

Abstract
The ligand-binding sites of many G protein-coupled receptors (GPCRs) are situated around and deeply embedded within the central pocket formed by their seven transmembrane-spanning α-helical domains. Generally, these binding sites are assumed accessible to endogenous ligands from the aqueous phase. Recent advances in the structural biology of GPCRs, along with biophysical and computational studies, suggest that amphiphilic and lipophilic molecules may gain access to these receptors by first partitioning into the membrane and then reaching the binding site via lateral diffusion through the lipid bilayer. In addition, several crystal structures of class A and class B GPCRs bound to their ligands offer unprecedented details on the existence of lipid-facing allosteric binding sites outside the transmembrane helices that can only be reached via lipid pathways. The highly organized structure of the lipid bilayer may direct lipophilic or amphiphilic drugs to a specific depth within the bilayer, changing local concentration of the drug near the binding site and affecting its binding kinetics. Additionally, the constraints of the lipid bilayer, including its composition and biophysical properties, may play a critical role in “pre-organizing” ligand molecules in an optimal orientation and conformation to facilitate receptor binding. Despite its clear involvement in molecular recognition processes, the critical role of the membrane in binding ligands to lipid-exposed transmembrane binding sites remains poorly understood and warrants comprehensive investigation. Understanding the mechanistic basis of the structure-membrane interaction relationship of drugs will not only provide useful insights about receptor binding kinetics but will also enhance our ability to take advantage of the apparent membrane contributions when designing drugs that target transmembrane proteins with improved efficacy and safety. In this minireview, we summarize recent structural and computational studies on membrane contributions to binding processes, elucidating both lipid pathways of ligand access and binding mechanisms for several orthosteric and allosteric ligands of class A and class B GPCRs.
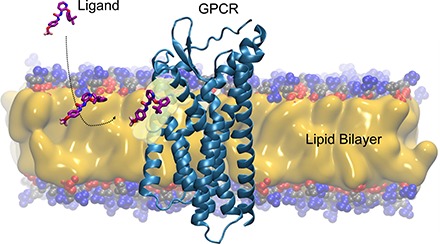
Introduction
The G protein-coupled receptor (GPCR) family represents one of the largest and most diverse families of proteins; proteins in this family are encoded by 1%–3% of the genes in our genome. These integral membrane proteins are involved in many physiologic and pathophysiological processes and continue to be the most privileged targets for approximately 33% of all small-molecule drugs currently on the market (Overington et al., 2006; Santos et al., 2017). GPCRs, also known as seven-transmembrane (7TM) receptors are highly dynamic (Kobilka, 2007; Kobilka and Deupi, 2007; Manglik and Kobilka, 2014; Manglik et al., 2015; Hilger et al., 2018) and exist in an equilibrium between multiple, functionally distinct conformational states. A broad spectrum of extracellular signals (e.g., photons, neurotransmitters, hormones, lipids, ions, and odorants) interact with GPCRs through their primary “orthosteric” binding site and transduce the signals by conformational rearrangement of the membrane-spanning 7TM region. This triggers intracellular signaling pathways through heterotrimeric G-proteins, β-arrestins, and other signaling proteins. Recent advances in the structural biology of GPCRs combined with biophysical, spectroscopic, and computational studies have provided new details (Latorraca et al., 2017) on the dynamics of the transmembrane domain critical for mediating downstream signals. Although the majority of the currently used drugs and probe compounds target endogenous agonist-binding orthosteric sites, substantial research efforts in recent years (Christopoulos, 2014) have been directed toward developing allosteric modulators that bind to sites that are topographically distinct, yet conformationally linked to the orthosteric sites. In principle, allosteric ligands can potentiate (positive allosteric modulators, PAMs) or inhibit (negative allosteric modulators, NAMs) signal transduction by altering the affinity for orthosteric ligands and by stabilizing active or inactive conformational states, respectively. They have also been found to modulate receptor interactions with intracellular signal-transducing molecules, or to have no net effect (neutral allosteric ligands, NALs) on orthosteric agonists. In some cases, the binding of allosteric ligands (Ago-PAMs) alone is adequate to induce receptor activation, though these compounds may increase the potency and/or efficacy of orthosteric ligands. The advantages of allosteric modulators (Gentry et al., 2015; Changeux and Christopoulos, 2017; Foster and Conn, 2017) include the prospect of greater receptor subtype selectivity, biased signaling (Rankovic et al., 2016; McCorvy et al., 2018), and the potential to “fine tune” endogenous agonism while maintaining the natural signaling patterns. Furthermore, allosteric modulators exhibit very slight amounts of positive or negative cooperativity that result in a “ceiling level” for the allosteric effect with a possibility of on-target safety in overdose situations (Keov et al., 2011).
All GPCRs share common structural features (Cherezov et al., 2007; Dawaliby et al., 2016) that include 1) an extracellular N terminus; 2) seven transmembrane helices (TMHs) forming a closed bundle; 3) three extracellular and three intracellular loops connecting the TMHs; and 4) an intracellular C terminus. Generally, endogenous ligands of GPCRs bind inside a crevice (the orthosteric binding site) formed by the TMH bundle. In case of class C GPCRs, the native ligands bind to an extracellular “Venus fly trap domain” connected to the N terminus, either in addition to or in place of the orthosteric site. However, recently solved experimental structures of GPCRs cocrystallized with their allosteric ligands revealed multiple (Christopoulos et al., 2014) spatially distinct binding sites at: 1) the extracellular vestibule of the receptor through which the orthosteric ligands enter; 2) the intracellular ends of the helices, partially overlapping with the binding surfaces of signaling molecules such as G proteins; and 3) the external surface of the receptor/lipid bilayer interface outside the 7TM helical bundle. A recent comprehensive review (Lu and Zhang, 2018) discusses the diverse locations and key receptor-ligand interactions of 18 crystal structures of classes A, B, C, and F GPCRs in complex with such small-molecule allosteric modulators. Crystal structures, in which the receptor-bound ligand molecules are significantly exposed to and buried within the membrane lipids, are extremely intriguing because of their binding-site locations that can be reached only via membrane pathways. Under these circumstances, partitioning of the ligands to a discrete bilayer depth, in addition to their suitable orientation and conformation within the membrane, becomes essential for subsequent access and binding to the intrabilayer receptor. The phospholipid bilayer can effectively increase the efficiency of ligand binding to an intrabilayer receptor site, even at low concentrations, by reducing the degrees of freedom of the drug and limiting it to a specific region of the bilayer. Although membrane interactions can result in improved binding kinetics (Sykes et al., 2019), excessive membrane accumulation of ligands can lead to toxicity through off-target interactions. Therefore, simply increasing ligand lipophilicity to gain efficacy for membrane-associated targets is often unfavorable in terms of the overall drug profile. Knowledge of membrane partitioning characteristics such as quantitative bilayer distribution, preferred location (depth), orientation, and conformation of drug molecules within the bilayer is essential for understanding their target binding kinetics, onset and duration of action, and disposition.
Although it is outside the scope of this review, it is important to recognize the fundamental role of the plasma membrane in proper localization, structure, and function of GPCRs (Ostrom and Insel, 2004; Phillips et al., 2009; Genheden et al., 2017). The lipid composition (including the presence of “lipid rafts”) and the biophysical properties of the membrane (such as thickness, curvature, fluidity, and surface tension) have been shown to affect structure, function, and agonist/antagonist binding and activation of GPCRs as well as the corresponding signaling proteins (Yang et al., 2005; Escribá et al., 2007; Periole et al., 2007; Dawaliby et al., 2016; Bruzzese et al., 2018). Most importantly, cholesterol and phospholipids can act as allosteric modulators of GPCRs by binding directly to receptor sites and/or by changing the membrane properties (Oates and Watts, 2011; Sengupta and Chattopadhyay, 2015; Gimpl, 2016; Guixà-González et al., 2017). In this review, we will focus mainly on allosteric binding sites that exist at the receptor/lipid bilayer interface, emphasizing the critical role of the membrane for ligand access and binding to these novel allosteric sites. We will also summarize some recent structural and computational studies that offer unprecedented insights into the atomistic details of plausible lipid pathways for orthosteric ligands.
The Structure-Membrane Interaction Relationship Affects Ligand Action and Disposition
The partitioning of drugs within the plasma membrane or the membranes of subcellular organelles is known to affect drug interactions with membrane-associated proteins, including G protein-coupled receptors (Sykes and Charlton, 2012; Sykes et al., 2014; Dickson et al., 2016), ion channels (Schmidt et al., 2012; Huang et al., 2017; Laverty et al., 2017; Poveda et al., 2017), cytochrome P450 enzymes (Berka et al., 2011; Paloncýová et al., 2014; Di Meo et al., 2016), and transporters (Melchior et al., 2012; Clay and Sharom, 2013), influencing their efficacy and fate in the body. In addition, evidence is accumulating for micropharmacokinetic-pharmacodynamic mechanisms (Swinney, 2006; Schoop and Dey, 2015; Swinney et al., 2015; Vauquelin, 2015) in which the so-called “nonspecific” membrane interactions of drugs can influence their receptor binding kinetics and affect affinity as well as residence time. According to one “microkinetic” model (Vauquelin and Packeu, 2009; Vauquelin, 2015, 2016), the surrounding lipid bilayer can influence the molecular recognition process (i.e., ligand molecules binding to transmembrane proteins) through the following mechanisms: 1) by acting as a reservoir for the drug, 2) by “preorganizing” the drug molecule in its appropriate orientation and conformation for binding, and 3) by altering the time the drug molecule needs to reach its receptor (see Fig. 2).
Fig. 2.

Multiple lipid bilayer roles in molecular recognition of transmembrane proteins. According to the “microkinetic” model, the cell membrane can govern drug-binding processes by: (A) acting as a local reservoir for the drug and thus prolonging the drug exposure of the target; (B) modulating the orientation and conformation of the drug for favorable receptor binding; and (C) reducing the time the drug needs to reach its receptor, thus facilitating the approach. The long-lasting bronchodilatory effect of salmeterol (a β2-adrenergic receptor partial agonist with a receptor-binding pharmacophore saligenin head, a hydrophobic phenylalkoxyalkyl tail, and an oxygen hinge), may be attributed to a combination of these effects. Adapted from Vauquelin (2016).
In a membrane-facilitated ligand binding process, the ligand molecule must first partition to a specific, energetically favorable location and adopt an appropriate orientation and conformation in the membrane bilayer before laterally diffusing to an intrabilayer receptor-binding site. The specific makeup and organization of the lipid bilayer may restrict lipophilic or amphiphilic drugs to a particular depth of penetration in the bilayer. Furthermore, the orientation of a drug relative to its binding site might also be optimized by the lipid bilayer structure, which may limit the drug’s degrees of rotational freedom (Sargent and Schwyzer, 1986; Schwyzer, 1991, 1995a,b). Drugs may exhibit entirely different conformational equilibria in membrane compared with those in aqueous bulk; a conformation fitting to the receptor binding site may exist only as a metastable state. Presumably, the drug enters the membrane, undergoes a conformational change, and then binds to a protein with a precise and metastable conformation. In addition, drugs that enter (wash in) rapidly into membranes attain relatively low concentrations in the membrane and leave rapidly, compared with slowly concentrating drugs that “load” into the membrane at greater concentrations, and then slowly wash out again. Thus, slowly accumulating drugs are available to interact with membrane-associated receptors over a longer period. For drug molecules targeting membrane-associated receptors, even subtle differences in structural features whose basis is the structure-membrane interaction relationship can result in altered drug-lipid interactions. This will affect the preferred location, orientation, conformation, and local concentration of the drug molecules in the immediate vicinity of the receptor binding site, and therefore directly influence observed pharmacology (Vauquelin and Packeu, 2009; Swinney et al., 2015; Vauquelin, 2015, 2016; Gherbi et al., 2018). Structural modifications designed to optimize drug interactions with membrane lipids can produce desired changes in the clinical half-life of drugs; for example, a short-acting parent compound can be rationally modified into a long-acting structural analog by the addition of a hydrophobic functional group at the proper location.
For compounds binding to a membrane-associated receptor, high receptor affinity may result from high intrinsic affinity for the receptor site (low value of dissociation rate, koff) or a high membrane partition coefficient (Kmem) that produces a change in the value of the measured association rate (kon′), or a combination of these two (see Fig. 1). Particularly, a compound with a high Kmem value, and thus higher local concentrations surrounding the receptor, may contribute to a longer duration of action (Vauquelin and Charlton, 2010), in part owing to altered kon, which directly affects receptor rebinding (Vauquelin, 2010). Increasing a drug’s lipophilicity to boost drug efficacy would not only increase the local concentration around the target but would also increase the likelihood of off-target pharmacology resulting from to nonspecific binding to other membrane-associated proteins, which in turn may result in an increased risk of toxicity. However, large increases in affinity obtained by changes in kon may be beneficial for highly selective ligands (Huggins et al., 2012) and/or where topical delivery is anticipated, such as β2-adrenergic receptor (β2-AR) agonists that are delivered to the lungs by inhalation. Agonists of β2-AR (Cazzola et al., 2011, 2005) are primarily used to treat asthma and other pulmonary disorders. They cause smooth muscle relaxation, dilation, and opening of the lung airways by β2-AR activation. These agents exhibit a wide range of clinical action duration (short, long, and ultra-long), arguably owing to their altered interactions with the membrane compartment surrounding the receptors. In a series of studies (Sykes and Charlton, 2012; Sykes et al., 2014; Dickson et al., 2016), researchers from Novartis demonstrated that for several clinically relevant β2-AR agonists and antagonists, the degree of phospholipid interaction affects the drug’s binding kinetics profile and thus the observed affinity. For drugs possessing a high membrane affinity, the high local concentration of the drug in the membrane surrounding the receptor (calculated from the membrane partition coefficient, log KIAM, measured using immobilized artificial membranes) was shown to affect its association rate (kon′) and thus its observed binding affinity (Kd′). For example, despite sharing the same pharmacophore (the adrenaline-mimicking saligenin pharmacophore), the measured affinity of salmeterol is three orders of magnitude higher than salbutamol (Sykes and Charlton, 2012), driven by the large difference in their association rates, attributable solely to membrane binding (Szczuka et al., 2009) (see Fig. 2). Upon correction of this increased membrane affinity (Mason et al., 1991; Vauquelin and Packeu, 2009), both agonists have comparable intrinsic binding affinity (Kd) values for the inactive form of β2-AR. Oftentimes, for membrane-embedded targets, the role of the membrane is neglected when the affinity of a drug is measured; however, this membrane contribution can affect the “observed” or “apparent” binding affinity (Kd′), making the results difficult to interpret. The intrinsic or “true” affinity (Kd) is the affinity of the drug for the receptor, excluding the contribution from the membrane. It has been shown that for salmeterol, the membrane contribution affects the overall experimental binding affinity, and after uncoupling the membrane’s contribution, the intrinsic dissociation affinity constant (Kd) is much more comparable to the affinities of other structurally analogous β2-AR agonists (Dickson et al., 2016). The lipid bilayer may serve as a reservoir for lipophilic/amphiphilic ligands (such as salmeterol), which in turn helps to concentrate the ligands near the binding site of the receptor, thereby affecting the observed association rate without necessarily changing interactions with binding site residues. The difference in affinity between salmeterol and salbutamol has been attributed (Rhodes et al., 1985; Green et al., 1996) to a possible interaction of the lipophilic phenylalkoxyalkyl tail of salmeterol with phospholipids in the bilayer. As this review was being prepared, an X-ray crystal structure (Masureel et al., 2018) of β2-AR bound to salmeterol with a resolution of 2.9 Å was published, revealing an exosite that is close to the orthosteric binding site wherein the lipophilic phenylalkoxyalkyl chain interacts with several hydrophobic residues. It may be possible that the receptor association and dissociation of salmeterol occur through lipid pathways via the transmembrane helices; but this possibility warrants further investigation.
Fig. 1.

| (1) |
| (2) |
| (3) |
where li and lo are the membrane wash-in and washout rates of the ligand, respectively. In this scenario, the ligand concentration near the receptor is different from its concentration in the aqueous bulk. On the basis of their lipophilicity and amphiphilicity profiles and their ionization states, ligand molecules may reside at discrete locations within the membrane compartment: 1) at the surface, interacting with hydrated polar head groups (LMH); 2) at the interface between the polar head groups and the hydrophobic tails, interacting with both strata (LMI); or 3) at the hydrophobic lipid core (LMC). For compounds with high lipophilicity and/or amphiphilicity, the equilibrium concentration near the binding site can be affected by the magnitude of individual transfer rates (see part A) representing adsorption and desorption (subscripts “a” and “d” respectively) at the headgroup, interface, and core regions of the membrane.
| (4) |
| (5) |
| (6) |
Here, Kd′ and Kd represent the “observed” or “apparent” dissociation affinity constant (determined in bulk experimental studies) and “intrinsic” dissociation affinity constant of the ligand calculated after correcting for the membrane contribution, respectively. The most challenging scenario arises when the binding site is deeply embedded within the membrane and ligands can reach their binding sites only through lipid pathways (see part B). In such cases, the drug concentration near the binding site (LBS) is dependent on the location of the binding site within the membrane compartment relative to the drug’s preferred membrane location—for example, at the membrane core (LMC), interface (LMI) or hydrated headgroup (LMH)—as well as the diffusion coefficient of the ligand in question. It is important to recognize that Kmem-based adjustments may often underestimate the effect of the membrane partitioning of ligands, because the ligand concentration in the vicinity of the receptor may be different from the concentration in the bulk membrane owing to discrete localization effects. Also, the actual volume of the membrane occupied by the drug is substantially less than the total membrane volume.
In contrast to the association rate (kon), the dissociation rate (koff) is independent of the ligand concentration and depends solely on the specific molecular interactions of the ligands with the binding site residues. For ligands targeting membrane-facing binding sites, the participation of lipid molecules in the binding process (as one of the components of the binding site) needs different treatment, as these interactions would affect koff as well. The extent to which the membrane lipids are capable of engaging in the binding process can be appreciated by noting the significant membrane-exposed surface area of the ligands in the analyzed crystal structures (see Fig. 5; Table 1).
Lipophilic and Amphiphilic Ligands May Access Their Membrane-Associated Receptors through Lipid Pathways
For the majority of the G protein-coupled receptors, the orthosteric binding site is deeply embedded within and around the central pocket formed by the 7TM-spanning α-helical domains. Generally, the hydrophilic endogenous ligands are assumed to enter the pocket directly from the aqueous phase (Dror et al., 2011). Recent evidence for the β2-AR (Sykes and Charlton, 2012; Sykes et al., 2014), cannabinoid (Hurst et al., 2010), sphingosine-1-phosphate (Hanson et al., 2012), and opsin receptors (Hildebrand et al., 2009), along with ion channel ligands (Rhodes et al., 1985; Mason et al., 1991), suggests that amphiphilic and lipophilic molecules partitioned into the membrane gain access to these binding sites via lateral diffusion between the α-helical transmembrane domains of the receptors. The concentrating effects of membrane-associated drugs and lateral diffusion of drugs across the two-dimensional surface rather than along three-dimensional pathways in the aqueous bulk alone could significantly increase receptor association rates and the corresponding affinity values by the “reduction of dimensionality” rate enhancement (McCloskey and Poo, 1986). In this section, we discuss some recent structural and computational studies that offer details on ligand access/exit pathways and binding to transmembrane domains of GPCRs.
The Lipid Pathway for Retinal: 11-cis Retinal and All-trans-Retinal Access and Leave the Opsin-Binding Pocket through a Ligand Channel That Has Openings into the Membrane.
Rhodopsins are class A GPCRs (class A GPCRs form the largest subfamily, constituting approx. 90% of all GPCRs). Rhodopsin is composed of an apoprotein opsin that is covalently linked to a chromophoric ligand, 11-cis-retinal, that acts as an inverse agonist and keeps the receptor in its inactive state. Upon activation by light (a photon), 11-cis-retinal undergoes isomerization to all-trans-retinal, an agonist that triggers conformational changes in the protein and turns on the signaling pathway, which eventually leads to vision. Retinal epithelial cells are responsible for the synthesis of 11-cis-retinal; rhodopsin regeneration occurs when the all-trans-retinal on activated rhodopsin is replaced by a newly synthesized 11-cis-retinal. Structural, mutational, and computational studies support the existence of and provide extensive details on the dynamics of a continuous retinal channel (approx. 70-Å length) in the active state of the protein. This channel traverses parallel to the membrane plane and opens into the membrane layer through openings A (between TM1 and TM7) and B (between TM5 and TM6), respectively. Hildebrand et al. (2009) investigated the channel properties with respect to the passage and uptake of 11-cis-retinal and the release of all-trans-retinal using two active forms of the crystal structures of opsin, namely the ligand-free apoprotein opsin (PDB ID 3CAP) (Park et al., 2008) and opsin bound to a high-affinity peptide derived from the C terminus of the α-subunit of the G protein (PDB 3DQB) (Scheerer et al., 2008). Both membrane-facing openings A and B are lined with hydrophobic aromatic residues, and the central retinal binding pocket is polar. Using a skeleton search algorithm and molecular docking, the study by Hildebrand et al. found that the inner width of the channel ranges from 11.6 to 3.2 Å. Throughout the channel, there are four constriction points, including a 90° elbow-like kink near the polar residue Lys296. These constriction sites are narrow and require significant stretching to allow passage of the retinal β-ionone-ring. The absence of electron density for Lys296 in the studied crystal structures indicates that the sidechain of this residue could be highly flexible. The smallest constriction site—just after opening A, with the 90° kink at Lys296—seems to be selective for 11-cis-retinal with a possible bent conformation instead of for extended all-trans-retinal. The probability of retinal entering the binding site through opening A as 11-cis-retinal (with the β-ionone ring making the first contact with hydrophobic residues at the entrance) and exiting through opening B as all-trans-retinal (again with the β-ionone ring interacting first with the aromatic residues lining the opening) seems very high. Additional docking simulations, in which side-chain flexibility of the binding site residues was allowed, explored multiple low-energy binding poses within the channel; analysis of these docked orientations and conformations provides convincing evidence for ligand entry and exit through lipid pathways.
The Lipid Pathway for Sphingosine 1-Phosphate Receptor Ligands.
The sphingosine-1-phosphate receptor family comprises five members (S1P1–5) and the activation of these receptors by exogenous ligands can result in modulation of lymphocyte trafficking, endothelial development, heart rate, and vascular tone. A nonselective agonist prodrug, fingolimod, has been approved by the FDA for the treatment of relapsing-remitting multiple sclerosis (Brinkmann et al., 2010). The crystal structure of the S1P1 receptor in complex (Hanson et al., 2012) with a selective antagonist (R)-3-amino-(3-hexylphenylamino)-4-oxobutylphosphonic acid (ML056) shows an N-terminal domain that folds over the top of the receptor binding site and essentially prevents ligands from entering from the extracellular side. This structural constraint, coupled with the fact that many of the S1P1 ligands exhibit delayed saturation of receptor binding in the presence of excess ligand (Mandala et al., 2002), leads to the possibility that the ligands gain access to the binding site through the lipid bilayer, specifically between a gap in helices I and VII. Utilizing the crystal structure of S1P1R (PDB 3V2Y) embedded in a membrane composed of POPC and cholesterol, Stanley et al., (2016) investigated the potential lipid binding pathway for the antagonist, ML056, using unbiased all-atom molecular dynamics simulations totaling approx. 800 microseconds. The series of simulations revealed a clear binding pathway through the lipid bilayer with four distinct steps: 1) ML056 moves through the membrane and comes in contact with a transmembrane-facing binding pocket formed by amino acids located mainly on TMH 1 and 7; 2) the phosphonate group of ML056 interacts with R2927.34 and S38, and the protonated primary amine interacts with E2947.36 (the lipophilic tail of the ligand fits into a cavity formed by hydrophobic residues V511.38, F521.39, I551.42, V2987.40, and L3027.44); 3) the tail of the ligand inserts itself into the entrance channel to interact with L1022.61 and L2977.39; and 4) the hydrophobic tail of the ligand enters the orthosteric binding site first, followed by the polar head group. During the binding process, the N-terminal helix unfolds and there is an analogous increase in the size of the channel, which is thought to accommodate the ligand as it enters the binding site. It should be noted that although ML056 was placed in the bulk membrane around the receptor, it did not enter the binding site in every simulation.
The Lipid Pathway for Cannabinoid Ligands.
By using isothiocyanate covalent labeling studies, Makriyannis and coworkers (Pei et al., 2008) have shown that a cannabinoid, (−)-7′-isothiocyanto-11-hydroxy-1′,1′dimethylheptyl-hexahydrocannabinol (AM841), enters the cannabinoid CB2 receptor through the lipid bilayer. Inspired by this study, Hurst et al. (2010) investigated a potential lipid pathway entry and interaction for endogenous cannabinoid sn-2-arachidonoylglycerol (2-AG) with the CB2 receptor in a POPC bilayer using microsecond-timescale all-atom molecular dynamic simulations. This study demonstrated that 2-AG first partitioned out from the bulk lipid at the interface between transmembrane helices (TMH) 6 and 7 before entering into the binding site by crossing between TMH6 and TMH7. This ligand entry results in the breaking of an ionic lock between TMH3 and TMH6 at the intracellular end and causes the displacement of TMH6 away from TMH3. Subsequent to manually applied protonation at the D3.49/D6.30 positions, further access by 2-AG deep into the binding pocket results in a conformational change in the W6.48 “toggle switch” and a significant flow of water. Several biophysical studies indicate that the listed observations are similar to the signature events (Farrens et al., 1996; Lin and Sakmar, 1996; Javitch et al., 1997; Ghanouni et al., 2000, 2001; Ballesteros et al., 2001; Jensen et al., 2001; Nakanishi et al., 2006; Kobilka and Deupi, 2007; Grossfield et al., 2008) that occur following ligand-induced activation of rhodopsin and β2-adrenergic receptors. It should be noted that although 2-AG was placed around the receptor in various positions in the bilayer, the ligand did not enter the receptor in every simulation. However, it was observed to enter only through TMH6 and TMH7, suggesting a specific lipid access pathway for this endogenous ligand. The negative cases were used as controls to compare the conformational changes (in W6.48) upon 2-AG entry into the receptor.
The Lipid Pathway for Protease-Activated Receptor-1 Ligands.
A recent study (Bokoch et al., 2018) combining MD simulations and kinetic cell signaling experiments suggests a possible lipid bilayer pathway for vorapaxar, a small molecule antagonist of protease-activated receptor-1 (PAR1). As previously reported, an unbiased long timescale (≤10 microseconds) molecular dynamic study (Zhang et al., 2012) did not result in spontaneous dissociation of vorapaxar owing to the slow experimental off rate (approx. 20 hours) of vorapaxar. Hence, the dissociation of the ligand from the binding pocket was investigated by temperature-accelerated molecular dynamics, an enhanced sampling method used to accelerate the dissociation process. In 8 out of 13 simulations, vorapaxar dissociates and exits the binding site via the lipid bilayer, passing between TMH4 and TMH5 (three times) and between TMH6 and TMH7 (five times), with multiple “metastable states” along the dissociation pathway forming reproducible sets of receptor and lipid interactions. Most remarkably, a qualitative analysis of contacts between the nonhydrogen atoms of vorapaxar and the receptor-membrane system in all metastable states revealed that POPC lipid molecules interacted with the ligand more frequently than any other residues from the receptor. In the PAR1-vorapaxar crystal structure (Zhang et al., 2012), the ethyl carbamate group of the ligand protrudes through a tunnel between TMH6 and TMH7 and points toward the lipid bilayer. In all of the trajectories involving the TMH6-TMH7 tunnel, the same ethyl carbamate group was found to leave the pocket first and interact with the outer leaflet of the lipid bilayer on the way out of the binding pocket. Further chemical biology experiments with alkyl derivatives of vorapaxar (hexyl vorapaxar and N-boc-hexyl vorapaxar) using functional calcium flux assays demonstrated that the addition of a bulky lipophilic group to vorapaxar did not decrease its antagonistic efficacy and had a relatively small effect on the inhibition kinetics. This is possible only if vorapaxar and its derivatives can enter and exit PAR1 via the TMH6-TMH7 tunnel; therefore, these experiments provide convincing evidence that these hexyl and N-box-hexyl tails can extend through the TMH6-TMH7 tunnel, adopting the same binding pose as the parent molecule in the crystal structure. This study suggests that the contribution of membrane lipids can be exploited to selectively alter on and off rates by modifying ligand-lipid interactions.
Membrane-Facilitated Ligand Access and Binding to Extrahelical Allosteric Binding Site of P2Y1 Receptor.
A recent study (Yuan et al., 2018) investigated the plausible membrane-facilitated molecular mechanism underlying the binding of allosteric antagonist 1-(2-(2-(tert-butyl) phenoxy) pyridine-3-yl)-3-(4-(trifluoromethoxy) phenyl) urea (BPTU) to the transmembrane extrahelical allosteric site of the P2Y1 receptor (P2Y1R), a class A GPCR that is a promising target for antithrombotic drugs. This computational study used a combination of conventional molecular dynamics simulations and multiple enhanced-sampling methods (Gervasio et al., 2005; Provasi et al., 2009; Limongelli et al., 2010; Saladino et al., 2012; Jämbeck and Lyubartsev, 2013; Schneider et al., 2015), including umbrella sampling (Kästner, 2011; Bochicchio et al., 2015; Lee et al., 2016), well-tempered metadynamics (Barducci et al., 2008; Yuan et al., 2018), and funnel-metadynamics (Troussicot et al., 2015; Comitani et al., 2016; Shang et al., 2016). The results obtained included: 1) the preferred location of BPTU in the POPC bilayer without the receptor; 2) the dynamics of BPTU as it transitions from the aqueous bulk phase into the lipid bilayer and subsequently binds to the receptor site by two-dimensional lateral diffusion; 3) a quantitative, well defined free energy surface; and 4) an accurate estimation of the BPTU-P2Y1R binding free energy. The free energy profile for the partitioning of BPTU in the POPC bilayer indicated a preferred (energetically favorable) location, which is approximately 12 Å from the center of the bilayer, above its receptor binding site (approx. 5 Å from the center of the bilayer). The analysis of the angle between the long axis of BPTU and the bilayer normal (z-axis) as a function of the distance of BPTU from the center of bilayer revealed that the interaction with lipids at this bilayer depth favored BPTU adoption of a preferential orientation and conformation to facilitate reaching its binding site (see Fig. 3). The strong H-bond interactions of BPTU’s urea group with the polar head groups of lipids, and the subsequent π-π stacking and hydrophobic interactions with I118 and F119 of extracellular loop 2 as first contacts with the extra-helical site, enabled BPTU to move from three-dimensional diffusion in the aqueous phase to two-dimensional diffusion in the membrane. The lowest energy conformation of BPTU from the free energy surface, obtained through well tempered metadynamics, was approximately equivalent to the conformation obtained in the experimental crystal structure (Zhang et al., 2015) (PDB ID 4XNV). It is critical to note that in this bound position, the ligand molecule is only partially bound to the shallow binding pocket, while the rest of it is buried in the lipid hydrophobic region. Though polar headgroups of lipids play a critical role in the dynamics of the binding process, only hydrophobic tails seem to be necessary for the final bound pose, at least in this case. The excellent agreement between the experimental binding free energies and the values calculated using these state-of-the-art simulation techniques appears to support the described binding mechanism.
Fig. 3.

Membrane-facilitated ligand access and binding. BPTU, a small-molecule antagonist (negative allosteric modulator) partitions within the membrane first, and reaches the transmembrane extrahelical binding site of the P2Y1 receptor through a lipid pathway. The preferred orientation and conformation of BPTU within the POPC bilayer near the binding site, obtained by funnel metadynamics simulations, is strikingly similar to that of the bound ligand in the crystal structure, indicating the plausible role of membrane lipids in “pre-organizing” the ligand for receptor binding. BPTU and P2Y1 receptor molecules are represented in stick and secondary structures, respectively. The approximate positions of the upper and lower leaflets of a hypothetical lipid bilayer are marked by two horizontal lines perpendicular to the bilayer normal. The binding site residues are depicted as transparent surface models in the color yellow. Picture adapted from Yuan et al. (2018).
Structural Evidence for Lipid-Facilitated Binding to Transmembrane Allosteric Sites of Class A and Class B GPCRs
Recent progress in crystallography and protein engineering has resulted in an exponential increase in the number of membrane protein structures, especially GPCRs (Lu and Zhang, 2018), available in multiple conformations (active, inactive, and intermediate metastable states), which in turn has provided remarkable details of structure-activation-function relationships (Granier and Kobilka, 2012; Shoichet and Kobilka, 2012; Kobilka, 2013; Manglik and Kobilka, 2014; Hilger et al., 2018). Here, we summarize several X-ray structures of class A and B GPCRs cocrystallized with their allosteric modulators (PAMs, agoPAMs, and NAMs), specifically bound at the extrahelical transmembrane binding sites that are embedded deep within the lipid bilayer (see Fig. 4; Table 1). Remarkably, these structures reveal that functionally important allosteric binding sites are spread across the entire bilayer depth, which can be reached only through lipid pathways. These allosteric ligands most probably partition into the membrane and concentrate at the vicinity of these binding sites before binding (Blanton et al., 1988). All but one of the ligands (TAK-875) have a calculated 1-octanol/water log P value above 3, which means they prefer a lipid phase 1000× more than an aqueous phase and probably first partition into the membrane, after which they can diffuse to their respective binding sites at the lipid bilayer/receptor interface (Table 1). The calculated distances between the center of mass (COM) of the bound ligands and COM of phosphorus atoms of the upper leaflet of the membrane lipids (POPC) indicate the diversity in bilayer depth of these lipid-facing sites (Table 1).
Fig. 4.

Transmembrane allosteric sites in GPCRs span the entire bilayer normal (z-axis). X-ray structures (PDB IDs are given in the brackets) of class A and class B GPCRs and their ligands bound to transmembrane allosteric sites embedded within and facing the lipid bilayer. The receptors and bound ligands are present in secondary structure (gray) and stick (blue) representations, respectively. CRF1R, corticotropin-releasing factor receptor 1; GCGR, glucagon receptor; GLP-1, glucagon-like peptide-1 receptor; GPR40, free fatty acid receptor; P2Y1, P2Y purinoreceptor 1; PAR2, protease activated receptor 2. The approximate positions of the upper and lower leaflets of a hypothetical lipid bilayer are marked by two horizontal lines perpendicular to the bilayer normal. The Ballesteros and Weinstein amino acid numbering system (Ballesteros and Weinstein, 1995) used in the main text as a superscript of the absolute sequence number, begins with the TMH number and is followed by a locant (0.50 assigned to most highly conserved amino acid in a TMH and other residues numbered relative to it).
TABLE 1.
X-ray structures of class A and B GPCRs in which the allosteric ligands are bound to transmembrane lipid-facing binding sites, which can be accessed only through lipid pathways
| GPCR Class | Receptor | Allosteric Ligand Name | PDB ID | Bilayer Depth (Å)a | Major Species at pH 7.4 | Solvent-Accessible Surface Area (SASA)b |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ligand Lipophilicity | Unbound Ligand |
Bound Ligand |
||||||||||
| C log P | log D7.4 | Total (Å2) | Polar (%) | Nonpolar (%) | MESA (%) | BSA (%) | ||||||
| A | GPR40 | TAK-875c | 4PHU | 16.1 | 1.30 | 1.23 | Negative | 656.5 | 49.8 | 50.2 | 31 | 69 |
| GPR40 | MK-8666c | 5TZR | 16.4 | 3.75 | 1.62 | Negative | 913.4 | 16.8 | 83.2 | 34 | 66 | |
| GPR40 | AP8d | 5TZY | 30.5 | 5.23 | 4.05 | Zwitterionic | 923.9 | 15.6 | 84.4 | 34 | 66 | |
| P2Y1 | BPTUe | 4XNV | 22.7 | 7.50 | 5.53 | Neutral | 739.8 | 34.8 | 65.2 | 40 | 60 | |
| PAR2 | AZ3451e | 5NDZ | 18.0 | 7.96 | 6.37 | Neutral | 773.1 | 18.1 | 81.9 | 43 | 57 | |
| B | CRF1R | CP-376395e | 4K5Y | 23.6 | 7.61 | 5.7 | Positive | 658.1 | 49.4 | 50.6 | 39 | 61 |
| GCGR | MK-0893e | 5EE7 | 31.6 | 4.88 | 3.42 | Negative | 971.6 | 9.9 | 90.1 | 44 | 56 | |
| GLP1R | PF-06372222f | 5VEW | 34.7 | 4.61 | −0.53 | Negative | 908.3 | 13.3 | 86.7 | 30 | 70 | |
| GLP1R | NNC0640f | 5VEX | 31.0 | 4.08 | 1.28 | Positive | 976.2 | 41.5 | 58.5 | 45 | 55 | |
CRF1R, corticotropin-releasing factor 1 receptor; GLP1R, glucagon-like peptide 1 receptor; GPR40, free fatty acid receptor 1; PAR2, protease-activated receptor 2.
Binding site location (bilayer depth) within the membrane, measured as the distance between center of mass (COM) of the ligand and that of the phosphorus atoms in the upper leaflet, measured along the bilayer normal.
Calculated using NACCESS (Hubbard and Thornton, 1993) with a probe of radius 1.4 Å; C log P: 1-octanol/water partition coefficients calculated using Bio-Loom for Windows version 1.5 (2006); log D7.4: partition coefficient at pH 7.4 that was calculated using ACD/Percepta (Advanced Chemistry Development, Inc., Toronto, ON, 2015). For bilayer depth calculation, the membrane-embedded crystal structures were obtained from the OPM database (Lomize et al., 2012). The average bilayer thickness was 36.92 Å for all the studied receptors.
Partial agonist.
Full allosteric agonists, AgoPAMs.
Antagonist.
Negative allosteric modulator.
Desolvation of Ligands While Binding to Membrane-Exposed Receptor Sites.
Binding of a ligand to its target receptor site involves changes in the solvent-accessible surface area (SASA) for both species. The extent to which the ligand and its binding site are desolvated and the amount of buried SASA provide direct insights into the contributions of solvents and hydrophobic interactions in the molecular recognition process. A strong correlation between the experimental binding affinities of several protein-ligand complexes and their SASA quantities has been reported (Li et al., 2018). Interestingly, for membrane-exposed binding sites, the binding of ligands often does not involve the removal of any water molecules from the binding sites. In contrast, the lipid molecules surrounding these sites are displaced during the ligand-binding event. Nevertheless, a significant portion of these ligands remains exposed to the lipid molecules surrounding the binding sites. Although it is well known that the membrane partitioning of ligand molecules significantly affects their local concentrations and subsequently may contribute to their kon rates, the extent of lipid-ligand interactions at the receptor site may also influence their koff rates. It is reasonable to consider these lipid molecules as part of the binding site given the magnitude of their interactions that contribute to the overall binding affinity as well as residence time of these ligands. This unconventional but logical treatment of the membrane as part of the receptor in membrane-exposed binding sites needs a careful, revised treatment of the koff rates, thus incorporating the effects of the membrane-partitioning characteristics into the overall dissociation rate constants.
In this paper, we calculated the extent to which the SASA of the bound ligands (presented in the X-ray crystal structures discussed here, and summarized and represented in Figs. 4 and 5; Table 1, respectively) are either buried in the receptor sites, denoted by “buried surface area” (BSA), or exposed to the lipid molecules, denoted by “membrane-exposed surface area” (MESA). The SASA values for the bound receptor-ligand complexes, as well as for the unbound receptors and the unbound ligand molecules, were calculated using the NACCESS program (Hubbard and Thornton, 1993; https://xip.uclb.com/i/software/naccess.html?item=naccess) with a probe radius of 1.4 Å. The X-ray structures of the receptor-ligand complexes were treated in MOE (Molecular Operating Environment 2013.08, 2018) (all water, nonprotein, and nonligand molecules were removed; missing residues and atoms were added wherever necessary). In principle, the solvent-accessible area of a ligand in its receptor-bound form provides the MESA value, whereas the difference between the SASA of an unbound ligand and its MESA value provides the BSA value, the proportion of the ligand surface area that is buried in the receptor. The majority of these ligands are highly lipophilic and characterized by up to 90% nonpolar SASA (see Fig. 5A; Table 1). Interestingly, for all these ligands, approximately 40%–55% of their nonpolar SASA remains exposed to the membrane lipids in their bound forms (see Fig. 5, B and C; Table 1). However, large quantities (50%–95%) of their polar SASA are still buried within the binding sites (see Fig. 5D).
Fig. 5.

Box and whisker plots of solvent-accessible surface area (in percent) characteristics of the allosteric GPCR ligands (discussed in Table 1) in free (A) and receptor-bound states (B–D). The majority of the ligands are highly lipophilic with larger nonpolar SASA (A). In their receptor-bound states, significant (approx. 40%) portions of their total SASA (B) remain exposed to membrane lipids, denoted as membrane-exposed surface area, whereas ≥60% of their total SASA are buried within the receptor binding sites, denoted as buried surface area (BSA). Nonpolar SASA (C) of these ligands have BSA and MESA distributions very similar to those of their total SASA. However, the polar SASAs are mostly buried into the receptor sites (D). Lines inside the boxes denote medians, lower and upper ends of the boxes denote 25th and 75th percentiles respectively, and whiskers denote the lowest and highest observations.
Class A GPCRs
Although all GPCRs share common structural features (e.g., 7TM helices, along with the ability to interact with G-proteins), substantial differences between them allow them to be classified into various groups. The diversity among the GPCR superfamily has allowed for the A-F classification system developed by Kolakowski in 1994 (Lagerström and Schiöth, 2008). Class A (rhodopsin-like) GPCRs make up the largest member of the family, which are further divided into four groups (α, β, γ, δ). These different groups bind various ligands, including peptides, lipids, amines, and purines, and there does not seem to be a clear correlation between the phylogenetic location of the receptor and the endogenous ligand. Clusters exist, with the amine-binding ligands primarily in the α-group and the majority of peptide-binding ligands in the β-group. The nucleotide-binding receptors and the glycoproteins are mainly confined to the δ-group (Fredriksson et al., 2003). There is great diversity in the composition and size of the N terminus and the extracellular loops for these receptors (Venkatakrishnan et al., 2013).
Free Fatty-Acid Receptor 1 Bound to Partial Agonists TAK-875 and MK-8666, and to Ago-PAM AP8.
GPR40, also known as the free fatty-acid receptor 1, has been shown to be a promising target for novel treatment of type-2 diabetes mellitus (McGarry and Dobbins, 1999; Flodgren et al., 2007). Natural ligands of the receptor include full agonists such as saturated C12-C16 and unsaturated C18-C20 free fatty acids, which mediate insulin secretion and glucagon-like peptide 1 secretion. The receptor was crystallized (Srivastava et al., 2014) with a small-molecule partial agonist, TAK-875 (PDB ID 4PHU), that stimulates glucose-dependent insulin secretion. The structure revealed a unique binding mode for TAK-875 at the transmembrane extrahelical allosteric binding site located in between TMH3 and TMH5, at a bilayer depth of approx. 16.1 Å (see Fig. 4; Table 1). The most striking feature of the bound ligand, aside from the position of the binding site at the receptor-lipid interface, was that more than 58% of the nonpolar SASA was sticking out and exposed to the membrane, specifically toward the head group region of the bilayer. In contrast, more than 95% of the bound ligand’s polar SASA was buried within the binding pocket, with the carboxyl group of TAK-875 forming H-bonds with multiple arginine (Arg1835.39 and Arg2587.35) and tyrosine (Tyr913.37 and Tyr2406.51) residues of the receptor. The spatial and topological location of the binding site with respect to the bilayer and the 380-fold higher affinity of the ligand for the lipophilic phase over the aqueous phase together suggest the first step of preferential partitioning into the bilayer around the receptor, followed by a lateral movement in between TMH3 and TMH5.
Two additional crystal structures for GPR40 have been published (Lu et al., 2017). One structure features another partial agonist, MK-8666 (PDB ID 5TZR), bound at the same TAK-875 binding pocket; the other features an AgoPAM AP8 (PDB ID 5TZY) and MK-8666, which are both bound at the same time. MK-8666s BSA and MESA profiles are similar to that of TAK-875 (66% and 34%, respectively), except that it has a larger nonpolar SASA (83% vs. 50%) implying more lipophilic nature of MK-8666 (see Table 1). The binding site of the AgoPAM revealed that it occupied a well defined pocket at a bilayer depth of approx. 30.5 Å (see Fig. 4; Table 1); this pocket was formed by transmembrane helices 3–5 and intracellular loop 2 (ICL2) outside of the helical bundle and away from the orthosteric binding site. The lipid-facing binding site is unique and composed of three distinct regions: 1) toward the extracellular side, the top of the binding site is formed by the intersection of TMH3, TMH4, and TMH5 with residues Ile1304.49, Leu1334.52, Val1344.53, and Leu1905.46, which collectively form a hydrophobic pocket to accommodate the terminal trifluoromethoxyphenyl moiety of AP8; 2) toward the intracellular side, the bottom portion of the binding site is formed by a polar cavity (Tyr442.42 and Ser1234.42) that interacts with the carboxylate group of AP8; 3) the middle region is formed by residues located on ICL2 and its surrounding area (Leu1063.52, Phe1103.56, Tyr114ICL2, Phe117ICL2, and Tyr1224.41), form stabilizing interactions with the AP8 methyl and cyclopropyl moieties, sealing the AP8 pocket from the intracellular environment.
P2Y1-P2Y Purinoreceptor 1 Bound to Antagonist BPTU.
The purinergic receptors are divided into two subfamilies, Gq-coupled P2Y1R and Gi-coupled P2Y12R. Both subfamilies are involved in thrombosis formation, and the blockade of either receptor significantly decreases ADP-induced platelet aggregation (Jin and Kunapuli, 1998; Jacobson et al., 2011). Though many currently available antithrombotic drugs act on P2Y12R, P2Y1R has been suggested as a promising alternative that offers the advantage of a reduced bleeding effect. Recently, Zhang et al. (2015) crystallized the human purinergic Gq-coupled P2Y1 receptor, in complex (PDB ID 4XNV) with a novel antagonist, BPTU, that reduces platelet aggregation with a minimal bleeding effect, for the treatment of thrombosis (Chao et al., 2013). BPTU is highly lipophilic (log D7.4 = 5.53) and binds at a relatively shallow binding pocket (located at a bilayer depth of approx. 27 Å; see Fig. 4; Table 1) that faces the lipid bilayer and is formed by several aromatic residues located on TMH1, TMH2, and TMH3 and extracellular loop 1. The interactions of BPTU with the binding site residues are predominantly hydrophobic in nature. At one end, the trifluoromethoxyphenyl group of BPTU interacts with F621.43 and F661.47; at the other end, the phenoxy moiety attached to the tert-butyl group engages in nonpolar interactions with a hydrophobic cavity formed by the residues from TMH2 and TMH3, including T1032.56, M1233.24, L1263.27, and Q1273.28. Though the tert-butyl group seems to be in close contact with the alkyl side chain of L1032.55, it clearly protrudes into the lipid bilayer. Two H-bonds are formed between the amido–NH groups of BPTU’s urea group and the carbonyl oxygen of L1022.55. Around 60% of the ligand is buried inside the protein, exposing the remaining approx. 40% (see Table 1) of the ligand to the membrane environment. A recent computational study (Yuan et al., 2018) investigated the plausible lipid pathway and the role of the membrane in “preorganizing” the molecule for receptor binding, the details of which are discussed elsewhere in this article.
Protease-Activated Receptor 2 in Complex with Antagonist AZ3451.
The protease-activated receptors (PARs) are involved in a variety of diseases, including cancer and various types of inflammation (Adams et al., 2011; Ramachandran et al., 2012). The structure of PAR1 in complex with the orthosteric small-molecule antagonist vorapaxar has been reported before; however, despite the sequence homology between PAR1 and PAR2, attempts to identify the PAR2 antagonists have been less fruitful. Recently the PAR2 receptor was crystallized (Cheng et al., 2017) in complex with an allosteric modulator (NAM), AZ3451, at a resolution of 3.6 Å (PDB ID 5NDZ), and it was suggested that the binding of this ligand probably prevents the structural realignment necessary for receptor activation and signaling. AZ3451 is a highly lipophilic molecule, and the location of the allosteric binding site (at the bilayer depth of 18 Å; see Fig. 4; Table 1) differs from allosteric sites observed for the P2Y1 and the glucagon receptors (Jazayeri et al., 2016). The 1,3-benzodioxole ring of AZ3451 fits into a hydrophobic pocket formed by residues Ala1202.49, Leu1232.52, Phe1543.31, Ala1573.34 (Gly1573.34 in wild type), Cys1613.38, Trp1994.50, and Ile2024.53. Tyr2104.61 forms a weak hydrogen bond (3.4 Å) to N1 of the benzimidazole. Mutations of Gly1573.34 into cysteine or of Tyr2104.61 into leucine resulted in the decreased efficacy of AZ3451, indicating their contribution to selectivity. The cyclohexyl ring of the ligand lies at the membrane interface of the receptor and engages with Leu1232.52, and the benzonitrile moiety points toward the extracellular aqueous interface, making π-stacking interactions with Y2104.61. The significant nonpolar interactions (MESA of approx. 43%, see Table 1) between the ligand and the surrounding lipid molecules in the receptor-bound state might be a critical factor in imparting the receptor binding affinity.
Class B GPCRs
Class B GPCRs (secretin-like receptors) all bind peptide hormones and contain an extracellular hormone binding domain. The majority of this family of receptors share conserved cysteine residues in the N-terminal domain that form a network of three cysteine bridges (Grauschopf et al., 2000; Hofmann et al., 2001; Bazarsuren et al., 2002). Class A and class B GPCRs differ in their transmembrane helices, featuring distinct patterns of conserved residues in each class respectively (Siu et al., 2013). In general, the class B GPCRs feature the same overall architecture as class A GPCRs, but the available crystal structures of some class B GPCRs have revealed differences between the orthosteric binding pockets of the two classes. Class B GPCRs have an orthosteric binding pocket that is deeper and wider than that of any class A GPCR (Hollenstein et al., 2013) and development of nonpeptide small-molecule ligands targeting these sites has proved extremely challenging. Thus, allosteric modulation of these receptors offers a potential direction for developing low-molecular-weight activators and inhibitors as therapeutic agents (Hoare, 2007).
Corticotropin-Releasing Factor Receptor Type 1 Bound to Antagonist CP-376395.
The corticotropin-releasing factor receptor type 1 (CRF1R) is a class B GPCR that has been shown to mediate the stress response and has been considered as a target for managing depression and anxiety (Bale and Vale, 2004). The receptor binds corticotropin-releasing hormone, a 41 amino acid peptide that acts as a neurotransmitter and can influence a variety of responses, including appetite control, cardiovascular regulation, immune function, and behavior (Hemley et al., 2007). Antagonists of CRF1R have been evaluated for the potential to treat different stress-related indications, including depression, anxiety, and irritable bowel syndrome (Hemley et al., 2007). The X-ray crystal structure of CRF1R bound to antagonist CP-376395 (PDB ID 4K5Y) reveals an extrahelical allosteric binding site embedded within the transmembrane domain of the receptor (Zorrilla and Koob, 2010). This allosteric pocket is topologically distinct (18 Å away from the center of the orthosteric binding pocket) and is 13–23 Å away from the center of mass of the small-molecule ligands in the crystal structures of some class A GPCRs (Hollenstein et al., 2013). The binding pocket is formed mainly by the hydrophobic residues from TMH3, TMH5, and TMH6 of the receptor, which seem to open laterally through rearrangements, allowing the entry of the lipophilic ligand from within the membrane, possibly through lateral diffusion. In the bound state, the ligand molecule aligns perpendicularly to the membrane bilayer normal at a bilayer depth of approx. 23.1 Å (see Fig. 4; Table 1). The ligand forms an H-bond with Asn2835.50 through its pyridine nitrogen atom and the aryloxy moiety fits into a hydrophobic pocket formed by Phe2845.51, Leu2875.54, Ile2905.57, Tyr3166.42, Leu3196.45, and Leu3206.46. The exocyclic alkylamino group interacts with Gly3246.50, Phe2033.44, Leu2805.47, Leu3236.49, and Tyr3276.53. However, a significant part of the ligand (MESA of 39%, see Table 1) is sticking out into the membrane, exposed to the hydrophobic environment of the lipid tails.
Glucagon Receptor Bound to Antagonist MK-0893.
Glucagon is a 29 amino acid peptide that plays an integral role in glucose homeostasis; its action is caused by the G protein-coupled glucagon receptor (GCGR), a class B GPCR deemed a potential drug target in the treatment of diabetes. Small-molecule GCGR antagonists have shown promise in treating patients with type 2 diabetes, resulting in reduction of fasting and postprandial glucose concentrations (Bagger et al., 2011). The crystal structure of GCGR in complex with antagonist MK-0893 (PDB ID 5EE7) reveals an allosteric site at the interface between TMH6 and TMH7 that extends further into the membrane to a bilayer depth of approx. 31.6 Å proximal to the lower leaflet (Jazayeri et al., 2016) (see Fig. 4; Table 1). This lipophilic ligand (more than 90% of its total SASA is nonpolar) engages in several important hydrophobic interactions that include interactions of the methoxynapthalene moiety of MK-0893 with residues Leu3295.61, Phe3456.36, Leu3526.43, Thr3536.44, and with the alkyl chain of Lys3496.40 at the TMH5-TMH6 interface. Within the TMH6-TMH7 cleft, MK-0893 is surrounded by several polar contacts: The amide group forms H-bonds with Lys3496.40 and Ser3506.41; and the carboxyl group forms a salt bridge with Arg3466.37, in addition to interactions with Asn4047.61 and the backbone of Lys4057.62, plus water-mediated H-bonds with Ser3506.41 and Leu3997.56. The phenylethylpyrazole core of the ligand is in close contact with the Lys3496.40 and Thr3536.44 located on TMH6. Strikingly, the lipophilic dicholorophenyl moiety attached to the core pyrazole ring is entirely sticking out into the membrane, engaging in hydrophobic interactions with the lipid tails. The ligand atoms in its receptor-bound state remain significantly exposed to the membrane (MESA value of approx. 44%) indicating the apparent contribution of the membrane to the binding affinity.
Glucagon-Like Peptide Receptor Bound to Negative Allosteric Modulators PF-06372222 and NNC0640.
Glucagon-like peptide-1 receptor (GLP-1R) is a class B GPCR and is activated by endogenous GLP-1 peptides. Activation of the receptor leads to the inhibition of glucagon secretion and the stimulation of insulin secretion in a glucose-dependent manner (Cho et al., 2012); thus, GLP-1R agonists can be used for the treatment of type 2 diabetes mellitus. Antagonism of GLP-1R was also shown to be clinically useful in the treatment of acute and chronic stress as well as anxiety. Two X-ray structures of GLP-1R (Song et al., 2017), crystallized with negative allosteric modulators PF-06372222 (PDB ID 5VEW) and NNC0640 (PDB ID 5VEX), reveal that both NAMs bind at the same extrahelical site, located outside the TMH6 and TMH7 helices, facing the lipid membrane. Interestingly, these are also NAMs of GCGR, and the binding modes of these ligands are remarkably like that of MK-0893 bound to GCGR. In addition, the trifluoromethylpyrazole end of PF-06372222 has extensive nonpolar interactions with the hydrophobic pocket formed by residues I3285.58, V3315.61, V3325.62, L3355.65, and F3476.36, and the pyridine and phenyl rings of the ligand make hydrophobic interactions with the alkyl side chains of K3516.40 and L4017.56. Most interestingly, the dimethylcyclobutane ring of the ligand sticks out into the lipid bilayer while making nonpolar contacts with L4016.38 and M3976.42. The polar anionic carboxylic acid and amide groups of PF-06372222 make H-bonds with the side chains of S3526.41, N4067.61, and K3516.40 residues, respectively, and the aminopyridine moiety forms an H-bond with the side chain of the S3556.44 residue.
The binding mode and residue interactions of NNC0640 are notably similar to those of PF-06372222. The polar tetrazole group of the ligand makes H-bond interactions with the side chains of S3526.41 and N4067.61, whereas the amide group forms H-bonds with the side chains of S3526.41and K3516.40 and the urea amino group makes an H-bond with S3556.44. The methylsulfone phenyl end of the ligand molecule occupies the same hydrophobic binding pocket occupied by the trifluoromethylpyrazole moiety of PF-06372222, engaging in nonpolar interactions with F3245.54, I3285.58, F3476.36, and L3516.40 and the alkyl chain of K3516.40. As with the dimethylcyclobutane ring of PF-06372222, the phenylcyclohexyl group distinctly sticks out into the lipid bilayer while making extensive nonpolar interactions with the S3556.44, L3596.48, F3906.31, M3976.31, and L4016.42 residues. In these unique binding modes, the NAMs restrain the movement of TMH6 away from TMH7 and thus prevent G protein coupling. MESA values for PF-06372222 and NNC0640 are 30% and 45%, respectively. However, it should be noted that the nonpolar SASA is much higher (86.7%) for PF-06372222 than it is for NNC0640 (which is only 58.5%). Interestingly for PF-0637222, more than 90% of its polar SASA is buried in the protein (see Table 1), whereas for NNC0640, 60% of its polar SASA is buried inside the receptor binding pocket.
Conclusions and Future Perspectives
Recent advances in GPCR structural biology and growing interest in allosteric modulation and biased signaling as novel therapeutic strategies have created tremendous opportunities as well as numerous challenges for rational drug discovery (May et al., 2007; Burford et al., 2011; Conn et al., 2014; Langmead and Christopoulos, 2014). The discovery of multiple spatially distinct yet conformationally linked allosteric binding sites located at the transmembrane extrahelical-lipid interfaces presents an enriched yet challenging landscape for small-molecule therapeutics. Many challenges must be overcome before the actions of allosteric modulators can be reliably detected, validated, and quantified, and the fact that transmembrane extra-helical membrane-facing binding sites can only be reached via lipid pathways adds further complexity. The tissue-to-tissue variations in lipid composition and the biophysical properties of the plasma membrane necessitate a thorough understanding of drug-membrane interactions in order for full advantage to be taken of the multiple mechanisms (Sargent and Schwyzer, 1986; Vauquelin and Packeu, 2009) by which the membrane can facilitate protein-ligand recognition processes (Strasser et al., 2017). In addition, the remarkable extent to which ligands are exposed to the membrane environment while bound to these shallow, transmembrane binding pockets poses additional challenges for researchers in the molecular docking community. The challenge is how to effectively include membranes as one of the receptor components when making predictions about ligand binding poses and scoring the bound receptor-ligand complexes. In addition, the heterogeneity of the phospholipid headgroup, the presence of cholesterol, and the locations of binding sites with respect to the bilayer normal will need thorough consideration.
The ability to predict the quantitative bilayer distribution and the energetically favorable location (depth), orientation, and conformation of a given chemical within the lipid bilayer is a key to understanding the molecular recognition processes of ligands binding to membrane-facing sites of transmembrane proteins. Partition coefficients, either measured or calculated as log P using 1-octanol/buffer systems, have been shown to correlate well with a drug’s therapeutic efficacy, intestinal absorption, plasma protein binding, metabolism rate, nonspecific binding to off-targets, and toxicity (Hansch, 1969; Leo et al., 1971; Hansch and Zhang, 1993; Debnath et al., 1994; Hansch et al., 1995; Tarcsay and Keserű, 2013). Though log P values provide information about ligand lipophilicity, they do not account for the ability of the ligand to interact with the polar head groups of lipid membranes. Differences in drug partitioning into octanol/buffer versus membranes has been observed for a wide variety of drugs (Herbette, 1994; Sykes and Charlton, 2012; Sykes et al., 2014; Dickson et al., 2016). Most importantly, for ionized ligand molecules, the strong interactions of ionized species (bases and acids) with the zwitterionic phospholipid molecules are better captured by anisotropic lipid membranes than by the 1-octanol/water system. The anisotropic structure of the lipid bilayer has very different physical and chemical characteristics across the bilayer normal. Partitioning of drugs within the membrane and drug interactions with lipid molecules appear to exploit these differences to achieve an energetically favorable location, orientation, and conformation. The membrane-drug interactions mentioned above can also be quantified as KIAM, a partition coefficient that essentially describes the degree to which a ligand concentrates from aqueous bulk phase into the phospholipid membrane (Taillardat-Ertschinger et al., 2002; Droge et al., 2017). KIAM quantifies a ligand’s degree of phospholipophilicity using column chromatography, consisting of immobilized artificial membrane (monolayers of phospholipid immobilized on a silica surface), to determine the extent of intermolecular interaction between ligand and lipid. Phospholipophilicity is a term used to describe membrane retention of chemicals and comprises hydrophobicity, polarity, and ionic (electrostatic) interactions (Tsopelas et al., 2017). However, the probability of utilizing this quantity to develop a meaningful structural optimization strategy seems slim. Furthermore, an in silico continuum solvent model that quantifies the percentage distribution of chemicals in various bilayer strata was developed using a novel surrogate system containing hexadecane and hydrated diacetylphosphatidylcholine as the core and headgroup, respectively (Natesan et al., 2014). This model mimics realistic membrane-drug interactions and provides approximate orientations of chemicals within the bilayer. Experimental approaches (such as X-ray, NMR, surface plasmon resonance, and fluorescence quenching) for obtaining “native” drug structures in biologic membranes are extremely time-consuming and not adaptable to high-throughput screening in early-stage drug discovery. Increases in computing speed, owing to the combination of graphical processing units, improved algorithms, multiscale modeling methods, and more accurate force field developments (Bassolino-Klimas et al., 1993, 1995; Alper and Stouch, 1995; Park et al., 2003; Bemporad et al., 2005; Pastor and MacKerell, 2011; Tejwani et al., 2011a,b; Jämbeck and Lyubartsev, 2012a,b; Dickson et al., 2014; Lee et al., 2016), have made molecular dynamic simulations an essential and complementary tool for investigating the partitioning characteristics of small molecules within the membrane compartment and their subsequent recognition by transmembrane proteins at the atomic level.
The current practice of structure-affinity relationship analysis in drug design, which is mainly based on the protein-ligand interactions, needs modification for drugs targeting membrane-associated proteins such as GPCRs (Congreve et al., 2017). This is critical, especially when the ligand is expected to take a lipid pathway to reach its binding site and remains significantly exposed to membrane lipids in its receptor-bound form. Deconvolution of membrane- and protein-ligand contributions to the overall binding affinity will certainly be helpful in optimizing drug structures for better receptor affinity. Nevertheless, knowledge of the structure-membrane interaction relationships of drugs, which captures the “specifics” of the so-called nonspecific membrane interactions, will undoubtedly enable us to optimize rationally the structural features of drugs to exploit the many unique advantages of a membrane environment, resulting in improved receptor affinity, binding kinetics, and selectivity.
Abbreviations
- BPTU
1-(2-(2-(tert-butyl) phenoxy) pyridine-3-yl)-3-(4-(trifluoromethoxy) phenyl) urea
- BSA
buried surface area
- GCGR
G protein-coupled glucagon receptor
- GPCR
G protein-coupled receptor
- Kd
intrinsic binding affinity
- Kd′
observed binding affinity
- Kmem
membrane partition coefficient
- kon′
measured association rate
- log KIAM
partition coefficient measured using immobilized artificial membrane
- MESA
membrane-exposed surface area
- MK-0893
3-({4-[(1S)-1-[3-(3,5-dichlorophenyl)-5-(6-methoxynaphthalen-2-yl)-1H-pyrazol-1-yl]ethyl]phenyl}formamido)propanoic acid
- NAM
negative allosteric modulator
- NNC0640
4-[[(4-cyclohexylphenyl)-[(3-methylsulfonylphenyl)carbamoyl]amino]methyl]-N-(2H-tetrazol-5-yl)benzamide
- P2Y1R
P2Y1 receptor
- PAR
protease-activated receptor
- PF-06372222
3-[[4-[(R)-(3,3-dimethylcyclobutyl)-[[6-[4-(trifluoromethyl)imidazol-1-yl]pyridin-3-yl]amino]methyl]benzoyl]amino]propanoic acid
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- PAM
positive allosteric modulator
- SASA
solvent-accessible surface area
- 7TM
seven-transmembrane
- TMH
transmembrane helix
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Szlenk, GC, Natesan.
Footnotes
This work was supported in part by the National Institutes of Health, National Institute of General Medical Sciences [Grant R15 GM131293-01].
References
- ACD Percepta (2015) Advanced Chemistry Development, Inc., Toronto, ON, Canada. [Google Scholar]
- Adams MN, Ramachandran R, Yau M-K, Suen JY, Fairlie DP, Hollenberg MD, Hooper JD. (2011) Structure, function and pathophysiology of protease activated receptors. Pharmacol Ther 130:248–282. [DOI] [PubMed] [Google Scholar]
- Alper HE, Stouch TR. (1995) Orientation and diffusion of a drug analog in biomembranes: molecular dynamics simulations. J Phys Chem 99:5724–5731. [Google Scholar]
- Bagger JI, Knop FK, Holst JJ, Vilsbøll T. (2011) Glucagon antagonism as a potential therapeutic target in type 2 diabetes. Diabetes Obes Metab 13:965–971. [DOI] [PubMed] [Google Scholar]
- Bale TL, Vale WW. (2004) CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol 44:525–557. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Jensen AD, Liapakis G, Rasmussen SGF, Shi L, Gether U, Javitch JA. (2001) Activation of the β 2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J Biol Chem 276:29171–29177. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. (1995) Integrated methods for the construction of three dimensional models and computational probing of structure function relations in G protein-coupled receptors, in Methods in Neurosciences (Sealfon SC, Conn PM. eds) pp 366–428, Academic Press, San Diego, CA. [Google Scholar]
- Barducci A, Bussi G, Parrinello M. (2008) Well-tempered metadynamics: a smoothly converging and tunable free-energy method. Phys Rev Lett 100:020603. [DOI] [PubMed] [Google Scholar]
- Bassolino-Klimas D, Alper HE, Stouch TR. (1993) Solute diffusion in lipid bilayer membranes: an atomic level study by molecular dynamics simulation. Biochemistry 32:12624–12637. [DOI] [PubMed] [Google Scholar]
- Bassolino-Klimas D, Alper HE, Stouch TR. (1995) Mechanism of solute diffusion through lipid bilayer membranes by molecular dynamics simulation. J Am Chem Soc 117:4118–4129. [DOI] [PubMed] [Google Scholar]
- Bazarsuren A, Grauschopf U, Wozny M, Reusch D, Hoffmann E, Schaefer W, Panzner S, Rudolph R. (2002) In vitro folding, functional characterization, and disulfide pattern of the extracellular domain of human GLP-1 receptor. Biophys Chem 96: 305–318. [DOI] [PubMed] [Google Scholar]
- Bemporad D, Luttmann C, Essex JW. (2005) Behaviour of small solutes and large drugs in a lipid bilayer from computer simulations. Biochim Biophys Acta 1718:1–21. [DOI] [PubMed] [Google Scholar]
- Berka K, Hendrychová T, Anzenbacher P, Otyepka M. (2011) Membrane position of ibuprofen agrees with suggested access path entrance to cytochrome P450 2C9 active site. J Phys Chem A 115:11248–11255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bio-Loom for Windows version 1.5 (2006) BioByte Corp., Claremont, CA.
- Blanton M, McCardy E, Gallaher T, Wang HH. (1988) Noncompetitive inhibitors reach their binding site in the acetylcholine receptor by two different paths. Mol Pharmacol 33:634–642. [PubMed] [Google Scholar]
- Bochicchio D, Panizon E, Ferrando R, Monticelli L, Rossi G. (2015) Calculating the free energy of transfer of small solutes into a model lipid membrane: comparison between metadynamics and umbrella sampling. J Chem Phys 143:144108. [DOI] [PubMed] [Google Scholar]
- Bokoch MP, Jo H, Valcourt JR, Srinivasan Y, Pan AC, Capponi S, Grabe M, Dror RO, Shaw DE, DeGrado WF, et al. (2018) Entry from the lipid bilayer: a possible pathway for inhibition of a peptide G protein-coupled receptor by a lipophilic small molecule. Biochemistry 57:5748–5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, Aradhye S, Burtin P. (2010) Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov 9:883–897. [DOI] [PubMed] [Google Scholar]
- Bruzzese A, Gil C, Dalton JAR, Giraldo J. (2018) Structural insights into positive and negative allosteric regulation of a G protein-coupled receptor through protein-lipid interactions. Sci Rep 8:4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burford NT, Watson J, Bertekap R, Alt A. (2011) Strategies for the identification of allosteric modulators of G-protein-coupled receptors. Biochem Pharmacol 81:691–702. [DOI] [PubMed] [Google Scholar]
- Cazzola M, Calzetta L, Matera MG. (2011) β(2) -adrenoceptor agonists: current and future direction. Br J Pharmacol 163:4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzola M, Matera MG, Lötvall J. (2005) Ultra long-acting beta 2-agonists in development for asthma and chronic obstructive pulmonary disease. Expert Opin Investig Drugs 14:775–783. [DOI] [PubMed] [Google Scholar]
- Changeux J-P, Christopoulos A. (2017) Allosteric modulation as a unifying mechanism for receptor function and regulation. Diabetes Obes Metab 19(Suppl 1):4–21. [DOI] [PubMed] [Google Scholar]
- Chao H, Turdi H, Herpin TF, Roberge JY, Liu Y, Schnur DM, Poss MA, Rehfuss R, Hua J, Wu Q, et al. (2013) Discovery of 2-(phenoxypyridine)-3-phenylureas as small molecule P2Y1 antagonists. J Med Chem 56:1704–1714. [DOI] [PubMed] [Google Scholar]
- Cheng RKY, Fiez-Vandal C, Schlenker O, Edman K, Aggeler B, Brown DG, Brown GA, Cooke RM, Dumelin CE, Doré AS, et al. (2017) Structural insight into allosteric modulation of protease-activated receptor 2. Nature 545:112–115. [DOI] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi H-J, Kuhn P, Weis WI, Kobilka BK, et al. (2007) High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 318:1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YM, Merchant CE, Kieffer TJ. (2012) Targeting the glucagon receptor family for diabetes and obesity therapy. Pharmacol Ther 135:247–278. [DOI] [PubMed] [Google Scholar]
- Christopoulos A. (2014) Advances in G protein-coupled receptor allostery: from function to structure. Mol Pharmacol 86:463–478. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Changeux J-P, Catterall WA, Fabbro D, Burris TP, Cidlowski JA, Olsen RW, Peters JA, Neubig RR, Pin J-P, et al. (2014) International Union of Basic and Clinical Pharmacology. XC. multisite pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol Rev 66:918–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay AT, Sharom FJ. (2013) Lipid bilayer properties control membrane partitioning, binding, and transport of p-glycoprotein substrates. Biochemistry 52:343–354. [DOI] [PubMed] [Google Scholar]
- Comitani F, Limongelli V, Molteni C. (2016) The free energy landscape of GABA binding to a pentameric ligand-gated ion channel and its disruption by mutations. J Chem Theory Comput 12:3398–3406. [DOI] [PubMed] [Google Scholar]
- Congreve M, Oswald C, Marshall FH. (2017) Applying structure-based drug design approaches to allosteric modulators of GPCRs. Trends Pharmacol Sci 38:837–847. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Lindsley CW, Meiler J, Niswender CM. (2014) Opportunities and challenges in the discovery of allosteric modulators of GPCRs for treating CNS disorders. Nat Rev Drug Discov 13:692–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawaliby R, Trubbia C, Delporte C, Masureel M, Van Antwerpen P, Kobilka BK, Govaerts C. (2016) Allosteric regulation of G protein-coupled receptor activity by phospholipids. Nat Chem Biol 12:35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath AK, Shusterman AJ, Lopez de Compadre RL, Hansch C. (1994) International Commission for Protection Against Environmental Mutagens and Carcinogens. The importance of the hydrophobic interaction in the mutagenicity of organic compounds. Mutat Res 305:63–72. [DOI] [PubMed] [Google Scholar]
- Dickson CJ, Hornak V, Velez-Vega C, McKay DJJ, Reilly J, Sandham DA, Shaw D, Fairhurst RA, Charlton SJ, Sykes DA, et al. (2016) Uncoupling the structure–activity relationships of β2 adrenergic receptor ligands from membrane binding. J Med Chem 59:5780–5789. [DOI] [PubMed] [Google Scholar]
- Dickson CJ, Madej BD, Skjevik AA, Betz RM, Teigen K, Gould IR, Walker RC. (2014) Lipid14: the amber lipid force field. J Chem Theory Comput 10:865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Meo F, Fabre G, Berka K, Ossman T, Chantemargue B, Paloncýová M, Marquet P, Otyepka M, Trouillas P. (2016) In silico pharmacology: drug membrane partitioning and crossing. Pharmacol Res 111:471–486. [DOI] [PubMed] [Google Scholar]
- Droge STJ, Hermens JLM, Gutsell S, Rabone J, Hodges G. (2017) Predicting the phospholipophilicity of monoprotic positively charged amines. Environ Sci Process Impacts 19:307–323. [DOI] [PubMed] [Google Scholar]
- Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, Xu H, Shaw DE. (2011) Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc Natl Acad Sci USA 108:13118–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escribá PV, Wedegaertner PB, Goñi FM, Vögler O. (2007) Lipid-protein interactions in GPCR-associated signaling. Biochim Biophys Acta 1768:836–852. [DOI] [PubMed] [Google Scholar]
- Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. (1996) Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science 274:768–770. [DOI] [PubMed] [Google Scholar]
- Flodgren E, Olde B, Meidute-Abaraviciene S, Winzell MS, Ahrén B, Salehi A. (2007) GPR40 is expressed in glucagon producing cells and affects glucagon secretion. Biochem Biophys Res Commun 354:240–245. [DOI] [PubMed] [Google Scholar]
- Foster DJ, Conn PJ. (2017) Allosteric modulation of GPCRs: new insights and potential utility for treatment of schizophrenia and other CNS disorders. Neuron 94:431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson R, Lagerström MC, Lundin L-G, Schiöth HB. (2003) The G-Protein-Coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharm 63:1256–1272. [DOI] [PubMed] [Google Scholar]
- Genheden S, Essex JW, Lee AG. (2017) G protein coupled receptor interactions with cholesterol deep in the membrane. Biochim Biophys Acta Biomembr 1859:268–281. [DOI] [PubMed] [Google Scholar]
- Gentry PR, Sexton PM, Christopoulos A. (2015) Novel allosteric modulators of G protein-coupled receptors. J Biol Chem 290:19478–19488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervasio FL, Laio A, Parrinello M. (2005) Flexible docking in solution using metadynamics. J Am Chem Soc 127:2600–2607. [DOI] [PubMed] [Google Scholar]
- Ghanouni P, Schambye H, Seifert R, Lee TW, Rasmussen SGF, Gether U, Kobilka BK. (2000) The effect of pH on β(2) adrenoceptor function. Evidence for protonation-dependent activation. J Biol Chem 275:3121–3127. [DOI] [PubMed] [Google Scholar]
- Ghanouni P, Steenhuis JJ, Farrens DL, Kobilka BK. (2001) Agonist-induced conformational changes in the G-protein-coupling domain of the β 2 adrenergic receptor. Proc Natl Acad Sci USA 98:5997–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherbi K, Briddon SJ, Charlton SJ. (2018) Micro-pharmacokinetics: quantifying local drug concentration at live cell membranes. Sci Rep 8:3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimpl G. (2016) Interaction of G protein coupled receptors and cholesterol. Chem Phys Lipids 199:61–73. [DOI] [PubMed] [Google Scholar]
- Granier S, Kobilka B. (2012) A new era of GPCR structural and chemical biology. Nat Chem Biol 8:670–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grauschopf U, Lilie H, Honold K, Wozny M, Reusch D, Esswein A, Schäfer W, Rücknagel KP, Rudolph R. (2000) The N-Terminal fragment of human parathyroid hormone receptor 1 constitutes a hormone binding domain and reveals a distinct disulfide pattern. Biochemistry 39:8878–8887. [DOI] [PubMed] [Google Scholar]
- Green SA, Spasoff AP, Coleman RA, Johnson M, Liggett SB. (1996) Sustained activation of a G protein-coupled receptor via “anchored” agonist binding. Molecular localization of the salmeterol exosite within the 2-adrenergic receptor. J Biol Chem 271:24029–24035. [DOI] [PubMed] [Google Scholar]
- Grossfield A, Pitman MC, Feller SE, Soubias O, Gawrisch K. (2008) Internal hydration increases during activation of the G-protein-coupled receptor rhodopsin. J Mol Biol 381:478–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guixà-González R, Albasanz JL, Rodriguez-Espigares I, Pastor M, Sanz F, Martí-Solano M, Manna M, Martinez-Seara H, Hildebrand PW, Martín M, et al. (2017) Membrane cholesterol access into a G-protein-coupled receptor. Nat Commun 8:14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansch C. (1969) Quantitative approach to biochemical structure-activity relationships. Acc Chem Res 2:232–241. [Google Scholar]
- Hansch C, Hoekman D, Leo A, Zhang L, Li P. (1995) The expanding role of quantitative structure-activity relationships (QSAR) in toxicology. Toxicol Lett 79:45–53. [DOI] [PubMed] [Google Scholar]
- Hansch C, Zhang L. (1993) Quantitative structure-activity relationships of cytochrome P-450. Drug Metab Rev 25:1–48. [DOI] [PubMed] [Google Scholar]
- Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, Reinhart G, Desale H, Clemons B, Cahalan SM, Schuerer SC, et al. (2012) Crystal structure of a lipid G protein-coupled receptor. Science 335:851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemley CF, McCluskey A, Keller PA. (2007) Corticotropin releasing hormone--a GPCR drug target. Curr Drug Targets 8:105–115. [DOI] [PubMed] [Google Scholar]
- Herbette LG. (1994) Membrane pathways for drug ion channel interactions: molecular basis for pharmacokinetic properties. Drug Dev Res 33:214–224. [Google Scholar]
- Hildebrand PW, Scheerer P, Park JH, Choe H-W, Piechnick R, Ernst OP, Hofmann KP, Heck M. (2009) A ligand channel through the G protein coupled receptor opsin. PLoS One 4:e4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilger D, Masureel M, Kobilka BK. (2018) Structure and dynamics of GPCR signaling complexes. Nat Struct Mol Biol 25:4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoare SR. (2007) Allosteric modulators of class B G-protein-coupled receptors. Curr Neuropharmacol 5:168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann BA, Sydow S, Jahn O, van Werven L, Liepold T, Eckart K, Spiess J. (2001) Functional and protein chemical characterization of the N-terminal domain of the rat corticotropin-releasing factor receptor 1. Protein Sci 10: 2050–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenstein K, Kean J, Bortolato A, Cheng RKY, Doré AS, Jazayeri A, Cooke RM, Weir M, Marshall FH. (2013) Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature 499:438–443. [DOI] [PubMed] [Google Scholar]
- Huang X, Chen H, Shaffer PL. (2017) Crystal structures of human GlyRα3 bound to ivermectin. Structure 25:945–950.e2. [DOI] [PubMed] [Google Scholar]
- Huggins DJ, Sherman W, Tidor B. (2012) Rational approaches to improving selectivity in drug design. J Med Chem 55:1424–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst DP, Grossfield A, Lynch DL, Feller S, Romo TD, Gawrisch K, Pitman MC, Reggio PH. (2010) A lipid pathway for ligand binding is necessary for a cannabinoid G protein-coupled receptor. J Biol Chem 285:17954–17964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Deflorian F, Mishra S, Costanzi S. (2011) Pharmacochemistry of the platelet purinergic receptors. Purinergic Signal 7:305–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jämbeck JP, Lyubartsev AP. (2012a) Derivation and systematic validation of a refined all-atom force field for phosphatidylcholine lipids. J Phys Chem B 116:3164–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jämbeck JP, Lyubartsev AP. (2012b) An extension and further validation of an all-atomistic force field for biological membranes. J Chem Theory Comput 8:2938–2948. [DOI] [PubMed] [Google Scholar]
- Jämbeck JP, Lyubartsev AP. (2013) Exploring the free energy landscape of solutes embedded in lipid bilayers. J Phys Chem Lett 4:1781–1787. [DOI] [PubMed] [Google Scholar]
- Javitch JA, Fu D, Liapakis G, Chen J. (1997) Constitutive activation of the β2 adrenergic receptor alters the orientation of its sixth membrane-spanning segment. J Biol Chem 272:18546–18549. [DOI] [PubMed] [Google Scholar]
- Jazayeri A, Doré AS, Lamb D, Krishnamurthy H, Southall SM, Baig AH, Bortolato A, Koglin M, Robertson NJ, Errey JC, et al. (2016) Extra-helical binding site of a glucagon receptor antagonist. Nature 533:274–277. [DOI] [PubMed] [Google Scholar]
- Jensen AD, Guarnieri F, Rasmussen SGF, Asmar F, Ballesteros JA, Gether U. (2001) Agonist-induced conformational changes at the cytoplasmic side of transmembrane segment 6 in the β 2 adrenergic receptor mapped by site-selective fluorescent labeling. J Biol Chem 276:9279–9290. [DOI] [PubMed] [Google Scholar]
- Jin J, Kunapuli SP. (1998) Coactivation of two different G protein-coupled receptors is essential for ADP-induced platelet aggregation. Proc Natl Acad Sci USA 95:8070–8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kästner J. (2011) Umbrella sampling. Wiley Interdiscip Rev Comput Mol Sci 1:932–942. [Google Scholar]
- Keov P, Sexton PM, Christopoulos A. (2011) Allosteric modulation of G protein-coupled receptors: a pharmacological perspective. Neuropharmacology 60:24–35. [DOI] [PubMed] [Google Scholar]
- Kobilka B. (2013) The structural basis of G-protein-coupled receptor signaling (Nobel Lecture). Angew Chem Int Ed Engl 52:6380–6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka BK. (2007) G protein coupled receptor structure and activation. Biochim Biophys Acta 1768:794–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka BK, Deupi X. (2007) Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci 28:397–406. [DOI] [PubMed] [Google Scholar]
- Lagerström MC, Schiöth HB. (2008) Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov 7:339–357. [DOI] [PubMed] [Google Scholar]
- Langmead CJ, Christopoulos A. (2014) Functional and structural perspectives on allosteric modulation of GPCRs. Curr Opin Cell Biol 27:94–101. [DOI] [PubMed] [Google Scholar]
- Latorraca NR, Venkatakrishnan AJ, Dror RO. (2017) GPCR dynamics: structures in motion. Chem Rev 117:139–155. [DOI] [PubMed] [Google Scholar]
- Laverty D, Thomas P, Field M, Andersen OJ, Gold MG, Biggin PC, Gielen M, Smart TG. (2017) Crystal structures of a GABAA-receptor chimera reveal new endogenous neurosteroid-binding sites. Nat Struct Mol Biol 24:977–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CT, Comer J, Herndon C, Leung N, Pavlova A, Swift RV, Tung C, Rowley CN, Amaro RE, Chipot C, et al. (2016) Simulation-based approaches for determining membrane permeability of small compounds. J Chem Inf Model 56:721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo A, Hansch C, Elkins D. (1971) Partition coefficients and their uses. Chem Rev 71:525–616. [Google Scholar]
- Li Y, Su M, Liu Z, Li J, Liu J, Han L, Wang R. (2018) Assessing protein-ligand interaction scoring functions with the CASF-2013 benchmark. Nat Protoc 13:666–680. [DOI] [PubMed] [Google Scholar]
- Limongelli V, Bonomi M, Marinelli L, Gervasio FL, Cavalli A, Novellino E, Parrinello M. (2010) Molecular basis of cyclooxygenase enzymes (COXs) selective inhibition. Proc Natl Acad Sci USA 107:5411–5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SW, Sakmar TP. (1996) Specific tryptophan UV-absorbance changes are probes of the transition of rhodopsin to its active state. Biochemistry 35:11149–11159. [DOI] [PubMed] [Google Scholar]
- Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL. (2012) OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res 40 (Database issue):D370–D376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Byrne N, Wang J, Bricogne G, Brown FK, Chobanian HR, Colletti SL, Di Salvo J, Thomas-Fowlkes B, Guo Y, et al. (2017) Structural basis for the cooperative allosteric activation of the free fatty acid receptor GPR40. Nat Struct Mol Biol 24:570–577. [DOI] [PubMed] [Google Scholar]
- Lu S, Zhang J. (2018) Small molecule allosteric modulators of G-protein-coupled receptors: drug–target interactions. J Med Chem 62:24–45. [DOI] [PubMed] [Google Scholar]
- Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, Thornton R, Shei G-J, Card D, Keohane C, et al. (2002) Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 296:346–349. [DOI] [PubMed] [Google Scholar]
- Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, Lerch MT, Kobilka TS, Thian FS, Hubbell WL, et al. (2015) Structural insights into the dynamic process of β2-adrenergic receptor signaling. Cell 161:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kobilka B. (2014) The role of protein dynamics in GPCR function: insights from the β2AR and rhodopsin. Curr Opin Cell Biol 27:136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason RP, Rhodes DG, Herbette LG. (1991) Reevaluating equilibrium and kinetic binding parameters for lipophilic drugs based on a structural model for drug interaction with biological membranes. J Med Chem 34:869–877. [DOI] [PubMed] [Google Scholar]
- Masureel M, Zou Y, Picard L-P, van der Westhuizen E, Mahoney JP, Rodrigues JPGLM, Mildorf TJ, Dror RO, Shaw DE, Bouvier M, et al. (2018) Structural insights into binding specificity, efficacy and bias of a β2AR partial agonist [published correction appears in Nat Chem Biol (2019) 15(2):205]. Nat Chem Biol 14:1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May LT, Leach K, Sexton PM, Christopoulos A. (2007) Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol 47:1–51. [DOI] [PubMed] [Google Scholar]
- McCloskey MA, Poo MM. (1986) Rates of membrane-associated reactions: reduction of dimensionality revisited. J Cell Biol 102:88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCorvy JD, Wacker D, Wang S, Agegnehu B, Liu J, Lansu K, Tribo AR, Olsen RHJ, Che T, Jin J, et al. (2018) Structural determinants of 5-HT2B receptor activation and biased agonism. Nat Struct Mol Biol 25:787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry JD, Dobbins RL. (1999) Fatty acids, lipotoxicity and insulin secretion. Diabetologia 42:128–138. [DOI] [PubMed] [Google Scholar]
- Melchior DL, Sharom FJ, Evers R, Wright GE, Chu JW, Wright SE, Chu X, Yabut J. (2012) Determining P-glycoprotein-drug interactions: evaluation of reconstituted P-glycoprotein in a liposomal system and LLC-MDR1 polarized cell monolayers. J Pharmacol Toxicol Methods 65:64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE) 2013.08 (2018) Chemical Computing Group ULC, Montreal, QC, Canada. [Google Scholar]
- Nakanishi J, Takarada T, Yunoki S, Kikuchi Y, Maeda M. (2006) FRET-based monitoring of conformational change of the β2 adrenergic receptor in living cells. Biochem Biophys Res Commun 343:1191–1196. [DOI] [PubMed] [Google Scholar]
- Natesan S, Lukacova V, Peng M, Subramaniam R, Lynch S, Wang Z, Tandlich R, Balaz S. (2014) Structure-based prediction of drug distribution across the headgroup and core strata of a phospholipid bilayer using surrogate phases. Mol Pharm 11:3577–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oates J, Watts A. (2011) Uncovering the intimate relationship between lipids, cholesterol and GPCR activation. Curr Opin Struct Biol 21:802–807. [DOI] [PubMed] [Google Scholar]
- Ostrom RS, Insel PA. (2004) The evolving role of lipid rafts and caveolae in G protein-coupled receptor signaling: implications for molecular pharmacology. Br J Pharmacol 143:235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overington JP, Al-Lazikani B, Hopkins AL. (2006) How many drug targets are there? Nat Rev Drug Discov 5:993–996. [DOI] [PubMed] [Google Scholar]
- Paloncýová M, DeVane R, Murch B, Berka K, Otyepka M. (2014) Amphiphilic drug-like molecules accumulate in a membrane below the head group region. J Phys Chem B 118:1030–1039. [DOI] [PubMed] [Google Scholar]
- Park JH, Scheerer P, Hofmann KP, Choe H-W, Ernst OP. (2008) Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 454:183–187. [DOI] [PubMed] [Google Scholar]
- Park S, Khalili-Araghi F, Tajkhorshid E, Schulten K. (2003) Free energy calculation from steered molecular dynamics simulations using Jarzynski’s equality. J Chem Phys 119:3559–3566. [Google Scholar]
- Pastor RW, Mackerell AD., Jr (2011) Development of the CHARMM force field for lipids. J Phys Chem Lett 2:1526–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Y, Mercier RW, Anday JK, Thakur GA, Zvonok AM, Hurst D, Reggio PH, Janero DR, Makriyannis A. (2008) Ligand-binding architecture of human CB2 cannabinoid receptor: evidence for receptor subtype-specific binding motif and modeling GPCR activation. Chem Biol 15:1207–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periole X, Huber T, Marrink SJ, Sakmar TP. (2007) G protein-coupled receptors self-assemble in dynamics simulations of model bilayers. J Am Chem Soc 129:10126–10132. [DOI] [PubMed] [Google Scholar]
- Phillips R, Ursell T, Wiggins P, Sens P. (2009) Emerging roles for lipids in shaping membrane-protein function. Nature 459:379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poveda JA, Marcela Giudici A, Lourdes Renart M, Morales A, González-Ros JM. (2017) Towards understanding the molecular basis of ion channel modulation by lipids: mechanistic models and current paradigms. Biochim Biophys Acta Biomembr 1859 (9 Pt B):1507–1516. [DOI] [PubMed] [Google Scholar]
- Provasi D, Bortolato A, Filizola M. (2009) Exploring molecular mechanisms of ligand recognition by opioid receptors with metadynamics. Biochemistry 48(42):10020–10029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Noorbakhsh F, Defea K, Hollenberg MD. (2012) Targeting proteinase-activated receptors: therapeutic potential and challenges. Nat Rev Drug Discov 11:69–86. [DOI] [PubMed] [Google Scholar]
- Rankovic Z, Brust TF, Bohn LM. (2016) Biased agonism: an emerging paradigm in GPCR drug discovery. Bioorg Med Chem Lett 26:241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DG, Sarmiento JG, Herbette LG. (1985) Kinetics of binding of membrane-active drugs to receptor sites. Diffusion-limited rates for a membrane bilayer approach of 1,4-dihydropyridine calcium channel antagonists to their active site. Mol Pharmacol 27:612–623. [PubMed] [Google Scholar]
- Saladino G, Gauthier L, Bianciotto M, Gervasio FL. (2012) Assessing the performance of metadynamics and path variables in predicting the binding free energies of p38 inhibitors. J Chem Theory Comput 8:1165–1170. [DOI] [PubMed] [Google Scholar]
- Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Karlsson A, Al-Lazikani B, Hersey A, Oprea TI, et al. (2017) A comprehensive map of molecular drug targets. Nat Rev Drug Discov 16:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent DF, Schwyzer R. (1986) Membrane lipid phase as catalyst for peptide-receptor interactions. Proc Natl Acad Sci USA 83:5774–5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe H-W, Hofmann KP, Ernst OP. (2008) Crystal structure of opsin in its G-protein-interacting conformation. Nature 455:497–502. [DOI] [PubMed] [Google Scholar]
- Schmidt D, del Mármol J, MacKinnon R. (2012) Mechanistic basis for low threshold mechanosensitivity in voltage-dependent K+ channels. Proc Natl Acad Sci USA 109:10352–10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider S, Provasi D, Filizola M. (2015) The dynamic process of drug–GPCR binding at either orthosteric or allosteric sites evaluated by metadynamics. Methods Mol Biol 1335:277–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoop A, Dey F. (2015) On-rate based optimization of structure-kinetic relationship--surfing the kinetic map. Drug Discov Today Technol 17:9–15. [DOI] [PubMed] [Google Scholar]
- Schwyzer R. (1991) New principle in QSAR: membrane requirements. J Recept Res 11:45–57. [DOI] [PubMed] [Google Scholar]
- Schwyzer R. (1995a) 100 years lock-and-key concept: are peptide keys shaped and guided to their receptors by the target cell membrane? Biopolymers 37:5–16. [DOI] [PubMed] [Google Scholar]
- Schwyzer R. (1995b) In search of the ‘bio-active conformation’--is it induced by the target cell membrane? J Mol Recognit 8:3–8. [DOI] [PubMed] [Google Scholar]
- Sengupta D, Chattopadhyay A. (2015) Molecular dynamics simulations of GPCR-cholesterol interaction: an emerging paradigm. Biochim Biophys Acta 1848(9):1775–1782. [DOI] [PubMed] [Google Scholar]
- Shang Y, Yeatman HR, Provasi D, Alt A, Christopoulos A, Canals M, Filizola M. (2016) Proposed mode of binding and action of positive allosteric modulators at opioid receptors. ACS Chem Biol 11:1220–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoichet BK, Kobilka BK. (2012) Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol Sci 33:268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu FY, He M, de Graaf C, Han GW, Yang D, Zhang Z, Zhou C, Xu Q, Wacker D, Joseph JS, Liu W, Lau J, Cherezov V, Katritch V, Wang MW, Stevens RC. (2013) Structure of the human glucagon class B G-protein-coupled receptor. Nature 499:444–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song G, Yang D, Wang Y, de Graaf C, Zhou Q, Jiang S, Liu K, Cai X, Dai A, Lin G, et al. (2017) Human GLP-1 receptor transmembrane domain structure in complex with allosteric modulators. Nature 546:312–315. [DOI] [PubMed] [Google Scholar]
- Srivastava A, Yano J, Hirozane Y, Kefala G, Gruswitz F, Snell G, Lane W, Ivetac A, Aertgeerts K, Nguyen J, et al. (2014) High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature 513:124–127. [DOI] [PubMed] [Google Scholar]
- Stanley N, Pardo L, Fabritiis GD. (2016) The pathway of ligand entry from the membrane bilayer to a lipid G protein-coupled receptor. Sci Rep 6:22639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A, Wittmann H-J, Seifert R. (2017) Binding kinetics and pathways of ligands to GPCRs. Trends Pharmacol Sci 38:717–732. [DOI] [PubMed] [Google Scholar]
- Swinney DC. (2006) Can binding kinetics translate to a clinically differentiated drug? From theory to practice. Lett Drug Des Discov 3:569–574. [Google Scholar]
- Swinney DC, Haubrich BA, Van Liefde I, Vauquelin G. (2015) The role of binding kinetics in GPCR drug discovery. Curr Top Med Chem 15:2504–2522. [DOI] [PubMed] [Google Scholar]
- Sykes DA, Charlton SJ. (2012) Slow receptor dissociation is not a key factor in the duration of action of inhaled long-acting β2-adrenoceptor agonists. Br J Pharmacol 165:2672–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes DA, Parry C, Reilly J, Wright P, Fairhurst RA, Charlton SJ. (2014) Observed drug-receptor association rates are governed by membrane affinity: the importance of establishing “micro-pharmacokinetic/pharmacodynamic relationships” at the β2-adrenoceptor. Mol Pharmacol 85:608–617. [DOI] [PubMed] [Google Scholar]
- Sykes DA, Stoddart LA, Kilpatrick LE, Hill SJ. (2019) Binding kinetics of ligands acting at GPCRs. Mol Cell Endocrinol 485:9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczuka A, Wennerberg M, Packeu A, Vauquelin G. (2009) Molecular mechanisms for the persistent bronchodilatory effect of the β 2-adrenoceptor agonist salmeterol. Br J Pharmacol 158:183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taillardat-Ertschinger A, Galland A, Carrupt PA, Testa B. (2002) Immobilized artificial membrane liquid chromatography: proposed guidelines for technical optimization of retention measurements. J Chromatogr A 953:39–53. [DOI] [PubMed] [Google Scholar]
- Tarcsay Á, Keserű GM. (2013) Contributions of molecular properties to drug promiscuity. J Med Chem 56:1789–1795. [DOI] [PubMed] [Google Scholar]
- Tejwani RW, Davis ME, Anderson BD, Stouch TR. (2011a) An atomic and molecular view of the depth dependence of the free energies of solute transfer from water into lipid bilayers. Mol Pharm 8:2204–2215. [DOI] [PubMed] [Google Scholar]
- Tejwani RW, Davis ME, Anderson BD, Stouch TR. (2011b) Functional group dependence of solute partitioning to various locations within a DOPC bilayer: a comparison of molecular dynamics simulations with experiment. J Pharm Sci 100:2136–2146. [DOI] [PubMed] [Google Scholar]
- Troussicot L, Guillière F, Limongelli V, Walker O, Lancelin J-M. (2015) Funnel-metadynamics and solution NMR to estimate protein-ligand affinities. J Am Chem Soc 137:1273–1281. [DOI] [PubMed] [Google Scholar]
- Tsopelas F, Giaginis C, Tsantili-Kakoulidou A. (2017) Lipophilicity and biomimetic properties to support drug discovery. Expert Opin Drug Discov 12:885–896. [DOI] [PubMed] [Google Scholar]
- Vauquelin G. (2010) Rebinding: or why drugs may act longer in vivo than expected from their in vitro target residence time. Expert Opin Drug Discov 5:927–941. [DOI] [PubMed] [Google Scholar]
- Vauquelin G. (2015) On the ‘micro’-pharmacodynamic and pharmacokinetic mechanisms that contribute to long-lasting drug action. Expert Opin Drug Discov 10:1085–1098. [DOI] [PubMed] [Google Scholar]
- Vauquelin G. (2016) Cell membranes… and how long drugs may exert beneficial pharmacological activity in vivo. Br J Clin Pharmacol 82:673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vauquelin G, Charlton SJ. (2010) Long-lasting target binding and rebinding as mechanisms to prolong in vivo drug action. Br J Pharmacol 161:488–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vauquelin G, Packeu A. (2009) Ligands, their receptors and ... plasma membranes. Mol Cell Endocrinol 311:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. (2013) Molecular signatures of G-protein-coupled receptors. Nature 494:185–194. [DOI] [PubMed] [Google Scholar]
- Yang Q, Alemany R, Casas J, Kitajka K, Lanier SM, Escribá PV. (2005) Influence of the membrane lipid structure on signal processing via G protein-coupled receptors. Mol Pharmacol 68:210–217. [DOI] [PubMed] [Google Scholar]
- Yuan X, Raniolo S, Limongelli V, Xu Y. (2018) The molecular mechanism underlying ligand binding to the membrane-embedded site of a g-protein-coupled receptor. J Chem Theory Comput 14:2761–2770. [DOI] [PubMed] [Google Scholar]
- Zhang C, Srinivasan Y, Arlow DH, Fung JJ, Palmer D, Zheng Y, Green HF, Pandey A, Dror RO, Shaw DE, et al. (2012) High-resolution crystal structure of human protease-activated receptor 1. Nature 492:387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Gao Z-G, Zhang K, Kiselev E, Crane S, Wang J, Paoletta S, Yi C, Ma L, Zhang W, et al. (2015) Two disparate ligand-binding sites in the human P2Y1 receptor. Nature 520:317–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorrilla EP, Koob GF. (2010) Progress in corticotropin-releasing factor-1 antagonist development. Drug Discov Today 15:371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]