucispirillum schaedleri, a core member of the murine gut microbiota. mSystems. 2017;2:e00171–16. doi: 10.1128/mSystems.00171-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rooks MG, Veiga P, Wardwell-Scott LH, Tickle T, Segata N, Michaud M, et al. Gut microbiome composition and function in experimental colitis during active disease and treatment-induced remission. ISME J. 2014;8:1403–17. doi: 10.1038/ismej.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Berry D, Schwab C, Milinovich G, Reichert J, Ben Mahfoudh K, Decker T, et al. Phylotype-level 16S rRNA analysis reveals new bacterial indicators of health state in acute murine colitis. ISME J. 2012;6:2091–106. doi: 10.1038/ismej.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Saengkerdsub S, Anderson RC, Wilkinson HH, Kim WK, Nisbet DJ, Ricke SC. Identification and quantification of methanogenic Archaea in adult chicken ceca. Appl Environ Microb. 2006;73:353–6. doi: 10.1128/AEM.01931-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hansen EE, Lozupone CA, Rey FE, Wu M, Guruge JL, Narra A, et al. Pan-genome of the dominant human gut-associated archaeon, Methanobrevibacter smithii, studied in twins. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4599–606. doi: 10.1073/pnas.1000071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Samuel BS, Hansen EE, Manchester JK, Coutinho PM, Henrissat B, Fulton R, et al. Genomic and metabolic adaptations of Methanobrevibacter smithii to the human gut. Proc Natl Acad Sci USA. 2007;104:10643–8. doi: 10.1073/pnas.0704189104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stams AJ. Metabolic interactions between anaerobic bacteria in methanogenic environments. Antonie Van Leeuwenhoek. 1994;66:271–94. doi: 10.1007/BF00871644. [DOI] [PubMed] [Google Scholar]
- 89.Yang Y, Mu C, Luo Z, Zhu W. Bromochloromethane, a methane analogue, affects the microbiota and metabolic profiles of the rat gastrointestinal tract. Appl Environ Microb. 2016;82:778–87. doi: 10.1128/AEM.03174-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tomkins NW, Colegate SM, Hunter RA. A bromochloromethane formulation reduces enteric methanogenesis in cattle fed grain-based diets. Anim Prod Sci. 2009;49:1053–8. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.