

Abstract
The functionalization of C–H bonds is an essential reaction in biology and chemistry. Metalloenzymes that often exhibit this type of reactivity contain metal-oxido intermediates which are directly involved the initial cleavage of the C–H bonds. Regulation of the cleavage process is achieved, in part, by hydrogen bonds that are proximal to the metal–oxido units, yet our understanding of their exact role(s) is still emerging. To gain further information into the role of H-bonds on C–H bond activation, a hybrid set of urea-containing tripodal ligands has been developed in which a single H-bond can be adjusted through changes in the properties of one ureayl N–H bond. This modularity is achieved by appending a phenyl ring with different para-substituents from one ureayl NH group. The ligands have been used to prepare a series of MnIII–oxido complexes and a Hammett correlation was found between the pKa values of the complexes and the substituents on the phenyl ring that was explained within the context of changes to the H-bonds involving the MnIII–oxido unit. The complexes were tested for their reactivity toward 9,10-dihydroanthracene (DHA) and a Hammett correlation was found between the second-order rate constants for the reactions and the pKa values. Studies to determine activation parameters and the kinetic isotope effects are consistent with a mechanism in which rate-limiting proton transfer is an important contributor. However, additional reactivity studies with xanthene found a significant increase in the rate constant compared to DHA, even though the substrates have the same pKa(C—H) values. These results suggest do not support a discrete proton-transfer/electron transfer process, but rather an asynchronous mechanism in which the proton and electron are transferred unequally at the transition state.
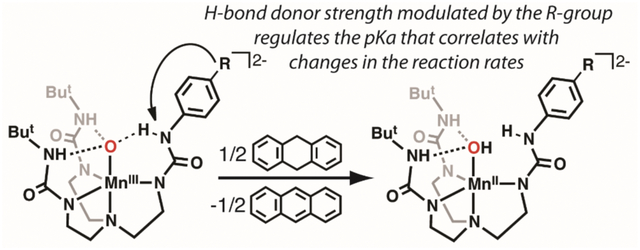
Graphical Abstract
Introduction
Metal–oxido complexes have long been implicated as key intermediates in a variety of chemical transformations.1–6 Examples include FeIV–oxido species found within the active sites of proteins which are proposed to homolytically cleave unactivated C–H bonds from external substrates.7–13 Similarly, there are several examples of synthetic MnIV/V–oxido complexes that have the ability to functionalize substrates via C–H bond activation.6,14–20 These reactions are often thermodynamically challenging because C–H bonds have relatively large bond dissociation free energies (BDFEs) that can approach 100 kcal/mol. Because of this difficulty, it is usually thought that reagents for C–H bond functionalization have to be strong oxidants; this concept is particularly relevant to M–oxido species in which the 4+ formal oxidation state at the metal center is common. However, strong oxidants are often unselective and are not useful for inclusion in late-stage synthetic preparations. Moreover, housing a powerful oxidant within a protein or a synthetic ligand is problematic because they can lead to intramolecular oxidative damage and ultimately, dysfunction.21,22
One way to further evaluate the reactivity of metal-oxido complexes with C–H bonds is to consider their ground state thermodynamic properties (Figure 1).23,24 This approach follows from the Bell-Evans-Poyanyi principle25,26 and requires that the BDFEs for the C–H bond broken be comparable to that of the M(O–H) bond formed during a reaction (Figure 1B). Within the context of the M–oxido thermodynamic scheme (Figure 1A), three mechanistic cases are often described: 1) concerted proton-electron transfer (CPET); 2) a two-step electron transfer-proton transfer path (ET-PT); and 3) a two-step proton transfer-electron transfer path (PT-ET). The scheme also illustrates that redox potentials alone are not always sufficient to describe the reactivity of a M–oxido towards C–H bonds: the basicity of the oxido ligand can also influence reactivity and can be evaluated via the pKa value of the conjugate acid of the M–oxido complex (that is, its M–OH analog). Moreover, this approach has been used to argue that complexes with a relatively basic oxido ligand can have low redox potentials and still cleavage strong C–H bonds.17,22,27,28
Figure 1.

Thermodynamic square schemes for M–oxido complexes (A) and substrates with C–H bonds (B).
These thermodynamic concepts have been used to explain the reactivity in cytochrome P450 enzymes (P450s), a class of monooxygenases that are central to the production of hormones and the metabolism of xenobiotics.29 Additional studies on synthetic systems have supported the pivotal role that the basicity of the oxido ligand can have on function.5,17,30 We have contributed to this area by examining a series of MIII–and MIV–oxido (M = Fe, Mn) complexes with the tripodal ligand [H3buea]3- and have found that even the low-valent MIII–oxido species are capable of cleaving C–H bonds (Figure 2A).4,28,31–33 For instance, MnIII–oxido complex [MnIIIH3buea(O)]2- can cleave the C–H bond in 9,10-dihyroanthracene (DHA, BDFEC—H = 75 kcal/mol) even though it has a redox potential of less than −2.0 V vs [FeIII/IICp2]+/0.28 This complex has a basic oxido ligand as gauged from the pKa value of 28 for its conjugate acid, [MnIIIH3buea(OH)]−, that was experimentally measured in DMSO. We made use of this information to argue that this reactivity was driven by the highly basic oxido ligand in [MnIIIH3buea(O)]2- in which proton transfer was part of the rate determining step.
Figure 2.

Previous reported MnIII–oxido (A, B) and newly design systems used in this study (C, D).
These thermodynamic insights further suggest that the basicity of the oxido ligand can be used as a tunable parameter to modulate the reactivity of metal–oxido complexes towards C–H bonds. We sought to develop a system that would allow us to test this premise by systematically modulating the basicity of the MnIII–oxido unit and then determining whether these changes would affect the rate of C–H bond cleavage. The system that we designed modulated the basicity by changing a single H-bond to the MnIII–oxido unit. We have re-designed [H3buea]3- into the new hybrid ligands [H3bpuea-R]3- (R = OMe, H, F, Cl, CF3, 5F) in which one of the tripodal arms contains a pheny lurea unit (Figure 2).34 Notice that two of the tripodal arms are the same in each ligand with urea groups that contain appended tert-butyl groups as in [H3buea]3-. However, the third arm is modified to influence the remaining H-bond. The placement of different substituents at the para position of the phenyl ring allowed us to change the acidity of the HNurea group. This change modulates the H-bond donor strength to the MnIII–oxido unit which, in turn, alters the basicity of the oxido ligand (Figure 2C–D). We report the preparation of the MnIII–oxido complexes for the series of [H3bpuea-R]3- ligands and describe how the change in this single H-bond within the secondary coordination sphere affects their structural and physical properties. The reactivity of the complexes with external substrates was examined and a correlation was established between oxido ligand basicity and the second-order rate constant for C–H bond cleavage.
Experimental
Reagents and Materials.
Unless otherwise stated, all manipulations were performed under an argon atmosphere in a Vacuum Atmospheres, Co. drybox. N, N-dimethylacetamide (DMA) was purchased from Sigma-Aldrich and further dried as follows; prior to use, the liquid was stirred over BaO for two days, refluxed for 1 h, and then vacuum distilled. The DMA obtained was further dried by storing over molecular sieves (3Å). 1-tert-Butyl-3‒(2‒chloroethyl)urea, N-tert-butoxycarbonyl‒1,2‒diaminoethane, (N’-tert-butyoxycarbonyl)-N-ethyl]-bis[(N’-tert-butylureayl), 1,1’-(((2‒aminoethyl)azanediyl)bis(ethane-2,1-diyl))bis(3‒(tert-butyl)urea) were prepared by following literature methods.35–38 The synthetic routes for the pre-ligands [H6bpuea-R] are found in the Supporting Information. The metal precursor MnII(OAc)2 was obtained from Sigma-Aldrich and was used as received. Potassium hydride (KH) as a 30% dispersion in mineral oil was filtered with a glass frit and washed with 20 mL pentane and Et2O 5 times, dried under vacuum, and stored under an argon atmosphere. 9,10-Dihydroanthracene (DHA) was purchased from Sigma-Aldrich and crystallized from ethanol three times, washed with pentane, and dried under vacuum. d4-DHA and d2-xanthene were synthesized by following literature procedure.28,39 4-Aminopyridine was purchased from Sigma-Aldrich in ≥ 99% purity, crystallized from toluene, washed with Et2O, and dried under vacuum. 2‒Aminopyrimidine was purchased from Sigma-Aldrich in 97% purity, crystallized from EtOH three times, washed with Et2O, and dried under vacuum. Silica gel (40–60 μm) was used for column chromatography to purify the pre-ligands.
Physical Methods.
Electronic absorption spectra for kinetics experiments were recorded in a 1 cm cuvette on an 8453E Agilent UV-vis spectrophotometer equipped with an Unisoku Unispeks cryostat. Room temperature electronic absorption spectra for determining extinction coefficients were recorded in a 1 cm cuvette on a Cary 50 spectrophotometer. Room temperature electronic absorption spectra used for measuring the pKa values of the Mn–OH complexes were recorded in a 1 cm cuvette on a Cary 60 spectrophotometer that was housed within a N2 atmosphere glove box; connections were made using fiber optic cables. Electron paramagnetic resonance (EPR) spectra were recorded using a X-band Bruker EMX spectrometer equipped with an ER041XG microwave bridge, an Oxford Instrument liquid-helium quartz cryostat, and a dual mode cavity (ER4116DM). 1H and 13C nuclear magnetic resonance (NMR) spectroscopies were conducted using a Bruker DRX500 spectrometer. Cyclic voltammetry experiments were conducted using a CHI600C electrochemical analyzer. A 2.0 mm glassy carbon electrode was used as the working electrode at scan velocities of 50 Mv·s−1. A ferrocenium/ferrocene couple (FeCp2+/FeCp2) was used to monitor the Ag wire reference electrode, and all potentials are referenced to the [FeCp2]+/0 couple. The synthetic procedures for the H6bpuea-R pre-ligands, the K2[MnIIH3buea(OH)] salts needed for the determination of BDFE values, the methods for determining of the pKa values of the [MnIIIH3buea(OH)]− complexes, and the methods for measuring the second-order rate constant for the reactions involving DHA and the [MnIII (H3bpuea-R)O]2- complex are found in the Supporting Information.
Preparative Methods.
K2[MnIIIH3bpuea-OMe(O)] (1). H6bpuea-OMe (0.152 g, 0.307 mmol) was dissolved in 3 mL DMA and solid KH (0.0516 g, 1.29 mmol) was added in one portion. The mixture was stirred for 45 min which was sufficient time for gas evolution to cease. The solution was treated with solid MnII(OAc)2 (0.0545 g, 0.315 mmol) and stirred for an additional 2 h to produce a light yellow heterogenous mixture. After transferring to a Schlenk flask and sealing with a rubber septum, the mixture was treated with dry O2 (3.4 mL, 0.15 mmol) that produced an immediate color change to brown. The mixture was further allowed to stir for 2 h and then degassed under vacuum for 5 min. The flask was transferred back to a drybox, after which the reaction mixture was filtered through a medium-porosity glass filter and the brown filtrate was layered with 10 mL Et2O. After 1-day, a brown solid was collected on a medium-porosity glass filter and washed with 20 mL of MeCN and 5 mL of Et2O to give 0.159 g of the desired purple solid (81%). SSingle crystals suitable for X-ray diffraction was obtained by vapor diffusion of Et2O to a DMF solution of the salt. λmax/nm (DMSO, ε, M−1 cm−1): 714, (211); 500, (419). EPR (2:1, DMF:THF, 10 K): g = 8.03, Az = 277 MHz). HRMS (ES+): [C24H40K2MnN7O5 + H+], 640.1824; Found, 640.1826. Anal. Calcd (found) for C24H40K2MnN7O5•H2O: C, 43.82 (43.87); H, 6.44 (6.53); N, 14.91 (15.50). FTIR (ATR, cm−1): 2961, 2896, 2853, 2831, 1661, 1595, 1579, 1526, 1505, 1447, 1407, 1382, 1352, 1327, 1315, 1249, 1223, 1201, 1177, 1139, 1092, 1061, 1034, 915, 850, 828, 787, 758, 722, 682, 660.
K2[MnIIIH3bpuea-H(O)] (2) was prepared following the same procedure as described above for K2[MnIIIH3bpuea-OMe(O)] using KH (0.0501 g, 1.25 mmol), H6bpuea-H (0.143 g, 0.308 mmol), MnII(OAc)2 (0.0534 g, 0.309 mmol), and dry O2 (3.5 mL, 0.16 mmol). K2[MnIIIH3LH(O)] was isolated by layering Et2O over a DMA solution of the salt. The isolated solid was washed with 20 mL of MeCN and 20 mL of Et2O to give 0.089 g of the desired purple solid (47%). λmax/nm (DMSO, ε, M−1 cm−1): 710, (240); 498, (438). EPR (DMF:THF, 10 K): g = 7.95, Az = 278 MHz). MS (ES+): [C23H38K2MnN7O4 + 3K+], 648.13; Found, 648.00. Anal. Calcd (found) for C23H38K2MnN7O4•DMA: C, 46.54 (46.46); H, 6.80 (6.98); N, 16.08 (15.59). FTIR (ATR, cm−1): 2960, 2899, 2856, 2811, 1641, 1592, 1574, 1532, 1497, 1478, 1449, 1412, 1384, 1354, 1331, 1262, 1249, 1219, 1169, 1139, 1098, 1034, 914, 834, 778, 767, 748, 693, 617.
K2[MnIIIH3bpuea-F(O)] (3) was prepared following the same procedure as described above for K2[MnIIIH3bpuea-OMe(O)] using KH (0.039 g, 0.96 mmol), H6bpuea-F (0.113 g, 0.235 mmol), MnII(OAc)2 (0.0412 g, 0.238 mmol), and dry O2 (2.6 mL, 0.12 mmol). K2[MnIIIH3bpuea-F(O)] was isolated by layering Et2O over a DMA solution of the salt. The precipitate was washed with 20 mL MeCN and 20 mL of Et2O to give 0.063 g of the desired purple solid (43%). λmax/nm (DMSO, ε, M−1 cm−1): 706, (242), 498, (443). EPR (DMF:THF, 10 K): g = 7.95, Az = 278 MHz). MS (ES+): [C23H37K2FMnN7O4 + 3K+], 666.1; Found, 666.0. Anal. Calcd (found) for C23H37FK2MnN7O4•DMA: C, 45.37 (45.25); H, 6.49 (6.71); N, 15.68 (15.70). FTIR (ATR, cm−1): 2959, 2897, 2856, 1627, 1589, 1530, 1504, 1447, 1411, 1396, 1383, 1351, 1326, 1257, 1250, 1223, 1198, 1087, 1057, 1036, 1013, 912, 831, 785, 767, 681, 590.
K2[MnIIIH3bpuea-Cl(O)] (4) was prepared following the same procedure as described above for K2[MnIIIH3bpuea-OMe(O)] using KH (0.0348 g, 0.867 mmol), H6bpuea-Cl (0.108 g, 0.216 mmol), MnII(OAc)2 (0.038 g, 0.22 mmol), and dry O2 (2.6 mL, 0.11 mmol). K2[MnIIIH3LCl(O)] was isolated by layering Et2O over a DMA solution of the salt. The precipitate was washed with ~25 mL MeCN and then dried under vacuum to get 0.052 g of the salt (37%). λmax/nm (DMSO, ε, M−1 cm−1): 697, (296); 500 (428). EPR (2:1, DMF:THF, 10 K): g = 8.05, Az = 279 MHz). MS (ES+): [C23H37ClK2MnN7O4 + H+], 644.13; Found, 644.03, [C23H37ClMnN7O4 + 3K+], 682.09; Found, 681.98. Anal. Calcd (found) for C, 42.88 (42.48); H, 5.79 (6.23); N, 15.22 (15.67). FTIR (ATR, cm−1): 2965, 2899, 2860, 1590, 1529, 1488, 1448, 1384, 1360, 1332, 1330, 1251, 1219, 1199, 1166, 1139, 1090, 1034,1002, 918, 821, 785, 685, 640, 590.
K2[MnIIIH3bpuea-CF3(O)] (5) was prepared following the same procedure as described above for K2[MnIIIH3bpuea-OMe(O)] using KH (0.040 g, 1.0 mmol), H6bpuea-CF3 (0.134 g, 0.252 mmol), MnII(OAc)2 (0.043 g, 0.252 mmol), and dry O2 (3.0 mL, 0.13 mmol). K2[MnIIIH3bpuea-CF3(O)] was isolated by layering Et2O over a DMA solution of the salt. The precipitate was washed with ~12 mL MeCN and then dried under vacuum to get 0.048 g of the salt (28%). λmax/nm (DMSO, ε, M−1 cm−1): 650 (300). EPR (DMF:THF, 10 K): g = 8.16, Az = 268 MHz). MS (ES+): [C24H37F3MnN7O4 + 2K+ + H+], 678.16; Found, 678.0. Anal. Calcd (found) for C24H37F3K2MnN7O4•3H2O: C, 39.39 (39.68); H, 5.92 (4.99); N, 13.40 (12.73). FTIR (ATR, cm−1): 2960, 2847, 1640, 1595, 1505, 1436, 1400, 1355, 1310, 1270, 1212, 1180, 1156, 1106, 1062, 1017,964, 919, 842, 785, 740, 720, 691, 6667, 618.
K2[MnIIIH2bpuea-5F(OH)] (6) was prepared following the same procedure as described above for K2[MnIIIH3bpuea-OMe(O)] using KH (0.0920 g, 2.29 mmol), H6bpuea-5F (0.316 g, 0.570 mmol), MnII(OAc)2 (0.0998 g, 0.577 mmol), and dry O2 (6.4 mL, 0.29 mmol). The salt was isolated as a green powder after diffusion of Et2O into the DMA solution. The green precipitate was collected on a medium-porosity glass filter and washed with 10 mL of MeCN and 20 mL of Et2O to give 0.265 g (66%) of the desired green solid (66%). Single crystals of (NMe4)2[MnIIIH2bpuea-5F(OH)] were obtained by adding 2 equiv of NMe4OAc to the solution of K2[MnIIIH2bpuea-5F(OH)] in MeCN, filtering through a medium porous-glass filter and allowing Et2O to diffuse into the solution. λmax/nm (DMSO, ε, M−1 cm−1): 390 (1350), 677 (274). EPR (DMF:THF, 10 K): g = 7.97, Az = 262 MHz). Anal. Calcd (found) for C23H33F5K2MnN7O4•0.5Et2O: C, 40.76 (40.52); H, 5.20 (5.53); N, 13.31 (13.27).
Molecular Structure Determination.
For molecular structure determination (X-ray diffraction; XRD), Bruker SMART APEX II diffractometer was employed. Data collection and the unit-cell parameters determination was performed by APEX240 program package. The raw data was processed with SAINT41 and SADABS42 to get the reflection data file. The SHELXTL43 program was used for subsequent calculations. There were no systematic absences nor any diffraction symmetry other than the Friedel condition. For 1, structure was solved by dual space methods and refined on F2 by full-matrix least-squares techniques. The analytical scattering factors44 for neutral atoms were used throughout the analysis. Hydrogen atoms were included using a riding model. Hydrogen atoms associated with O(14) and O(15) could not be located and were not included in the refinement. There were several high residuals present in the final difference-Fourier map. It was not possible to determine the nature of the residuals. The SQUEEZE45 routine in the PLATON46 program package was used to account for the electrons in the solvent accessible voids. The structure of 6 was solved by direct space methods and refined on F2 by full-matrix least-squares techniques. Hydrogen atoms associated with O1, N6, and N7 were located from a difference-Fourier map and refined (x,y,z and Uiso) and the remaining hydrogen atoms were included using a riding model.
Results and Discussion
Design Considerations and Preparative Methods.
The design of complexes 1-6 relies on modifications within the secondary coordination sphere with minimal changes within the primary coordination sphere. We reasoned that using para-R-phenyl groups would minimize the effects on the primary coordination sphere because the major changes within the series of complexes would be at the HNurea group which is not coordinated to the Mn ion. This more remote HNurea group is, however, involved in forming an intramolecular H-bond; therefore, as the primary coordination sphere around the Mn center remains relatively constant, a change in one H-bond within the secondary coordination sphere would occur that affects the basicity of the oxido ligand. Moreover, as the phenyl ring becomes more electron-withdrawing the possibility for intramolecular proton transfer can occur to protonate the oxido ligand and afford the species MnIII–OH⋯−NR (Scheme 1).
Scheme 1.

The [H6bpuea-R] pre-ligands were prepared using 1,1’-(((2‒aminoethyl)azanediyl)-bis(ethane-2,1-diyl))bis(3‒(tert-butyl)urea), whose primary amine was functionalized to a urea upon treatment with the appropriate para-R-phenyl isocyanate (Scheme S1). The formation of the MnIII–oxido complexes followed our established procedure that employed [MnII(H3bpuea-R)OAc]2- and dioxygen as the source of the oxido ligand (Scheme S2).47 We also needed the related MnII–OH analogs to complete the necessary thermodynamic analysis discussed below and these complexes as their dipotassium salts were synthesized from water using a protocol that we have used previously (Scheme S3). All salts of the MnIII–oxido and MnII–OH complexes were isolated as stable solids at room temperature.
EPR and Absorbance Properties.
Changes in the R-groups of the [H3bpuea-R]3- ligands modulated the electronic absorbance and EPR properties of their MnIII complexes. EPR spectra for each [MnIII (H3bpuea-R)O]2- complex were obtained to probe their spin states. We have previously demonstrated that parallel-mode at X-band is an effective method to probe MnIII–O(H) complexes in trigonal symmetry.33,48,49 Following the same method, all the [MnIII(H3bpuea-R)O]2- complexes produced EPR spectra that are consistent with high spin, mononuclear MnIII complexes (Figure S1, Table 1) with g-values ~ 8. However, a trend was observed in the six-line hyperfine splitting (A) pattern among some of the complexes. For 1-4, A-values were found to range from 277 to 279 MHz, which are similar to the 280 MHz splitting observed for [MnIIIH3buea(O)]2-.33 The A-values decrease below 270 MHz for 5 and 6, and are values comparable to those we reported for the related MnIII–OH, [MnIII H3buea(OH)]−.49 We have also used EPR spectroscopy to examine the related [MnII(H3bpuea-R)OH]2- complexes and they all have spectral properties that are consistent with S = 5/2 spin ground states (Figure S2).
Table 1.
Spectroscopic Properties for 1-6.
| Complex | λmaxa (εM)b | g value | Azc |
|---|---|---|---|
| 1 | 714 (210) 500 (420) |
8.03 | 277 |
| 2 | 710 (240) 498 (440) |
7.95 | 278 |
| 3 | 706 (240) 498 (440) |
7.95 | 278 |
| 4 | 697 (300) 500 (430) |
8.05 | 279 |
| 5 | 650 (300) | 8.16 | 268 |
| 6 | 677 (270) | 7.97 | 262 |
in DMSO,
M−1cm−1,
MHz
Electronic absorbance spectra further support that 1-4 are MnIII–oxido complexes (Figure S3A, Table 1). This set of complexes have two ligand-field bands with energies and intensities that match those found for [MnIIIH3buea(O)]2-. The higher energy band for all the complexes is found at λmax = 500 nm with nearly identical extinction coefficients. There is a red shift of the lower energy band (λmax = 714–697 nm) as more electron withdrawing R-groups are appended to the phenyl ring. This small shift corresponds to an energy difference of only ~340 cm−1 suggesting there is little change in the ligand-field splitting for these complexes. The electronic absorbance spectra for 5 and 6 differ from that of [MnIIIH3buea(OH)]− because they contain broad shoulders at λmax = 650 and 677 nm (Figure S3B, Table 1). These spectral features resemble those found for [MnIIIH2bupa(O)(H)]− (Figure 3B, λmax = 675 nm (sh)), a MnIII complex in which a strong intramolecular H-bond between MnIII–oxido and an appended carboxyamido group prevented exact determination of its structure (that is, whether MnIII–O⋯HNR or MnIII–OH⋯NR was present).50 Taken together, these results indicate that 1-4 can be best described as a monomeric MnIII–oxido species. The spectral properties of 5 and 6 also support the presence of a single MnIII center but do not conclude if the oxido ligand is protonated or strongly H-bonded.
Figure 3.

Thermal ellipsoid plots of 1 and 6. Thermal ellipsoids are at 50% probability level. Only ureayl and hydroxide hydrogen atoms are shown for clarity.
Structural Properties of 1 and 6.
The molecular structures of salts K2[1]·DMA·H2O and (NMe4)2[6].2MeCN were determined by X-ray diffraction (XRD) methods (Figure 3, Tables 2 and S1). XRD analysis revealed that the asymmetric unit of K2[1]·DMA·H2O consists of two independent, but chemically identical anions, with similar metrical parameters — only one of them will be discussed. Both 1 and 6 have N4O donor sets arranged in a trigonal bipyramidal coordination geometry in which the trigonal planes are defined by the three deprotonated ureayl N-atoms. The axial coordination sites are occupied by the O1-atom and N1-atom from the tripodal ligands with O1-Mn1-N1 angles of 177.0(1) and 178.6(2)°. Differences are observed in the Mn–O1 bond distances between the two complexes to support our suggestion that the oxido ligands in these complexes have different protonation states. For instance, the Mn1–O1 bond length of 1.771(3) Å in 1 is the same as that found in [MnIIIH3buea(O)]2- (1.771(5) Å) and is significantly longer than the Mn1–O1 bond distance of 1.819(2) Å in 6; this longer distance is similar to the Mn1–O1 bond distances of 1.875(2) Å and 1.822(4) Å reported for [MnIIIH3buea(OH)]− and [MnIIIH2bupa(O)(H)]−. A difference was also found in the Mn1–N1 bond distances which for 1 is 2.095(4) Å and contracts to 2.058(5) Å in 6 — this change is consistent with an oxido ligand having a stronger trans influence than a hydroxido ligand.47
Table 2.
Selected Structural Parameters for 1 and 6.
| Bond Distances (Å) and Angles (°) | 1a | 6 |
|---|---|---|
| Mn1-O1 | 1.771(3) | 1.819(4) |
| Mn1-N1 | 2.095(4) | 2.058(5) |
| Mn1-N2 | 2.087(3) | 2.017(5) |
| Mn1-N3 | 2.084(3) | 2.069(5) |
| Mn1-N4 | 2.039(3) | 2.059(5) |
| N5·⋯O1 | 2.613(5) | 2.613(6) |
| N6⋯O1 | 2.701(5) | 2.798(7) |
| N7⋯O1 | 2.732(5) | 2.760(7) |
| O1-Mn1-N1 | 177.01(14) | 178.6(2) |
| O1-Mn1-N2 | 96.38(15) | 96.25(19) |
| O1-Mn1-N3 | 97.71(13) | 99.3(2) |
| O1-Mn1-N4 | 101.02(14) | 98.25(19) |
| N1-Mn1-N2 | 81.22(15) | 82.39(19) |
| N1-Mn1-N3 | 81.74(13) | 81.7 (2) |
| N1-Mn1-N4 | 81.72(13) | 82.24(19) |
| N2-Mn1-N3 | 114.22(14) | 117.6(2) |
| N2-Mn1-N4 | 117.97(14) | 124.3(2) |
| N3-Mn1-N4 | 121.45(14) | 112.5(2) |
Data for the second complex in the asymmetric unit are listed in the Table S2.
Three intramolecular H-bonds involving the O1-atom are also present in 1 and 6. In each complex, two N—H⋯O1 H-bonds are formed with the N6H6 and N7H7 units of the [H3bpueaR]3- ligands. However, a noticeable difference was found in the third H-bond formed with N5 of the phenyl urea. In 1, the O1-atom serves as an H-bond acceptor to produce the N5—H5⋯O1, a result that further supports O1 being an oxido ligand. In 6, O1 is protonated and serves as an H-bond donor to form the N5⋯H1—O1 H-bond. The H1-atom in 6 was located from a difference-Fourier map and refined to give an O1—H1 bond distance of 0.85 Å. Moreover, the difference-Fourier map showed no residual electron density within bonding distance to N5 to further suggest that it has been deprotonated. We were also able to find H6 and H7 in the difference-Fourier map of 6 and after refinement found N—H bond distances of 0.90 Å for each. These structural data thus support that 1 has a MnIII–oxido unit while 6 has a MnIII–OH unit that is H-bonded to the deprotonated N5-atom of [H3bpuea-5F]3-.
pKa and Redox Values.
To gain insights into the effects of H-bonds on the basicity of MnIII–O(H) complexes, pKa values were determined spectrophotometrically from titrations with organic acids (Supporting Information). Treating 1-4 with 2-aminopyrimidine (pKa = 25.3)51 produced the protonated analogs, which was monitored by measuring changes in intensity of the band at λmax= 700 nm (Figures S4, S5). The pKa values ranged from 24.4 for 1 to 23. 4 for 4. We were unsuccessful in determining the pKa values for 5 and 6 via spectrophotometric titrations but used a series of acids to bracket their values (see SI and Figure S6–S8). From these studies, a bracketed pKa range between 22–23 was found for 5 and between 19–20 for 6. The data show a correlation between the pKa values and R-groups on the phenyl ring of [H3bpuea-R]3- with more electron withdrawing groups lowering of the pKa values. This trend can be seen in a Hammett analysis (Figure S9) and can be explained by a change in the intramolecular H-bond involving the tripodal arm containing the phenyl urea group. A consequence of the R-groups becoming more electron withdrawing is that the adjacent HNurea groups would form a stronger H-bond with the MnIII–oxido unit. This single change within the secondary coordination sphere thus modulates the pKa values of the complexes since a stronger H-bond would decrease the basicity of the oxido ligand (Scheme 1).
Cyclic voltammetry (CV) studies on 1 ‒ 6 revealed one-electron responses which were assigned as the [MnIVH3bpuea-R(O)]−/[MnIIIH3bpuea-R(O)]2- redox processes and these values are listed in the experimental section for completeness (Figure S10). For our thermodynamic analyzes (see below), we needed the redox potential for the related [MnIIIH3bpuea-R(OH)]−/[MnIIH3bpuea-R(OH)]2- couple, which prompted us to prepare the MnII–OH complexes of the [H3bpuea-R]3- ligands. CV analysis produced an oxidative response for each [MnIIH3bpuea-R(OH)]2- complex with E1/2 values that only change by 0.090 V (−1.29 to −1.38 V vs. [FeCp2]+/0, (Figure S11, Table 3). The related E1/2 value for the MnII–OH of [H3bpuea-5F]3- was not obtained because we were unable to prepare this complex.
Table 3.
Thermodynamic and Kinetic Parameters for 1–5.a
| Complex | E1/2b | pKa | BDFEc | kd,e |
|---|---|---|---|---|
| 1 | −1.38 | 24.4(1) | 73 | 0.038(3) |
| 2 | −1.39 | 23.7(1) | 72 | 0.022(1) |
| 3 | −1.41 | 23.8(1) | 71 | 0.019(2) |
| 4 | −1.29 | 23.4(1) | 73 | 0.012(1) |
| 5 | −1.33 | 22 – 23 | 71 – 72 | 0.0020(2) |
in DMSO,
versus [FeIII/IICp2]+/0,
kcal/mol;
M−1s−1,
k = k2/4
Thermodynamic Evaluations.
The accumulated data (Table 3) allowed us to determine the BDFEO–H using the approach described in the Introduction (Figure 1). The specific square scheme used is shown in Scheme S4 and numerical values were obtained using eq 1.23,52,53
| (1) |
where C accounts for the thermodynamic properties of the hydrogen atom in solution and has a value of 71.1 kcal/mol for the conditions used in our studies.23 The evaluations found that the values for the BDFEO–H for the series of [MnIIH3bpuea-R(OH)]2- complexes did not change within experimental error and had an average free energy of 72 kcal/mol. It appears that the small changes in redox potential were offset by the changes in the pKa values within the series. We note that the BDFEO–H for these complexes is comparable to that for the previously reported [MnIIH3buea(OH)]2- (BDFEO–H = 75 kcal/mol).
Reactivity Studies. 9,10-Dihydroanthracene (DHA).
The [MnIIIH3bpuea-R(O)]2- complexes were investigated for their ability to cleave C–H bonds. We used 9,10-dihydroanthracene (DHA) as a test substrate and found under anaerobic conditions that all the complexes except 6 activated the C‒H bonds in DHA (Scheme 2). Monitoring the reactions spectrophotometrically showed the disappearance of the spectral features of the MnIII–O(H) complexes with concomitant appearance of bands associate with anthracene (Figure S12). We further probed this reactivity via kinetic studies with 1-5, which were performed under pseudo first-order conditions (greater than 10-fold excess of substrate). Again, the reactions were followed by the disappearance of the absorbance feature for MnIII‒O(H) complexes at λmax ~ 500 nm for 1-4 and at 650 nm for 5 (6 the reaction was too slow to determine a rate constant). Under pseudo first-order conditions with excess DHA, the values of kobs were found to be independent of complex concentrations and the rates of the reactions were linearly dependent on complex concentrations (Figure S13). In addition, the pseudo-first-order rate constant (kobs) for 1-5 varied linearly with the concentration of DHA (Figure 4). Based on these results the second-order rate law was utilized (eq 2). The values for k2 were obtained from the slope of kobs vs. [DHA] plots for 1-5 that were corrected for the number of activatable C–H bonds in the substrate to give the reported second-order rate constant k (Figure 4 and Table 4, see SI). For 1, we found that the addition of 18-crown-6 was needed to sequester the potassium ion which appeared to interfere with the reaction—see Supporting Information for details (Figure S14). All other complexes did not show an effect with the addition of crowns.
| (2) |
Scheme 2.

Figure 4.

Plots of kobs vs. [DHA] for 1-5.
Table 4.
Activation Parameters and KIE Values for the reaction of 1, 2, 4, and 5 with DHA.a
| Complex | ΔH‡,b | ΔS‡,c | kH/kD |
|---|---|---|---|
| 1 | 15(1) | −15(2) | 5.8 |
| 2 | 15(1) | −16(2) | 5.1 |
| 4 | 15(1) | −15(2) | 4.4 |
| 5 | 13(1) | −25(3) | 5.7 |
in DMSO,
kcal/mol;
eu
The second-order rate constant for the [MnIIIH3bpuea-R(O)]2- complexes varied by over an order of magnitude (Table 3) with 1 having the largest value at k = 0.038(3) M−1s−1 and 5 the smallest at 0.0020(2) M−1s−1. This relative trend does not correlate with the BDFEO–H which remained constant throughout the series of complexes (Table 3). However, the trend in rate constants do show a correlation with changes in the basicity of the complexes: the most basic complex (1) has the largest rate constant (Figure 5A). As discussed above, the regulation of the basicity in these complexes is proposed to be from a single H-bond whose properties can be modulated by substituent effects on the distal phenyl ring. Consistent with this premise, a Hammett correlation was observed for this reaction: the plot of log(k/kH) versus σp values of the para-substituents on the phenyl ring of the [H3bpuea-R]3- ligand revealed a linear relationship in which ρ = − 1.57 (Figure 5B). This type of analysis is rare for the cleavage of C–H bonds by metal-oxido species and highlights that H-bonds can have significant effects on both structural and functional properties of metal complexes.
Figure 5.

A plot of log k vs. pKa for the oxidation of DHA (A) and a Hammett analysis for the oxidation of DHA (B) by 1-5.
The temperature-dependency of the rate constants were determined to further investigate the changes in the relative rate of C–H bond cleavage by the [MnIIIH3bpuea-R(O)]2- complexes: we studied these effects with complexes 1, 2, 4, and 5. Each complex produced linear plots of kobs vs [DHA] for each temperature (Figure S15) and activation parameters were calculated from Eyring analysis (Figure S16, Table 4). The enthalpies of activation for the complexes were the same within experimental error and agree with those reported for [MnIIIH3buea(O)]2-. The entropies of activation were also similar to each other with only that for 5 being slightly lower. For all the complexes ΔH‡ is greater than −TDS‡ which suggests a comparable reaction profile for C–H bond cleavage in which the enthalpy of activation predominates over the activation entropy. The kinetic isotope effects (KIE) for 1, 2, 4, and 5 were measured at 20°C using d4-DHA (Figures S17–S20) and have values that are indicative of primary isotope effects with C–H bond breakage being involved in the rate-limiting step. The values are statistically different for each complex and decrease from 5.8 for 1 to 4.4 for 4. These values are significantly larger than the KIE of 2.6 determined for [MnIIIH3buea(O)]2-.
Reactivities with other Substrates.
To gain more insights into C–H bond cleavage by the [MnIIIH3bpuea-R(O)]2- complexes, we explored the reactivity of 2 and 4 with other substrates having different pKa and BDFE values for their C–H bonds. For instance, treating each complex with fluorene resulted in acid-base chemistry in which the initial products were [MnIIIH3bpuea-Cl(OH)]− and the fluorene anion (Figure S21). This finding shows that 2 and 4 are only capable of deprotonating fluorene which is significantly more acidic than DHA: the pKa value of DHA and fluorene are 30.1 and 22.6, respectively. In addition, the resulting fluorene anion has a potential of −1.01 V which is not reducing enough to form [MnIIH3bpuea-Cl(OH)]2- (Table 5) and the fluorene radical. The reactivity of the complexes with xanthene produced a kinetic profile that was significantly faster than with DHA: for 2, 4, and 5 measured k2 values of 0.37, 0.17, 0.014 M−1s−1 were found, which are significantly larger than those obtained with DHA (Figures S22–S24, Table 5). The pKa values for the two substrates are the same (~30) but xanthene has a lower BDFE value of 73.3 kcal/mol. The 3 kcal/mol difference between the two substrates is attributed to the redox potential for the xanthene anion/xanthene radical being more negative by −0.12 V (−1.69 V compared to −1.57 V for [DHA]−/[DHA]•). The temperature-dependency of the rate constant for the reaction of 4 with xanthene in DMSO was examined (Figure S25) and an Eyring analysis gave activation parameters of ΔH‡ = 14(1) kcal/mol and ΔS‡ = −13(2) eu — these values are within experimental error from those found for the reaction of 4 with DHA (Table 4). Moreover, we determined a KIE = 3.9 at 20°C in DMSO for 4 and d2-xanthene which is also similar to that found for the reaction of 4 with DHA (Figure S26, Table 4). These data suggest that the reaction of 4 with DHA and xanthene follow a similar mechanistic route. We also examined the reactivity of 4 with triphenylmethane (Ph3CH) which has a similar pKa value (30.6) to DHA but has a larger BDFE (78.8 kcal/mol). This reaction was slow and we were unable to determine a rate constant.
Table 5.
Thermodynamic Parameters for Various Substrates and Reaction Results with 2, 4, and 5.a
| Substrate | pKab | E(ox)c,d | k(2)e,f | k(4)e,f | k(5)e,f |
|---|---|---|---|---|---|
| DHA | 30.1 | −1.57 | 0.022(1) | 0.012(1) | 0.0020(1) |
| fluorene | 22.6 | −1.07 | g | g | g |
| xanthene | 30 | −1.69 | 0.37(2) | 0.17(1) | 0.014(1) |
| Ph3CH | 30.6 | −1.49 | no reaction | no reaction | no reaction |
in DMSO,
ref. 14;
[substrate anion]−/[substrate]•;
V versus [FeIII/IICp2]+/0;
second-order rate constant for C–H bond cleavage;
M−1s−1;
deprotonation of substrate.
Mechanistic Considerations.
The results with DHA suggest that the pKa values, rather than the BDFEO–H, of the [MnIIH3bpuea-R(OH)]2- complexes are the important determinant in their reactivity toward C–H bonds. The significance of the pKa parameter is further shown in the reactivity of 2, 4, and 5 with fluorene in which only deprotonation of the substrate was observed. However, it does not appear that a straightforward PT-ET mechanism is operative in our series of MnIII–oxido complex. The reactivity of 2, 4, and 5 with xanthene produced a large increase in the second-order rate constant compared to that for DHA, even though each substrate has the same pKa value. Moreover, the values of ΔS‡ are relatively large (Tables 4) which is consistent with charge delocalized transition states.54 Taken together, these results suggest an asynchronous transition state that is dominated by proton transfer with partial electron transfer. A similar premise has recently been reported for a CoIII–oxido complexes30 and found from theoretical analyzes of [FeIIIH3buea(O)]2- and FeIV–oxido complexes.55,56
Complex 6 has not been included in our mechanistic experiments because of its diminished reactivity with DHA. We are unsure as to the exact reason behind this observation, but it is undoubtedly caused, in part, to the change in the H-bonding network that produced a MnIII–OH species. Complex 6 is the least basic species within the series that should contribute to the low reactivity. However, it is also possible that a change in mechanism has occurred because the oxido ligand is now protonated. The structurally differences in 6 could result in the proton and electron being electronically decoupled because the basic site is now on the [H2bpuea-5F]4- ligand – in the other complexes the proton and electron are more strongly coupled when they react directly with the Mn–oxido unit. These differences within our series of complexes highlight the complexity associate with understanding the mechanism of C–H bond activation at a M–oxido site.
Summary and Conclusions
Hydrogen bonds within the secondary coordination sphere have often been invoked as important structural interactions within the active site of metalloproteins. They have also been shown to regulate physical properties of metal complexes, such as modulating redox potential in proteins and synthetic complexes. We have further demonstrated their utility by developing a system in which a single H-bond can control the basicity of an Mn–oxido unit. The design of a new hybrid tripodal ligand [H3bpuea-R]3- allowed for the systematic changes in the H-bond donation of a lone HNurea group within the secondary coordination sphere. Variations in the pKa(OH) values for the [MnIIIH3bpuea-R(OH)]2- were determined experimentally and had a Hammett correlation. This result supports the premise that the H-bond can regulate the basicity of the corresponding MnIII–oxido species. Moreover, 6 illustrated that the species MnIII–OH⋯−Nurea can be formed when the HNurea donor becomes sufficiently acidic to promote intramolecular proton transfer.
The [MnIIIH3bpuea-R(O)]2- complexes react with DHA and more than 10-fold decrease in the second-order rate constant for the reaction was observed between 1-5. A Hammett correlation was observed that showed changes at a remote site from the metal center can alter the C–H cleavage for an external substrate. In addition, a correlation between the pKa(OH) and the second-order rate constants suggests that tuning of the observed reactivity is directly linked to changes in H-bonding networks. Further kinetic investigations with 1-5 and DHA pointed toward a two-step mechanism in which proton-transfer is rate limiting. However, studies with other substrates having similar pKa(C─H) values as DHA, but different BDFEC─H values, do not support this premise. Results from the reactivity of 2, 4, and 5 with xanthene and DHA showed that xanthene is significantly faster even though both substrates have the same pKa(C─H) values.
Mechanisms for C–H bond cleavage by M–oxido complexes are often considered within the context of the three limiting mechanisms depicted in Figure 1A: CPET, ET-PT, and PT-ET. Results from our experimental investigations indicate that our systems do not follow any of these mechanisms which prompted us to consider other possibilities. Asynchronous processes55,56 in which the proton and electron are transferred unequally to a M–oxido unit at the transition state have also been touted as relevant pathways for C–H bond activation. Our results with 1-5 fit this type of description and offer support of the premise that asynchronous mechanistic routes are important in the activation of C–H bond activation by M-oxido complexes.
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge the NIH (GM050781) for funding. We thank the reviewers for helpful comments.
Footnotes
Supporting Information
Preparative routes and characterization of the pre-ligands and MnII–OH complexes; EPR, FTIR, and optical spectra, and cyclic voltammograms for the Mn complexes; details for the determinations of the pKa values, the rate constants, activation parameters, and KIE values; a scheme showing specific thermodynamic cycle used for the Mn–O(H) complexes, and tables of XRD data for 1 and 6 are provided free of charge at http://pubs.acs.org.
REFERENCES
- (1).Baglia RA; Zaragoza JPT; Goldberg DP Biomimetic Reactivity of Oxygen-Derived Manganese and Iron Porphyrinoid Complexes. Chem. Rev 2017, 117, 13320–13352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Gunay A; Theopold KH C-H Bond Activations by Metal Oxo Compounds. Chem. Rev 2010, 110, 1060–1081. [DOI] [PubMed] [Google Scholar]
- (3).McDonald AR; Que L Jr. High-Valent Nonheme Iron-Oxo Complexes: Synthesis, Structure, and Spectroscopy. Coord. Chem. Rev 2013, 257, 414–428. [Google Scholar]
- (4).Borovik AS Role of Metal-Oxo Complexes in the Cleavage of C-H Bonds. Chem. Soc. Rev 2011, 40, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sacramento JJD; Goldberg DP Factors Affecting Hydrogen Atom Transfer Reactivity of Metal-Oxo Porphyrinoid Complexes. Acc. Chem. Res 2018, 51, 2641–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rice DB; Massie AA; Jackson TA Manganese-Oxygen Intermediates in O-O Bond Activation and Hydrogen-Atom Transfer Reactions. Acc. Chem. Res 2017, 50, 2706–2717. [DOI] [PubMed] [Google Scholar]
- (7).Shan X; Que L Jr. High-Valent Nonheme Iron-Oxo Species in Biomimetic Oxidations. J. Inorg. Biochem 2006, 100, 421–433. [DOI] [PubMed] [Google Scholar]
- (8).Chanda A; Shan X; Chakrabarti M; Ellis WC; Popescu DL; de Oliveira FT; Wang D; Lawrence Que J; Collins TJ; Munck E; Bominaar EL (TAML)FeIV=O Complex in Aqueous Solution: Synthesis and Spectroscopic and Computational Characterization. Inorg. Chem 2008, 47, 3669–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Company A; Feng Y; Guell M; Ribas X; Luis Josep M; Que LJ; Costas M Olefin-Dependent Discrimination between Two Nonheme HO-Fev=O Tautomeric Species in Catalytic H2O2 Epoxidations. Chem.-Eur. J 2009, 15, 3359–3362. [DOI] [PubMed] [Google Scholar]
- (10).Nam W High-Valent Iron(IV)–Oxo Complexes of Heme and Non-Heme Ligands in Oxygenation Reactions. Acc. Chem. Res 2007, 40, 522–531. [DOI] [PubMed] [Google Scholar]
- (11).Nam W; Lee Y-M; Fukuzumi S Tuning Reactivity and Mechanism in Oxidation Reactions by Mononuclear Nonheme Iron(IV)-Oxo Complexes. Acc. Chem. Res 2014, 47, 1146–1154. [DOI] [PubMed] [Google Scholar]
- (12).Cho K; Leeladee P; McGown AJ; DeBeer S; Goldberg DP A High-Valent Iron–Oxo Corrolazine Activates C–H Bonds via Hydrogen-Atom Transfer. J. Am. Chem. Soc 2012, 134, 7392–7399. [DOI] [PubMed] [Google Scholar]
- (13).Groves JT; Gross Z; Stern MK Preparation and Reactivity of Oxoiron(IV) Porphyrins. Inorg. Chem 1994, 33, 5065–5072. [Google Scholar]
- (14).Yin G; Danby AM; Kitko D; Carter JD; Scheper WM; Busch DH Oxidative Reactivity Difference among the Metal Oxo and Metal Hydroxo Moieties: PH Dependent Hydrogen Abstraction by a Manganese(IV) Complex Having Two Hydroxide Ligands. J. Am. Chem. Soc 2008, 130, 16245–16253. [DOI] [PubMed] [Google Scholar]
- (15).Shi S; Wang Y; Xu A; Wang H; Zhu D; Roy SB; Jackson TA; Busch DH; Yin G Distinct Reactivity Differences of Metal Oxo and Its Corresponding Hydroxo Moieties in Oxidations: Implications from a Manganese(IV) Complex Having Dihydroxide Ligand. Angew. Chem., Int. Ed. Engl 2011, 50, 7321–7324. [DOI] [PubMed] [Google Scholar]
- (16).Garcia-Bosch I; Company A; Cady CW; Styring S; Browne WR; Ribas X; Costas M Evidence for a Precursor Complex in C-H Hydrogen Atom Transfer Reactions Mediated by a Manganese(IV) Oxo Complex. Angew. Chem., Int. Ed. Engl 2011, 50, 5648–5653. [DOI] [PubMed] [Google Scholar]
- (17).Prokop KA; de Visser Sam P; Goldberg DP Unprecedented Rate Enhancements of Hydrogen-Atom Transfer to a Manganese(V)-Oxo Corrolazine Complex. Angew. Chem., Int. Ed. Engl 2010, 49, 5091–5095. [DOI] [PubMed] [Google Scholar]
- (18).Baglia RA; Krest CM; Yang T; Leeladee P; Goldberg DP High-Valent Manganese-Oxo Valence Tautomers and the Influence of Lewis/Brönsted Acids on C-H Bond Cleavage. Inorg. Chem 2016, 55, 10800–10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Baglia RA; Prokop-Prigge KA; Neu HM; Siegler MA; Goldberg DP Mn(V)(O) versus Cr(V)(O) Porphyrinoid Complexes: Structural Characterization and Implications for Basicity Controlling H-Atom Abstraction. J. Am. Chem. Soc 2015, 137, 10874–10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Massie AA; Sinha A; Parham JD; Nordlander E; Jackson TA Relationship between Hydrogen-Atom Transfer Driving Force and Reaction Rates for an Oxomanganese(IV) Adduct. Inorg. Chem 2018, 57, 8253–8263. [DOI] [PubMed] [Google Scholar]
- (21).Stone KL; Borovik A Lessons from Nature: Unraveling Biological CH Bond Activation. Curr. Opin. Chem. Biol 2009, 13, 114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Green MT CH Bond Activation in Heme Proteins: The Role of Thiolate Ligation in Cytochrome P450. Curr. Opin. Chem. Biol 2009, 13, 84–88. [DOI] [PubMed] [Google Scholar]
- (23).Warren JJ; Tronic TA; Mayer JM Thermochemistry of Proton-Coupled Electron Transfer Reagents and Its Implications. Chem. Rev 2010, 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Darcy JW; Koronkiewicz B; Parada GA; Mayer JM A Continuum of Proton-Coupled Electron Transfer Reactivity. Acc. Chem. Res 2018, 51, 2391–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bell RP The Theory of Reactions Involving Proton Transfers. Proc. R. Soc. London. Ser. A 1936, 154, 414–429. [Google Scholar]
- (26).Evans MG; Polanyi M Inertia and Driving Force Of Chemical Reactions. Trans. Faraday Soc. 1938, 34, 11–23. [Google Scholar]
- (27).Green MT; Dawson JH; Gray HB Oxoiron(IV) in Chloroperoxidase Compound II Is Basic: Implications for P450 Chemistry. Science 2004, 304, 1653–1656. [DOI] [PubMed] [Google Scholar]
- (28).Parsell TH; Yang M-Y; Borovik AS C–H Bond Cleavage with Reductants: Re-Investigating the Reactivity of Monomeric MnIII/IV−Oxo Complexes and the Role of Oxo Ligand Basicity. J. Am. Chem. Soc 2009, 131, 2762–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Poulos TL Heme Enzyme Structure and Function. Chem. Rev 2014, 114, 3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Goetz MK; Anderson JS Experimental Evidence for p K a -Driven Asynchronicity in C-H Activation by a Terminal Co(III)-Oxo Complex. J. Am. Chem. Soc 2019, 141, 4051–4062. [DOI] [PubMed] [Google Scholar]
- (31).Lacy DC; Gupta R; Stone KL; Greaves J; Ziller JW; Hendrich MP; Borovik AS Formation, Structure, and EPR Detection of a High Spin FeIV—Oxo Species Derived from Either an FeIII—Oxo or FeIII—OH Complex. J. Am. Chem. Soc 2010, 132, 12188–12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Cook SA; Borovik AS Molecular Designs for Controlling the Local Environments around Metal Ions. Acc. Chem. Res 2015, 48, 2407–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gupta R; Taguchi T; Lassalle-Kaiser B; Bominaar EL; Yano J; Hendrich MP; Borovik AS High-Spin Mn–Oxo Complexes and Their Relevance to the Oxygen-Evolving Complex within Photosystem II. Proc. Natl. Acad. Sci 2015, 112, 5319–5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Surendhran R; D’Arpino AA; Sciscent BY; Cannella AF; Friedman AE; MacMillan SN; Gupta R; Lacy DC Deciphering the Mechanism of O2 Reduction with Electronically Tunable Non-Heme Iron Enzyme Model Complexes. Chem. Sci 2018, 9, 5773–5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lucas RL; Zart MK; Murkerjee J; Sorrell TN; Powell DR; Borovik AS A Modular Approach toward Regulating the Secondary Coordination Sphere of Metal Ions: Differential Dioxygen Activation Assisted by Intramolecular Hydrogen Bonds. J. Am. Chem. Soc 2006, 128, 15476–15489. [DOI] [PubMed] [Google Scholar]
- (36).Douat-Casassus C; Marchand-Geneste N; Diez E; Gervois N; Jotereau F; Quideau S Synthetic Anticancer Vaccine Candidates: Rational Design of Antigenic Peptide Mimetics That Activate Tumor-Specific T-Cells. J. Med. Chem 2007, 50, 1598–1609. [DOI] [PubMed] [Google Scholar]
- (37).Lucas RL Development of Methods for Varying the Hydrogen Bond Network Around Metal Ions: Site-Directed Modification of the Secondary Coordination Sphere, University of Kansas, 2005. [Google Scholar]
- (38).Jones JR; Ziller JW; Borovik AS Modulating the Primary and Secondary Coordination Spheres within a Series of Co(II)-OH Complexes. Inorg. Chem 2017, 56, 1112–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Gao H; Groves JT Fast Hydrogen Atom Abstraction by a Hydroxo Iron(III) Porphyrazine. J. Am. Chem. Soc 2017, 139, 3938–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Bruker AXS Inc, APEX2 Version 2014.11–0. Madison, WI: 2014. [Google Scholar]
- (41).Bruker AXS Inc, SAINT Version 8.34a. Madison, WI: 2013. [Google Scholar]
- (42).Sheldrick GM SADABS. Bruker AXS, Inc: Madison: 2014. [Google Scholar]
- (43).Sheldrick GM SHELXTL. Bruker AXS, Inc: Madison: 2014. [Google Scholar]
- (44).Wilson AJC; Geist V International Tables for Crystallography. Volume C: Mathematical, Physical and Chemical Tables. Kluwer Academic Publishers, Dordrecht/Boston/London 1992 (Published for the International Union of Crystallography), 883 Seiten, ISBN 0-792-3-16-38X. Cryst. Res. Technol 1993, 28, 110–110. [Google Scholar]
- (45).Spek AL PLATON SQUEEZE: A Tool for the Calculation of the Disordered Solvent Contribution to the Calculated Structure Factors. Acta Crystallogr. Sect. C Struct. Chem 2015, 71, 9–18. [DOI] [PubMed] [Google Scholar]
- (46).Spek AL Structure Validation in Chemical Crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr 2009, 65, 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).MacBeth CE; Gupta R; Mitchell-Koch KR; Young VG; Lushington GH; Thompson WH; Hendrich MP; Borovik AS Utilization of Hydrogen Bonds To Stabilize M–O(H) Units: Synthesis and Properties of Monomeric Iron and Manganese Complexes with Terminal Oxo and Hydroxo Ligands. J. Am. Chem. Soc 2004, 126, 2556–2567. [DOI] [PubMed] [Google Scholar]
- (48).Taguchi T; Gupta R; Lassalle-Kaiser B; Boyce DW; Yachandra VK; Tolman WB; Yano J; Hendrich MP; Borovik AS Preparation and Properties of a Monomeric HighSpin MnV–Oxo Complex. J. Am. Chem. Soc 2012, 134, 1996–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Gupta R; Taguchi T; Borovik AS; Hendrich MP Characterization of Monomeric Mn(II/III/IV)-Hydroxo Complexes from X- and Q-Band Dual Mode Electron Paramagnetic Resonance (EPR) Spectroscopy. Inorg. Chem 2013, 52, 12568–12575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Shook RL; Peterson SM; Greaves J; Moore C; Rheingold AL; Borovik AS Catalytic Reduction of Dioxygen to Water with a Monomeric Manganese Complex at Room Temperature. J. Am. Chem. Soc 2011, 133, 5810–5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Bordwell FG Equilibrium Acidities in Dimethyl Sulfoxide Solution. Acc. Chem. Res 1988, 21, 456–463. [Google Scholar]
- (52).Bordwell FG; Cheng J; Ji GZ; Satish AV; Zhang X Bond Dissociation Energies in DMSO Related to the Gas Phase Values. J. Am. Chem. Soc 1991, 113, 9790–9795. [Google Scholar]
- (53).Parker VD; Handoo KL; Roness F; Tilset M Electrode Potentials and the Thermodynamics of Isodesmic Reactions. J. Am. Chem. Soc 1991, 113, 7493–7498. [Google Scholar]
- (54).Mader E. a; Davidson ER; Mayer JM Large Ground-State Entropy Changes for Hydrogen Atom Transfer Reactions of Iron Complexes. J. Am. Chem. Soc 2007, 129, 5153–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Usharani D; Lacy DC; Borovik AS; Shaik S Dichotomous Hydrogen Atom Transfer vs Proton-Coupled Electron Transfer During Activation of X–H Bonds (X = C, N, O) by Nonheme Iron–Oxo Complexes of Variable Basicity. J. Am. Chem. Soc 2013, 135, 17090–17104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Bím D; Maldonado-Domínguez M; Rulíšek L; Srnec M Beyond the Classical Thermodynamic Contributions to Hydrogen Atom Abstraction Reactivity. Proc. Natl. Acad. Sci 2018, 115, E10287–E10294. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.