

Abstract
Exposure of the gastrointestinal (GI) tract to ionizing radiation can cause acute and delayed injury. However, critical cellular targets that regulate the development of radiation-induced GI injury remain incompletely understood. Here, we investigated the role of vascular endothelial cells in controlling acute and delayed GI injury after total-abdominal irradiation (TAI). To address this, we used genetically engineered mice in which endothelial cells are sensitized to radiation due to the deletion of the tumor suppressor p53. Remarkably, we found that VE-cadherin-Cre; p53FL/FL mice, in which both alleles of p53 are deleted in endothelial cells, were not sensitized to the acute GI radiation syndrome, but these mice were highly susceptible to delayed radiation enteropathy. Histological examination indicated that VE-cadherin-Cre; p53FL/FL mice that developed delayed radiation enteropathy had severe vascular injury in the small intestine, which was manifested by hemorrhage, loss of microvessels and tissue hypoxia. In addition, using dual-energy CT imaging, we showed that VE-cadherin-Cre; p53FL/FL mice had a significant increase in vascular permeability of the small intestine in vivo 28 days after TAI. Together, these findings demonstrate that while sensitization of endothelial cells to radiation does not exacerbate the acute GI radiation syndrome, it is sufficient to promote the development of late radiation enteropathy.
INTRODUCTION
Abdominal and pelvic radiation therapy are frequently delivered to patients with gastrointestinal (GI) cancers, retroperitoneal sarcomas, prostate cancers and gynecological tumors. One of the dose-limiting structures for safely delivering a curative radiation dose in these patients is the small intestine (1). Single-fraction high-dose irradiation of the small intestine can lead to the potentially fatal acute radiation syndrome, however, with fractionated radiotherapy the dose-limiting toxicity is the late effect termed radiation enteropathy (1–3). Indeed, with increasing cancer survivorship there are now more patients living with late radiation-induced intestinal toxicity than ulcerative colitis and Crohn’s disease combined (4). Delayed radiation enteropathy is a chronic condition. It is characterized by loss of mucosa, fibrosis and vascular changes that result in malabsorption and intestinal dysmotility (5). These features of delayed radiation toxicity are generally irreversible and often progressive.
Numerous published studies have demonstrated that GI epithelial cells are a critical cellular target for acute GI radiation injury (6–9). Upon high-dose irradiation to the small intestine the stem cells in the intestinal crypts are lost, resulting in insufficient epithelial cell production and an inability to maintain the villous structures and the mucosal barrier (10, 11). In contrast, the mechanisms governing the radiation-induced cellular damage and ensuing tissue injury that lead to late GI toxicity remain elusive. It has been proposed that pathogenesis of radiation enteropathy is strongly associated with endothelial dysfunction and death (5). However, the relative contributions of endothelial cell damage to the development of acute versus delayed GI radiation injury remain unclear.
We previously demonstrated that one key gene that regulates the radiosensitivity of endothelial cells is the tumor suppressor p53 (7, 12). For example, deletion of p53 sensitizes cardiac endothelial cells to radiation in vitro and in vivo (12). Thus, to investigate the role of endothelial cells in regulating GI toxicity after abdominal radiation, we utilized genetically engineered vascular endothelial (VE) cadherin–Cre (VECre) mice to delete p53 specifically in endothelial cells (12). Using this mouse model, we show that radiosensitization of endothelial cells by deleting p53 does not exacerbate the acute GI radiation syndrome, but it is sufficient to promote late GI toxicity.
MATERIALS AND METHODS
Animals and Irradiation
All animal procedures for this study were approved by the Institutional Animal Care and Use Committee (IACUC) at Duke University. The p53FL/FL and VECre mice have been described elsewhere (12). Experiments were performed with both male and female mice that were between 8 and 12 weeks old and were on mixed genetic backgrounds. Age-matched, littermate controls that retained one allele of wild-type p53 were utilized to minimize the effect of genetic background. Therefore, potential genetic modifiers of the response to radiation were randomly distributed among the experimental and control groups.
Total-abdominal irradiation (TAI) was performed using a small-field biological irradiator, the X-RAD 225Cx (Precision X-Ray Inc., North Branford, CT). Mice were irradiated with parallel-opposed anterior and posterior fields, which encompassed the small and large intestines, using a collimating cone that produces a square radiation field of 40 mm at treatment isocenter. The average dose rate was 298.8 cGy/min at target depth with a 225 kVp, 13 mA beam and a 0.3-mm copper filter. The dose rate was measured with an ion chamber by members of the Radiation Safety Division at Duke University.
GI Epithelium Permeability Assay
Fluorescein isothiocyanate (FITC)-dextran (4 kD; Sigma-Aldrich® LLC, St. Louis, MO) was dissolved in sterile water at 40 mg/ml. Mice were gavaged before irradiation or at various time points postirradiation with 0.6 mg/g body weight of the FITC-dextran solution 4 h before sacrifice when serum was collected. The levels of FITC-dextran in serum were quantified following methods described elsewhere (13).
Histology
To prepare paraffin-embedded tissues, tissue specimens were fixed in 10% neutralized formalin overnight and preserved in 70% ethanol. The small intestines were bundled for embedding to obtain the ideal orientation of the crypts (11). To prepare frozen tissues, tissue specimens were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 2 h at 4°C and transferred to 30% sucrose in PBS overnight at 4°C. The small intestines were bundled and immersed in OCT compound (Sakura Finetek), snap frozen in dry ice/isopentane slurry and stored at –80°C. Immunofluorescent staining for wheat germ agglutinin (WGA), Griffonia simplicifolia isolectin B4 (GS-IB4) and 2-nitroimidazole EF5 were performed following methods described elsewhere (12). Images were quantified, using MATLAB® (MathWorks® Inc., Natick, MA), by a single observer (SH) blinded to genotypes and radiation treatment. The radiation injury score was assessed by a veterinary pathologist (LB) blinded to genotypes and radiation treatment according to a published scoring system (14, 15).
Isolation of Intestinal Epithelial Cells
The intestinal epithelium was isolated according to a method described elsewhere (6). Small intestines were cut longitudinally and washed with ice-cold PBS without Mg2+/Ca2+ (PBS0) and then incubated in PBS0 supplemented with 2 mM EDTA, 1 mM DTT, 50 mM gentamicin (Sigma-Aldrich) and 1× penicillin/streptomycin antibiotics (Gibco®, Grand Island, NY) at 4°C for 60 min on a rotating platform. The intestines were then gently transferred to another tube containing cold PBS0 supplemented with 2 mM EDTA and 1 mM DTT and shaken vigorously to detach the epithelial cells. The muscularis layers were then removed and the cells were centrifuged to obtain an epithelial isolate. The cell fraction was then washed one more time in cold PBS0. Cell pellet was snap frozen in liquid nitrogen and stored at –80°C.
Quantitative Reverse Transcription-PCR
Mouse small intestinal epithelial cell frozen pellets were thawed and resuspended in 1 ml of TRI Reagent (Invitrogen™, Grand Island, NY). Total RNA was extracted and quantified. cDNA was generated from 1 μg of RNA using iScript™ cDNA Synthesis Kit (Bio-Rad® Laboratories Inc., Hercules, CA). Relative expression levels were determined using qPCR assays performed on the QuantStudio™ 6 Flex Real-Time PCR System with PowerUp™ SYBR® Green Master Mix and specific primer sets (16) or TaqMan™ Fast Advanced Master Mix and specific Taqman probes (Thermo Fisher Scientific™ Inc., Waltham, MA). Target gene quantification levels were normalized to the housekeeper gene, Gapdh.
Mdr1 (Abcb1b): Taqman probe Mm00440736_m1; Tff3: Taqman probe Mm00495590_m1;
Gapdh: Taqman probe Mm99999915_g1;
Tjp1/ZO-1: 5’-CCACCTCTGTCCAGCTCTTC-3’; 5’-CACCG GAGTGATGGTTTTCT-3’
Claudin-2: 5’-GTCATCGCCCATCAGAAGAT-3’; 5’-ACTGTTG GACAGGGAACCAG-3’.
Computed Tomography (CT) Scan
To assess the effects of radiation on the GI tract we used an in-house dual source micro-CT system specifically developed for dynamic and spectral applications (17, 18). The system incorporates two imaging chains capable of independently triggered acquisitions. The system contains two G-297 X-ray tubes (Varian Medical Systems, Palo Alto, CA) with 0.3/0.8-mm focal spot size, two Epsilon high-frequency X-ray generators (EMD Technologies; Saint-Eustache, Canada), and two CCD-based detectors with a Gd2O2S phosphor (XDI-VHR 2 Photonic Science, East Sussex, UK) with 22-μm pixels, which we bin to 88 μm. Dual-energy acquisition is controlled via custom sequencing applications written in LabVIEW (National Instruments™, Austin, TX).
A total of 360 projection images per imaging chain were acquired. Dual-energy settings were configured as follows: 1. 40 kVp, 250 mA, 16 ms per exposure; and 2. 80 kVp, 160 mA, 10 ms per exposure. The radiation dose for each micro-CT involving 720 projections with the described exposure parameters is approximately 245 mGy.
To assess the vascular permeability, mice were administered gold nanoparticles (AuNps) via retro-orbital injection (www.nanoprobes.com) at a dose of 0.004 ml/g. As a result of radiation damage to the endothelial cells in the GI tract, the gold nanoparticles accumulated via the enhanced permeability and retention effect (19). Three days later the mice were re-injected with a liposomal iodinated contrast agent Lip-I (20) (dose 0.012 ml/g). The use of both nanoparticle agents for this application has been described elsewhere (21) and enables the visualization of both vascular permeability in the damaged GI tract via AuNps (which extravasate from vessels and accumulate in the injured tissue) and the vessels (via Lip-I which remains within the vessels). Micro-CT imaging resulted in two separate 3D datasets for each scan, which were acquired at the two different energies with a voxel size of 88 μm. The size of the reconstructed volume was 512 μm3 voxels spanning 45 mm3.
For dual-energy micro-CT, material decomposition of iodine and gold was performed after reconstruction and Hounsfield unit (HU) conversion, as described elsewhere (22, 23). Briefly, image-based decomposition used paired temporal reconstructions at two different kVps (40 kVp, 80 kVp) and solved a linear system at each voxel,
| (1) |
where C is the least-squares solution for the concentrations of iodine and gold in mg/ml in the voxel under consideration, A is a constant sensitivity matrix measured in Hounsfield units per contrast agent concentration (HU/mg/ml) for iodine and gold at 40 and 80 kVp, respectively, and b is the intensity of the voxel at the two energies, in Hounsfield units.
After the solution of the matrix equation was determined, a nonnegativity constraint was enforced by setting voxels with negative concentrations of both materials to zero. Voxels with a negative concentration of one material and a positive concentration of the other material were orthogonally projected onto the subspace of positive concentrations. Values for the coefficients of the sensitivity matrix at each energy were determined empirically using a calibration phantom.
Statistical Analysis
For all experiments, measurements are presented as mean ± SEM. Each data point represents one mouse. Two-way analysis of variance (ANOVA) followed by Bonferroni post hoc test was performed to examine the interaction between genotype and radiation treatment. Two-tailed Student’s t test (parametric) or Mann-Whitney test (nonparametric) was performed to compare the data between two groups. If the normality test failed, data were log-transformed prior to applying statistical tests. For the GI radiation injury study, Kaplan-Meier estimate was performed followed by the log-rank test. We considered the evidence against a null hypothesis to be significant if the unadjusted P value for the corresponding test was less than 0.05. GraphPad Prism version 8 (GraphPad Software Inc., La Jolla, CA) was used to perform these statistical analyses.
RESULTS
Deletion of p53 Specifically in Endothelial Cells Promotes Delayed, but not Acute, Radiation-Induced Intestinal Injury
To sensitize endothelial cells to radiation, we deleted the floxed p53 allele (p53FL) specifically in endothelial cells using VECre (12). VECre; p53FL/FL mice, in which both alleles of p53 are deleted in endothelial cells, as well as their VECre; p53FL,/+ littermates, which retain one allele of p53 in endothelial cells, received 15 Gy TAI. After irradiation, the percentage of mice that became moribund within 10 days as a result of the acute GI radiation syndrome (11) was not significantly different between the VECre; p53FL/FL cohort (35%, 7 out 20) and the VECre; p53FL/+ cohort (45%, 10 out of 22) (Fig. 1A). One hallmark of the acute GI radiation syndrome is breakdown of the GI mucosal barrier due to extensive crypt destruction (11). Therefore, we examined the integrity of the GI mucosal barrier by delivering FITC- dextran solution through oral gavage and then detected the level of FITC-dextran in serum 4 h later (13). Compared to nonirradiated mice, the concentration of FITC-dextran in serum was highly elevated 5 days after 15 Gy TAI in both VECre; p53FL/FL and VECre; p53FL/+ mice (Fig. 1B). Together, these results suggest that deletion of p53 in endothelial cells does not promote acute GI radiation injury.
FIG. 1.

Deletion of p53 in endothelial cells sensitizes mice to delayed, but not acute, radiation-induced GI injury. Panel A: Kaplan-Meier survival analysis of VECre; p53FL/FL mice (p53 deleted in endothelial cells) and VECre; p53FL/+ mice (p53 retained in endothelial cells) after a single dose of 15 Gy TAI. Panel B: Quantification of FITC-dextran in serum collected from VECre; p53FL/ + (blue) and VECre; p53FL/FL (red) mice at various time points after 15 Gy TAI. Mice were given FITC-dextran via oral gavage 4 h before sacrifice. Serum was collected from each mouse and the concentration of FITC-dextran in serum was quantified. *P < 0.05 by Student’s t test compared to nonirradiated mice. Each dot represents the averaged data from one mouse. Panel C: Change in body weight of VECre; p53FL/ + and VECre; p53FL/FL mice after 15 Gy TAI. *P < 0.05 by two-way ANOVA with Bonferroni post hoc test. Panel D: The expression of tight junction genes Tjp1 (ZO-1) and Claudin-2 mRNA in intestinal epithelial cells from unirradiated mice or at day 30 after 15 Gy TAI. P value was calculated by two-way ANOVA with Bonferroni post hoc test. *P < 0.05. Each dot represents one mouse.
Although deletion of p53 in endothelial cells did not sensitize mice to the acute GI radiation syndrome, VECre; p53FL/FL mice that survived acute radiation injury became moribund starting at approximately one month after 15 Gy TAI (Fig. 1A) and showed a significant decrease in body weight (Fig. 1C). In contrast to the mice that developed acute GI radiation injury, both VECre; p53FL/FL and VECre; p53FL/ + mice, at 30 days after 15 Gy TAI, did not exhibit a significant increase in the concentration of FITC-dextran in serum (Fig. 1B), suggesting that intestinal crypts were not extensively depleted. To validate the results of the FITC- dextran assay, we isolated intestinal epithelial cells from VECre; p53FL/FL and VECre; p53FL/ + mice at 0 and 30 days after 15 Gy TAI to examine mRNA expression of tight junction genes Tjp1 (ZO-1) and Claudin-2 (16). Tjp1 (ZO-1) is a tight junction protein expressed ubiquitously in all tight junctions that binds directly to the cytoplasmic tails of claudins and occludin (24, 25). It has been shown that the mRNA and protein of Tjp1 (ZO-1) is markedly decreased in the small intestine at days 1 and 7 after 13 Gy TAI (26). However, our results indicated that Tjp1 mRNA in the intestinal epithelium was not significantly decreased in either VECre; p53FL/FL or VECre; p53FL/+ mice 30 days after 15 Gy TAI compared to nonirradiated controls (Fig. 1D). In contrast, we unexpectedly found that intestinal epithelial cells collected from VECre; p53FL/+ mice and VECre; p53FL/FL mice 30 days after 15 Gy TAI significantly overexpressed Claudin-2. Of note, Claudin-2 overexpression was substantially greater in irradiated VECre; p53FL/FL mice compared to irradiated VECre; p53FL/ + mice (P = 0.01 by two-way ANOVA) (Fig. 1D). Claudin-2 is a pore-forming tight-junction protein that is highly upregulated in patients with inflammatory bowel diseases including ulcerative colitis and Crohn’s disease (27–30). Collectively, our findings indicate that sensitization of endothelial cells to radiation exacerbates delayed, but not acute, intestinal injury.
Delayed Radiation-Induced Intestinal Injury is Characterized by Increased Vascular Permeability and Tissue Hypoxia
Examination of the gross histology indicated that, compared to the small intestines from irradiated VECre; p53FL/+ mice, the small intestines from irradiated VECre; p53FL/FL mice that developed delayed intestinal injury were shorter and hemorrhagic (Fig. 2A). To quantify the severity of tissue injury, we examined intestinal tissue sections to calculate the radiation injury score according to a published scoring system (14, 15). This analysis revealed that irradiated VECre; p53FL/FL mice that succumbed to delayed intestinal injury (32 to 38 days after 15 Gy) showed a significantly higher injury score compared to irradiated VECre; p53FL/+ mice that were sacrificed at approximately the same time period (32 to 73 days after 15 Gy) (Fig. 2B and Supplementary Table S1; http://dx.doi.org/10.1667/RR15371.1.S1). The small intestines of VECre; p53FL/FL mice that developed delayed intestinal injury 32 days after irradiation exhibited hemorrhage of blood vessels with secondary epithelial damage and inflammation. Throughout the sections the lamina propria and submucosa were moderately expanded by edema and hemorrhage, which was characterized by a pale eosinophilic material mixed with small scattered aggregates of extravasated red blood cells (Fig. 2C).
FIG. 2.
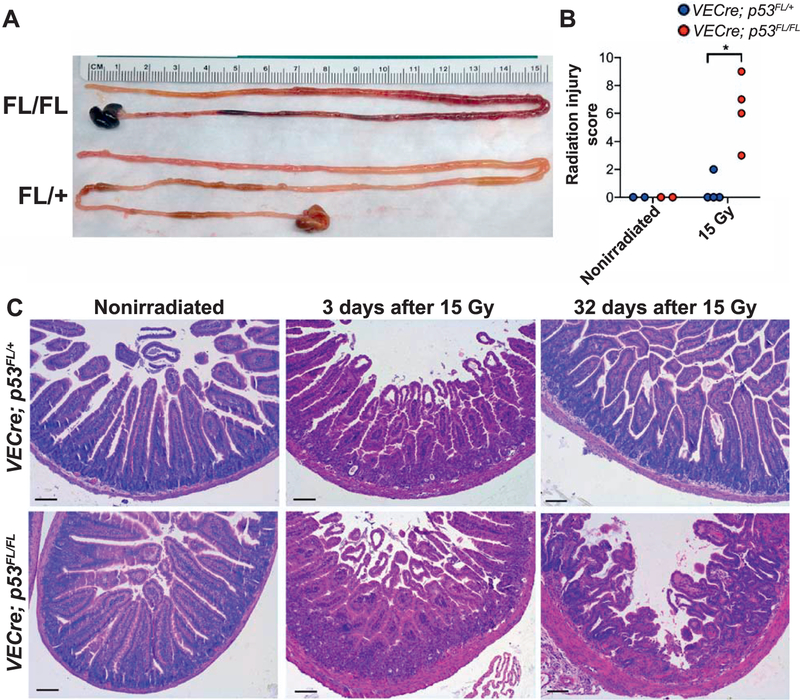
Histological examination of delayed radiation-induced intestinal injury. Panel A: Gross histology of small intestines collected from VECre; p53FL/FL and VECre; p53FL/+ mice at day 32 after 15 Gy TAI. Panel B: Quantification of tissue injury score of the small intestine. Tissues were collected from irradiated VECre; p53FL/FL mice that succumbed to delayed intestinal injury and irradiated VECre; p53FL/+ mice that were sacrificed at approximately the same time frame after 15 Gy TAI. Nonirradiated VECre; p53FL/FL and VECre; p53FL/ + mice were used as controls. *P < 0.05 by Mann-Whitney test. Each dot represents one mouse. Panel C: Representative H&E-stained sections of the small intestines collected from VECre; p53FL/+ and VECre; p53FL/FL unirradiated mice as well as at days 3 and 32 after 15 Gy TAI. Notably, at day 3 after 15 Gy TAI both VECre; p53FL/FL and VECre; p53FL/+ mice showed almost complete depletion of intestinal crypts. In contrast, at day 32 after 15 Gy TAI the VECre; p53FL/FL mice that developed delayed intestinal injury still retained some intestinal crypts even in regions where vascular injury was present. Scale bar: 100 μm.
We next examined vascular injury of the small intestines collected from VECre; p53FL/FL and VECre; p53FL/+ littermates 30 days after 15 Gy TAI by staining with GS-IB4 to label endothelial cells and an antibody against EF5 to identify regions of hypoxia (12). We found that while irradiated VECre; p53FL/+ mice retained vasculature within the villi and lacked regions of hypoxia, irradiated VECre; p53FL/FL mice exhibited destruction of GS-IB4+ microvessels in the villi and displayed focal lesions of tissue hypoxia (Fig. 3A). Quantification of tissue sections indicated that, compared to irradiated VECre; p53FL/+ mice, irradiated VECre; p53FL/FL mice showed a decrease in the percentage of GS-IB4+ fraction area (P = 0.057) and a significant increase in EF5+ fraction area per high-power field. We also assessed mRNA expression of hypoxia-responsive genes Tff3 (31) and Mdr1 (Abcbl) (32) in intestinal epithelial cells of VECre; p53FL/FL and VECre; p53FL/+ mice at day 30 after 15 Gy TAI. Our results showed that, compared to nonirradiated mice, the expression of Mdr1 was modestly increased in irradiated VECre; p53FL/+ mice, but it was highly elevated in a subset of irradiated VECre; p53FL/FL mice (Fig. 3D).
FIG. 3.

Delayed radiation-induced intestinal injury is characterized by microvessel damage and tissue hypoxia. Panel A: Representative tissue sections of one VECre; p53FL/+ mouse and two VECre; p53FL/FL mice at day 30 after 15 Gy TAI. The small intestines of irradiated VECre; p53FL/FL mice showed a decrease in GS-IB4+ endothelial cells within the lamina propria of the villi. In addition, the small intestines of irradiated VECre; p53FL/FL mice, but not the VECre; p53FL/+ mouse, exhibited areas of hypoxia as shown by EF5 staining. Note that in irradiated VECre; p53FL/FL mice, even in regions with loss of microvessels in the villi and associated hypoxia, the adjacent crypts (yellow arrows) are largely intact. Scale bar: 100 μm. Panels B and C: Quantification of the area stained positive for GS-IB4 or EF5 per high-power field 30 days after 15 Gy TAI. P value was calculated using Student’s t test. Each dot represents one mouse. Panel D: The expression of hypoxia-responsive genes Tff3 and Mdr1 (Abcb1) in intestinal epithelial cells from unirradiated mice or at day 30 after 15 Gy TAI. *P < 0.05 (two-way ANOVA with Bonferroni post hoc test).
To quantify vascular permeability of the small intestine in vivo, we utilized dual-energy-micro-CT imaging to detect the leakage of gold nanoparticles from blood vessels (21). Our previously published studies using a mouse model of radiation-induced cardiac injury demonstrated accumulation of gold in the myocardium where endothelial cells were damaged (21). Dual-energy-micro-CT was performed using VECre; p53FL/FL and VECre; p53FL/+ mice that were not irradiated or irradiated at day 28 after 15 Gy TAI. These mice were injected intravenously with a PEGylated gold nanoparticles three days before imaging and a blood-pool Lip-I immediately before imaging to label the blood pool (21). Compared to VECre; p53FL/+ mice and VECre; p53FL/FL mice at day 28 after 15 Gy TAI had a marked accumulation of gold nanoparticles along the irradiated GI tract (Fig. 4A). Quantification of gold signals also indicated a significant increase in the amount and concentration of gold in the small intestine of VECre; p53FL/FL mice at day 28 after 15 Gy TAI (Fig. 4B and C). Collectively, our results demonstrate that radiosensitization of endothelial cells promotes delayed intestinal radiation injury that is manifested by increased vascular permeability and tissue hypoxia.
FIG. 4.

Assessing vascular permeability of the small intestine in vivo using dual-energy-micro-CT. Panel A: VECre; p53FL/+ and VECre; p53FL/FL mice were intravenously injected with gold nanoparticle (AuNp; green) contrast agent once on day 0. Three days later mice were injected with iodinated nanoparticle (Lip-I; red). The use of both types of nanoparticles enables the measurement of vascular permeability in the irradiated intestines (via AuNp, which extravasated after a few days and accumulated in the injured tissue), and the measurement of vascularity (via Lip-I which remains within the blood vessels). Accumulation of AuNp along the small intestine is indicated by yellow arrows. Panels B and C: Quantification of the mean gold mass and gold concentration in the small intestine at days 0 and 28 after 15 Gy TAI. *P < 0.05 (Student’s t test). Each dot represents one mouse.
DISCUSSION
Delayed radiation enteropathy is a chronic condition that affects many long-term cancer survivors. It is associated with significant morbidity and mortality with few therapeutic options. While new treatment techniques for delivering radiation more precisely to the tumor have decreased the risk of late GI toxicity for some patients, for many tumors some radiation exposure to the intestines is unavoidable. Thus, for certain patients, such as those with pancreatic cancer, GI normal tissue toxicity remains a critical dose-limiting obstacle to achieving local control. Defining the underlying mechanisms that drive the pathology of late GI enteropathy after irradiation is an important step toward developing preventative measures and treatment options. Advancing the management of radiation enteropathy would not only improve the outcomes for current patients, but might allow for the delivery of a higher radiation dose to tumors to increase local tumor control. Our data illustrate a clear mechanistic distinction in the pathophysiology of acute GI radiation syndrome and delayed radiation enteropathy. We showed that endothelial cell damage mediated by the loss of p53 is not a key mediator of the acute radiation syndrome, however, it is sufficient to cause delayed intestinal radiation injury (Fig.1). Therefore, these results suggest that developing treatment strategies to preserve endothelial cell function in the intestine after irradiation may ameliorate late effects from radiation.
The findings from our mouse model also provide mechanistic insight into the pathogenesis of delayed radiation enteropathy. Our data indicate that radiosensitization of endothelial cells causes an increase in vascular permeability and tissue hypoxia, which collectively contribute to secondary damage to intestinal epithelium. As shown by the FITC-dextran assay, the level of FITC-dextran in serum is much lower in mice during the delayed injury phase (day 30 after 15 Gy TAI) compared to mice that developed acute injury (day 5 after 15 Gy TAI) (Fig. 1B). Indeed, intestinal tissue sections of mice that developed delayed intestinal radiation injury did not show extensive destruction of intestinal crypts (Figs. 2C and 3A). In addition, the expression of the tight junction gene Tjp1 (ZO-1) is not significantly decreased in intestinal epithelial cells of either VECre; p53FL/FL or VECre; p53FL/+ mice at day 30 after 15 Gy TAI. On the other hand, intestinal epithelial cells from both VECre; p53FL/FL and VECre; p53FL/+ mice at day 30 after 15 Gy TAI showed a significant increase in Claudin-2 (Fig. 1D). Claudin-2 is a unique tight junction protein that increases the permeability of the epithelial barrier by forming a paracellular channel for small cations and water (33, 34). Multiple studies have shown that Claudin-2 is highly upregulated in patients with ulcerative colitis and Crohn’s disease (27–30). Intriguingly, mice that overexpress a Claudin-2 transgene in the GI epithelium are protected from experimental colitis, despite increased intestinal mucosal permeability (35). Therefore, further studies are warranted to investigate the role of Claudin-2 in regulating the pathogenesis of radiation enteropathy.
The association between early injury and late effects has remained an important question in GI radiation-induced pathophysiology (4). Prior studies have suggested a disassociation between acute and late GI toxicities. For example, rat models that exhibit extensive early epithelial cell injury develop only low levels of intestinal fibrosis at late timepoints (36, 37). Our data are consistent with a model where delayed intestinal radiation injury may arise from damage to a cell population that is distinct from the epithelial cells lost during acute radiation injury. Although others have maintained that early endothelial cell death is a key mechanism of GI crypt loss driving the acute GI radiation syndrome (38), our data are consistent with studies reporting minimal endothelial cell death contributing to the acute GI radiation syndrome (39, 40). Indeed, it is remarkable that in VECre; p53FL/FL mice at day 30 after 15 Gy TAI where the microvasculature is lost from villi with vascular injury documented by hypoxia (Fig. 3A) and leakiness of gold nanoparticles (Fig. 4), the adjacent crypts remain largely intact (Fig. 3A).
Endothelial cell damage has been shown to mediate other radiation-induced delayed effects. A reduction in microvasculature precedes organ damage in the heart in multiple models of radiation-induced myocardial injury (41–45). Indeed, we showed that p53 deletion in endothelial cells in Tie2Cre and VE-cadherin-Cre mice sensitized mice to myocardial injury after whole-heart irradiation due to increased endothelial cell death and reduced myocardial microvascular density (12). In the current study, we demonstrate that loss of endothelial cells after abdominal irradiation in VE-cadherin-Cre mice with p53 deletion in endothelial cells leads to reduced intestinal length and an increase in hypoxic regions. These data are consistent with a model where late radiation enteropathy can occur as a result of a decrease in blood supply over time, which leads to parenchymal destruction, tissue remodeling and replacement with fibrotic tissue (46). Our results with mice in which deletion of p53 specifically in endothelial cells sensitized endothelial cells to radiation provides strong genetic evidence that endothelial cell injury can initiate late radiation enteropathy. Moreover, these results suggest that therapies that maintain endothelial cell integrity and function after radiotherapy could limit late intestinal radiation injury.
Supplementary Material
ACKNOWLEDGMENTS
The content of this work was presented at the the CONTREC Conference, May 14–18, 2018, Winthrop Rockefeller Institute, Petit Jean Mountain, Morrilton, AR. This work was supported by National Institutes of Health [grant nos. R35CA197616 (DGK), U19AI067798 (DGK), R01CA196667 (CTB), U24CA220245 (CTB)] and by the Whitehead Scholar Award from Duke University School of Medicine (C-LL). All CT imaging was performed by the Duke Center for In Vivo Microscopy, an NIH/National Institute of Biomedical Imaging and Bioengineering (NIBIB) Biomedical Technology Resource Center (grant no. P41 EB015897). The authors have no conflicting financial interests with this work. DGK is a cofounder of and stockholder in XRAD Therapeutics, which is developing radiosensitizers. DGK is a member of the scientific advisory board for and owns stock in Lumicell, Inc, a company commercializing intraoperative imaging technology. He is an inventor of a handheld imaging device under U.S. patent 20140301950-A1 and is a co-inventor on a submitted patent on radiosensitizers. XRAD Therapeutics, Merck, Bristol Myers Squibb and Eli Lilly provide research support to DGK.
Footnotes
Editor’s note. The online version of this article (DOI: 10.1667/RR15371.1) contains supplementary information that is available to all authorized users.
REFERENCES
- 1.Shadad AK, Sullivan FJ, Martin JD, Egan LJ Gastrointestinal radiation injury: symptoms, risk factors and mechanisms. World J Gastroenterol 2013; 19:185–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Husebye E, Hauer-Jensen M, Kjorstad K, Skar V. Severe late radiation enteropathy is characterized by impaired motility of proximal small intestine. Dig Dis Sci 1994; 39:2341–9. [DOI] [PubMed] [Google Scholar]
- 3.Hauer-Jensen M, Poulakos L, Osborne JW Effects of accelerated fractionation on radiation injury of the small intestine: a new rat model. Int J Radiat Oncol Biol Phys 1988; 14:1205–12. [DOI] [PubMed] [Google Scholar]
- 4.Hauer-Jensen M, Denham JW, Andreyev HJ Radiation enteropathy–pathogenesis, treatment and prevention. Nat Rev Gastroenterol Hepatol 2014; 11:470–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang J, Boerma M, Fu Q, Hauer-Jensen M. Significance of endothelial dysfunction in the pathogenesis of early and delayed radiation enteropathy. World J Gastroenterol 2007; 13:3047–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taniguchi CM, Miao YR, Diep AN, Wu C, Rankin EB, Atwood TF, et al. PHD inhibition mitigates and protects against radiation- induced gastrointestinal toxicity via HIF2. Sci Transl Med 2014; 6:236ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kirsch DG, Santiago PM, di Tomaso E, Sullivan JM, Hou WS, Dayton T, et al. p53 controls radiation-induced gastrointestinal syndrome in mice independent of apoptosis. Science 2010: 327:593–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leibowitz BJ, Wei L, Zhang L, Ping X, Epperly M, Greenberger J, et al. Ionizing irradiation induces acute haematopoietic syndrome and gastrointestinal syndrome independently in mice. Nat Commun 2014; 5:3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metcalfe C, Kljavin NM, Ybarra R, de Sauvage FJ Lgr5+ stem cells are indispensable for radiation-induced intestinal regeneration. Cell Stem Cell 2014; 14:149–59. [DOI] [PubMed] [Google Scholar]
- 10.Olcina MM, Giaccia AJ Reducing radiation-induced gastrointestinal toxicity - the role of the PHD/HIF axis. J Clin Invest 2016; 126:3708–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Booth C, Tudor G, Tudor J, Katz BP, MacVittie TJ Acute gastrointestinal syndrome in high-dose irradiated mice. Health Phys 2012; 103:383–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee CL, Moding EJ, Cuneo KC, Li Y, Sullivan JM, Mao L, et al. p53 functions in endothelial cells to prevent radiation-induced myocardial injury in mice. Sci Signal 2012; 5:ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee CL, Oh P, Xu ES, Ma Y, Kim Y, Daniel AR, et al. Blocking cyclin-dependent kinase 4/6 during single dose versus fractionated radiation therapy leads to opposite effects on acute gastrointestinal toxicity in mice. Int J Radiat Oncol Biol Phys 2018; 102:1569–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langberg CW, Sauer T, Reitan JB, Hauer-Jensen M. Tolerance of rat small intestine to localized single dose and fractionated irradiation. Acta Oncol 1992; 31:781–7. [DOI] [PubMed] [Google Scholar]
- 15.Langberg CW, Sauer T, Reitan JB, Hauer-Jensen M. Influence of fractionation schedule on development of intestinal complications following localized irradiation. An experimental study in the rat. Acta Oncol 1994; 33:403–8. [DOI] [PubMed] [Google Scholar]
- 16.Volynets V, Rings A, Bardos G, Ostaff MJ, Wehkamp J, Bischoff SC Intestinal barrier analysis by assessment of mucins, tight junctions, and alpha-defensins in healthy C57BL/6J and BALB/cJ mice. Tissue Barriers 2016; 4:e1208468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Badea C, Johnston S, Johnson B, De Lin M, Hedlund LW, Johnson GA A dual micro-CT System for small animal imaging. Proc SPIE Int Soc Opt Eng 2008; 6913:691342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holbrook M, Clark DP, Badea CT Low-dose 4D cardiac imaging in small animals using dual source micro-CT. Phys Med Biol 2018; 63:025009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul 2001; 41:189–207. [DOI] [PubMed] [Google Scholar]
- 20.Mukundan S Jr, Ghaghada KB, Badea CT, Kao CY, Hedlund LW, Provenzale JM, et al. A liposomal nanoscale contrast agent for preclinical CT in mice. AJR Am J Roentgenol 2006; 186:300–7. [DOI] [PubMed] [Google Scholar]
- 21.Lee CL, Min H, Befera N, Clark D, Qi Y, Das S, et al. Assessing cardiac injury in mice with dual energy-microCT, 4D-microCT, and microSPECT imaging after partial heart irradiation. Int J Radiat Oncol Biol Phys 2014; 88:686–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clark DP, Ghaghada K, Moding EJ, Kirsch DG, Badea CT In vivo characterization of tumor vasculature using iodine and gold nanoparticles and dual energy micro-CT. Phys Med Biol 2013: 58:1683–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clark DP, Lee CL, Kirsch DG, Badea CT Spectrotemporal CT data acquisition and reconstruction at low dose. Medical Phys 2015; 42:6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J Cell Biol 1999: 147:1351–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Furuse M, Itoh M, Hirase T, Nagafuchi A, Yonemura S, Tsukita S, et al. Direct association of occludin with ZO-1 and its possible involvement in the localization of occludin at tight junctions. J Cell Biol 1994; 127:1617–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H, Sun RT, Li Y, Yang YF, Xiao FJ, Zhang YK, et al. HGF gene modification in mesenchymal stem cells reduces radiation- induced intestinal injury by modulating immunity. PLoS One 2015; 10:e0124420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luettig J, Rosenthal R, Barmeyer C, Schulzke JD Claudin-2 as a mediator of leaky gut barrier during intestinal inflammation. Tissue Barriers 2015; 3:e977176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weber CR, Nalle SC, Tretiakova M, Rubin DT, Turner JR Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab Invest 2008; 88:1110–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeissig S, Burgel N, Gunzel D, Richter J, Mankertz J, Wahnschaffe U, et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007; 56:61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prasad S, Mingrino R, Kaukinen K, Hayes KL, Powell RM, MacDonald TT, et al. Inflammatory processes have differential effects on claudins 2, 3 and 4 in colonic epithelial cells. Lab Invest 2005; 85:1139–62. [DOI] [PubMed] [Google Scholar]
- 31.Hernandez C, Santamatilde E, McCreath KJ, Cervera AM, Diez I, Ortiz-Masia D, et al. Induction of trefoil factor (TFF)1, TFF2 and TFF3 by hypoxia is mediated by hypoxia inducible factor-1: implications for gastric mucosal healing. Br J Pharmacol 2009; 156:262–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res 2002; 62:3387–94. [PubMed] [Google Scholar]
- 33.Amasheh S, Meiri N, Gitter AH, Schoneberg T, Mankertz J, Schulzke JD, et al. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J Cell Sci 2002; 115:4969–76. [DOI] [PubMed] [Google Scholar]
- 34.Rosenthal R, Milatz S, Krug SM, Oelrich B, Schulzke JD, Amasheh S, et al. Claudin-2, a component of the tight junction, forms a paracellular water channel. J Cell Sci 2010; 123:1913–21. [DOI] [PubMed] [Google Scholar]
- 35.Ahmad R, Chaturvedi R, Olivares-Villagomez D, Habib T, Asim M, Shivesh P, et al. Targeted colonic claudin-2 expression renders resistance to epithelial injury, induces immune suppression, and protects from colitis. Mucosal Immunol 2014; 7:1340–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng H, Wang J, Hauer-Jensen M. Role of mast cells in early and delayed radiation injury in rat intestine. Radiat Res 2000; 153:533–9. [DOI] [PubMed] [Google Scholar]
- 37.Wang J, Zheng H, Kulkarni A, Ou X, Hauer-Jensen M. Regulation of early and delayed radiation responses in rat small intestine by capsaicin-sensitive nerves. Int J Radiat Oncol Biol Phys 2006; 64:1528–36. [DOI] [PubMed] [Google Scholar]
- 38.Paris F, Fuks Z, Kang A, Capodieci P, Juan G, Ehleiter D, et al. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science 2001; 293:293–7. [DOI] [PubMed] [Google Scholar]
- 39.Schuller BW, Rogers AB, Cormier KS, Riley KJ, Binns PJ, Julius R, et al. No significant endothelial apoptosis in the radiation-induced gastrointestinal syndrome. Int J Radiat Oncol Biol Phys 2007; 68:205–10. [DOI] [PubMed] [Google Scholar]
- 40.Schuller BW, Binns PJ, Riley KJ, Ma L, Hawthorne MF, Coderre JA Selective irradiation of the vascular endothelium has no effect on the survival of murine intestinal crypt stem cells. Proc Natl Acad Sci U S A 2006; 103:3787–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fajardo LF, Stewart JR Pathogenesis of radiation-induced myocardial fibrosis. Lab Invest 1973; 29:244–57. [PubMed] [Google Scholar]
- 42.Fajardo LF, Stewart JR Experimental radiation-induced heart disease. I. Light microscopic studies. Am J Pathol 1970; 59:299–316. [PMC free article] [PubMed] [Google Scholar]
- 43.Lauk S, Kiszel Z, Buschmann J, Trott KR Radiation-induced heart disease in rats. Int J Radiat Oncol Biol Phys 1985; 11:801–8. [DOI] [PubMed] [Google Scholar]
- 44.Yeung TK, Lauk S, Simmonds RH, Hopewell JW, Trott KR Morphological and functional changes in the rat heart after X irradiation: strain differences. Radiat Res 1989; 119:489–99. [PubMed] [Google Scholar]
- 45.Seemann I, Gabriels K, Visser NL, Hoving S, Te Poele JA, Pol JF, et al. Irradiation induced modest changes in murine cardiac function despite progressive structural damage to the myocardium and microvasculature. Radiother Oncol 2012; 103:143–50. [DOI] [PubMed] [Google Scholar]
- 46.Wynn TA Cellular and molecular mechanisms of fibrosis. J Pathol 2008; 214:199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.