

Abstract
Longitudinal human gut microbiome datasets generated using community-level, sequence-based approaches often report a sub-set of long-lived “resident” taxa that rarely, if ever, are lost. This result contrasts with population-level turnover of resident clones on the order of months to years. We hypothesized that the disconnect between these results is due to a relative lack of simultaneous discrimination of the human gut microbiome at both the community and population-levels. Here, we present results of a small, longitudinal cohort study (n = 8 participants) of healthy human adults that identifies static and dynamic members of the gut microbiome at the clone level based on cultivation/genetic discrimination and at the operational taxonomic unit/amplified sequence variant levels based on 16S rRNA sequencing. We provide evidence that there is little “stability” within resident clonal populations of the common gut microbiome bacterial family, Enterobacteriaceae. Given that clones can vary substantially in genome content and that evolutionary processes operate on the population level, these results question the biological relevance of apparent stability at higher taxonomic levels.
Subject terms: Microbiome, Microbial ecology, Population dynamics, Population genetics
Introduction
Fundamental aspects of the adult human gut ecosystem remain understudied, including why and/or how some microbes colonize and persist while others do not. Estimates of residency (the length of time an organism occupies a niche) in the gut are scarce even for bacteria easily isolated on selective and/or differential media. The few cultivation-based, longitudinal studies of human adults suggest that strain-level turnover in the gut is common over relatively short time scales (i.e., months) [1–4]. Given the variability in genome content between bacterial strains of the same species [5], this variation could potentiate important functional dynamics. In contrast, the few studies that examined longitudinal gut microbiome dynamics using 16S rRNA [6–9] and shotgun metagenomic sequencing [10] suggest that a significant sub-set of the microbiome is highly stable, remaining present perhaps for an individual’s entire adult life [11, 12].
At least some of the above discrepancy is likely due to the scarcity of published studies. However, the discriminatory power of sequencing-based versus cultivation-based approaches is also likely to lead to different conclusions. For example, 16S sequencing is notoriously inconsistent or, depending on one’s view, inaccurate at identifying bacterial species [13], and is by design blind to population-level dynamics. Furthermore, sequencing-based studies rarely discuss or quantify lower limits of detection (e.g., concentrations down to ~106 CFU/gram feces for 16S sequencing [14]), making it difficult to understand sampling effort. Shotgun metagenomic sequencing can also be limited by genome coverage (i.e., the amount of the genome needed to accurately differentiate between strains), which limits accurate strain-level identification. It seems at least possible that -omic sequencing approaches skew important attributes of microbial populations in the gut and in other environments. In this study, we sought to simultaneously quantify and compare community and population-level diversity to better clarify which elements of the gut microbiome are static versus dynamic.
Enterobacteriaceae is the most taxonomically diverse bacterial family [15] recognized by the International Committee on Systematic Bacteriology [16]. Most research on Enterobacteriaceae in humans has narrowly focused on the epidemiology, pathogenesis, virulence, and/or antibiotic resistance of pathogenic strains. Similarly, despite the ability to cultivate all (or nearly all) of the currently named Enterobacteriaceae species on a single selective, differential medium (MacConkey agar), there are few longitudinal studies from healthy human adults. As a result, it is difficult to extrapolate fundamental aspects of non-pathogenic Enterobacteriaceae ecology (e.g., prevalence and diversity) from studies of pathogenic and/or antibiotic resistant strains from hospitalized patients [17].
For this study, we collected semi-weekly stool samples from a cohort of eight healthy human adults and quantified the abundance and distribution of resident and transient operational taxonomic units (OTUs) and amplified sequence variants (ASVs) using 16S rRNA sequencing. From the same samples, we generated a large collection of Enterobacteriaceae isolates (n = 32,470) from MacConkey agar plates and identified residents at the individual clone level. The dynamics observed with cultured Enterobacteriaceae isolates are at odds with community-level stability and suggest that the adult human gut microbiome is not as stable as previously reported. Finally, with respect to longitudinal studies of healthy human adults, this is the first study to report statistical evidence that certain Enterobacteriaceae clones are more likely to be resident compared to others.
Methods
Sample collection and processing
This study was approved by the institutional review board of Montana State University. All participants were enrolled with informed consent, and a total of 9 volunteers (3 female, 6 male) were recruited, ranging in age from 25 to 40 years of age. No information beyond sex and age range was recorded, as correlating host factors with temporal dynamics was not the focus of this study. Although not asked directly, none of the participants co-habitated to our knowledge. Stool samples were requested once every 2 weeks from March 2016 to January 2018 (Table 1). Samples (~5 g of stool) were self-collected using disposable (plastic) commodes (FisherbrandTM Commode Specimen Collection System, Thermo Fisher Scientific, Inc.) into sterile 50 mL conical screw-cap tubes using a sterile tongue depressor. If samples could not be processed immediately, they were refrigerated at 4 °C for no longer than 2 hours prior to processing. Samples were processed inside an anaerobic chamber (Coy Laboratory Products, Inc., Grass Lake, MI, USA) to help preserve anaerobic bacteria for future investigations. Each sample was mixed by shaking with ~30 mL of pre-reduced, sterile phosphate buffered saline. Aliquots (1 mL) were then mixed with 0.2 mL of 80% glycerol in sterile cryogenic vials containing gaskets (Neptune® 1.5 mL CryoTube, VWR International part of Avantor®, Radnor, PA, USA) at a final glycerol concentration of 15%. In parallel, additional aliquots (0.2 mL of stool) were made directly into DNeasy Power Soil DNA extraction tubes (Qiagen, Hilden, Germany) for bulk DNA extraction prior to 16S rRNA sequencing (see below). All aliquots were immediately frozen at −80 °C.
Table 1.
Summary of samples, sampling days, number of OTUs, and residence time according to operationally defined estimates of time (see Methods)
| Minimum | Average | Maximum | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Participant | Samples | Days | OTUs | Resident (%) | Transient (%) | Always resident (%) | Resident (%) | Transient (%) | Always resident (%) | Resident (%) | Transient (%) | Always resident (%) |
| 1 | 76 | 722 | 220 | 170 (77) | 50 (23) | 63 (29) | 196 (89) | 24 (11) | 76 (35) | 215 (98) | 5 (2) | 92 (42) |
| 2 | 43 | 342 | 244 | 181 (74) | 63 (26) | 90 (37) | 206 (84) | 38 (16) | 99 (41) | 223 (91) | 21 (9) | 105 (43) |
| 3 | 24 | 425 | 189 | 151 (80) | 38 (20) | 65 (34) | 179 (95) | 10 (5) | 80 (42) | 185 (98) | 4 (2) | 95 (50) |
| 4 | 29 | 480 | 186 | 145 (78) | 41 (22) | 51 (27) | 163 (88) | 23 (12) | 57 (31) | 178 (96) | 8 (4) | 97 (52) |
| 5 | 38 | 412 | 179 | 137 (77) | 42 (23) | 62 (35) | 158 (88) | 21 (12) | 77 (43) | 177 (99) | 2 (1) | 92 (51) |
| 6 | 48 | 412 | 181 | 111 (61) | 70 (39) | 56 (31) | 125 (69) | 56 (31) | 63 (35) | 145 (80) | 36 (20) | 82 (45) |
| 7 | 42 | 399 | 242 | 196 (81) | 46 (19) | 80 (33) | 213 (88) | 29 (12) | 90 (37) | 231 (95) | 11 (5) | 107 (44) |
| 8 | 24 | 245 | 181 | 142 (78) | 39 (22) | 83 (46) | 158 (87) | 23 (13) | 94 (52) | 171 (94) | 10 (6) | 106 (59) |
| Average | 41 | 430 | 203 | 154 (76) | 49 (24) | 69 (34) | 175 (86) | 28 (14) | 80 (39) | 191 (94) | 12 (6) | 97 (48) |
Sampling and characterization of resident clones
Fecal slurries were thawed to room temperature, serially diluted in sterile PBS, and plated onto MacConkey agar plates, followed by overnight incubation at 37 °C. A target of 95 single colonies per sample were picked from plates using sterile toothpicks and transferred into lysogeny broth (LB) in a 96 deep-well format. One well served as an uninoculated control. If lactose fermenting and non-fermenting colonies were present, each type was picked at roughly the ratio present on the plate. In addition, care was taken to select colonies that differed in morphology, such that rare, odd-looking colonies were present in downstream characterizations. This approach was used to maximize the probability of observing diverse Enterobacteriaceae species as opposed to precisely estimating true abundances. LB cultures were incubated overnight (37 °C), and freezer stocks were made in 15% glycerol and immediately frozen at −80 °C.
Diluted (1:10) cultures were used for PCR-based genotyping; first using the E. coli phylogrouping algorithm of Clermont et al. [18] (half-reaction volumes were used with 1.6 µL of diluted culture; Supplementary Fig. 1). Negative PCRs were repeated twice. If the PCR failed a second time, the isolate was presumed to belong to a species other than E. coli. Residents were defined as described in the Results section. Resident clones were identified as having identical GTG5 rep-PCR fingerprints [19] (Supplementary Fig. 2). Non-E. coli residents were characterized biochemically (API 20E test strips) to identify their likely species designation. In one case, a cryptic Escherichia clade was confirmed by multilocus sequence typing (Sanger sequencing) following a published protocol [20]. Resident clones belonging to phylogroups A and B2.3 were clonotyped using methods described by Tchesnokova et al. [21].
16S rRNA sequencing and analysis
DNA was extracted from fecal slurries (DNeasy PowerSoil kit, QIAGEN) and the V4 region of 16S rRNA encoding gene was amplified and sequenced as described [22] at the University of Michigan Center for Microbial Systems using Illumina MiSeq 2 × 250 bp paired-end sequencing. Reads were generated on two separate sequencing runs in February and June of 2018.
Raw reads were processed using mothur v.1.39.5 [23]. Low-quality reads were removed following the mothur SOP [22] (accessed on May 7, 2017). Briefly, forward and reverse reads were assembled into 253 bp long contigs. Contigs containing ambiguous bases or homopolymers >8 bp were discarded. Identical sequences were combined, and the remaining sequences were aligned against the SILVA database (version 128) trimmed to the V4 region. A pre-cluster step was used to combine rare contigs with more abundant members in the dataset if they differed at 3 or fewer nucleotide sites. Chimeras were identified and removed in mothur using UCHIME [24]. Singletons and doubletons were removed prior to clustering. Sequences sharing 97% similarity were binned into Operational Taxonomic Units (OTUs) using VSEARCH [25], and classified within mothur using the Ribosomal Database Project’s Bayesian classifier (training set 10) [26]. OTUs represented by less than 100 sequences in the dataset were removed to guard against spurious reads. In parallel, quality filtered reads were assembled into amplicon sequencing variants (ASVs) using the DADA2 v1.10.1 pipeline [27]. Trimming and chimera removal were done following the DADA2 Pipeline Tutorial (1.8), and ASVs were classified using the SILVA database (version 128). All OTUs/ASVs that classified to mitochondria, chloroplast, Eukaryota, or remained unclassified at the domain level were removed. Of the original 9 350 936 reads, 7 055 736 and 7 109 813 were assigned to OTUs and ASVs, respectively.
Reads from each sample were rarefied to 10,000 and all downstream analyses were conducted using R version 3.5.1 as follows: PERMANOVA was conducted using the Adonis function in vegan 2.5-3 and 1 000 permutations [28], partitioning around mediods (PAM) clustering was conducted using cluster 2.0.7-1 [29], and ecological diversity estimates and ordinations were generated with Labdsv 1.8-0 [30], and custom R and Python scripts [30, 31, 32].
Results
Study participants and stool samples
Nine healthy volunteers between 24 and 40 years of age (3 females, 6 males) were enrolled with informed consent. No other information was collected (height, weight, BMI, etc.), but all were well enough to participate and presumed to represented healthy adults in this geographic location. Only a single enrollee did not participate beyond three sampling dates and was excluded. From the remaining eight, a total of 392 stool samples were collected, averaging 49 samples per individual (range = 25–97), spanning an average of 512 days (range = 245–849 days), and a median of 8 days apart (Supplementary Fig. 3). All samples were plated directly onto MacConkey agar plates for evaluation of population-level dynamics, and a sub-set of samples (n = 324) were used to evaluate community-level dynamics by 16S rRNA sequencing.
16S rRNA sequencing reveals similar microbiomes within participants over time
Stool samples from eight participants were sequenced using dual-index barcoding and paired-end Illumina MiSeq sequencing (V4 region), resulting in 293 OTUs among all eight participants. Non-metric multidimensional scaling (NMDS) grouped samples according to participant, and PERMANOVA (f = 176.46, p < 0.001) supported statistically different groups (Fig. 1a). PAM clustering was used to identify the most statistically supported number of clusters by evaluating the average silhouette width for 2 to 100 clusters. Seven clusters had the highest support; six of which were comprised of participants 1, 3, 5, 6, 7, 8 and a seventh comprised of participants 2 and 4 with highly similar microbiomes (Supplementary Fig 4). Beta diversity (Bray-Curtis dissimilarity) within participant 2 or 4 was statistically lower than between these participants (Wilcoxon rank sum test p-value < 2.2e-16). However, 150 of the 1 247 (12%) between-participant comparisons yielded lower beta diversity than the median within-participant diversity, highlighting the high degree of similarity between these microbiomes.
Fig. 1.

16S rRNA sequencing-based OTU analysis. Non-metric multidimensional scaling (NMDS) of microbiomes from different participants (a). Each sample is represented by a dot and colors correspond to participants. Bray-Curtis dissimilarity between consecutive samples was plotted through time (b). Differences in Bray-Curtis dissimilarities shown in panel B were tested for significant participant-wise differences (c; Kruskal-Wallis test; p < 0.0001; error bars represent median and 95% confidence limits). Significant differences (p < 0.05) following correction for multiple comparisons (p < 0.05; Dunn’s test) between groups are summarized above plots by letters. Participants that share letters were not significantly different
The change in beta diversity from one sampling date to the next was typically small for all participants (Fig. 1b and error bars in Fig. 1c), and some participants (4 and 5) displayed significantly lower overall diversity compared to others (Fig. 1c). We also evaluated whether beta diversity increased with time and a significant positive correlation (Pearson correlation) was found for all but Participant 2 (Supplementary Fig. 5). However, both the correlation coefficients (range = 0.056–0.377) and the effect size of beta diversity change (range = 0.04–0.13) were small, suggesting that microbiomes sampled close in time were nearly as similar as those sampled over long periods of time.
ASV-based analysis identified 453 ASVs among participants and overall, NMDS (Supplementary Fig. 6), PERMANOVA (f = 207.69, p < 0.001), and PAM clustering (Supplementary Fig. 4) were highly similar to OTU-based results. Interestingly, Procrustes analysis of OTU and ASV-based ordinations (Supplementary Fig. 7) revealed very different sum of squared distances (SSDs) across participants (Participant 2 had 20-fold greater SSDs compared to Participant 7), suggesting that ASVs and OTUs produce nearly identical results for some but not all participants. With respect to PAM clustering, larger mean silhouette widths were generated using ASVs, resulting in well-supported clusters corresponding to all eight participants. Because more ASVs were identified compared to OTUs, estimates of alpha (Inverse Simpson) and beta diversity were expectedly lower with OTUs. However, ASV and OTU estimates of both alpha and beta were highly correlated (Pearson correlation; coefficients for alpha range = 0.9097–0.9860; coefficients for beta range = 0.8638–0.9903) in each participant (Supplementary Fig. 8 and 9).
OTU/ASV residency varies significantly
Residency was defined based on human gut transit time. We reasoned that bacteria entering the gut would be lost at the rate of gut transit unless they were able to become established and colonize. Based on a previously published gut transit time (0.7–4 days) for a dietary residue in healthy human adults [33], we set a threshold of 14 days, meaning that to be considered a resident, OTUs/ASVs had to overcome at least 3 times the maximum transit time. We also allowed for gaps, such that an OTU/ASV could be absent for no more than 30 days before assuming it was lost. If the OTU/ASV reappeared, it would have to again fulfill a 14-day threshold before being considered a resident again. Thus, our definition allowed for both a binary (yes/no) evaluation of residency and quantification of residence time (days). Finally, because not every stool sample produced by each participant was collected, we included flexibility into the way that time was estimated such that a minimum residency period (Min) was the difference between the first and last dates that an OTU/ASV was observed; the maximum (Max) residency period was the difference between the date prior to the first observation and the date immediately following last observation; and the average (Ave) residency period was the difference between the date of the first observation and the date immediately following the last observation (Supplementary Fig. 10).
The most stable sub-set of the microbiome was those OTUs/ASVs that were resident for the entire study period (Always Resident), corresponding to an average of 34% (Min), 39% (Ave), or 48% (Max) of OTUs and 27% (Min), 32% (Ave), or 41% (Max) of ASVs observed within participants (Table 1). To understand whether resident OTUs in one participant were residents in other participants (i.e., a “core resident microbiome”), we plotted the number of OTUs that were always resident in at least “x” number of participants (from 1 to 8, Supplementary Fig. 11). In such plots, lines would plateau at a non-zero threshold if at least some of OTUs were always observed. Exponential decay equations fit very well to curves according to each residency period estimation method (R square = 0.99 for each), but none of the modeled plateaus (i.e., predicted number of OTUs if an infinite number of participants were considered) significantly differed from 0 (p = 0.1976, 0.2502, 0.4157 for Min, Ave, and Max, respectively). Thus, there was little support for a “core resident microbiome”.
We next generated stacked bar charts of OTUs and ASVs according to percentile residency rank (Fig. 2, Supplementary Fig. 12). These plots showed that significant proportions of the microbiome were rarely resident and only an average of 55% (Min), 64% (Ave), or 72% (Max) of OTUs and 46% (Min), 55% (Ave), or 65% (Max) of ASVs were resident in participants for more than half (>50%) of the study period. Collectively, these results suggest that while the microbiome of participants maintained its uniqueness over time (i.e., NMDS), there appeared to be substantial flux in membership.
Fig. 2.

Stacked bar charts of microbiome residency using the average estimate. The number of observed OTUs (a) and ASVs (b) are shown with respect to percentile rank according to the percentage of time present in each participant using the average residency estimate. Black stacks in each bar correspond to transient OTUs and ASVs (i.e., never resident)
Culture-based evaluation of Enterobacteriaceae in the healthy human adult gut
A total of 32 470 single colonies were picked from MacConkey agar plates from the same stool samples (n = 324) used for 16S sequencing and the additional 68 stool samples that were available (total = 392). An average of 84 colonies per stool sample per participant (SD = 4.7) were characterized by PCR-based assays, and the clear majority (n = 28 156, 87%) belonged to one of ten E. coli sensu stricto phylogroups. The percentage of non-E. coli varied between participants from <1 to 37% (mean = 13.27%, SD = 13.28%). Since all members of the Enterobacteriaceae known to colonize the human gut should be cultivable on MacConkey agar and since we attempted to isolate all colony morphology types, these results suggest that E. coli is by far the most dominant Enterobacteriaceae in healthy humans. Interestingly, a significant number of E. coli (n = 7699, 27%) did not appear to ferment lactose when grown on MacConkey agar, suggesting that this commonly used biochemical marker of E. coli sensu stricto requires re-evaluation.
Enterobacteriaceae residency
GTG5 rep-PCR was used to identify distinct clonal lineages that met our residency definition and the presence of individual clones through time was plotted for each participant (Fig. 3). We also plotted the relative abundance of phylogroups within participants (Supplementary Fig. 13) and while we believe these plots reasonably represent true abundances, it should be noted that colonies were not necessarily selected at random (see Methods) and so some rarer members are likely overrepresented. It is also important to note that not all 32 470 isolates were fingerprinted to identify residents. Instead, we first identified potential residents using a step-wise algorithm (Supplementary Fig. 2), which allowed us to narrow down the number of isolates to a reasonable sub-set for rep-PCR. Some transients were identified using the combination of phylogrouping PCR results and residence time (≤14 days) and subsequently confirmed by rep-PCR. We did not fingerprint all transient isolates or try to identify all transient clones. For simplicity, we assumed that E. coli transients belonging the same phylogroup in the same sample also belonged to the same clone, which is a highly conservative assumption. Likewise, we did not attempt to identify all transient non-E. coli Enterobacteriaceae clones. Instead, we conducted rep-PCR on all non-E. coli Enterobacteriaceae from three of the eight participants (n = 116) and a reasonable random sub-set (22–68% of isolates) in the remaining participants (Supplementary Table 1). In total, 967 of the 3164 (31%) potentially resident non-E. coli Enterobacteriaceae were evaluated. Again, this approach provided an accurate estimate of the number of resident clones and a conservative estimate of the number of transient clones.
Fig. 3.

Presence of Enterobacteriaceae clones. Unique Enterobacteriaceae clones defined by PCR-based discriminatory assays (see Methods) where plotted according the day(s) observed. Day 0 in each panel corresponds to the first stool sample collected from both participants and ticks along the x-axis represent samples. Clones are ordered by order of appearance along the y-axis and colored according to either their phylogroup membership (E. coli) or species as defined by biochemical testing. The legend shown for Participant 2 is the same for all panels. No clones were shared between any participant (e.g., “Clone 1” in Participant 1 is not the same as “Clone 1” for any other participant)
A total of 120 Enterobacteriaceae clones were identified among all participants and 37 (31%) of these were resident. Note that because rep-PCR was not performed on all isolates, the number of transient clones was likely to be somewhat greater than 86. Of the 37 resident clones, only three were non-E. coli, and each belonged to a different “species” (Citrobacter freundii, Klebsiella pneumoniae, and cryptic Escherichia clade IV), suggesting that with respect to residency, E. coli was again the dominant species. Within E. coli sensu stricto, all phylogroups were observed with the single exception of phylogroup C. The most commonly observed phylogroup was B1 (n = 30 clones, 25%), followed by A.1 (n = 26, 22%), A (n = 17, 14%), B2.3 (n = 16, 13%), and F (n = 10, 8%). All other clones (B2.1, B2.2, D, and E) comprised ≤ 5% each and together accounted for only 15%. Residence time (log 10 transformed number of days observed) was significantly different between Enterobacteriaceae clones (Fig. 4a) using the Ave and Max estimates (ANOVA; Ave, p = 0.0037; Max, p = 0.0034) and approached significance using the Min estimate (ANOVA; p = 0.0735). Phylogroups A and B2.3 had the longest residence times, and following correction for multiple comparisons, both phylogroups resided significantly longer than phylogroup A.1 using both Ave (Fig. 4a) and Max (Supplementary Fig. 14) estimates. Phylogroup F clones also resided significantly longer than A.1 clones but using the Max estimate only (Supplementary Fig. 14). In addition to having the longest mean residence times, A, B2.3, and F clones were more often observed “beyond the study period” (Fig. 4b). Finally, A, B2.3, and F clones were identified as residents (as opposed to transients) more often (Fisher’s exact test, p = 0.024) using the Ave estimate (Fig. 4c). Given their greater overall residence times, likeliness to be present beyond the study period, and frequency as residents, these results suggest that E. coli belonging to A, B2.3, and F phylogroups are more likely to be human residents compared to all other members of the Enterobacteriaceae.
Fig. 4.
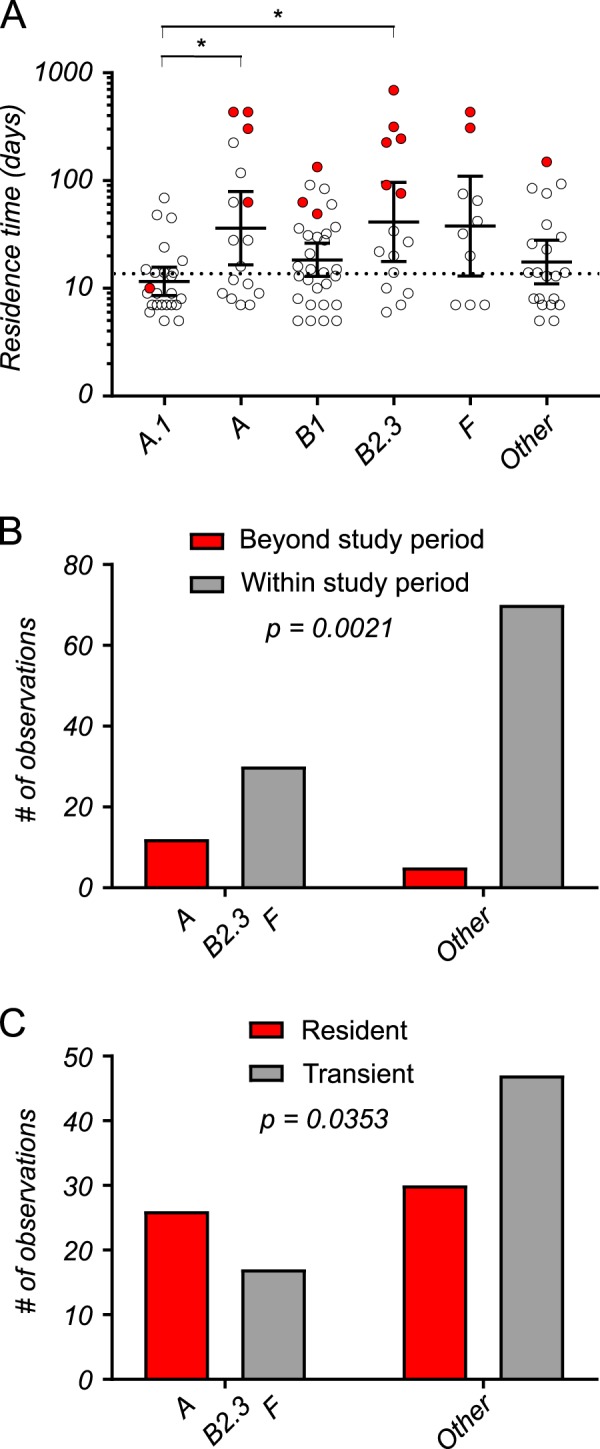
Enterobacteriaceae clone residency using the average estimate. All Enterobacteriaceae clones were plotted according to their log 10 transformed residence time in days (a). Clones were grouped according their phylogroup (E. coli) or into an “Other” category if ≤6 representative isolates were observed (Other = B2.1, n = 4; B2.2, n = 4; D, n = 6; E, n = 4; Cryptic Escherichia clade IV, n = 1; C. freundii, n = 1; K. pneumoniae, n = 1). Groups had significantly different residence time (ANOVA, F = 3.729, p = 0.0037) and following correction for multiple comparisons (Holm-Sidak’s test), groups A and B2.3 resided significantly longer than A.1 (p = 0.0338, p = 0.0171, respectively). Red dots identify clones that were present at the outset of the study, throughout the entire study period, or at the end of the study. These clones were presumed to have colonized participants beyond the study period. Contingency table analysis (Chi-square test) was used to test whether significantly more clones were observed beyond the study period in some groups versus others (b) or whether significantly more resident clones were present (c) among the longest lived phylogroups (A, B2.3, and F) compared to other groups. Both comparisons were significant and p-values are shown in each panel
16S rRNA sequencing is not diagnostic for Enterobacteriaceae or Escherichia/Shigella
Enterobacteriaceae and E. coli were cultured from 308 and 289 of the 324 (95% and 89%, respectively) stool samples that were evaluated by 16S sequencing. Only two OTUs classified as Enterobacteriaceae, and one of these classified to the Escherichia/Shigella genus. Likewise, five ASVs classified as Enterobacteriaceae and one of these classified to the Escherichia/Shigella genus. By comparing OTU/ASV presence-absence to culture-based presence-absence across samples, we evaluated the diagnostic potential of 16S to identify Enterobacteriaceae or Escherichia/Shigella in stool samples. Counts for false positives, true positives, false negatives, and true negatives as well as positive and negative predictive values and accuracy were generated (Supplementary Table 3). Overall, 16S sequencing had excellent positive predictive value, regardless of OTU or ASV-based analysis. However, both analyses had poor negative predictive values for both Enterobacteriaceae and Escherichia/Shigella (all <23%). OTU-based analysis was slightly more accurate than ASV-based analysis, but neither performed remarkably well (all accuracy estimates <63%). Thus, with respect to Enterobacteriaceae and Escherichia/Shigella, 16S sequencing and culture results did not correlate well.
Discussion
Few longitudinal studies have been conducted to identify short and long-lived members of the human gut microbiome, estimate rates of migration into and out of the gut ecosystem, or understand the functional impact of taxonomic turnover. Temporal variability is an important attribute of ecosystems affecting colonization dynamics [34]. In macrobiomes (e.g., animals), the decision to migrate or reside is a function of environmental condition-dependent fitness costs and benefits [34, 35]. It seems reasonable to hypothesize the same is true for human gut microbiome members and that evolutionary strategies leading to migration or residency result from realized fitness tradeoffs inside and outside the gut environment. Adaptation to one or both of these habitats, was previously proposed as an evolutionary strategy for E. coli [36], and as a model, seems applicable to most gut microbiome species. A primary goal of this study was to assemble a longitudinal collection of stool samples from healthy human adults and begin to examine gut microbiome residency.
Two previous longitudinal 16S rRNA sequencing studies of healthy human adults [6, 7] both considered two individuals for between 6 and 15 months. Thus, our study (n = 8 participants for an of ~17 months) more than doubles the size of current datasets. Other longitudinal 16S datasets have been published, but their comparatively short sampling periods (e.g., 3 months) [8], use of alternative sequencing methods [9, 11], evaluation of controlled diets [9] and/or living conditions [37, 38] make them somewhat difficult to compare. That said, these studies all report that the microbiome is “stable” over long periods, sometimes even predicting that the same bacteria reside for >50 years [11, 12]. Such statements are based on taxonomic levels well above the unit of natural selection (i.e. populations) making it difficult to know whether and to what extent this so-called stability is biologically relevant. An extreme example suggested that a core microbiome was maintained over the course of an entire adult lifetime [12]. Our results cast serious doubt on this level of stability. We and others [1, 3, 39–41] have shown that clonal E. coli populations in the gut are dynamic and turn over on the order of months to years. Indeed, clones observed in only two of our eight participants resided for the entire sampling period.
Our results do not address whether E. coli clone dynamics scale to the rest of the microbiome, but culture-based evidence suggests that it does. For example, Faith et al. cultured and genome sequenced resident (up to 1.3 years) “strains” of the Bacteroidetes, Firmicutes, Proteobacteria, and Actinobacteria phyla from longitudinally collected stool samples [11]. They defined the same “strains” as isolates with genome coverage score of >0.96, which from a population genetic perspective would not necessarily be considered clonal, so these results are very much consistent with E. coli resident turnover in our study. Another study cultured Bacteroides species from longitudinal stool samples of 15 adults and found that some individuals harbored different strains over the study period [4]. So, how important is strain-level differences to gut microbiome function? Considering that the first three completed genomes of E. coli shared <40% of their enzyme coding loci [5] and the fact that 16S cannot resolve this level of diversity, the potential for clonal turnover to change gut function—even when upper-level taxonomy is “stable”—is great.
Previous approaches to examine E. coli residency in human adults have been limited to some degree by both sampling depth (within samples) and breadth (between individuals). According to rare events calculation, the average of 84 isolates per sample that we achieved allowed for confident detection of representative clones at ~2% relative abundance (power = 0.8, expected event rate lambda = 0.02, critical tolerance limit = 1), which is by far the most comprehensive study to date. It is important to recognize, however, that our approach was meant to capture diversity and so while our identification of resident clones (Fig. 3) should be reasonably robust, estimates of relative abundance (Supplementary Fig. 13) may be somewhat biased. Regardless, our results provide clear evidence that E. coli was by far the most abundant and resident gut species of Enterobacteriaceae in at least this cohort, and that E. coli of the A, B2.3, and F phylogroups were more likely to reside over long periods of time compared to other lineages. Interestingly, phylogroup B2 may also be a common resident of infants [42]. A common observation from cross-sectional E. coli studies is that one clone is “dominant” in stool samples, or at a high relative abundance. One interpretation is that this clone represents the resident clone. Caution should be used when making this assumption as we observed dramatic shifts in phylogroup abundance through time (Supplementary Fig. 13). Future examination of seemingly adaptive associations between phylogenetically related clones and residence may provide a better understanding of the microbial factors underlying gut colonization and persistence. Whether the most important adaptations arose long ago as our results with E. coli suggest or whether they arose over much shorter time scales (as was recently suggested for Bacteroides fragilis [43]) requires more data from healthy participants.
If E. coli is so prevalent among humans, why is it often missing from gut microbiome datasets? A principle factor seems to be that the accuracy of 16S sequencing, regardless of analytical approach (OTU or ASV), is poor for Enterobacteriaceae and Escherichia/Shigella, which is consistent with analyses of simple mock communities (n ≤ 20 members) where E. coli cells or genomic DNA were mixed at relatively equal abundances with that of other gut bacteria [13, 44]. In addition, nearly all Enterobacteriaceae was E. coli in our cohort, which is not the case for other human gut microbiome families, such as the Bacteroidaceae, that can be comprised of at least 20 different species [4]. Thus, there seems to be ample evidence that 16S sequencing should not be used to estimate the absolute abundance of microbiome members nor should it be used to determine whether taxa are absent from a sample without first benchmarking a high correlation between these observations and the gold-standard of culture. This also means that 16S-based statements regarding the “dominance” of certain higher order taxa over others (e.g. Bacteroidaceae vs. Enterobacteriaceae) should be avoided, as they could mean very little at the individual population or species levels.
Regarding the use of OTU versus ASV-based analysis, we found these two approaches yielded nearly identical ordinations (a common proxy for community diversity) for five of eight participants but produced different results for the remaining three. This suggests that ASV-analysis differs in discriminatory power from sample-to-sample and that comparisons are sometimes (and perhaps unpredictably) made at different taxonomic levels (e.g., species vs. genera). In contrast, the 97% 16S OTU cut-off is a reasonable approximation of both the 95% average nucleotide identity of genomes and 70% DNA-DNA hybridization cut-offs that are commonly used to differentiate bacterial species [45, 46], thus ensuring that communities are compared equally with respect to taxonomic level. Also, it is unclear how variant 16S alleles (within genomes) contribute to ASV-based analyses, and whether, for example, ASVs that classify to the same species and are always observed together represent different alleles, populations, species, or higher taxa [47]. We showed that OTU-based analysis was the more accurate approach for detecting both Enterobacteriaceae and Escherichia/Shigella. That said, given the widespread use of both analyses, more diagnostic testing against the gold-standard of culture is needed to benchmark their biological relevance.
Although not the focus of this study, it is likely that both host and microbial factors are involved in microbiome residency. There is evidence from shorter-term longitudinal studies that host factors (e.g., antibiotic use [2] and travel [48]) are important as are socio-economic factors (e.g., phylogroup B2 E. coli were more prevalent among people from industrialized compared to developing countries [49, 50]). There is also evidence that GI distress (e.g., natural and induced diarrhea) does not necessarily purge resident strains from a host [51, 40]. Similarly, resident E. coli were remarkably stable in the face of enterotoxigenic E. coli infection and antibiotic treatment [52]. Microbial virulence factors [53] seem to be important in E. coli residency, and especially factors associated with uropathogenesis (e.g., adhesins, siderophores, and hemolysins [42, 53]). Consistent with this finding, clonotyping suggests that A and B2.3 residents in our study belong to sequence types commonly observed in urinary tract infections [21] (Supplementary Table 2). Microbial metabolism may also be important for residency [54, 55]. Freter’s nutrient niche hypothesis suggests that metabolism determines residency such that only clones that most efficiently utilize limiting nutrient(s) will persist [55, 56]. Finally, direct clone interference through bacteriocins or bacteriophage production may be important [40, 57]. Determining which of these factors predict residency will certainly be useful in microbiome-directed therapies.
Few studies have considered lactose non-fermenting (lac-) E. coli, but we found them to be quite common. For example, four long-term (>50 day) lac- resident clones belonging to three different phylogroups were observed among half (n = 4) of our cohort; lac- clones belonging to six of the nine observed phylogroups were identified; and lac- clones comprised 17% of the 120 E. coli clones observed. These results are not entirely without precedent as Sears et al. [51] and a published doctoral thesis [58] reported several lac- residents in healthy adults. Interestingly, lac- E. coli also appear to be associated with uropathogenesis [59, 60], which is consistent with the above mentioned overlap with virulence factors and gut residency. The combined evidence should compel future studies to consider all E. coli, regardless of lactose utilization.
Supplementary information
Acknowledgements
This work was supported with funds provided by the Office of Research and Economic Development at Montana State University (STW), the Undergraduate Research Program at Montana State University (GWP & HC), the National Institute of Health (National Institute of General Medical Sciences) under Award Number P20GM103474 (GWP), a Kopriva Graduate Student Fellowship at Montana State University (JNVM), a United States Department of Agriculture Hatch Project 1009600 (STW), and the National Institutes of Health (National Institute of General Medical Sciences and the National Cancer Institute) under Award Number R01CA215784 (STW).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41396-019-0435-7) contains supplementary material, which is available to authorized users.
References
- 1.Caugant DA, Levin BR, Selander RK. Genetic diversity and temporal variation in the E. Coli population of a human host. Genetics. 1981;98:467–90. doi: 10.1093/genetics/98.3.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jernberg C, Lofmark S, Edlund C, Jansson JK. Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J. 2007;1:56–66. doi: 10.1038/ismej.2007.3. [DOI] [PubMed] [Google Scholar]
- 3.Johnson JR, Clabots C, Kuskowski MA. Multiple-host sharing, long-term persistence, and virulence of Escherichia coli clones from human and animal household members. J Clin Microbiol. 2008;46:4078–82. doi: 10.1128/JCM.00980-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zitomersky NL, Coyne MJ, Comstock LE. Longitudinal analysis of the prevalence, maintenance, and IgA response to species of the order Bacteroidales in the human gut. Infect Immun. 2011;79:2012–20. doi: 10.1128/IAI.01348-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Welch RA, Burland V, Plunkett G, 3rd, Redford P, Roesch P, Rasko D, et al. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci U S A. 2002;99:17020–4. doi: 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caporaso JG, Lauber CL, Costello EK, Berg-Lyons D, Gonzalez A, Stombaugh J, et al. Moving pictures of the human microbiome. Genome Biol. 2011;12:R50. doi: 10.1186/gb-2011-12-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.David LA, Materna AC, Friedman J, Campos-Baptista MI, Blackburn MC, Perrotta A, et al. Host lifestyle affects human microbiota on daily timescales. Genome Biol. 2014;15:R89. doi: 10.1186/gb-2014-15-7-r89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flores GE, Caporaso JG, Henley JB, Rideout JR, Domogala D, Chase J, et al. Temporal variability is a personalized feature of the human microbiome. Genome Biol. 2014;15:531. doi: 10.1186/s13059-014-0531-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinez I, Muller CE, Walter J. Long-term temporal analysis of the human fecal microbiota revealed a stable core of dominant bacterial species. PLoS ONE. 2013;8:e69621. doi: 10.1371/journal.pone.0069621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mehta RS, Abu-Ali GS, Drew DA, Lloyd-Price J, Subramanian A, Lochhead P, et al. Stability of the human faecal microbiome in a cohort of adult men. Nat Microbiol. 2018;3:347–55. doi: 10.1038/s41564-017-0096-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, et al. The long-term stability of the human gut microbiota. Science. 2013;341:1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jayasinghe TN, Hilton C, Tsai P, Apple B, Shepherd P, Cutfield WS, et al. Long-term stability in the gut microbiome over 46 years in the life of Billy Apple(R) Hum Microbiome J. 2017;5-6:7–10. doi: 10.1016/j.humic.2017.09.001. [DOI] [Google Scholar]
- 13.Xue Z, Kable ME, Marco ML. Impact of DNA sequencing and analysis methods on 16S rRNA gene bacterial community analysis of dairy products. mSphere. 2018;3:e00410-18. [DOI] [PMC free article] [PubMed]
- 14.Lagier JC, Armougom F, Million M, Hugon P, Pagnier I, Robert C, et al. Microbial culturomics: paradigm shift in the human gut microbiome study. Clin Microbiol Infect. 2012;18:1185–93. doi: 10.1111/1469-0691.12023. [DOI] [PubMed] [Google Scholar]
- 15.Adeolu M, Alnajar S, Naushad S, S Gupta R. Genome-based phylogeny and taxonomy of the ‘Enterobacteriales’: proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Int J Syst Evol Microbiol. 2016;66:5575–99. doi: 10.1099/ijsem.0.001485. [DOI] [PubMed] [Google Scholar]
- 16.Bacteriology JCotICoS. Present standing of the family name Enterobacteriaceae Rahn 1937. Int J Syst Bacteriol. 1981;31:104. doi: 10.1099/00207713-31-1-104. [DOI] [Google Scholar]
- 17.Robin F, Beyrouthy R, Bonacorsi S, Aissa N, Bret L, Brieu N, et al. Inventory of extended-spectrum-beta-lactamase-producing Enterobacteriaceae in France as assessed by a multicenter study. Antimicrob Agents Chemother. 2017;61:e01911-16. [DOI] [PMC free article] [PubMed]
- 18.Clermont O, Christenson JK, Denamur E, Gordon DM. The Clermont Escherichia coli phylo-typing method revisited: improvement of specificity and detection of new phylo-groups. Environ Microbiol Rep. 2013;5:58–65. doi: 10.1111/1758-2229.12019. [DOI] [PubMed] [Google Scholar]
- 19.Mohapatra BR, Mazumder A. Comparative efficacy of five different rep-PCR methods to discriminate Escherichia coli populations in aquatic environments. Water Sci Technol. 2008;58:537–47. doi: 10.2166/wst.2008.424. [DOI] [PubMed] [Google Scholar]
- 20.Walk ST, Alm EW, Gordon DM, Ram JL, Toranzos GA, Tiedje JM, et al. Cryptic lineages of the genus Escherichia. Appl Environ Microbiol. 2009;75:6534–44. doi: 10.1128/AEM.01262-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tchesnokova V, Avagyan H, Billig M, Chattopadhyay S, Aprikian P, Chan D, et al. A novel 7-single nucleotide polymorphism-based clonotyping test allows rapid prediction of antimicrobial susceptibility of extraintestinal Escherichia coli directly from urine specimens. Open Forum Infect Dis. 2016;3:ofw002. doi: 10.1093/ofid/ofw002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013;79:5112–20. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–41. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rognes T, Flouri T, Nichols B, Quince C, Mahe F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42(Database issue):D633–42. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oksanen J, Guillaume Blanchet F, Friendly M, Kindt R, Legendre P, McGlinn D, et al. Vegan: community ecology package. R package version 2.5-2. 2018.
- 29.Maechler M, Rousseeuw P, Struyf A, Hubert M, Hornik K. Cluster analysis basics and extensions. R package version 2.0.7-1. 2018.
- 30.Roberts DW. labdsv: Ordination and multivariate analysis for ecology. 1.8-0. 2016.
- 31.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’hara R, et al. Package ‘vegan’. Version 2. 2013.
- 32.Maechler M, Rousseeuw P, Struyf A, Hubert M. Hornik KJRpv. Cluster: cluster analysis basics and extensions. 2012;1:56. [Google Scholar]
- 33.Cummings JH, Jenkins DJ, Wiggins HS. Measurement of the mean transit time of dietary residue through the human gut. Gut. 1976;17:210–8. doi: 10.1136/gut.17.3.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacArthur R. On the breeding distribution pattern of North American migrant birds. Auk. 1959;76:318–25. doi: 10.2307/4081809. [DOI] [Google Scholar]
- 35.Lack D. The natural regulation of animal numbers. Oxford, UK: Oxford University Press; 1954. The significance of migration; pp. 243–54. [Google Scholar]
- 36.Savageau MA. Escherichia-coli habitats, cell-types, and molecular mechanisms of gene-control. Am Nat. 1983;122:732–44. doi: 10.1086/284168. [DOI] [Google Scholar]
- 37.Hao Z, Li L, Fu Y, Liu H. The influence of bioregenerative life-support system dietary structure and lifestyle on the gut microbiota: a 105-day ground-based space simulation in Lunar Palace 1. Environ Microbiol. 2018;20:3643–56. doi: 10.1111/1462-2920.14358. [DOI] [PubMed] [Google Scholar]
- 38.Turroni S, Rampelli S, Biagi E, Consolandi C, Severgnini M, Peano C, et al. Temporal dynamics of the gut microbiota in people sharing a confined environment, a 520-day ground-based space simulation, MARS500. Microbiome. 2017;5:39. doi: 10.1186/s40168-017-0256-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kauffmann F. Perch BJAPMS. Uber die Coliflora des gesunden Menschen. 1943;20:201–20. [Google Scholar]
- 40.Sears HJ, Brownlee I, Uchiyama JK. Persistence of individual strains of Escherichia coli in the intestinal tract of man. J bacteriol. 1950;59:293. doi: 10.1128/jb.59.2.293-301.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wallick H, Stuart CA. Antigenic Relationships of Escherichia coli Isolated from One Individual. J Bacteriol. 1943;45:121–6. doi: 10.1128/jb.45.2.121-126.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nowrouzian FL, Wold AE, Adlerberth I. Escherichia coli strains belonging to phylogenetic group B2 have superior capacity to persist in the intestinal microflora of infants. J Infect Dis. 2005;191:1078–83. doi: 10.1086/427996. [DOI] [PubMed] [Google Scholar]
- 43.Zhao S, Lieberman TD, Poyet M, Kauffman KM, Gibbons SM, Groussin M, et al. Adaptive evolution within gut microbiomes of healthy people. Cell Host & Microbe. 2019. [DOI] [PMC free article] [PubMed]
- 44.Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE. 2011;6:e27310. doi: 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim M, Oh HS, Park SC, Chun J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol. 2014;64(Pt 2):346–51. doi: 10.1099/ijs.0.059774-0. [DOI] [PubMed] [Google Scholar]
- 46.Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci USA. 2005;102:2567–72. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vetrovsky T, Baldrian P. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS ONE. 2013;8:e57923. doi: 10.1371/journal.pone.0057923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rogers BA, Kennedy KJ, Sidjabat HE, Jones M, Collignon P, Paterson DL. Prolonged carriage of resistant E. coli by returned travellers: clonality, risk factors and bacterial characteristics. Eur J Clin Microbiol Infect Dis. 2012;31:2413–20. doi: 10.1007/s10096-012-1584-z. [DOI] [PubMed] [Google Scholar]
- 49.Bailey JK, Pinyon JL, Anantham S, Hall RM. Distribution of human commensal Escherichia coli phylogenetic groups. J Clin Microbiol. 2010;48:3455–6. doi: 10.1128/JCM.00760-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lescat M, Clermont O, Woerther PL, Glodt J, Dion S, Skurnik D, et al. Commensal Escherichia coli strains in Guiana reveal a high genetic diversity with host-dependant population structure. Environ Microbiol Rep. 2013;5:49–57. doi: 10.1111/j.1758-2229.2012.00374.x. [DOI] [PubMed] [Google Scholar]
- 51.Sears HJ, Brownlee I. Further observations on the persistence of individual strains of Escherichia coli in the intestinal tract of man. J Bacteriol. 1952;63:47–57. doi: 10.1128/jb.63.1.47-57.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Richter TKS, Michalski JM, Zanetti L, Tennant SM, Chen WH, Rasko DA. Responses of the human gut Escherichia coli population to pathogen and antibiotic disturbances. mSystems. 2018;3:e00047-18. [DOI] [PMC free article] [PubMed]
- 53.Wold AE, Caugant DA, Lidin-Janson G, de Man P, Svanborg C. Resident colonic Escherichia coli strains frequently display uropathogenic characteristics. J Infect Dis. 1992;165:46–52. doi: 10.1093/infdis/165.1.46. [DOI] [PubMed] [Google Scholar]
- 54.Conway T, Cohen PS. Commensal and pathogenic Escherichia coli metabolism in the gut. Microbiol Spectr. 2015;3. 10.1128/microbiolspec.MBP-0006-2014. [DOI] [PMC free article] [PubMed]
- 55.Freter R, Brickner H, Fekete J, Vickerman MM, Carey KE. Survival and implantation of Escherichia coli in the intestinal tract. Infect Immun. 1983;39:686–703. doi: 10.1128/iai.39.2.686-703.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pereira FC, Berry D. Microbial nutrient niches in the gut. Environ Microbiol. 2017;19:1366–78. doi: 10.1111/1462-2920.13659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Branche WC, Jr., Young VM, Robinet HG, Massey ED. Effect of colicine production on Escherichia coli in the normal human intestine. Proc Soc Exp Biol Med. 1963;114:198–201. doi: 10.3181/00379727-114-28624. [DOI] [PubMed] [Google Scholar]
- 58.Anantham S. Analysis of persistent and antibiotic resistant commensal Escherichia coli from healthy adults. 2013. PhD thesis. University of Sydney, Sydney, Australia.
- 59.Chang J, Yu J, Lee H, Ryu H, Park K, Park YJ. Prevalence and characteristics of lactose non-fermenting Escherichia coli in urinary isolates. J Infect Chemother. 2014;20:738–40. doi: 10.1016/j.jiac.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 60.Johnson TJ, Elnekave E, Miller EA, Munoz-Aguayo J, Flores Figueroa C, Johnston B, et al. Phylogenomic analysis of extraintestinal pathogenic Escherichia coli sequence type 1193, an Emerging Multidrug-Resistant Clonal Group. Antimicrob Agents Chemother. 2019;63:e01913-18. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.