

Abstract
Microbial community succession is a fundamental process that affects underlying functions of almost all ecosystems; yet the roles and fates of the most abundant colonizers are often poorly understood. Does early abundance spur long term persistence? How do deterministic and stochastic processes influence the ecological contribution of colonizers? We performed a succession experiment within a hypersaline ecosystem to investigate how different processes contributed to the turnover of founder species. Bacterial and eukaryotic colonizers were identified during primary succession and tracked through a defined, 79-day biofilm maturation period using 16S and 18S rRNA gene sequencing in combination with high resolution imaging that utilized stable isotope tracers to evaluate successional patterns of primary producers and nitrogen fixers. The majority of the founder species did not maintain high abundance throughout succession. Species replacement (versus loss) was the dominant process shaping community succession. We also asked if different ecological processes acted on bacteria versus Eukaryotes during succession and found deterministic and stochastic forces corresponded more with microeukaryote and bacterial colonization, respectively. Our results show that taxa and functions belonging to different kingdoms, which share habitat in the tight spatial confines of a biofilm, were influenced by different ecological processes and time scales of succession.
Subject terms: Microbial ecology, Environmental microbiology
Introduction
While each distinct microbial community is subjected to specific environmental limitations and opportunities, some established principles guide community assembly and succession [1]. For example, the arrival or disappearance of species may exert feed-back responses onto the local habitat. The niches available for microbial life strategies can change during community succession for every new species and/or function recruited [2]. This is especially true for both primary producers and nitrogen fixing autotrophs, which will eventually compete with heterotrophs occupying niches they helped create. Another principle is that almost all microbial communities studied to date, tend toward species abundance distributions where the majority of the taxa are found in low relative abundance [3–5]. Some evidence suggests that the minority of taxa found in high abundance correlate better with environmental factors than complete microbiomes [6]. These findings prompt the following question: do the most abundant members apply a great enough influence on their environment to direct the recruitment and selection of other taxa—i.e., exert priority effects [7]. The ecological processes underlying the fate of colonizers found in high abundance (founder species) is often unexplored. Does early abundance relate to persistence or are founder species turned over as a microbiome matures and establishes new selection forces?
In this context, turnover is the outcome of ecological processes that replace existing taxa with new species that have successfully immigrated into the community. This definition of turnover implies that new taxa are introduced into a given spatial domain via dispersal [8], and that some taxa are lost either because they were deterministically selected against or due to random demographic events leading to stochastic local extinction. Ecological processes that drive changes in community composition can also result in nestedness. This occurs when species are lost without replacement, which may also result from deterministic selection against some taxa or stochastic local extinction [9]. A key difference between processes leading to turnover versus nestedness is the lack of successful immigration from the regional species pool. Together, these ecological processes collectively influence the succession patterns and biogeochemical functioning of microbial communities [10, 11].
Due to the extreme diversity and swift succession of microbial communities, predictive microbial models remain elusive. Much remains unknown about the roles that colonizing taxa play in influencing the future states of their respective microbiomes and microbial community succession remains an active area of research [12–15]. We do know, however, that these processes can be influenced by both deterministic and stochastic factors [1, 16, 17]. Deterministic ecological processes include selection and environmental filtering, which are governed by the limitations and opportunities imposed by both the environment and niches created by predecessors. If founder species play key roles in establishing the trajectory for succession, then we hypothesized that there will be a strong relationship between high abundance organisms and deterministic processes during colonization. However, stochastic processes may also be strong influencers during colonization and throughout succession and this is especially true when selection and/or environmental filtering processes are weak [18, 19]. We further hypothesized that if the combined effects of the founder species are able to strongly influence their localized environment through niche modification processes such as primary productivity or nitrogen fixation, then they will more strongly direct recruitment of new taxa during community succession [7]. This effect is expected to be manifested in a high degree of phylogenetic relatedness in the successful incoming species. This postulate comes with an assumption that related taxa are more likely to be recruited together because they are expected to have similar habitat associations. While the validity of this assumption can be debated, multiple studies have shown that relationships between phylogenetic relatedness, habitat selection, and functional traits are observable [20–23].
Because organisms from different domains of life often have different ecological roles [24, 25], we investigated whether selective pressures or patterns of succession can be observed between bacterial and microeukaryotic members recruited to the community. We know that different kingdoms have unique sensitivities to environmental cues and have tendencies to fill different niches, however we hypothesize that these differences also translate into temporal dynamics of succession. This biofilm succession study was designed to address these questions and to provide new knowledge about how microbes belonging to different kingdoms are influenced by different ecological processes and timescales of community succession.
We performed a microbial community succession study in a standardized 79-day colonization experiment and placed specific emphasis on the founder species as the community matured. This was done in situ within the unique Hot Lake ecosystem, which is a MgSO4-dominated hypersaline microbial mat located in north-central Washington State (USA). The microbial ecosystem was chosen because of a priori knowledge of seasonal community development presented in a previous study that ascertained a distinct mat-development period during summer months (June–August) [26]. The microbiome characterized from the mat during this growth period remained remarkably similar, in terms of members and relative abundance of taxa, across multiple related studies [25, 27–29]. Our first goal was to determine the structure and function (autotrophy and diazotrophy) of the community from the colonization period through to the season’s typical maturation period. We hypothesized that heterotrophic cells would be as likely as photoautotrophs to act as pioneering founder species, but that nitrogen fixation would be important for at least a subset of colonizers. This hypothesis was based on a priori knowledge of high dissolved organic carbon levels in Hot Lake [30, 31]. The potential for autotrophy and nitrogen fixation was assayed via stable isotope tracers and nano-SIMS imaging. We found that while carbon fixing cells were present during colonization, the majority were not labeled with a 13C dissolved inorganic carbon tracer indicating that the recruitment of new cells to the biofilm did not depend on niches created by primary producers. Active nitrogen fixation was more prevalent during the colonization period showing the potential for priority effects driven by niche formation. Next, we quantified the relative contributions of species loss and replacement to the observable beta diversity across this same time scale. We used a high degree of sample replication (n = 20) in combination with ecological null models commonly used to infer the relative influences of stochastic and deterministic processes at each period of succession. Lastly, we identified the founder species and determined how their abundances changed from the colonization through the maturation periods. We found relatively few highly abundant colonizers that were defined as founder species and of these, most were outcompeted in later stages of succession. We then determined that turnover (versus nestedness) was the dominant process that shaped the mature stages of community succession. Along with turnover and nestedness, the relative contributions of deterministic and stochastic ecological processes varied with respect to the bacterial and eukaryotic components of the microbiome. Null model analyses indicated that the bacterial components of the community trended towards homogenous selection and that the bacterial founder species likely modified their localized environment in ways that mediated environmental filtering of other bacteria. In contrast, colonization of eukaryotic founder species corresponded with deterministic yet variable selection (inferred as non-homogenous) and trended towards stochastic processes [32]. This study highlighted the fact that although species from different kingdoms are often tightly entangled within a biofilm, the respective forces and time scales that shape successional dynamics can be distinct.
Materials and methods
In situ incubation and sampling
We deployed sterile, glass substrata in Hot Lake, which is a MgSO4 dominated, hypersaline lake in north-central Washington State (USA); 48.973062°N, 119.476876°W (Supplemental Fig. S1) [28, 30, 31]. The surfaces included plain borosilicate microscope slides (75 × 25 mm) and round class coverslips (1 in. diameter) placed in large glass petri dishes (15 cm diameter). Each dish contained either 5 slides or 15 coverslips; 55 dishes were deployed totaling 205 slides and 210 coverslips. The slides and cover slips were provided as identical substrates for colonization. Both were used because cover slips are more amenable to imaging and slides are more amenable to biomass removal. Incubations were initiated at 13:00, 20 May 2015 when all surfaces were place on the bottom of the lake at an initial depth of 43–50 cm. One day prior to deployment, the native mat was denuded from the incubation site. The first sample was taken at 16:00, 20 May 2015 (2 h of incubation); the following samples were extracted on near 12:00: 28 May, 3 June, 24 June, 15 July, and 6 August (2015). Samples (at least 30 slides and coverslips per time point) were removed from the lake using an inflatable raft to avoid disturbing the sediment; lake water was simultaneously collected from the sampling position; each slide was dipped briefly in the native water to remove loose sediment using sterile forceps. Samples were fixed in the field for the following: gDNA extraction (20 slides per time point; stored on dry ice); confocal imaging (five slides per time point; fixed in 4% paraformaldehyde; stored at 4 °C); stable isotope probing for nano-SIMS imaging (15 coverslips per time point; fixed in 4% paraformaldehyde stored at 4 °C).
Amplicon sequencing
gDNA was extracted using the MoBio PowerSo DNA isolation kit (Qiagen, Carlsbad, CA) in accordance with the Earth Microbiome Project (EMP) protocols [33]. Sequencing was performed on an Illumina MiSeq instrument (Illumina, San Diego, CA). Twenty separate 16S and 18S rRNA gene amplification reactions were performed on template from each extraction. The extraction and amplification were unsuccessful for all 20 slides harvested after the first 2-h of incubation and only successful four slides from day-1 (24 h); only one 16S and one 18S sample sequenced deeply from the 20 replicates harvested at day-1. We therefore, chose to begin our analysis at day-8, where initial colonization of both bacterial and eukaryotic taxa could be quantified. DNA extraction, amplification and sequencing were successful on five slides from day-8 and all 20 slides for every other sample taken after. The primers targeted the V4 hypervariable region of the 16S SSU rRNA gene using the V4 forward primer (515F) and V4 reverse primer (806R) with 0–3 random bases and the Illumina sequencing primer binding site [34]. The V9 hypervariable region of the 18S SSU rRNA was also targeted with the V9 forward primer (1391F) and V9 reverse primer (EukBr), as outlined by the Earth Microbiome project and modified from Amaral-Zettler et al. [35].
Amplicon analysis
Illumina reads were processed with VSEARCH 2.0.2, an open-source tool for search and clustering [34, 36]; overlapping 16S reads were paired, filtered to a maximum expected error of 1 bp per read, and labeled. Reads were pooled, de-replicated, and chimera-checked with the VSEARCH followed by UCHIME-ref using the RDP Gold database [37]. After discarding chimeras and singletons, reads were clustered into OTUs at 97% similarity and an OTU table was constructed by mapping all labeled reads to these clusters. Taxonomy was assigned to each OTU centroid using the SILVA database v132 [38] and a last common ancestor approach as implemented in QIIME 1.9.1 [39]. The same pipeline was used to process 18S rRNA genes, with a few changes; reads were filtered to a maximum expected error of 0.1 bp per read and no reference-based chimera checking was used.
Diversity analysis
Downstream analysis was completed in R [40], using the ‘phyloseq’ [41] and ‘vegan’ packages [42]. All reads not assigned to their expected kingdom (bacteria and Eukarya, respectively) were filtered along with all mitochondria and chloroplast assignments, and samples were rarified to an even depth of 13,000 reads per sample. Counts of unique OTUs (species richness) and Simpson’s evenness were used to characterize alpha diversity [43]. We note that the traditional 97% similarity threshold may overestimate species richness but also that it is appropriate for this study´s goal of comparing changes between time points during community succession. A metric for the successful immigration of taxa was calculated as the fraction of the community composed of OTUs that were previously unobserved on all prior sampling days. This metric is equivalent to taking the finite difference of the sum of observed microbes (i.e., gamma diversity) over time. Beta diversity was measured by calculating unweighted Jaccard distances between samples, and then partitioning those distances into contributes from nestedness (species loss) and turnover (species gain) using the ‘betapart’ R package [44], followed by the adonis function in the ‘vegan’ package to test to measure changes associated with sampling day (Supplemental Table S1).
Confocal microscopy
Confocal fluorescence microscopy was used to confirm the attachment and composition of the biofilm. The slides were stained with 10 μg mL−1 Hoechst 33342 (Life Technologies, Carlsbad, CA) for 10 min to target DNA. Images were acquired at 1.5 μm z-steps on a Zeiss LSM 710 scanning head confocal microscope equipped with a Zeiss a Plan-Apochromat 63 × /1.40 Oil DIC M27 objective. Excitation lasers were 405 and 633 nm for the blue and red emission channels, respectively. DNA staining was detected at 410–559 nm, and chlorophyll autofluorescence was detected at 647–721 nm. Laser dwell times were 0.64 μs for both channels. Image analysis was conducted using Volocity (PerkinElmer, Waltham, MA).
Stable isotope probing
Incubations of colonized glass cover slips were performed in sterile HLA-400 medium [45] with stable isotope tracers were performed with 13C labeled HCO3- and 15N labeled N2 gas. A control incubation was established to account for the natural abundance of 13C by adding 1.5 mL of a 0.75 M NaHCO3 solution. The labeled incubations were prepared by mixing 0.75 mL of this natural abundance solution with 0.75 mL of a 0.74 M 13C labeled NaHCO3 solution. The 15N labeled N2 incubations were performed in the HLA-400 medium (minus NH4Fe(III) citrate) containing 1 mM NaHCO3; 100 mL glass bottles were charged with 20 mL of degassed medium and one coverslip sample. A more detailed report of these methods is provided in the supplementary material.
NanoSIMS imaging
High-resolution secondary ion intensity and isotope ratio maps were generated using a NanoSIMS 50L (CAMECA, Gennevilliers, France) at the Environmental Molecular Sciences Laboratory (EMSL) at Pacific Northwest National Laboratory (PNNL). Biofilm-coated coverslips were mounted on one-inch-diameter aluminum pin stubs and coated with 20 nm of high-purity gold to improve conductivity. A more detailed report of these methods is provided in the supplementary material.
Statistics and null models
This study made use of the R package ‘vegan’ to partition binary Jaccard distance matrices as described above using the adonis function [46]. Generalized linear models were fit to richness, evenness, and within-day βNTI using sampling day as a fixed effect, and using Poisson, gamma, and Gaussian distributions respectively. The ‘multcomp’ package was used to perform post hoc multiple comparison testing using Tukey’s HSD [47]. We employed a null-model analysis to compare the beta Mean Nearest Taxon Index (βNTI) for all pairwise comparisons within, but not between, the time-points of community development to assess if the phylogenetic similarity between two samples (beta diversity) was significantly higher or lower than expected by chance. The analysis relied on a previously reported convention [32] that reports βNTI values as a z-score with the following interpretation: scores greater than +2 standard deviations from the mean indicate variable selection pressures; scores near zero indicate no significant selection (dominance of stochastic processes); and scores less than −2 standard deviations indicate homogeneous selective pressures. This method has proven robust in a range of ecosystems [10, 18, 19]. The ‘picante’ package and previously described custom scripts [48] were used to calculate βNTI in order to measure changes associated with niche-based processes and selective pressure towards homogenous or heterogeneous phylogenetic structure.
Data repository and reproducible analyses
Genetic sequencing data is available on the Open Science Framework (osf.io) for both 16S and 18S rRNA gene amplicons as part of this project: https://osf.io/a48vj/. Feature abundance tables of amplicons, along with environmental measurements and scripts used for analysis and graphing are available on GitHub: https://github.com/pnnl/brislawn-2018-founders-species
Results
The components and taxonomy of the developing community
Microbial cells colonized the sterile glass substrate and formed a dynamic, multi-kingdom community over a 79-day succession period. Rich microbial communities were established within 8-days of colonization (Fig. 1a). By this early time point, representatives were identified across 33 bacterial and 13 eukaryotic phyla from 999 and 238 bacterial (16S) and eukaryotic (18S) OTUs, respectively. Species richness decreased strongly as the community developed. After a 79-day incubation, the mature microbiome had lost, on average, more than 59 and 15% of the total respective bacterial and eukaryotic OTUs observed from the initial 8-day (colonization) sample. These decreases were statistically significant as determined using post hoc general linear hypothesis testing with Tukey’s all-pair comparisons, which yielded p-values < 0.03. The colonizing community was very uneven, as measured by Simpson’s evenness. This metric ranges between zero and one, with zero representing a community dominated by a single member. The median evenness of 0.02 and 0.03 for 16S and 18S rRNA gene sequences, respectively, emphasizes that a relatively small number of abundant microbes dominated during the development of this community. Bacterial evenness did not mirror the reduction in species richness; evenness only decreased on day-56 (all p-values < 0.001) but rebounded by day-79. No statistically significant changes were observed in eukaryotic evenness during the development of this community.
Fig. 1.
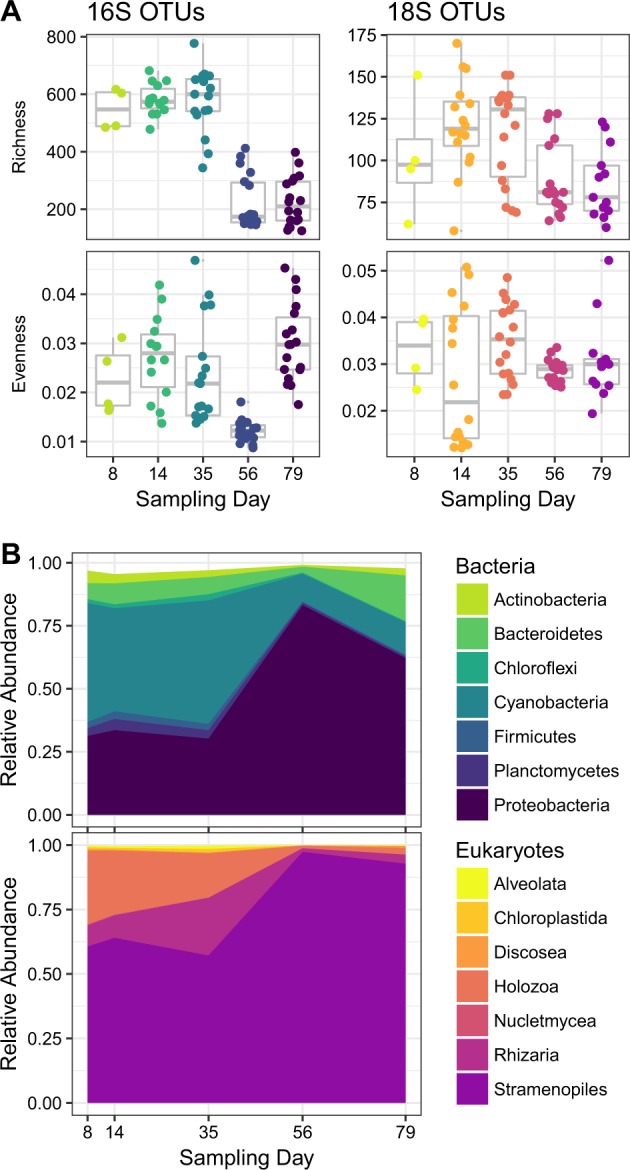
The bacterial and eukaryotic composition of the biofilm community changed during the observed succession period. a Changes in alpha diversity as described by species richness (unique OTUs) and Simpson’s evenness. b Changes in the relative abundance of common bacterial and eukaryotic taxa identified from 16S and 18S OTUs
Cyanobacteria and Proteobacteria were the dominate bacterial phyla over the observed maturation period (Fig. 1b). Some of the most common bacteria had greater relative abundances during the early (colonizing) time periods but then decreased with time. These included: Actinobacteria, Firmicutes, and Planctomycetes. The most abundant Eukaryotes, through all phases of community development, belonged to the Stramenopiles supergroup. Interestingly, this broad phylogenetic group was mostly composed of Bacillariophyceae, which increased in relative abundance as the microbiome matured (Fig. 1b), while the remaining eukaryotic taxa decreased in relative abundance.
Biofilm morphology and basic functional recruitment across succession
The morphology and spatial distribution patterns of dissolved inorganic carbon and N2 incorporation also changed during the 79-day succession. Confocal micrographs showed that diatom-like cell structures became more frequent as the community matured (Fig. 2a) (Supplemental Fig. S2). Interestingly, a shift in morphology was observed; larger diatom-like cells common to the 14- and 35-day samples were replaced by a more dominant and smaller diatom-like cell structures in the final sampling points. This imaging-based result corresponded to the molecular evidence for the apparent takeover of Stramenopiles in the latest stages of observed succession (Fig. 1b). The magnitude and distribution of 13C and 15N incorporation potential was measured by using 13C-labelled bicarbonate and 15N-labelled N2 tracers in combination with nano-SIMS imaging (Fig. 2b) (Supplemental Fig. S3, S4 and S5). These results show that autotrophy was minimal during the early recruitment stages of the biofilm community but became more prevalent throughout maturation. Increased 13C enrichment in cell-like structures also corresponded with higher occurrence of diatom-like structures. Hence, while we infer that primary producers were present, the recruitment of new cells to the biofilm did not depend on the localized metabolic activity of primary producers. This inference is further supported by reports from related studies that also uncovered evidence of heterotrophs acting as the founding colonizers of more complex biofilm communities [49, 50]. Relatively frequent occurrences of N2 fixation were observed during all phases of community development indicating that at least a subset of the founding species was diazotrophic and capable of actively fixing N2.
Fig. 2.

The community changed morphology frequency of autotorphic and diazotrophic cells as members were recruited during succession. a Confocal micrographs showing changes in biofilm community morphology as visualized by the DNA stain (Hoechst; blue) and chlorophyll autofluorescence (red); scale bars represent 20 µm. b Magnitude and distribution of 13C and 15N incorporation in representative biofilm communities showing incorporation of labelled substrates over different incubation periods. Top row: δ13C distribution after incubation with 13C-labelled bicarbonate. Bottom row: δ15N distribution after incubation with 15N-labelled N2. The full suite of replicate nanoSIMS images are presented in a supplementary file
Which ecological processes drive community structure changes?
Changes in the community over the maturation period were attributed to both nestedness and turnover, though turnover was the dominant process in most cases. In this context, turnover is made possible by successful immigration and governed by the dispersal of regional taxa, followed by attachment and growth. Dispersal is the movement of microbes across space, specifically, those recruited to the biofilm. In order to measure taxonomic immigration, we employed a relatively simple metric which counts the fraction of the community represented by OTUs that were undetected at any previous time point (Fig. 3a). This analysis showed that the recruitment of new OTUs diminished 50% by day-14. The successful immigration of bacteria and eukaryotic species continued to decrease during the maturation period and was negligible by day-56 (only 3.4% of the observed OTUs were new). One limitation to this result is the underlying assumption that the appearance of new OTUs was actual due to dispersal and not caused by blooms of taxa residing below detection.
Fig. 3.

Different ecological processes contributed to the observed beta diversity depending on the stage of community succession and faction of the microbiome (bacteria or Eukarya). a The successful immigration of taxa over time as estimated by introduction of new OTUs. b, c Binary Jaccard distance between samples, partitioned by contributions from nestedness (species loss) and turnover (species replacement). Lighter colors represent samples that have few OTUs in common, highlighting changes in community structure over time. The total change (βjac) is the sum of changes due to nestedness (βjne) and turnover (βjtu). d Inferred changes in degree to which variable selection (βNTI > +2), stochastic processes (−2 < βNTI < +2) and homogenous selection (βNTI < −2) contribute to different factions of the community (bacteria and Eukarya) at each observed stage of succession
While the patterns of new OTU recruitment are informative, they do not directly account for how much temporal variation in community structure was due to the gain and/or loss of OTUs. For this, we employed a more rigorous analysis of beta diversity by accounting for the additive contributions of nestedness (species loss through death or emigration) and turnover (species replacement through dispersal) to the binary Jaccard distance (total beta diversity) calculated between all samples taken at all time points [8]. Turnover was a much greater contributor to the observed beta diversity than nestedness with respect to both the bacterial and eukaryotic components of the community. However, within the bacterial samples, more than 50% of the variance observed from nestedness could be attributed to sampling day (Fig. 3). In comparison, only 20% of turnover and 34% of total Jaccard could be attributed to differences observed between sampling days. Hence, while bacterial turnover was high, it was relatively constant between each successional period. Bacterial nestedness was more variable between samples and changes to the bacterial parts of the community coincided more with changes to nestedness. Specifically, there was an increase during the drop in alpha diversity on day-56. We observed the opposite result with respect to changes in eukaryotic beta diversity assessed between sampling days, which was attributed more to turnover. Eukaryotic nestedness was essentially constant and showed little association with time (Fig. 3b, c). Observations from the bacterial components of the microbiome were consistent with the temporal diminishment of successful immigration and showed that the structure of the community was driven by the OTUs gained in the early phases of development.
Another core question from our study was whether bacterial and eukaryotic succession patterns were respectively influenced more by environmental selection or stochastic processes. We know that if we assume dispersal to be a stochastic process—which is a reasonable assumption for this benthic ecosystem—then the earliest stages of succession were at least partially influenced by stochastic processes. However, this insight does not tell us if stochastic processes such as dispersal outweighed selection and/or environmental filtering. If the founder species exerted strong homogenous selection pressures onto their environment, then taxa recruited in the subsequent stages of succession would be expected to be more phylogenetically related than would occur by chance [19, 21]. In contrast, if stochastic process dominated, then the effect of abundant colonizers would be damped such that no phylogenetic pattern in successive OTU recruitment would be expected. By measuring how phylogenetic similarity (given as the beta-Mean Nearest Taxon Index, βNTI) differs from what would be expected by chance, we were able to infer the strength of deterministic processes (selection and environmental filtering) and stochastic processes (dispersal and drift) [10, 32]. The phylogenetic relatedness of the community during colonization was not significantly different than what could be expected by chance (− < βNTI < +2 at early time points). Hence, stochastic processes were found to dominate bacterial colonization but trended towards deterministic, homogenous selection (βNTI < −2) in the latest stages of succession (Fig. 4d and Table S4). In contrast, the Eukaryotes appeared to be influenced by an opposite temporal ordering of stochastic and deterministic process during the observed succession period. The within time-point βNTI values showed that eukaryotic colonization processes were initially influenced by deterministic yet variable selection (βNTI > +2) but then proceeded to weak selection with a significant degree of stochastic processes at work as the community matured (+2 > βNTI > −2) (Fig. 3c).
Fig. 4.

Identification of founder species and their abundances during maturation. a, b Ranked abundances of common bacterial and eukaryotic families, color-coded by phyla. A threshold relative abundance value was set at 5% and the most abundant OTUs belonging to taxa that exceed the threshold by day-8, which was chosen as the representative time for colonization, are defined as founder species. The persistence of founder species is compared to the relative abundances of other taxa that were able to exceed the threshold by day-79, the end point for this study. c, d Temporal changes in abundances of specific founder species (solid lines) compared to other highly abundant OTUs that emerge over the microbiome’s maturation
Who were the founder species and how well did they persist?
We determined which taxa had the highest relative abundances (>5%) from the initial period of colonization defined by the 8-day sampling point. As mentioned, these abundant colonizers were defined as the founder species (Fig. 4). An identical analysis was performed on the end point 79-day samples. This threshold identified only four and five bacterial and eukaryotic taxa ranked at the Family level. Of these nine Families, only three maintained high relative abundance through the observed succession period. As the community matured, the remaining majority of the founder species (six of nine) were replaced by taxonomic Families that were found only at low relative abundance at the 8-day colonization sampling point. The most abundant taxa observed in the community did not bloom until the 56-day sampling point indicating a clear pattern of succession (Fig. 4c, d).
The most abundant founder species were annotated as an uncultured Cyanobacterium Nodosilineaceae, the diatom Pinnularia and a Metazoa represented by the order Podocopida. Interestingly, they both maintained high relative abundances for the first 2–4 weeks, but were ultimately replaced by Rhodobacter, Saccarospirillum and a diatom from the genus Nitzschia, which comprised nearly 90% of the microeukaryotes by day-56. All founder species showed similar dynamics; initial success (in terms of relative abundance) followed by replacement. Yet, this replacement was not absolute; the cyanobacteria (represented by Phormidesmailes, Nodosilneaceae) remained abundant over the observed succession period. This persistence throughout months of consistently high turnover (Fig. 3b) demonstrates the ability to invoke priority effects; even as the selective pressure towards a homogenous community increased (Fig. 3d). The uncultured Nodosilneaceae was the lone bacterial founder species able to persist at high relative abundance. Interestingly, the type of selection that could be inferred from this study differed between kingdoms. Initially, bacterial founder species experienced a minimal amount of phylogenetic selective pressure, and selection towards related microbes slowly increased (Supplemental Fig. S6 and Table S2). Eukaryotes experienced different selective pressures, where all founder species were turned over and largely replaced by the diatom Nitszchia in the later phases of community development, when selection based on niche was minimal and stochastic processes dominated (Supplemental Fig. S7 and Table S3).
Discussion
This study was designed to discover the processes by which early abundance of colonizers (founder species) influence successional trajectories of a multi-kingdom microbiome. We found that only one of seven identified founder species maintained high relative abundances during the observed succession. Hence most of the abundant colonizers forfeited their priority effects that would lead to high relative abundance in later stages of succession. We also investigated the degree to which deterministic and/or stochastic processes dictate the roles that founder species play in their mature communities. We postulated that if a founder species was able to influence community succession by imposing strong selection, then we would see a greater degree of phylogenetic relatedness of new taxa recruited in the next stages of community succession. The null models indicated that colonization of the bacteria was likely not influenced by strong selection but rather by stochastic processes. Likely the initial selective forces were overwhelmed by dispersal and a high degree of taxonomic sampling from the contiguous benthic habitat and sedimentation from overlaying water column. However, the null models indicated that successive stages in the bacterial community development trended towards homogenous selection. Hence, niche modification was likely performed by bacterial founder species in ways that mediated environmental filtering—i.e., exerted priority effects. Homogenous selection appeared to act more strongly on the bacterial components of the community in later stages of succession when the biofilm community had presumably sampled the majority of taxa from the nearby environment. We note that founder species—as well as other abundant taxa identified in this study—were identical or highly related to some of the most common taxa reported by previous studies performed in the Hot Lake ecosystem in different years [26–29]. This indicates that, although the phyla are shown to decrease during this 79-day succession period, these taxa are likely stable and persistent within the ecosystem.
Interestingly, the ecological processes acting on bacterial succession were found to be in sharp contrast to the results obtained for the eukaryotic colonizers. This was evident by comparing the relative contributions of nestedness to diversity through time, which is a metric of species loss without replacement [9]. It was also evident by comparing the initial and ending βNTI values, which reflect the relative contribution of deterministic processes (selection and environmental filtering) and stochastic processes (dispersal and drift) [32]. We concluded that deterministic yet variable selection forces were initially imposed onto the Eukaryotes followed by a mixture of stochastic and deterministic processes during later stages of succession. One possibility that must be considered is that by day-79, bacteria ended the known succession period that was linked to previously identified seasonal assembly [26], but that the recruitment and succession of Eukaryotes operated on different time scales. This would explain why the bacterial and eukaryotic βNTI curves exhibited negative slopes indicating a trend towards convergent, homogenous selective pressures through time. This also implies that the dominant eukaryotic blooms occur after bacterial recruitment had stabilized. Perhaps bacterial taxa found in high abundance in the later stages of succession established the niches required to recruit diatoms and other abundant Eukaryotes. The microscopy provided qualitative evidence of different kingdom-specific time scales as diatom-like structures became common only in the later weeks of the season.
Differences in the relative contribution of turnover and nestedness—attributed to inter-sample diversity—also supported the idea that eukaryotic succession not only operated on a different time scale than the bacteria but also by different sets of ecological processes. We note that the eukaryotic components of the Hot Lake microbiome have not been characterized as thoroughly as the bacteria. However, the abundant Eukarya identified here were consistent with one other study of the Hot Lake ecosystem, performed at a similar sampling location and on a different year [25]. Another important consideration for our interpretation of turnover and nestedness is the fact that these measurements (as applied here) only accounted for presence/absence of taxa. We know that the majority of OTUs are found in low abundance, but we do not know how many taxa are undiscovered and below detection limits of our amplicon analyses. The low abundance and undiscovered taxa can serve as a seed bank, blooming to replace others as conditions change [51]. Those that may have escaped detection could create artifacts in our interpretations. However, within the limitations of our current measurement technologies we are able to determine that turnover and nestedness are important to microbial community succession dynamics and drive shifts in taxonomic abundance.
Turnover was the dominant ecological process observed, yet species loss also played a role in shaping the successional dynamics of this biofilm community. This was shown by decreases in species richness and the diminishing rate of new OTU recruitment. Similar observations have been made in other aquatic biofilm succession studies [52, 53] that also started from a bare substratum. This reoccurring observation presents the possibility of a broader principle in that the conditions that favor recruitment in biofilm rich ecosystems select for those taxa that have the ability to adhere to surfaces [54]. It also expected that that new niches are created during the initial stages of biofilm community succession as these surfaces are filled [55]. The biofilms observed from this current study exhibited clear distinctions between the succession patterns of bacteria and Eukaryotes. This was observed from both molecular and imaging evidence. Diatoms were recruited during the mid-development phase after bacteria-like cell structures and nitrogen fixing cells had attached. Unfortunately, these current results are not sufficient for determining inter-kingdom relationships; however, we infer from their tight spatial associations that they are coupled and that they influence each other’s recruitment to the biofilm.
Diatoms were by far the most abundant Eukaryotes identified, yet the microscopy analyses did not reveal diatom-like structures to be common until the mid-development stages of this community (beginning on day-35). The images also qualitatively show uptake of 13C-label increases with the arrival of frequent diatom-like cell structures. While it is not surprising that microalgae act as primary producers, the finding adds context to this unique hypersaline ecosystem where previous reports have mostly attributed primary production by cyanobacteria [26, 28, 29]; but see [25]. The importance of diatoms to the ecosystem is also supported by our identification of founder species, and observation that they made up at least 30% of 18S OTUs in any given sample (>90% by day-56). This is not to imply that autotrophic bacteria were not contributing to overall primary productivity, as 16S OTUs belonging to the uncultured cyanobacterium Nodosilineaceae (an OTU previously identified to Pseudoanabaenaceae) were highly abundant in the current study and previous reports from the Hot Lake ecosystem [25, 29]. It seems plausible that the diazotrophy could be more important than autotrophy as a pioneer function for creating new niches required for species recruitment; many of the abundant cyanobacteria, including those belonging to Pseudanabaenales, are either taxonomically related to known nitrogen fixers or implicated by a previous metagenomics study [27, 56]. A precedent for this idea has been described by microbial succession studies in other ecosystems from relatively extreme environments including acid mine drainage and glacial forelands [57–59]. This concept is partially supported by the eukaryote-like cell structures were found to be in close physical proximity to diazotrophic cells signifying tightly associated food webs with respect to multi-kingdom nitrogen cycling.
Another goal of this study was to elucidate the ecological processes motivating changes in structure and basic function of the microbiome from colonization to maturation over the observed succession period. The basic functions interrogated were limited to autotrophy and nitrogen fixation based on previous research that indicated that potentially nitrogen fixing, filamentous cyanobacteria dominated the Hot Lake microbial mat [26, 27]. To capture this, we have assembled a narrative to comparatively interpret which ecological processes underpin community development by dividing the community maturation period into three distinct stages: colonization (defined by the 8-day sample), development (defined between the 14- and 56-day samples) and mature (defined by the 79-day sample). The colonization stage was characterized by the arrival of bacteria-like cell structures. Many of which were found to fix N2 and were not exclusively autotrophic as shown by the isotope tracer incubations and nanoSIMS images. At least some of the colonizing species were heterotrophic, which indicates that primary producers are not necessarily required for recruitment of other cells. This observation is somewhat contrary to other, similar succession studies performed in situ [50, 60] but is not surprising based on our knowledge of high abundances of dissolved organic carbon in Hot Lake [30, 31]. The development stage was characterized by the arrival and increased abundance of diatom-like cell structures diminished rates of successful immigration of taxa from the surrounding environment. The maturation stage was characterized by the effective loss or turnover of most founder species and a contrasting shift in the stochastic versus deterministic ecological processes as compared to colonization; specifically, bacteria shifted from stochastic towards homogenous selective and eukarya shifted from deterministic yet variable selection towards significant mixtures of stochastic and deterministic processes.
Conclusions
The identity and roles of the most abundant colonizers in microbial communities—i.e., founder species—are often overlooked. In this succession study, we found that both the bacterial and eukaryotic founder species were mostly turned over. Diatoms were recruited during later stages of succession which corresponded to the observation of widespread autotrophic cells inhabiting the biofilm. Cells that were capable of active nitrogen fixation were present in the early stages of colonization and throughout succession, indicating that diazotrophic bacteria may influence succession more than the early primary producers. We also found that that each kingdom appeared to be influenced by different ecological processes and time scales of succession. Specifically, bacteria shifted from stochastic towards homogenous selective and Eukarya shifted from deterministic yet variable selection towards stochastic processes. Successive stages in the bacterial community development trended towards homogenous selection. Hence, the bacterial founder species likely modified their localized environment in ways that mediated environmental filtering where eukaryotic founder species may not have played as great of a role in the later stages of succession.
Supplementary information
Supplemental Material for - Forfeiting the priority effect: turnover defines biofilm community succession
Acknowledgements
The authors would like to thank Joe Brown and James Fredrickson for valuable discussions and technical assistance. The authors would also like to acknowledge the U.S. Bureau of Land Management, Wenatchee Field Office, for their assistance in authorizing this research and providing access to the Hot Lake Research Natural Area.
Funding
This work was supported by the U.S. Department of Energy (DOE), Office of Biological and Environmental Research (BER), as part of BER’s Genomic Science Program (GSP). This contribution originates from the GSP Foundational Scientific Focus Area (FSFA) at the Pacific Northwest National Laboratory (PNNL). A portion of this study was supported by PNNL’s institutional computing resource (PIC). A portion of the research was performed using EMSL, a DOE Office of Science User Facility sponsored by BER under user proposal number 49356. PNNL is operated for DOE by Battelle Memorial Institute under contract DE-AC05-76RL01830.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
James J. Moran, Email: James.Moran@pnnl.gov
Hans C. Bernstein, Email: Hans.C.Bernstein@uit.no
Supplementary information
The online version of this article (10.1038/s41396-019-0396-x) contains supplementary material, which is available to authorized users.
References
- 1.Nemergut DR, Schmidt SK, Fukami T, O’Neill SP, Bilinski TM, Stanish LF, et al. Patterns and processes of microbial community assembly. Microbiol Molec Biol Rev. 2013;77:342–56. doi: 10.1128/MMBR.00051-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones CG, Lawton JH, Shachak M. Positive and negative effects of organisms as physical ecosystem engineers. Ecology. 1997;78:1946–57. doi: 10.1890/0012-9658(1997)078[1946:PANEOO]2.0.CO;2. [DOI] [Google Scholar]
- 3.Curtis TP, Sloan WT, Scannell JW. Estimating prokaryotic diversity and its limits. Proc Nat Acad Sci USA. 2002;99:10494–9. doi: 10.1073/pnas.142680199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delgado-Baquerizo M, Oliverio AM, Brewer TE, Benavent-González A, Eldridge DJ, Bardgett RD, et al. A global atlas of the dominant bacteria found in soil. Science. 2018;359:320–5. doi: 10.1126/science.aap9516. [DOI] [PubMed] [Google Scholar]
- 5.Magurran AE. Measuring biological diversity. John Wiley & Sons, The Blackwell Publishing Company, MA, USA; 2013.
- 6.Gobet A, Quince C, Ramette A. Multivariate Cutoff Level Analysis (MultiCoLA) of large community data sets. Nucleic Acids Res. 2010;38:e155. doi: 10.1093/nar/gkq545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukami T, Dickie IA, Paula Wilkie J, Paulus BC, Park D, Roberts A, et al. Assembly history dictates ecosystem functioning: evidence from wood decomposer communities. Ecol Lett. 2010;13:675–84. doi: 10.1111/j.1461-0248.2010.01465.x. [DOI] [PubMed] [Google Scholar]
- 8.Baselga A. The relationship between species replacement, dissimilarity derived from nestedness, and nestedness. Glob Ecol Biogeogr. 2012;21:1223–32. doi: 10.1111/j.1466-8238.2011.00756.x. [DOI] [Google Scholar]
- 9.Baselga A. Partitioning the turnover and nestedness components of beta diversity. Glob Ecol Biogeogr. 2010;19:134–43. doi: 10.1111/j.1466-8238.2009.00490.x. [DOI] [Google Scholar]
- 10.Graham EB, Crump AR, Resch CT, Fansler S, Arntzen E, Kennedy DW, et al. Deterministic influences exceed dispersal effects on hydrologically‐connected microbiomes. Environ Microbiol. 2017;19:1552–67. doi: 10.1111/1462-2920.13720. [DOI] [PubMed] [Google Scholar]
- 11.Stegen JC, Johnson T, Fredrickson JK, Wilkins MJ, Konopka AE, Nelson WC, et al. Influences of organic carbon speciation on hyporheic corridor biogeochemistry and microbial ecology. Nat Commun. 2018;9:585. doi: 10.1038/s41467-018-02922-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castle SC, Sullivan BW, Knelman J, Hood E, Nemergut DR, Schmidt SK, et al. Nutrient limitation of soil microbial activity during the earliest stages of ecosystem development. Oecologia. 2017;185:513–24. doi: 10.1007/s00442-017-3965-6. [DOI] [PubMed] [Google Scholar]
- 13.Knelman JE, Graham EB, Prevéy JS, Robeson MS, Kelly P, Hood E, et al. Interspecific plant interactions reflected in soil bacterial community structure and nitrogen cycling in primary succession. Front Microbiol. 2018;9:128. doi: 10.3389/fmicb.2018.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kolinko S, Wu YW, Tachea F, Denzel E, Hiras J, Gabriel R, et al. A bacterial pioneer produces cellulase complexes that persist through community succession. Nat Microbiol. 2018;3:99. doi: 10.1038/s41564-017-0052-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zegeye EK, Brislawn CJ, Farris Y, Fansler SJ, Hofmockel KS, Jansson JK, et al. Selection, succession and stabilization of soil microbial consortia. Biorxiv. 2019;533604. [DOI] [PMC free article] [PubMed]
- 16.Vellend M. Conceptual synthesis in community ecology. Quart Revi Biol. 2010;85:183–206. doi: 10.1086/652373. [DOI] [PubMed] [Google Scholar]
- 17.Zhou J, Ning D. Stochastic community assembly: does it matter in microbial ecology? Microbiol Molec Biol Rev. 2017;81:e00002–17. doi: 10.1128/MMBR.00002-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chase JM, Myers JA. Disentangling the importance of ecological niches from stochastic processes across scales. Philos T Roy Soc B. 2011;366:2351–63. doi: 10.1098/rstb.2011.0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stegen JC, Lin X, Konopka AE, Fredrickson JK. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012;6:1653. doi: 10.1038/ismej.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bryant JA, Lamanna C, Morlon H, Kerkhoff AJ, Enquist BJ, Green JL. Microbes on mountainsides: contrasting elevational patterns of bacterial and plant diversity. Proc Natl Acad Sci USA. 2008;105:11505–11. doi: 10.1073/pnas.0801920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andersson AF, Riemann L, Bertilsson S. Pyrosequencing reveals contrasting seasonal dynamics of taxa within Baltic Sea bacterioplankton communities. ISME J. 2010;4:171–81. doi: 10.1038/ismej.2009.108. [DOI] [PubMed] [Google Scholar]
- 22.Martiny AC, Treseder K, Pusch G. Phylogenetic conservatism of functional traits in microorganisms. ISME J. 2013;7:830. doi: 10.1038/ismej.2012.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J, Shen J, Wu Y, Tu C, Soininen J, Stegen JC, et al. Phylogenetic beta diversity in bacterial assemblages across ecosystems: deterministic versus stochastic processes. ISME J. 2013;7:1310. doi: 10.1038/ismej.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bock C, Salcher MM, Jensen M, Pandey RV, Boenigk J. Synchrony of Eukaryotic and Prokaryotic Planktonic Communities in three seasonally sampled Austrian Lakes. Front Microbiol. 2018;9:1290. doi: 10.3389/fmicb.2018.01290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernstein HC, Brislawn CJ, Dana K, Flores-Wentz T, Cory AB, Fansler SJ, et al. Primary and heterotrophic productivity relate to multi-kingdom diversity in a hypersaline mat. FEMS Microbiol Ecol. 2017;93:fix121. 10.1093/femsec/fix121. [DOI] [PMC free article] [PubMed]
- 26.Lindemann SR, Moran JJ, Stegen JC, Renslow RS, Hutchison JR, Cole JK, et al. The epsomitic phototrophic microbial mat of Hot Lake, Washington: community structural responses to seasonal cycling. Front Microbiol. 2013;4:323. doi: 10.3389/fmicb.2013.00323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mobberley JM, Lindemann SR, Bernstein HC, Moran JJ, Renslow RS, Babauta J, et al. Organismal and spatial partitioning of energy and macronutrient transformations within a hypersaline mat. FEMS Microbiol Ecol. 2017;93:fix028, 10.1093/femsec/fix028. [DOI] [PMC free article] [PubMed]
- 28.Moran JJ, Doll CG, Bernstein HC, Renslow RS, Cory AB, Hutchison JR, et al. Spatially tracking 13C‐labelled substrate (bicarbonate) accumulation in microbial communities using laser ablation isotope ratio mass spectrometry. Environ Microbiol Rep. 2014;6:786–91. doi: 10.1111/1758-2229.12211. [DOI] [PubMed] [Google Scholar]
- 29.Bernstein HC, Brislawn C, Renslow RS, Dana K, Morton B, Lindemann SR, et al. Trade-offs between microbiome diversity and productivity in a stratified microbial mat. ISME J. 2017;11:405–14. doi: 10.1038/ismej.2016.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anderson G. Some limnological features of a shallow saline meromictic lake. Limnol Oceanogr. 1958;3:259–70. doi: 10.4319/lo.1958.3.3.0259. [DOI] [Google Scholar]
- 31.Zachara JM, Moran JJ, Resch CT, Lindemann SR, Felmy AR, Bowden ME, et al. Geo-and biogeochemical processes in a heliothermal hypersaline lake. Geochim Cosmochim Ac. 2016;181:144–63. doi: 10.1016/j.gca.2016.02.001. [DOI] [Google Scholar]
- 32.Dini-Andreote F, Stegen JC, van Elsas JD, Salles JF. Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc Natl Acad Sci USA. 2015;112:E1326–32. doi: 10.1073/pnas.1414261112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gilbert JA, Meyer F, Antonopoulos D, Balaji P, Brown CT, Brown CT, et al. Meeting report: the terabase metagenomics workshop and the vision of an Earth microbiome project. Stand Genom Sci. 2010;3:243. doi: 10.4056/sigs.1433550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amaral-Zettler LA, McCliment EA, Ducklow HW, Huse SM. A method for studying protistan diversity using massively parallel sequencing of V9 hypervariable regions of small-subunit ribosomal RNA genes. PloS ONE. 2009;4:e6372. doi: 10.1371/journal.pone.0006372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rognes T, Mahé F, Flouri T, McDonald D, Schloss P, Woodcroft BJ. vsearch: VSEARCH 1.9.10: GitHub; 2016.
- 37.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–6. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–8. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Team RC. R language definition. Vienna, Austria: R foundation for statistical computing; 2000. [Google Scholar]
- 41.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS ONE. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’hara R, et al. Package ‘vegan’. Community Ecology Package, Version 2. 2013.
- 43.Hamady M, Lozupone C, Knight R. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 2010;4:17–27. doi: 10.1038/ismej.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baselga A, Orme CDL. betapart: an R package for the study of beta diversity. Methods Ecol Evol. 2012;3:808–12. doi: 10.1111/j.2041-210X.2012.00224.x. [DOI] [Google Scholar]
- 45.Cole JK, Hutchison JR, Renslow RS, Kim YM, Chrisler WB, Engelmann HE, et al. Phototrophic biofilm assembly in microbial-mat-derived unicyanobacterial consortia: model systems for the study of autotroph-heterotroph interactions. Front Microbiol. 2014;5:109. doi: 10.3389/fmicb.2014.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McArdle BH, Anderson MJ. Fitting multivariate models to community data: a comment on distance‐based redundancy analysis. Ecology. 2001;82:290–7. doi: 10.1890/0012-9658(2001)082[0290:FMMTCD]2.0.CO;2. [DOI] [Google Scholar]
- 47.Hothorn T, Bretz F, Westfall P. Simultaneous inference in general parametric models. Biom J. 2008;50:346. doi: 10.1002/bimj.200810425. [DOI] [PubMed] [Google Scholar]
- 48.Stegen JC, Lin X, Fredrickson JK, Chen X, Kennedy DW, Murray CJ, et al. Quantifying community assembly processes and identifying features that impose them. ISME J. 2013;7:2069. doi: 10.1038/ismej.2013.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leefmann T, Heim C, Lausmaa J, Sjövall P, Ionescu D, Reitner J, et al. An imaging mass spectrometry study on the formation of conditioning films and biofilms in the subsurface (Äspö Hard Rock Laboratory, SE Sweden) Geomicrobiol J. 2015;32:197–206. doi: 10.1080/01490451.2014.910570. [DOI] [Google Scholar]
- 50.Roeselers G, Van Loosdrecht M, Muyzer G. Heterotrophic pioneers facilitate phototrophic biofilm development. Microb Ecol. 2007;54:578–85. doi: 10.1007/s00248-007-9238-x. [DOI] [PubMed] [Google Scholar]
- 51.Shoemaker William R., Lennon Jay T. Evolution with a seed bank: The population genetic consequences of microbial dormancy. Evolutionary Applications. 2018;11(1):60–75. doi: 10.1111/eva.12557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Besemer K, Singer G, Limberger R, Chlup AK, Hochedlinger G, Hödl I, et al. Biophysical controls on community succession in stream biofilms. Appl Environ Microbiol. 2007;73:4966–74. doi: 10.1128/AEM.00588-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lyautey E, Jackson CR, Cayrou J, Rols JL, Garabétian F. Bacterial community succession in natural river biofilm assemblages. Microb Ecol. 2005;50:589–601. doi: 10.1007/s00248-005-5032-9. [DOI] [PubMed] [Google Scholar]
- 54.Niederdorfer R, Peter H, Battin TJ. Attached biofilms and suspended aggregates are distinct microbial lifestyles emanating from differing hydraulics. Nat Microbiol. 2016;1:16178. doi: 10.1038/nmicrobiol.2016.178. [DOI] [PubMed] [Google Scholar]
- 55.Niederdorfer R, Besemer K, Battin TJ, Peter H. Ecological strategies and metabolic trade-offs of complex environmental biofilms. NPJ Biofilms Micro. 2017;3:21. doi: 10.1038/s41522-017-0029-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stal LJ. Is the distribution of nitrogen‐fixing cyanobacteria in the oceans related to temperature? Environ Microbiol. 2009;11:1632–45. doi: 10.1111/j.1758-2229.2009.00016.x. [DOI] [PubMed] [Google Scholar]
- 57.Brankatschk R, Töwe S, Kleineidam K, Schloter M, Zeyer J. Abundances and potential activities of nitrogen cycling microbial communities along a chronosequence of a glacier forefield. ISME J. 2011;5:1025–37. doi: 10.1038/ismej.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Duc L, Noll M, Meier BE, Bürgmann H, Zeyer J. High diversity of diazotrophs in the forefield of a receding alpine glacier. Microb Ecol. 2009;57:179–90. doi: 10.1007/s00248-008-9408-5. [DOI] [PubMed] [Google Scholar]
- 59.Huang LN, Tang FZ, Song YS, Wan CY, Wang SL, Liu WQ, et al. Biodiversity, abundance, and activity of nitrogen-fixing bacteria during primary succession on a copper mine tailings. FEMS Microbiol Ecol. 2011;78:439–50. doi: 10.1111/j.1574-6941.2011.01178.x. [DOI] [PubMed] [Google Scholar]
- 60.Beam JP, Bernstein HC, Jay ZJ, Kozubal MA, deM Jennings R, Tringe SG, et al. Assembly and succession of iron oxide microbial mat communities in acidic geothermal springs. Front Microbiol. 2016;7:25. doi: 10.3389/fmicb.2016.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material for - Forfeiting the priority effect: turnover defines biofilm community succession