

Abstract
The syntheses of new conformationally locked North and South bicyclo[3.1.0]hexene nucleosides is reported. The North analogues were synthesized by a convergent approach from the known (1S,2R,5R)-5-[(tert-butyldiphenylsilyloxy)methyl]bicyclo[3.1.0]hex-3-en-2-ol (7) via Mitsunobu coupling with the nucleobases. The South analogues were synthesized from their bicyclo[3.1.0]hexane nucleoside precursors by the selective protection of the primary hydroxyl group, conversion of the secondary alcohol into a good leaving group, and base-catalyzed elimination to generate the olefin. The transformation of a bicyclo[3.1.0]hexane nucleoside into a bicyclo[3.1.0]hexene nucleoside flattens the five-membered ring of the bicyclic system and rescues anti-HIV activity for North-D4T (4a), North-D4A (4c), and South-D4C (6d). The relationship between planarity and the anti/syn disposition of the nucleobase that is favored by a particular pseudosugar platform are proposed as key parameters in controlling biological activity.
Keywords: nucleosides, carbocycles, conformationally locked, antiviral agents, pseudorotational cycle
Introduction
Recognition by cellular kinases and the viral enzyme, reverse transcriptase (RT), are essential prerequisites for a nucleoside drug to display good anti-HIV activity. Provided that the drug is efficiently metabolized to the 5′-triphosphate, the ensuing incorporation of the 5′-monophosphate into the DNA primer and interference with the proper functioning of HIV-1 RT– either by direct or delayed chain termination – stops viral replication. For HIV-1 RT, we have provided evidence that the conformation of the sugar moiety preferred by the polymerase is proximate to the North (3T2, P = 0°) conformation.[1, 2] However, in the case of AZT (1), the strong gauche effect between the 3′-azido group and the ribose O4’oxygen forces the nucleoside and the corresponding 5′-triphosphate (AZTTP) to instead adopt the antipodal South (3T2, P = 180°) conformation (Figure 1A).[3] This conformational preference is opposite of that favored by the polymerase, which explains why AZTTP is incorporated less efficiently than the natural substrate TTP during DNA- or RNA-directed DNA synthesis.[4, 5] The ability of HIV-1 RT to distinguish between the antipodal North and South conformations of AZTTP was studied in our laboratory with two bicyclo[3.1.0]hexane analogues of AZT built on a platform that, contrary to the normal riboside ring of nucleosides, is not in a dynamic N ⇆ S equilibrium (Figure 1B). Those results showed conclusively that HIV-1 RT prefers the North conformation.[1]
Figure 1.
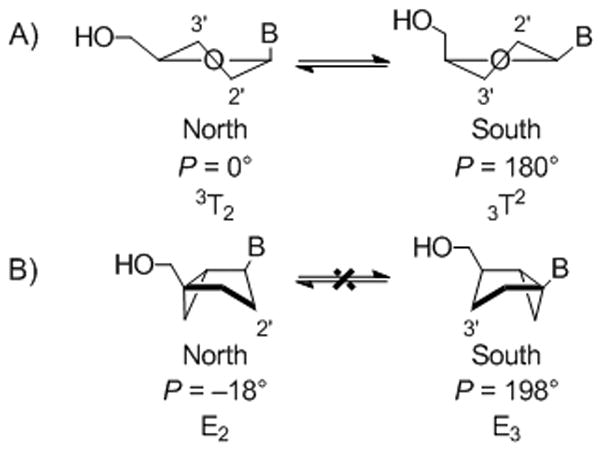
(A) Rapidly interconverting North and South conformations in conventional nucleosides defined by changes in the pseudorotational angle (P). (B) Bicyclo[3.1.0]hexane nucleosides locked in North and South conformations located 18° away from the ideal P values of 0° and 180°.
Intriguingly, the 5′-triphosphate of a nucleoside with a pseudoribose ring that has virtually no puckering, stavudine (2, D4T), was shown to be incorporated as efficiently as the natural substrate TTP during both DNA- and RNA-dependent DNA polymerization.[6] However, counter to the ability of D4TTP to interact efficiently with HIV-1 RT is the finding that phosphorylation of D4T by the critical cellular thymidine kinase (TK) is 500-fold less efficient than for AZT.[7, 8] Indeed, the South conformation of AZT, which is detrimental for its interaction with HIV-1 RT, facilitates its interaction with the cellular thymidine kinase (TK) by displaying an affinity for the enzyme equivalent to that of thymidine itself.[7, 8] This means that South nucleoside conformers are preferred by cellular kinases and that either North or planar sugar conformations with no puckering are better substrates for the polymerases.
During the course of our investigations with bicyclo[3.1.0]hexane models of 2′-deoxynucleosides, we found that while chemically synthesized 5′-triphosphates that were restricted in the North conformation (3a–3d) were readily incorporated by HIV-1 RT and other polymerases, none of the nucleosides were effective anti-HIV agents due to inefficient cellular phosphorylation.[1, 2] On the other hand, the conformational constraint in South bicyclo[3.1.0]hexane nucleosides, which influences the nucleobase to adopt the biologically unsuitable syn conformation that is favored by intramolecular hydrogen bonding in a hydrophobic environment — such as the enzyme’s binding site— resulted in compounds (5a–d) that were not phosphorylated by cellular kinases despite their excellent substrate recognition by the more tolerant, viral HSV-1 kinase.[9]
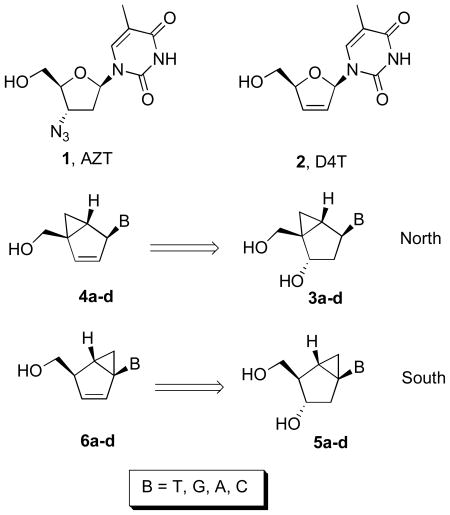
The incorporation of the main structural feature of D4T, which is its planarity, into the structure of the very potent anti-HSV compound, North-methanocarbathymidine (N-MCT, 3a, B = T)[10] resulted in the very effective anti-HIV thymidine analogue (4a, B = T) with a novel bicyclo[3.1.0]hexene pseudosugar that included the critical double bond.[11] The 4 to 10-fold decrease in potency of 4a relative to D4T was tentatively correlated with the nearly 10-fold reduction in the level of planarity (νmax = 6.81°) of the embedded five-membered ring segment of the bicyclo[3.1.0]hexene template relative to the nearly flat pseudosugar ring of D4T (νmax = 0.61°). Interestingly, in this particular case, it was planarity that converted the inactive 3a into an active anti-HIV agent by allowing a significant level of recognition by the cellular TK resulting in the production of higher levels of the requisite 5′-triphosphate metabolite.
Because each of the cellular kinases that perform the critical first phosphorylation step have different preferences for the various nucleobases, we decided to complete our study by incorporating the rest of the nucleobases (A, C, and G) to the same bicyclo[3.1.0]hexene template (Table 1, 4a–d) derived from the conformationally locked North-bicyclo[3.1.0]hexane nucleosides (3a–d). It is important to remember that in terms of sugar conformation, once the amplitude of the puckering measured by νmax reaches a small value, such as ~6° in 4a–d, the designation of North and South loses its significance. Based on that argument, we decided to investigate the synthesis and anti-HIV activities of the complementary set of bicyclo[3.1.0]hexene nucleosides (Table 1, 6a–d) derived from the antipodal, conformationally locked, South-bicyclo[3.1.0]hexane nucleosides (5a–d) bearing the four natural nucleobases.
Table 1.
Structures of North-derived (4a–d) and South-derived (6a–d) bicyclo[3.1.0]hexene nucleosides.
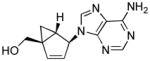
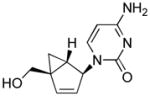
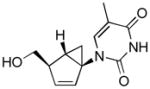
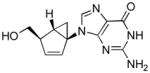
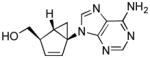
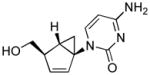
Results and Discussion
Synthesis of “North-derived” bicyclo[3.1.0]hexene nucleosides
The syntheses of the thymidine (4a) and guanosine (4b) nucleosides were derived from the syntheses of their respective conformationally locked North conformers, which have been published.[11, 12] In a similar manner, the adenine (4c) and cytosine (4d) analogues were assembled from the bicyclo-cyclopentenol 7, which was prepared as described previously.[11] Reaction of 7 with 6-chloropurine under Mitsunobu conditions gave 8 with the expected inversion of configuration. Subsequent displacement of the chloride with NH4OH/dioxane in a pressure bomb gave the corresponding amine 9, which after fluoride-catalyzed deprotection of the silyl group gave the target 4c (Scheme 1). The characteristic UV λmax at 262 nm confirmed that the adenine ring in 4c was connected to the pseudosugar through the N9 nitrogen. In a similar manner, compound 10 was assembled from the same bicyclo-cyclopentenol 7 after coupling with N-benzoyluracil[13] under Mistunobu conditions. Subsequent deprotection of the benzoyl group was followed by formation of the 4-(1,2,4-triazol-1-yl) intermediate 12 according to the method of Reese.[14] Ammonolysis of this intermediate provided the penultimate cytosine analogue which required only deprotection of the silyl ether to give the target compound 4d (Scheme 2).
Scheme 1.
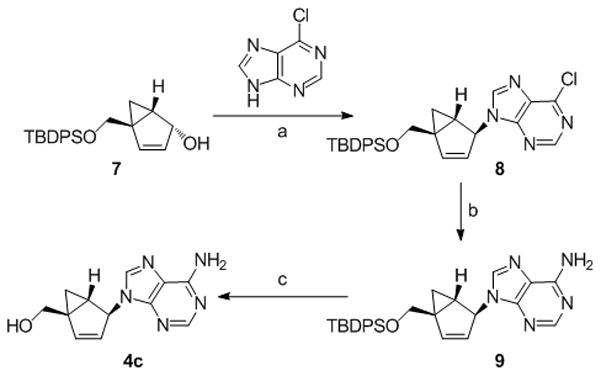
Reagents and conditions: (a) diisopropyl azodicarboxylate (DIAD), PPh3, THF, 0 °C → rt; (b) NH4OH, dioxane, 70 °C; (c) Et3N•3HF, MeCN, Δ.
Scheme 2.
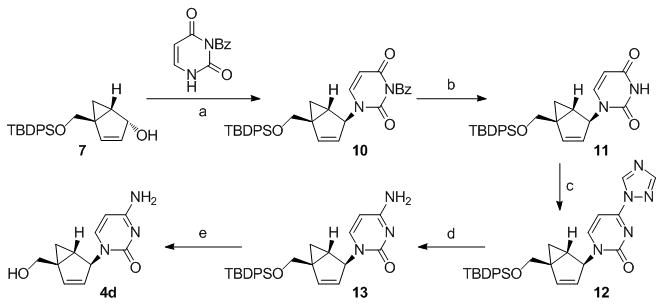
Reagents and conditions: (a) diisopropyl azodicarboxylate (DIAD), PPh3, THF, 0 °C - rt; (b) NH4OH, MeOH, rt; (c) POCl3, 1,2,4-triazole, Et3N, MeCN; (d) NH4OH, dioxane, rt; (e) Et3N•HF, MeCN.
Synthesis of “South-derived” bicyclo[3.1.0]hexene nucleosides
The construction of carbocyclic nucleosides (6a–d) derived from their respective conformationally locked South conformers is reported here for the first time. The syntheses of the precursors to generate these South conformers were performed as described previously.[15] The same methodology was used for the entire series and involved the selective protection of the primary hydroxyl group and conversion of the secondary alcohol into a good leaving group, such as the methanesulfonate ester, by standard chemistry. This was followed by base-catalyzed elimination, which in most cases occurred with the simultaneous deprotection of the silyl group, to give the desired target compounds 6b–d. In the case of the thymine analogue (6a), the base-catalyzed elimination did indeed result in the desired olefin but an additional step using n-tetrabutylammonium fluoride (TBAF) was required to remove the silyl protecting group.
X-ray Structures and Conformational Analysis
We have shown earlier that in the case of the North-derived D4T analogue (4a), the nearly planar structure of its fused cyclopentene ring nullifies the relevance of its location in the pseudorotational cycle.[11] The puckering amplitude (νmax) of the cyclopentene ring in 4a was only 6.81° with a mean deviation from planarity of 0.025 Å. As expected, the compound was superimposed nicely with D4T with an RMS deviation of only 0.039 Å and its activity paralleled that of D4T.[11] Unfortunately, we were unable to grow good quality crystals of the corresponding South-derived D4T analogue (6a), but the guanine analogue (6b) provided good crystals for X-ray analysis. The crystal structure (Figure 2) validated all of the spectral assignments for this compound and the oher targets derived from the South template.
Figure 2.

Displacement ellipsoid plot of 6b drawn at the 50% probability level.
While both compounds 4a and 6b are similarly planar (νmax ~ 7°, Table 2), as we expected, some of the pseudorotational parameters are quite different (Table 2). According to the value of P, 4a is in the North hemisphere (1To) and 6b is in the South (4E) hemisphere, although both conformations are in reality closer to the West end of the pseudorotational cycle. As mentioned before, their very planar structures with a νmax ~ 7°, which corresponds to the radius of the pseudorotational cycle, makes the differences in P irrelevant. However, a significant divergence is found in the value of χ. The purine ring of 6b occupies the syn region as opposed to the pyrimidine ring in 4a, which prefers the anti region. This penchant for the syn conformation is an interesting remnant from the saturated bicyclo[3.1.0]hexane scaffold in the South hemisphere which tends to favor the orientation of the base in the syn conformation.[9]
Table 2.
Pseudorotational parameters (in degrees) for North-derived 4a and South-derived 6ba.
| ν0 | ν1 | ν2 | ν3 | ν4 | P | νmax | χ | γ | type | |
|---|---|---|---|---|---|---|---|---|---|---|
| 4a | 6.56 | −5.38 | 2.67 | 1.68 | −5.20 | 293.14 | 6.81 | −119.12 | 56.17 | 1TO (N) |
| 6b | 4.05 | −0.14 | −4.05 | 6.38 | −6.05 | 233.11 | 6.75 | 48.43 | 52.58 | 4E (S) |
ν0–ν4 represent the torsion angles of the embedded cyclopentene ring.
Anti-HIV Activity
The anti-HIV activity was determined using an HIV-1 based vector containing wild-type (wt) HIV-1 RT that lacks a functional Env coding region and contains a luciferase reporter gene in the nef coding region.[16–18] This vector is pseudotyped with the vesicular stomatitis virus G protein (VSV-G) and is limited to a single replication cycle. The vector was used to infect conventional human osteosarcoma (HOS) cells or a modified HOS cell line containing and expressing the herpes simplex virus 1 thymidine kinase (HSV-1 TK).[19] Because HSV-1 TK is able to phosphorylate a wider range of nucleosides, cells that express HSV-1 TK can be used to analyze the anti-HIV activity of nucleosides that are not readily phosphorylated.[2] The HOS cell line expressing the HSV-1 TK has been designated HOS-313. Either HOS or HOS-313 cells were plated in 96 well luminescence cell culture plates and treated with each compound at different concentrations. After 48 h, anti-HIV activity was determined by measuring the reduction in the luciferase activity in infected cells. Only cells, which contain integrated viral DNA, will produce luciferase in the infectivity assay. Cytotoxicity, in the absence of virus, was determined by measuring the amount of ATP in the cells using a luciferase-based assay.[20] The amount of light produced by the reaction of the ATP from the cells with added luciferase and D-luciferin is proportional to the amount of ATP. ATP is required for vital cellular processes and the ATP concentration declines rapidly when cells undergo necrosis or apoptosis. Thus, cellular ATP levels serve as a sensitive indicator of cytotoxicity. Viral infectivity and cellular cytotoxicity is expressed on the Y-axis of the graphs as “Relative Infectivity (or Cell Health)”, respectively. In both assays, the results were normalized to a value of 1.0 representing the luciferase signal in the absence of compound. Among the bicyclo[3.1.0]hexene nucleosides (4a–d) derived from North templates, we have already reported that North-D4T (4a) was active, whereas North-D4G (4b) was not.[11, 12]
In three independent experiments using a regression analysis with a 4 parameter decay model, the North-D4A analogue (4c) was active in HOS cells (Figure 3A, EC50(HOS) = 16.7 μM). Regression analysis on cytotoxicity data was performed using a simple exponential decay model and indicated a 25-fold difference in cytotoxicity versus infectivity (Figures 3A, CC50(HOS) = 423 μM). The compound was slightly more potent in HOS-313 cells (Figure 3B, EC50(HOS-313) = 11.1 μM and CC50(HOS-313) = 246 μM) with a similar selectivity index of 24.
Figure 3.


Cell-based assay of North-D4A (4c) on the replication of an HIV-1 vector containing wt HIV-1 RT in HOS cells [(A) ● = infectivity; ○ = cytotoxicity) and HOS-313 cells [(B), ▲ = infectivity; △ = cytotoxicity). Luciferase activity in the absence of compound was considered to be 100 and the luciferase activity in the presence of different concentrations of 4c was normalized to this value to give relative infectivity or cytotoxicity in the absence of the vector.
The North-D4C (4d) analogue was inactive and relatively non-toxic (data not shown) suggesting the possibility that neither HOS cells nor the HSV-1 TK transfected HOS-313 cells were capable of efficiently phosphorylating the compound.
With the exception of South-D4C (6d), all South analogues were inactive and non-toxic (data not shown). As seen in Figure 4, South-D4C had an EC50 = 26.8 μM in HOS cells, which was reduced to 3.0 μM in HOS-313 infected cells. Selectivity indices for the compound in HOS and HOS-313 cells were 15.8 and 130, respectively.
Figure 4.

Cell-based assay of South-D4C (6d) on the replication of an HIV-1 vector containing wt HIV-1 RT in HOS-313 cells and HOS cells. Luciferase activity in the absence of compound was considered to be 100 and the luciferase activity in the presence of different concentrations of 6d was normalized to this value to give relative infectivity or cytotoxicity in the absence of the vector (● = HOS infectivity; ▲ = HOS-313 infectivity; ○ = HOS cytotoxicity; △ = HOS-313 cytotoxicity).
Conclusions
The transformation of a bicyclo[3.1.0]hexane nucleoside into a bicyclo[3.1.0]hexene nucleoside by the addition of a double bond flattens the five-membered ring of the bicyclic system and rescues anti-HIV activity for North-D4T (4a), North-D4A (4c), and South-D4C (6d). North-D4A (4c) and South-D4C (6d), both of which were active in non-transfected HOS cells, demonstrate the existence of a significant level of substrate recognition by both cellular kinases and HIV-1 RT. Based on the known anti-HIV activity of North-D4T (4a)[11] we expected that other related North analogues would also be active. However, only the North-D4A was active; the rest of this series of North analogues (G[12] and C) failed to show activity. Only one of the compounds in the South series, South-D4C (6d), was active. This was surprising in view of the lack of activity of North-D4C (4d), particularly when one assumes that the cyclopentene ring would be equally flat in both compounds. Other structural elements, such as the location of the fused cyclopropane ring and the rotational freedom around the C–N bond may be contributing factors.
Experimental Section
General Techniques
All reagents and solvents purchased were of the highest commercial quality and used without further purification unless otherwise stated. Column chromatography was performed on silica gel 60, 230–400 mesh (E. Merck). 1H and 13C NMR spectra were recorded on a Varian Unity Inova instrument at 400 and 100 MHz, respectively. Spectra are referenced to the solvent in which they were run (7.24 ppm for CDCl3). Infrared spectra were recorded on a Jasco model 615 FT-IR instrument. Positive-ion fast-atom bombardment mass spectra (FAB MS) were obtained on a VG 70-SE double-focusing mass spectrometer operated at an accelerating voltage of 8 kV under the control of a MASPEC-II32 data system for Windows (MasCom GmbH, Bremen, Germany). Either glycerol or 3-nitrobenzyl alcohol was used as the sample matrix, and ionization was effected by a beam of xenon atoms generated in a saddle-field ion gun at 8.0 ± 0.5 kV. The FAB mass spectra of all analyzed compounds indicated a molecular species ([M+H]+ or [M+Na]+) as well as fragment ions highly indicative of structure. Nominal mass MS were obtained at a resolution of 1500, and matrix-derived ions were background subtracted during data system processing. Accurate mass analyses (HR-FAB-MS) were carried out under data system control at a resolution of approximately 7000 employing a limited-range V/E scan. For these analyses, matrix-derived ions were utilized as the internal mass references for accurate mass determination. Both 1H and 13C NMR data, in conjunction with the observed isotopic distribution of [M+H]+, were used to set constraints for the calculation of all possible elemental compositions within 20 ppm of the measured accurate mass. In all cases, a unique molecular formula could be determined by consideration of the molecular ion species and appropriate fragment ions. Optical rotations were recorded on a Jasco P-1010 polarimeter. Elemental analyses were performed by Atlantic Microlab, Inc., Atlanta, GA.
9-((1′S, 2′S, 5′R)-5-((tert-Butyldiphenylsilyloxy)methyl)bicyclo[3.1.0]hex-3-en-2-yl)-6-chloro-9H-purine (8)
Under argon, DIAD (0.53 mL, 2.67 mmol) was added to a 0 °C solution of triphenylphosphine (718 mg, 1.16 mmol) in THF (25 mL). The resultant yellow solution continued to stir at 0 °C for 20 minutes and was then added to a 0 °C mixture of 7 (423 mg, 1.16 mmol) and 6-chloropurine (287 mg, 1.86 mmol) in THF (25 mL). The resulting solution was stirred at 0 °C for 1 h and then warmed to room temperature overnight. Concentration in vacuo and purification of the residue by silica gel flash chromatography (CH2Cl2 and 10% Et2O/CH2Cl2, followed by 10%MeOH/CH2Cl2) gave an impure product. Further purification by silica gel flash chromatography (5% and 20% EtOAc/hexanes) gave 8 (88 mg, 17% yield) as a sticky foam; 1H NMR (400 MHz, CDCl3): δ = 8.74 (s, 1 H, H-2), 8.23 (s, 1 H, H-8), 7.59 (m, 4 H, Ph), 7.36 (m, 6 H, Ph), 6.46 (d, J = 4.4 Hz, 1 H, H-4′), 5.54 (m, 2 H, H-2′ and H-3′), 4.16 (d, J = 11.2 Hz, 1 H, CH2a), 3.58 (d, J = 11.2 Hz, 1 H, CH2b), 1.69 (dd, J = 4.2, 8.4 Hz, 1 H, H-1′), 1.15 (dd, J = 4.5, 8.4 Hz, 1 H, H-6′a), 1.05 (s, 9 H, C(CH3)3), 0.52 (t, J = 4.4 Hz, 1 H, H-6′b); 13C NMR (100 MHz, CDCl3): δ = 151.73, 151.06, 150.82, 144.05, 143.39, 135.55, 135.44, 133.09, 133.08, 131.90, 129.83, 129.77, 127.73, 127.68, 124.89, 64.53, 60.09, 38.51, 28.35, 26.87, 24.25, 19.13; FAB MS: m/z (%) 501 (100) [M+H]+; Anal. calcd for C28H29ClN4O2: C 67.11, H 5.83, N 11.18, found: C 67.16, H 6.16, N 10.59.
9-((1′S, 2′S, 5′R)-5-((tert-Butyldiphenylsilyloxy)methyl)bicyclo[3.1.0]hex-3-en-2-yl)-9H-purin-6-amine (9)
A solution of 8 (54 mg, 0.11 mmol) in concentrated NH4OH (3 mL) and dioxane (3 mL) was heated to 70° C in a glass pressure bomb overnight. After cooling to room temperature, the reaction mixture was concentrated in vacuo. Purification of the residue by silica gel flash chromatography (2.5% and 5% MeOH/CHCl3) gave 9 (41 mg, 79% yield) as a colorless foam; 1H NMR (400 MHz, CDCl3): δ = 8.37 (s, 1 H, H-2), 7.92 (s, 1 H, H-8), 7.61 (m, 4 H, Ph), 7.35 (m, 6 H, Ph), 6.41 (dd, J = 1.4, 5.5 Hz, 1 H, H-4′), 6.05 (br s, 2 H, NH2), 5.54 (dt, J = 1.9, 5.4 Hz, 1 H, H-3′), 5.47 (br s, 1 H, H-2′), 4.13 (d, J = 11.1 Hz, 1 H, CH2a), 3.56 (d, J = 11.2 Hz, 1 H, CH2b), 1.69 (dd, J = 4.1, 8.1 Hz, 1 H, H-1′), 1.10 (irr dd, 1 H, H-6′a), 1.05 (s, 9 H, C(CH3)3), 0.48 (t, J = 4.4 Hz, 1 H, H-6′b); 13C NMR (100 MHz, CDCl3): δ = 155.57, 152.83, 149.32, 142.47, 139.29, 135.58, 135.47, 133.21, 129.79, 129.75, 127.72, 127.68, 125.58, 119.80, 64.80, 59.45, 38.34, 28.73, 26.86, 24.26, 19.16; FAB MS: m/z (%) 482 (57) [M+H]+, 136 (100); Anal. calcd for C28H31N5OSi•0.9H2O: C 67.55, H 6.64, N 14.07, found: C 67.36, H 6.25, N 13.83.
((1′R, 4′S, 5′S)-4-(6-Amino-9H-purin-9-yl)bicyclo[3.1.0]hex-2-en-1-yl)methanol (4c)
A mixture 9 (107 mg, 0.22 mmol) and triethylamine trihydrofluoride (0.22 mL, 1.33 mmol) in acetonitrile (10 mL) was refluxed overnight. After cooling to room temperature, the reaction mixture was diluted with water (10 mL) and stirred for 1 h at room temperature. Concentration in vacuo and purification of the residue by silica gel flash chromatography (2.5%, 5%, and 10% MeOH/CHCl3) followed by a second silica gel flash chromatography (2.5%, 5%, and 10% MeOH/CHCl3) purification gave 4c (41 mg, 76% yield) as a white solid. Treatment of this solid with methanol/diethyl ether gave 32 mg (59% yield) of analytically pure 4c; mp 201–203 °C; [α]D19 = +208.69 (c 0.17, CH3OH); UV (MeOH:H2O, 1:1) λmax = 262 nm (ε = 1.40 × 104 mol−1cm−1); 1H NMR (400 MHz, CD3OD): δ = 8.19 (s, 1 H, H-2), 8.11 (s, 1 H, H-8), 6.51 (dm, J = 5.4 Hz, 1 H, H-2′), 5.59 (dt, J = 1.9, 5.4 Hz, 1 H, H-3′), 5.43 (br t, J = 1.8 Hz, 1 H, H-4′), 4.14 (d, J = 11.8 Hz, 1 H, CH2a), 3.50 (d, J = 11.8 Hz, 1 H, CH2b), 1.93 (m, 1 H, H-5′), 1.27 (dd, J = 4.4, 8.4 Hz, 1 H, H-6′a), 0.58 (t, J = 4.4 Hz, 1 H, H-6′b); 13C NMR (100 MHz, CD3OD): δ = 157.52, 153.59, 149.98, 143.84, 141.47, 127.13, 120.54, 64.71, 62.32, 40.20, 29.45, 25.58; FAB MS: m/z (%) 244 (100) [M+H]+; Anal. calcd for C12H13N5O: C 59.25, H 5.39, N 28.79, found: C 59.05, H 5.28, N 28.79.
3-Benzoyl-1-((1′S, 2′S, 5′R)-5-((tert-butyldiphenylsilyloxy)methyl)bicyclo[3.1.0]hex-3-en-2-yl)pyrimidine-2,4(1H,3H)-dione (10)
Under argon, DIAD (0.54 mL, 2.70 mmol) was added to a 0 °C solution of triphenylphosphine (707 mg, 2.70 mmol) in THF (20 mL). The resultant yellow solution continued to stir at 0 °C for 30 min. A solution of 7 (393 mg, 1.08 mmol) in THF (30 mL) was added to the 0 °C solution immediately followed by N-3-benzoyl uracil (350 mg, 1.62 mmol) and the resultant solution was stirred at 0 °C for 1 h, and then warmed to room temperature overnight. Concentration in vacuo and purification of the residue by silica gel flash chromatography (CH2Cl2 and 5% Et2O/CH2Cl2) gave a crude product which was subjected to a second purification by silica gel flash chromatography (CH2Cl2 and 5% Et2O/CH2Cl2) to give 10 (187 mg, 31% yield) as a colorless foam; 1H NMR (400 MHz, CDCl3): δ = 7.95 (m, 2 H, H-6), 7.63 (m, 7 H, Ph), 7.44 (m, 8 H, Ph), 6.38 (d, J = 5.4 Hz, 1 H, H-4′), 5.54 (d, J = 8.1 Hz, 1 H, H-5), 5.49 (br s, 1 H, H-2′), 5.41 (m, 1 H, H-3′), 4.33 (d, J = 11.3 Hz, 1 H, CH2a), 3.39 (d, J = 11.3 Hz, 1 H, CH2b), 1.64 (dd, J = 4.0, 8.2 Hz, 1 H, H-1′), 1.07 (s, 9 H, C(CH3)3), 1.05 (m, 1H, H-6′a), 0.42 (t, J = 4.5 Hz, 1 H, H-6′b); 13C NMR (100 MHz, CDCl3): δ = 169.07, 162.24, 149.73, 143.32, 141.63, 135.59, 135.43, 134.98, 133.09, 132.86, 131.60, 130.48, 130.05, 129.98, 129.10, 127.87, 127.86, 125.96, 102.51, 65.43, 60.47, 38.46, 27.87, 26.91, 23.90, 19.31; FAB MS: m/z (%) 563 (43) [M+H]+, 105 (100); Anal. calcd for C34H34N2O4Si•0.3H2O: C 71.88, H 6.14, N 4.93, found: C 71.55, H 6.12, N 4.78.
1-((1′S, 2′S, 5′R)-5-((tert-Butyldiphenylsilyloxy)methyl)bicyclo[3.1.0]hex-3-en-2-yl)pyrimidine-2,4(1H,3H)-dione (11)
Concentrated NH4OH (4 mL) was added to a solution of 10 (182 mg, 0.32 mmol) in methanol (30 mL) and stirred for 2 h in a capped vessel at room temperature. Concentration in vacuo and purification of the residue by silica gel flash chromatography (CH2Cl2 and 5% MeOH/CH2Cl2, followed by a second silica gel flash chromatography 20% and 50% EtOAc/hexanes) gave 11 (134 mg, 90% yield) as a colorless foam; 1H NMR (400 MHz, CDCl3): δ = 8.75 (br s, 1 H, NH), 7.64 (q, J = 1.6, 1 H, Ph), 7.62 (m, 1 H, Ph), 7.60 (q, J = 1.5, 1 H, Ph), 7.58 (m, 1 H, Ph), 7.40 (m, 7 H, Ph and H-6), 6.34 (ddd, J = 0.7, 1.5, 5.5 Hz, 1 H, H-4′), 5.50 (br t, J = 1.7 Hz, 1 H, H-2′), 5.44 (dd, J = 2.3, 8.0 Hz, 1 H, H-5), 5.36 (dt, J = 1.9, 5.4 Hz, 1 H, H-3′), 4.30 (d, J = 11.3 Hz, 1 H, CH2a), 3.36 (d, J = 11.3 Hz, 1 H, CH2b), 1.57 (dd, J = 4.3, 8.2 Hz, 1 H, H-1′), 1.05 (s, 9 H, C(CH3)3), 1.01 (dd, J = 4.6, 8.5 Hz, 1 H, H-6′a), 0.41 (t, J = 4.5 Hz, 1 H, H-6′b); 13C NMR (100 MHz, CDCl3): δ = 163.15, 150.69, 142.99, 141.89, 135.59, 135.42, 133.12, 132.91, 130.00, 129.93, 127.84, 127.81, 126.11, 102.55, 65.41, 60.19, 38.40, 27.90, 26.88, 23.86, 19.29; FAB MS: m/z (%) 459 (100) [M+H]+; Anal. calcd for C27H30N2O3Si•1H2O: C 68.04, H 6.77, N 5.88, found: C 67.91, H 6.39, N 5.71.
1-((1′S, 2′S, 5′R)-5-((tert-Butyldiphenylsilyloxy)methyl)bicyclo[3.1.0]hex-3-en-2-yl)-4-(1H-1,2,4-triazol-1-yl)pyrimidin-2(1H)-one (12)
Phosphorous oxychloride (0.064 mL, 0.70 mmol) was added dropwise to a solution of 11 (132 mg, 0.29 mmol), 1,2,4-triazole (457 mg, 6.62 mmol) and triethylamine (0.93 mL, 6.62 mmol) in acetonitrile (10 mL) and stirred for 2 h at room temperature. The reaction mixture was then poured into a solution of chloroform (30 mL) containing triethylamine (2 mL), extracted with saturated aqueous NaHCO3 (3 ×10 mL), dried over MgSO4 and concentrated in vacuo. Purification of the residue by silica gel flash chromatography (20%, 50% and 75% EtOAc/hexanes) gave 12 (140 mg, 96% yield) as a colorless glass; 1H NMR (400 MHz, CDCl3): δ = 9.27 (s, 1 H, triazole-H), 8.10 (overlapping s and d, J = 7.1 Hz, 2 H, H-6, triazole-H), 7.60 (m, 3 H, Ph), 7.44 (m, 2 H, Ph), 7.37 (m, 5 H, Ph), 6.71 (d, J = 7.1, 1 H, H-5), 6.41 (ddd, J = 0.8, 1.4, 5.4 Hz, 1 H, H-4′), 5.78 (br s, 1 H, H-2′), 5.44 (dt, J = 2.0, 5.4 Hz, 1 H, H-3′), 4.34 (d, J = 11.3 Hz, 1 H, CH2a), 3.38 (d, J = 11.3 Hz, 1 H, CH2b), 1.65 (dd, J = 4.1, 8.4 Hz, 1 H, H-1′), 1.07 (m containing s at 1.07, 10 H, H6′a and C(CH3)3), 0.50 (t, J = 4.5 Hz, 1 H, H-6′b); 13C NMR (100 MHz, CDCl3): δ = 158.72, 155.14, 153.88, 148.88, 143.77, 135.56, 135.42, 133.12, 132.89, 130.07, 129.99, 127.88, 127.85, 126.42, 95.12, 65.37, 62.45, 38.65, 28.69, 26.94, 24.27, 19.33; FAB MS: m/z (%) 510 (100) [M+H]+; HRMS-FAB: m/z [M+H]+ calcd for C29H31N5O2Si: 510.2325, found: 510.2306.
4-Amino-1-((1′S, 2′S, 5′R)-5-((tert-butyldiphenylsilyloxy)methyl)bicyclo[3.1.0]hex-3-en-2-yl)pyrimidin-2(1H)-one (13)
A solution of 12 (136 mg, 0.27 mmol) and concentrated NH4OH (5 mL) in dioxane (20 mL) was stirred in a capped vessel overnight at room temperature. Concentration in vacuo and purification of the residue by silica gel flash chromatography (5% and 10% MeOH/CHCl3) gave 13 (134 mg, 90% yield) as a colorless foam; 1H NMR (400 MHz, CDCl3): δ = 7.63 (m, 1 H, Ph), 7.61 (m, 1 H, Ph), 7.59 (m, 1 H, Ph), 7.57 (m, 1 H, Ph), 7.52 (d, J = 7.3 Hz, 1 H, H-6), 7.39 (m, 6 H, Ph), 6.29 (ddd, J = 0.8, 1.6, 5.5 Hz, 1 H, H-4′), 5.67 (br s, 1 H, H-2′), 5.37 (m, 2 H, H-5 and H-3′), 4.26 (d, J = 11.2 Hz, 1 H, CH2a), 3.34 (d, J = 11.2 Hz, 1 H, CH2b), 1.56 (dd, J = 4.3, 8.4 Hz, 1 H, H-1′), 1.04 (s, 9 H, C(CH3)3), 0.96 (dd, J = 4.5, 8.5 Hz, 1 H, H-6′a), 0.38 (t, J = 4.4 Hz, 1 H, H-6′b); 13C NMR (100 MHz, CDCl3): δ = 165.16, 156.35, 143.56, 141.94, 135.60, 135.45, 133.37, 133.04, 129.91, 129.77, 127.80, 127.74, 127.24, 94.04, 65.62, 60.87, 38.19, 28.80, 26.90, 23.89, 19.30; FAB MS: m/z (%) 458 (100) [M+H]+; HRMS-FAB: m/z [M+H]+ calcd for C27H33N5O2Si: 458.2264, found: 458.2267.
4-Amino-1-((1′S, 2′S, 5′R)-5-(hydroxymethyl)bicyclo[3.1.0]hex-3-en-2-yl)pyrimidin-2(1H)-one (4d)
Under argon, triethylamine trihydrofluoride (0.177 mL, 1.09 mmol) was added to a mixture of 13 (83 mg, 0.18 mmol) in acetonitrile (10 mL) and refluxed overnight. After cooling to room temperature, the reaction mixture was diluted with water (10 mL) and stirred for 1 h at room temperature. Concentration in vacuo and purification of the residue by silica gel flash chromatography (15% MeOH/CHCl3) followed by recrystallization from (MeOH/CH2Cl2) gave 4d (30 mg, 75% yield) as a white solid; mp 250 °C (dec); [α]D19 = −21.91 (c 0.13, MeOH); 1H NMR (400 MHz, d6-DMSO): δ = 7.03 (d, J = 7.3 Hz, 1 H, H-6), 7.01 (br s, 1 H, NHH), 6.83 (br s, 1 H, NHH), 6.06 (dm, J = 5.4 Hz, 1 H, H-4′), 5.41 (d, J = 7.2 Hz, 1 H, H-5), 5.02 (dt, J = 1.7, 5.1 Hz, 1 H, H-3′), 4.49 (t, J = 5.2 Hz, 1 H, OH), 3.56 (dd, J = 4.7, 11.6 Hz, 1 H, CH2a), 3.03 (dd, J = 4.8, 11.6 Hz, 2 H, CH2b), 1.23 (dd, J = 4.0, 8.1 Hz, 1 H, H-1′), 0.77 (dd, J = 4.0, 8.4 Hz, 1 H, H-6′a), 0.01 (t, J = 4.1 Hz, 1 H, H-6′b); 13C NMR (100 MHz, d6-DMSO): δ = 164.62, 154.52, 142.59, 142.24, 126.60, 93.84, 62.00, 60.31, 38.52, 27.78, 23.66; FAB MS: m/z (%) 220 (100) [M+H+]; HRMS-FAB: m/z [M+H]+ calcd for C11H13N5O2: 220.1086, found: 220.1127; HRMS-FAB: m/z [M+Na]+ calcd for C11H13N5O2Na: 242.0905, found: 242.0906.
1-[(1′S, 3′S, 4′R, 5′S)-4-((tert-Butyldiphenylsilyloxy)methyl)-3-hydroxybicyclo-[3.1.0]hexan-1-yl]-5-methylpyrimidine-2,4(1H,3H)-dione (14a)
4-Dimethyl-aminopyridine (DMAP, 31 mg, 0.25 mmol) was added to a solution of 5a (126 mg, 0.5 mmol) in pyridine (5 mL) and TBDPSCl (0.52mL, 2.0 mmol) was added dropwise to this solution. After stirring overnight at room temperature, the mixture was concentrated in vacuo. Purification of the residue by silica gel flash chromatography (CHCl3, 2% and 5% MeOH/CHCl3) followed by a second silica gel flash chromatography (CHCl3, 2% and 5% MeOH/CHCl3) gave 14a (0.242g, 98 %) as a colorless foam; 1H NMR (400 MHz, CDCl3): δ = 7.91 (s, 1 H, NH), 7.66 (m, 4 H, Ph), 7.38 (m, 6 H, Ph), 7.08 (d, J = 1.3 Hz, 1 H, H-6), 4.36 (dd, J = 0.9, 6.9 Hz, 1 H, H-3′), 3.84 (d, J = 7.0 Hz, 2 H, CH2), 2.35 (ddd, J = 2.2, 7.1, 13.7 Hz, 1 H, H-2′a), 2.09 (m, 2 H, H-4′, H-2′b), 1.76 (d, J = 1.2 Hz, 3 H, CH3), 1.64 (ddd, J = 0.9, 4.8, 9.8 Hz, 1 H, H-5′), 1.48 (m, 1 H, H-6′a), 1.09 (m, 10 H, H-6′b and C(CH3)3); 13C NMR (100 MHz, CDCl3): δ = 163.69, 150.48, 149.81, 135.57, 135.48, 133.53, 133.45, 129.82, 129.81, 127.78, 110.27, 74.58, 65.11, 53.37, 48.25, 40.04, 27.35, 26.99, 19.40, 19.34, 12.12; FAB MS: m/z (%) 491 (41) [M+H]+, 243 (100); HRMS-FAB: m/z [M+H]+ calcd for C28H34N2O4Si: 491.2366, found: 491.2385.
General procedure A
Atmospheric water was azeotropically removed from a mixture of starting material (5b–5d) (1 equiv) and imidazole (2.2 equiv) by evaporation with DMF (2 × 10 mL). The residue was then re-dissolved in DMF and TBDPSCl (1.1 equiv) was added under argon. After stirring 3 h to overnight at room temperature, the reaction mixture was concentrated in vacuo. Purification of the residue by silica gel flash chromatography (2.5%, 5% and 10% MeOH/CHCl3) gave product (14b–14d).
2-Amino-9-((1′S, 3′S, 4′R, 5′S)-4-((tert-butyldiphenylsilyloxy)-methyl)-3-hydroxybicyclo-[3.1.0]hexan-1-yl)-1H-purin-6(9H)-one (14b)
According to general procedure A, 5b (150 mg, 0.54 mmol), imidazole (83 mg, 1.19 mmol) and TBDPSCl (200 μL, 0.77 mmol) were combined in DMF (10 mL) overnight to give 14b (247 mg, 86%) as a white solid, which was recrystallized from MeOH; mp >300 °C; 1H NMR (400 MHz, d6-DMSO): δ = 10.53 (br s, 1 H, NH), 7.66 (m, 4 H, Ph), 7.45 (m, 7H, H-8 and Ph), 6.28 (br s, 2 H, NH2), 4.78 (d, J = 3.2 Hz, 1 H, H-3′), 4.16 (br s, 1 H, OH), 3.82 (dd, J = 7.5, 10.4 Hz, 1 H, CH2a), 3.77 (dd, J = 6.7, 10.3 Hz, 1H, CH2b), 2.19 (m, 2 H, H-2′), 2.11 (t, J = 7.1 Hz, 2 H, H-4′), 1.85 (irr dd, 1 H, H-5′), 1.49 (t, J = 4.9 Hz, 1 H, H-6′a), 1.17 (dd, J = 5.0, 9.4 Hz, 1 H, H-6′b), 1.02 (s, 9 H, C(CH3)3); 13C NMR (100 MHz, d6-DMSO): δ = 156.67, 153.28, 152.22, 137.07, 137.02, 135.10, 133.11, 133.08, 129.85, 127.90, 117.07, 72.43, 51.97, 42.31, 40.96, 26.74, 25.14, 18.84, 17.65; FAB MS: m/z (%) 516 (100) [M+H]+; Anal. calcd for C28H33N5O3Si: C 65.21, H 6.45, N 13.58, found: C 65.03, H 6.44, N 13.55.
(1′S, 3′S, 4′R, 5′S)-1-(6-Amino-1H-purin-9(6H)-yl)-4-((tert-butyldiphenylsilyloxy)-methyl)bicyclo[3.1.0]hexan-3-ol (14c)
According to general procedure A, 5c (164 mg, 0.63 mmol), imidazole (94 mg, 1.38 mmol) and TBDPSCl (180 μL, 0.69 mmol) were combined in DMF (10 mL) to give 14c (265 mg, 82%) as a white solid; mp 189–191 °C; 1H NMR (400 MHz, d6-DMSO): δ = 8.00 (s, 1 H, H-2), 8.01 (s, 1 H, H-8), 7.67 (dd, J = 1.4, 7.9, 4 H, Ph), 7.45 (m, 6 H, H-8 and Ph), 7.15 (s, 2 H, NH2), 4.83 (d, J = 3.0 Hz, 1 H, OH), 4.19 (br dd, J = 2.4, 6.0 Hz, 1 H, H-3′), 3.97 (d, J = 7.7 Hz, 2 H, CH2), 2.44 (ddd, J = 1.3, 6.4, 13.0 Hz, 1 H, H-2′a), 2.19 (t, J = 7.5 Hz, 1 H, H-4′), 2.11 (d, J = 13.3 Hz, 1 H, H-2′b), 1.84 (irregular dd, 1 H, H-5′), 1.56 (t, J = 5.0 Hz, 1 H, H-6′a), 1.37 (ddd, J = 1.6, 4.9, 9.3 Hz, 1 H, H-6′b), 1.02 (s, 9 H, C(CH3)3); 13C NMR (100 MHz, d6-DMSO): δ = 155.94, 152.24, 150.23, 140.92, 135.08, 135.04, 133.30, 133.26, 129.78, 127.86, 127.82, 119.18, 71.89, 65.08, 52.14, 42.39, 40.43, 26.71, 25.53, 18.91, 16.71; FAB MS: m/z (%) 500 (100) [M+H]+; Anal. calcd for C28H33N5O2Si: C 67.30, H 6.66, N 14.02, found: C 67.06, H 6.65, N 14.01.
4-Amino-1-((1′S, 3′S, 4′R, 5′S)-4-((tert-butyldiphenylsilyloxy)methyl)-3-hydroxybicyclo-[3.1.0]hexan-1-yl)pyrimidin-2(1H)-one (14d)
According to general procedure A, 5d (241 mg, 1.02 mmol), imidazole (155 mg, 2.28 mmol) and TBDPSCl (290 μL, 1.11 mmol) were combined in DMF (20 mL) to give 14d (447 mg, 94%) as a colorless glass; mp 168–170 °C; 1H NMR (400 MHz, CD3OD): δ = 7.72 (m, 4 H, Ph), 7.65 (d, J = 7.5 Hz, 1 H, H-6), 7.44 (m, 6 H, Ph), 5.81 (d, J = 7.5 Hz, 1 H, H-5), 4.28 (d, J = 6.9 Hz, 1 H, H-3′), 3.90 (m, 2 H, CH2), 2.33 (irregular dd, 1 H, H-2′a), 2.20 (t, J = 7.1 Hz, 1 H, H-4′), 2.10 (d, J = 13.6 Hz, 1 H, H-2′b), 1.74 (dd, J = 4.8, 9.7 Hz, 1 H, H-5′), 1.52 (t, J = 5.2 Hz, 1 H, H-6′a), 1.16 (m, 1 H, H-6′b), 1.09 (s, 9 H, C(CH3)3); 13C NMR (100 MHz, CD3OD): δ = 164.80, 154.76, 149.54, 136.94, 136.87, 135.05, 134.95, 131.07, 129.01, 129.00, 95.30, 74.91, 66.58, 54.46, 50.95, 40.62, 28.50, 27.64, 20.29, 19.69; 476 FAB MS: m/z (%) 476 (100) [M+H]+; HRMS-FAB: m/z [M+H]+ calcd for C27H33N3O3Si: 476.2370, found: 476.2384.
(1′S, 3′S, 4′R, 5′S)-4-((tert-Butyldiphenylsilyloxy)methyl)-1-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)bicyclo[3.1.0]hexan-3-yl methanesulfonate (15a)
Methanesulfonyl chloride (252 μL, 3.26 mmol) was added dropwise to a solution of 14a (400 mg, 0.82 mmol) in pyridine (16 mL). After stirring at room temperature for 2 h, the mixture was concentrated in vacuo. Purification by silica gel flash chromatography (5% MeOH/CH2Cl2) gave 15a (426 mg, 92%) as a white solid; mp 93–95 °C; 1H NMR (400 MHz, CDCl3): δ = 8.27 (br s, 1 H, NH), 7.65 (m, 4 H, Ph), 7.40 (m, 6 H, Ph), 7.05 (d, J = 1.2 H z, 1 H, H-6), 5.14 (d, J = 6.9 Hz, 1 H, H-3′), 3.91 (m, 2 H, CH2), 2.90 (s, 3 H, CH3), 2.52 (irregular ddd, 1 H, H-2′a), 2.43 (m, 2 H, H-2′b and H-4′), 1.77 (d, J = 1.0 Hz, 3 H, CH3), 1.61 (m, 1 H, H-5′), 1.34 (dd, J = 5.1, 6.3 Hz, 1 H, H-6′a), 1.11 (m, 10 H, H-6′b and C(CH3)3); 13C NMR (100 MHz, CDCl3): δ = 163.60, 150.32, 140.56, 135.54, 135.50, 133.06, 133.03, 129.97, 129.95, 127.88, 127.86, 110.74, 83.50, 63.85, 51.73, 47.58, 38.36, 37.87, 27.00, 26.85, 19.31, 17.84, 12.14; FAB MS: m/z (%) 569 (81) [M+H]+, 217 (100); HRMS-FAB: m/z [M+H]+ calcd for C29H36N2O6Si: 569.2142, found: 569.2101.
(1′S, 3′S, 4′R, 5′S)-1-(2-Amino-6-oxo-1H-purin-9(6H)-yl)-4-((tert-butyldiphenylsilyloxy) methyl)bicyclo[3.1.0]hexan-3-yl methanesulfonate (15b)
Atmospheric water was azeotropically removed from 14a (79 mg, 0.15 mmol) by evaporation with pyridine (3 × 4 mL), and then 14a was dissolved in pyridine (5 mL) under argon. Methanesulfonyl chloride (24 μL, 0.31 mmol) was added dropwise and the reaction stirred at room temperature. After 1 h, additional methanesulfonyl chloride (12 μL, 0.15 mmol) was added and the reaction stirred for an additional 30 mins. The reaction mixture was then concentrated in vacuo and purified by silica gel flash chromatography (5% and 10% MeOH/CHCl3) to give 15b (80 mg, 88%) as a white solid; mp 218–220 °C; 1H NMR (400 MHz, CD3OD): δ = 10.58 (s, 1 H, NH), 7.68 (m, 4 H, Ph), 7.46 (m, 7 H, Ph and H-8), 6.27 (s, 2 H, NH2), 5.14 (d, J = 4.6 Hz, 1 H, H-3′), 3.93 (dd, J = 6.0, 10.6 Hz,1 H, CH2a), 3.84 (dd, J = 8.7, 10.6 Hz, 1H, CH2b), 3.16 (s, 3 H, CH3), 2.55 (m, 2 H, H-2′a), 2.50 (m, 2 H, H-2′b, H-4′), 1.97 (dd, J = 4.7, 9.4 Hz, 1 H, H-5′), 1.30 (dd, J = 6.0, 10.6 Hz, 1H, H-6′a), 1.21 (t, J = 5.7 Hz, 1H, H-6′b), 1.02 (s, 9H, C(CH3)3); 13C NMR (100 MHz, CD3OD): δ = 167.74, 159.19, 147.53, 136.97, 134.69, 134.65, 131.19, 129.12, 129.08, 96.05, 85.24, 65.58, 53.49, 50.25, 38.89, 38.75, 27.81, 27.69, 20.25, 18.77; FAB MS: m/z (%) 594 (100) [M+H]+; Anal. calcd for C29H35N5O5SSi: C 58.66, H 5.94, N 11.79, found: C 58.43, H 5.83, N 11.68.
(1′S, 3′S, 4′R, 5′S)-1-(6-Amino-1H-purin-9(6H)-yl)-4-((tert-butyldiphenylsilyloxy)-methyl)bicyclo[3.1.0]hexan-3-yl methanesulfonate (15c)
Atmospheric water was azeotropically removed from 14c (255 mg, 0.51 mmol) by evaporation with pyridine (2 × 10 mL), and then 14c was dissolved in pyridine (15 mL) under argon. Methanesulfonyl chloride (77 μL, 1.00 mmol) was added dropwise and the resulting yellow solution stirred at room temperature. After 1.25 h, additional methanesulfonyl chloride (35 μL, 0.45 mmol) was added and the reaction stirred for an additional 30 min. The reaction mixture was then concentrated in vacuo and purified by silica gel flash chromatography (5% and 10% MeOH/CHCl3) followed by a second purification (5% MeOH/CHCl3) to give 15c (291 mg, 98%) as a colorless foam; 1H NMR (400 MHz, d6-DMSO): δ = 8.05 (s, 1 H, H-2), 7.95 (s, 1 H, H-8), 7.69 (m, 4 H, Ph), 7.46 (m, 6 H, Ph), 7.20 (br s, 2 H, NH2), 5.21 (d, J = 6.7 Hz, 1 H, H-3′), 4.12 (irr t, 1 H, CH2a), 4.01 (dd, J = 6.5, 10.5 Hz, 1 H, CH2b), 3.19 (s, 3 H, CH3), 2.76 (dd, J = 6.1, 14.1 Hz, 1 H, H-2′a), 2.58 (dd, J = 6.6, 9.0 Hz, 1 H, H-4′), 2.50 (d, overlapped with DMSO, J = 14.1 Hz, 1 H, H-2′b), 1.88 (dd, J = 4.8, 9.2 Hz, 2 H, H-5′), 1.54 (m, 1 H, H-6′a), 1.27 (m, 1 H, H-6′b), 1.03 (s, 9 H, C(CH3)3); 13C NMR (100 MHz, d6-DMSO): δ = 155.96, 152.28, 150.16, 140.85, 135.12, 135.09, 132.88, 132.79, 129.90, 127.93, 127.88, 119.12, 82.69, 63.84, 50.76, 41.83, 38.21, 37.89, 26.64, 24.84, 18.81; FAB MS: m/z (%) 578 (100) [M+H]+. The unstable mesylate was used in the following elimination step without further purification.
(1′S, 3′S, 4′R, 5′S)-1-(4-Amino-2-oxopyrimidin-1(2H)-yl)-4-((tertbutyldiphenylsilyloxy) methyl)bicyclo[3.1.0]hexan-3-yl methanesulfonate (15d)
Atmospheric water was azeotropically removed from 14d (447 mg, 0.94 mmol) by evaporation with pyridine (2 × 20 mL), and then 14d was dissolved in pyridine (25 mL) under argon. Methanesulfonyl chloride (300 μL, 3.87 mmol) was added dropwise and the resulting solution stirred at room temperature for 1 h. The reaction mixture was then concentrated in vacuo and purified by silica gel flash chromatography (5% MeOH/CHCl3) to give 15d (428 mg, 82%) as a colorless glass. Treatment with 5% MeOH/Et2O gave colorless crystals; mp 120 °C; 1H NMR (400 MHz, CD3OD): δ = 7.73 (m, 4 H, Ph), 7.45 (m, 7 H, Ph and H-6), 5.75 (d, J = 7.3 Hz, 1 H, H-5), 5.19 (d, J = 6.8 Hz, 1 H, H-3′), 4.06 (irr t, 1 H, CH2a), 3.97 (dd, J = 5.6, 10.7 Hz, 1 H, CH2b), 3.02 (s, 3 H, CH3), 2.52 (m, 2 H, H-2′a, H-4′), 2.38 (d, J = 14.8 Hz, 1 H, H-2′b), 1.64 (dd, J = 4.7, 9.7 Hz, 1 H, H-5′), 1.30 (irr t, 1 H, H-6′a), 1.19 (m, 1 H, H-6′b), 1.11 (s, 9 H, C(CH3)3); 13C NMR (100 MHz, CD3OD): δ = 167.74, 159.19, 147.53, 136.97, 134.69, 134.65, 131.19, 129.12, 129.08, 96.05, 85.24, 65.58, 53.49, 50.25, 38.89, 38.75, 27.81, 27.69, 20.25, 18.77; FAB MS: m/z (%) 554 (100) [M+H]+; Anal. calcd for C28H35N3O2SSi: C 60.73, H 6.37, N 7.59, found: C 60.73, H 6.42, N 7.51.
1-((1′S, 4′R, 5′S)-4-((tert-Butyldiphenylsilyloy)methyl)bicyclo[3.1.0]hex-2-en-1-yl)-5-methylpyrimidine-2,4(1H,3H)dione (16a)
Under argon, DBU (0.50 mL, 3.21 mmol) was added to a solution of 15a (234 mg, 0.41 mmol) in toluene (10 mL) and heated to reflux overnight. After cooling to room temperature, the reaction mixture was diluted with EtOAc and washed with water (2 × 10 mL). The combined aqueous washings were extracted with EtOAc (2 × 10 mL) and the combined organic extracts were further washed with brine (2 × 10 mL), dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. Purification by silica gel flash chromatography (5%, 20%, 50% and 75% EtOAc/hexanes) gave 16a along with unreacted 15a, which was recycled through the same procedure and combined with the first batch to give 16a (93 mg, 53% yield) as a white solid; mp 175–177 °C; 1H NMR (400 MHz, CDCl3): δ = 8.04 (br s, 1 H, NH), 7.67 (m, 4 H, Ph), 7.39 (m, 6 H, Ph), 6.99 (q, J = 1.1 Hz, 1 H, H-6), 5.99 (ddd, J = 0.6, 1.9, 5.6 Hz, 1 H, H-3′), 5.46 (dt, J = 2.1, 5.6 Hz, 1 H, H-2′), 3.86 (dd, J = 5.3, 10.0 Hz, 1 H, CH2a), 3.72 (dd, J = 7.2, 10.0 Hz, 1 H, CH2b), 2.72 (m, 1 H, H-4′), 1.70 (d, J = 1.2 Hz, 3 H, CH3), 1.49 (dd, J = 5.2, 9.3 Hz, 1 H, H-5′), 1.08 (m, 10 H, H-6′a and C(CH3)3), 0.86 (t, J = 5.2 Hz, 1 H, H-6′b); 13C NMR (100 MHz, CDCl3): δ = 163.70, 150.79, 140.99, 135.57, 135.53, 133.95, 133.67, 133.54, 130.51, 129.79, 129.75, 127.75, 127.71, 110.36, 66.53, 52.25, 51.13, 26.99, 26.66, 24.88, 19.43, 12.02; FAB MS: m/z (%) 473 (86) [M+H]+, 135 (100); Anal. calcd for C28H32N2O3Si•0.1H2O: C 70.88, H 6.84, N 5.90, found: C 70.63, H 6.85, N 5.83.
4-Amino-1-((1′S, 4′R, 5′S)-4-(hydroxymethyl)bicyclo[3.1.0]hex-2-en-1-yl)pyrimidin-2(1H)-one (6d)
Atmospheric water was azeotropically removed from 15d (82 mg, 0.144 mmol) by evaporation with DMF (2 × 5 mL), and then 15d was dissolved in DMF (7 mL) under argon, treated with treated with DBU (0.44 mL, 2.88 mmol) and heated to 100 °C. After stirring at 100 °C for 4 days, the reaction mixture was concentrated in vacuo and the residue triturated with Et2O (3 × 25 mL). Purification by silica gel flash chromatography (10% and 15% MeOH/CHCl3) gave 6d (14 mg, 45% yield) as a white solid after a second purification (same conditions) and treatment with Et2O; mp 222–224 °C; [α]D20 = +1.37° (c 0.47, CH3OH); 1H NMR (400 MHz, CD3OD): δ = 7.67 (dd, J = 0.5, 7.3 Hz, 1 H, H-6), 6.05 (dd, J = 1.9, 5.6 Hz, 1 H, H-3′), 5.86 (dd, J = 0.5, 7.3 Hz, 1 H, H-5), 5.51 (dt, J = 2.0, 5.6 Hz, 1 H, H-3′), 3.77 (dd, J = 4.2, 10.9 Hz, 1 H, CH2a), 3.70 (dd, J = 5.3, 10.9 Hz, 1 H, CH2b), 2.72 (m, 1 H, H-4′), 2.05 (m, 1 H, H-5′), 1.48 (dd, J = 5.0, 9.2 Hz, 1 H, H-6′a), 0.80 (t, J = 5.0 Hz, 1 H, H-6′b); 13C NMR (100 MHz, CD3OD): δ = 167.86, 159.89, 147.85, 136.01, 130.93, 96.24, 65.65, 55.13, 52.40, 28.04, 25.27; FAB MS: m/z (%) 220 (100) [M+H]+; Anal. calcd for C11H13N3O2•0.2H2O: C 59.29; H 6.06, N 18.86, found: C 59.24, H 6.12, N 18.81.
2-Amino-9-((1′S, 4′R, 5′S)-4-(hydroxymethyl)bicyclo[3.1.0]hex-2-en-1-yl)-1H-purin-6(9H)-one (6b)
Under argon, DBU (1.1 mL, 7.2 mmol) was added to 15b (221 mg, 0.36 mmol) in DMF (15 mL) and heated to 100 °C for 3 days. After cooling to room temperature, the reaction mixture was concentrated in vacuo. Purification by silica gel flash chromatography (5%, 10% and 15% MeOH/CHCl3) gave 6b (41 mg, 44% yield) as a tan solid along with the 5′-protected analogue (19 mg, 10% yield) as a white solid. Recrystallization from MeOH/Et2O afforded pure 6b as tan crystals; mp 142–143 °C; [α]D19 = +21.74° (c 0.1, CH3OH); 1H NMR (400 MHz, d6-DMSO): δ = 10.55 (br d, J = 1.2, 1 H, NH), 7.63 (d, J = 0.6 Hz, 1 H, H-8), 6.38 (s, 2 H, NH2), 6.06 (dd, J = 1.9, 5.6 Hz, 1 H, H-3′), 5.50 (dt, J = 2.0, 5.5 Hz, 1 H, H-2′), 4.77 (t, J = 5.7 Hz, 1 H, OH), 3.65 (dt, J = 5.3, 10.6 Hz, 1 H, CH2a), 3.52 (m, 1 H, CH2b), 2.60 (m, 1 H, H-4′), 2.13 (br dd, J = 5.1, 8.2 Hz, 1 H, H-5′), 1.62 (dd, J = 4.7, 9.1 Hz, 1 H, H-6′a), 0.69 (t, J = 4.9 Hz, 1 H, H-6′b); 13C NMR (100 MHz, d6-DMSO): δ = 156.68, 153.41, 152.31, 137.87, 134.18, 130.25, 116.88, 63.99, 51.11, 46.28, 40.14, 39.93, 39.72, 39.51, 39.30, 39.19, 39.09, 38.88, 25.07, 23.10; FAB MS: m/z (%) 260 (100) [M+H]+; HRMS-FAB: m/z [M+H]+ calcd for C12H13N5O2: 260.1147, found: 260.1155.
((1′S, 2′R, 5′S)-5-(6-Amino-9H-purin-9-yl)bicyclo[3.1.0]hex-3-en-2-yl)methanol (6c)
Under argon, DBU (1.1 mL, 7.2 mmol) and 15c (210 mg, 0.36 mmol) were combined in DMF (20 mL) and heated to 100 °C for 4 days. After cooling to room temperature, the reaction mixture was concentrated in vacuo. Purification of the residue by silica gel flash chromatography (5% and 10% MeOH/CHCl3) followed by a second purification by silica gel flash chromatography (5% MeOH/CHCl3) gave 6c (97 mg, 87% yield) as an ivory solid along with the 5′-protected analogue (28 mg, 13% yield). An analytically pure sample of 6c was obtained by triturating with dichloromethane; mp 170–171 °C; [α]D19 = +1.32° (c 0.33, CH3OH); 1H NMR (400 MHz, d6-DMSO): δ = 8.13 (s, 1 H, H-2), 8.11 (s, 1 H, H-8), 7.21 (br s, 2 H, NH2), 6.09 (br dd, J = 1.8, 5.6 Hz, 1 H, H-3′), 5.54 (dt, J = 2.0, 5.5 Hz, 1 H, H-4′), 4.90 (dd, J = 4.8, 6.5 Hz, 1 H, OH), 3.70 (overlapping dt, J = 4.9, 10.1 Hz, 1 H, CH2a), 3.59 (dt, J = 6.8, 10.6 Hz, 1 H, CH2b), 2.66 (m, 1 H, H-2′), 2.15 (m, 1 H, H-1′), 1.76 (dd, J = 4.8, 9.1 Hz, 1 H, H-6′a), 0.77 (t, J = 4.9 Hz, 1 H, H-6′b); 13C NMR (100 MHz, d6-DMSO): δ = 155.98, 152.50, 150.51, 141.33, 134.16, 130.37, 118.95, 64.02, 51.03, 46.52, 25.21, 22.65. FAB MS: m/z (%) 244 (100) [M+H]+; HRMS-FAB: m/z [M+H]+ calcd for C12H13N5O: 244.1198, found: 244.1201.
1-((1′S, 4′R, 5′S)-4-(Hydroxymethyl)bicyclo[3.1.0]hex-2-en-1-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (6a)
TBAF (0.4 mL, 1 M in THF) was added to a solution of 16a (66O170) (87 mg, 0.18 mmol) in THF (5 mL). After stirring at room temperature for 1h, the reaction mixture was concentrated in vacuo. Purification by silica gel flash chromatography (5%, 20%, 50% and 75% EtOAc/hexanes) gave 6a (46 mg) as a colorless foam. Recrystallization from EtOAc followed by hexanes gave 6a (34 mg, 79% yield) as a white crystalline solid; mp 142–143 °C; [α]D19 = −190.82° (c 0.09, CH3OH); 1H NMR (400 MHz, CDCl3): δ = 8.30 (br s, 1 H, NH), 7.21 (q, J = 1.2 Hz, 1 H, H-6), 5.99 (ddd, J = 0.6, 2.0, 5.6 Hz, 1 H, H-3′), 5.53 (dt, J = 2.1, 5.6 Hz, 1 H, H-2′), 3.83 (d, J = 3.3, 2 H, CH2), 2.76 (m, 1 H, H-4′), 2.08 (m, 1 H, H-5′), 1.91 (d, J = 1.2 Hz, 3 H, CH3), 1.43 (dd, J = 4.8, 9.3 Hz, 1 H, H-6′a), 0.78 (t, J = 5.0 Hz, 1 H, H-6′b); 13C NMR (100 MHz, CDCl3): δ = 163.50, 151.69, 140.83, 133.88, 130.52, 111.31, 77.32, 77.00, 76.68, 64.56, 52.73, 50.77, 27.59, 23.92, 12.32. FAB MS: m/z (%) 235 (100) [M+H]+; Anal. calcd for C12H14N2O3•0.1H2O: C 61.06, H 6.06, N 11.87, found: C 60.80, H 5.99, N 11.53.
Cell-based Assays
The human embryonic kidney cell culture line 293T was obtained from the American Type Culture Collection (ATCC). The human osteosarcoma cell line, HOS, was obtained from Dr. Richard Schwartz (Michigan State University, East Lansing, MI). The HOS-313 cells expressing herpes simplex virus-1 thymidine kinase were previously prepared by infecting HOS cells with a previously described MLV-vector.[19,21] Cell lines were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) supplemented with 5% (v/v) fetal bovine serum, 5% newborn calf serum, and penicillin (50 units/mL) plus streptomycin (50 μg/mL) (Quality Biological, Gaithersburg, MD). VSV-g –pseudotyped HIV was produced by transfection of 293T cells. On the day prior to transfection, 293T cells were plated in 100 mm dishes at a density of 9×105 cells per plate and transfected with 10 μg of pNLNgoMIVR+ ΔEnv.LUC (wild-type or NRTI resistant mutant) and 3 μg of pHCMV-g (obtained from Dr. Jane Burns, University of California, San Diego) using calcium phosphate precipitation.[16, 18] After 48 hours, virus-containing supernatants were harvested, clarified by low-speed centrifugation and filtration and diluted 1-to-5 in preparation for infection assays. HOS cells were plated in 96 well luminescence cell culture plates at a density of 4000 cells in 100 μL per well the day prior to infection. On the day of infection, cells were pretreated with the target compounds for 3 h. Infections were carried out by adding 100 μL of virus-containing supernatants to each well and incubating for 48 h. Infectivity was determined using a luciferase reporter assay.[22] Luciferase activity was measured by adding 100 μL of steadylite plus reagent (PerkinElmer, Waltham, MA) directly to the cells and measuring luminescence using a microplate reader. Activity was normalized to infections done in the absence of target compounds for the appropriate NRTI variant. Regression analysis on the data was performed using a 4-parameter sigmoidal binding model, f(x) = a + b/(1+(x/c)d) and EC50 values were determined from the fit. Cytotoxic effects were determined by measuring the effects of the compounds on the ATP levels in the cells. After treating HOS (or HOS-313) cells with the target compounds ATP levels were determined by measuring the relative luciferase activity using the PerkinElmer ATPlite kit. Luminescence data was normalized to cell data in the absence of target compound. The data was fit as above and CC50 values were determined from the fit.
X-ray Crystal Structure of compound 6b
Single-crystal X-ray diffraction data on compound 6b were collected at 173 °K using CuKα radiation and a Bruker Proteum Diffractometer equipped with Helios optics and a Platinum 135 CCD area detector. A 0.237 × 0.167 × 0.019 mm3 crystal was prepared for data collection coating with high viscosity microscope oil (Paratone-N, Hampton Research). The oil-coated crystal was mounted on MicroMesh mount (MiTeGen, Ithaca, NY) and transferred to the cold stream (173 °K) on the diffractometer. The crystal was orthorhombic in space group P212121 with unit cell dimensions a = 10.7840(3) Å, b = 10.8458(3) Å, and c = 21.3305(6) Å. Corrections were applied for Lorentz, polarization, and absorption effects. Data were 92.9% complete to 25.00° θ (approximately 0.83 Å) with an average redundancy of 5.2. The structure was solved by direct methods and refined by full-matrix least squares on F2 values using the programs found in the SHELXTL suite (Bruker, SHELXTL v6.10, 2000, Bruker AXS Inc., Madison, WI). Parameters refined included atomic coordinates and anisotropic thermal parameters for all non-hydrogen atoms. Hydrogen atoms on carbons were included using a riding model [coordinate shifts of C applied to H atoms] with C-H distance set at 0.96 Å. The relative configuration was C1 = S, C4 = R, C5 = S. The asymmetric unit contains two molecules with the same relative configuration. Atomic coordinates for compound 6b have been deposited with the Cambridge Crystallographic Data Centre (deposition number 727856). Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK [fax: +44(0)-1223-336033 or e-mail: deposit@ccdc.cam.ac.uk
Scheme 3.
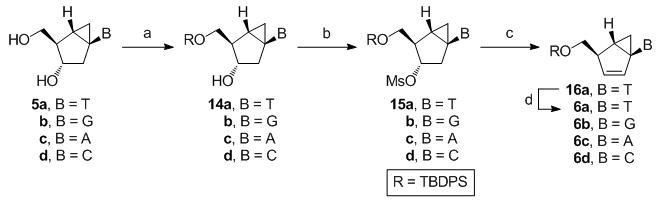
Reagents and conditions: (a) tert-butyldiphenylsilyl chloride (TBDPSCl), imidazole, DMF or TBDPSCl, DMAP, pyridine; (b) MsCl, pyridine; (c) 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), PhMe or DMF, Δ; (d) TBAF, THF.
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
References
- 1.Marquez VE, Ezzitouni A, Russ P, Siddiqui MA, Ford H, Feldman RJ, Mitsuya H, George C, Barchi JJ. J Am Chem Soc. 1998;120:2780–2789. [Google Scholar]
- 2.Boyer PL, Julias JG, Marquez VE, Hughes SH. J Mol Biol. 2005;345:441–450. doi: 10.1016/j.jmb.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 3.Vanroey P, Salerno JM, Chu CK, Schinazi RF. Proc Natl Acad Sci U S A. 1989;86:3929–3933. doi: 10.1073/pnas.86.11.3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kerr SG, Anderson KS. Biochemistry. 1997;36:14064–14070. doi: 10.1021/bi9713862. [DOI] [PubMed] [Google Scholar]
- 5.Vaccaro JA, Anderson KS. Biochemistry. 1998;37:14189–14194. doi: 10.1021/bi9810353. [DOI] [PubMed] [Google Scholar]
- 6.Vaccaro JA, Parnell KM, Terezakis SA, Anderson KS. Antimicrob Agents Chemother. 2000;44:217–221. doi: 10.1128/aac.44.1.217-221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balzarini J, Herdewijn P, De Clercq E. J Biol Chem. 1989;264:6127–6133. [PubMed] [Google Scholar]
- 8.Gao WY, Agbaria R, Driscoll JS, Mitsuya H. J Biol Chem. 1994;269:12633–12638. [PubMed] [Google Scholar]
- 9.Marquez VE, Ben-Kasus T, Barchi JJ, Green KM, Nicklaus MC, Agbaria R. J Am Chem Soc. 2004;126:543–549. doi: 10.1021/ja037929e. [DOI] [PubMed] [Google Scholar]
- 10.Marquez VE, Siddiqui MA, Ezzitouni A, Russ P, Wang JY, Wagner RW, Matteucci MD. J Med Chem. 1996;39:3739–3747. doi: 10.1021/jm960306+. [DOI] [PubMed] [Google Scholar]
- 11.Choi YS, George C, Comin MJ, Barchi JJ, Kim HS, Jacobson KA, Balzarini J, Mitsuya H, Boyer PL, Hughes SH, Marquez VE. J Med Chem. 2003;46:3292–3299. doi: 10.1021/jm030116g. [DOI] [PubMed] [Google Scholar]
- 12.Choi YS, Sun GY, George C, Nicklaus MC, Kelley JA, Marquez VE. Nucleosides Nucleotides & Nucleic Acids. 2003;22:2077–2091. doi: 10.1081/ncn-120026631. [DOI] [PubMed] [Google Scholar]
- 13.Cruickshank KA, Jiricny J, Reese CB. Tetrahedron Lett. 1984;25:681–684. [Google Scholar]
- 14.Divakar KJ, Reese CB. J Chem Soc, Perkin Trans 1. 1982:1171–1176. [Google Scholar]
- 15.Ezzitouni A, Marquez VE. J Chem Soc, Perkin Trans 1. 1997:1073–1078. [Google Scholar]
- 16.Oh J, McWilliams MJ, Julias JG, Hughes SH. Journal of Virology. 2008;82:719–727. doi: 10.1128/JVI.02611-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Julias JG, Boyer PL, McWilliams MJ, Alvord WG, Hughes SH. Virology. 2004;322:13–21. doi: 10.1016/j.virol.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 18.Julias JG, Ferris AL, Boyer PL, Hughes SH. Journal of Virology. 2001;75:6537–6546. doi: 10.1128/JVI.75.14.6537-6546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delviks KA, Hu WS, Pathak VK. Journal of Virology. 1997;71:6218–6224. doi: 10.1128/jvi.71.8.6218-6224.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strehler BL, McElroy WD. Methods Enzymol. 1957;3:871–873. [Google Scholar]
- 21.Boyer PL, Julias JG, Ambrose Z, Siddiqui MA, Marquez VE, Hughes SH. J Mol Biol. 2007;371:873–882. doi: 10.1016/j.jmb.2007.05.043. [DOI] [PubMed] [Google Scholar]
- 22.Petropoulos CJ, Parkin NT, Limoli KL, Lie YS, Wrin T, Huang W, Tian H, Smith D, Winslow GA, Capon DJ, Whitcomb JM. Antimicrob Agents Chemother. 2000;44:920–928. doi: 10.1128/aac.44.4.920-928.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]