

Abstract
Dihydrolipoamide dehydrogenase (LADH, E3) deficiency is a rare (autosomal, recessive) genetic disorder generally presenting with an onset in the neonatal age and early death; the highest carrier rate has been found among Ashkenazi Jews. Acute clinical episodes usually involve severe metabolic decompensation and lactate acidosis that result in neurological, cardiological, and/or hepatological manifestations. Clinical severity is due to the fact that LADH is a common E3 subunit to the alpha-ketoglutarate, pyruvate, alpha-ketoadipate, and branched-chain alpha-keto acid dehydrogenase complexes, and is also a constituent in the glycine cleavage system, thus a loss in LADH function adversely affects multiple key metabolic routes. However, the severe clinical pictures frequently still do not parallel the LADH activity loss, which implies the involvement of auxiliary biochemical mechanisms; enhanced reactive oxygen species generation as well as affinity loss for multienzyme complexes proved to be key auxiliary exacerbating pathomechanisms. This review provides an overview and an up-to-date molecular insight into the pathomechanisms of this disease in light of the structural conclusions drawn from the first crystal structure of a disease-causing hE3 variant determined recently in our laboratory.
Keywords: Dihydrolipoamide dehydrogenase, E3 deficiency, Alpha-ketoglutarate dehydrogenase complex, Pyruvate dehydrogenase complex, Pathogenic mutation, Reactive oxygen species (ROS), X-ray crystallography, Structure
E3 Deficiency—the Disease, Enzyme, and Affected Multienzyme Complex Functions
(Dihydro)lipoamide dehydrogenase (LADH, E3; gene: dld) deficiency is an often prematurely lethal rare autosomal recessive genetic disorder [1]; the highest carrier rate (1:94-1:110, G194C-hE3, h for human) has been found among Ashkenazi Jews with a disease frequency of 1:35,000–1:48,000 [2, 3]. The first and sole review on the molecular pathomechanisms of this disease was written by our laboratory [4]. This disorder involves mainly neurological, cardiological, and hepatological manifestations whose symptoms generally arise very early in life. The phenotypic spectrum includes hyperammonemia, failure to thrive, hypotonia, encephalopathy, seizure, hepatomegaly, liver dysfunction, lactate acidosis, hypoglycemia, Leigh syndrome, developmental delay, hypertrophic cardiomyopathy, vision impairment/optic atrophy, ataxia, and microcephaly, among others [1, 3, 5–16]. Potentially lethal hepatological consequences, often together with encephalopathy and coagulopathy, may present in isolation and in adulthood [3, 17–19]. The severity of the clinical outcomes is due to the simultaneous defects of the mitochondrial E3-harboring multienzyme dehydrogenase complexes for alpha-ketoglutarate (KGDHc), pyruvate (PDHc), alpha-ketoadipate (KADHc), and branched-chain alpha-keto acids (BCKDHc); interestingly, the glycine cleavage system (GCS), which also contains the LADH protein, remains unaffected in E3-deficiency [1, 4, 20–22]. In the above dehydrogenase complexes the common E3 subunit catalyzes the re-oxidation of the dihydrolipoate (DHLA) moieties covalently linked to the respective E2 components and the reduction of NAD+ to NADH (LADH activity, forward reaction). Pathogenic gene variants include missense or nonsense mutations, splice site variants, and ones with (small) deletions/insertions; 14 disease-causing enzyme variants have been reported to date in the clinical literature [4, 6, 8].
Auxiliary Exacerbating Pathomechanisms
Clinical severity, more often than not, does not parallel the loss in LADH activity [4, 6, 8, 13, 20, 23, 24]. Recent results suggest that the missing clues could be (i) enhanced reactive oxygen species (ROS) production by various pathogenic hE3 mutants [20, 23, 24], particularly in acidosis [24], (ii) liberation of selected hE3 variants from the E3-tethering multienzyme complexes [25–29], and (iii) ROS generation by the E1–E2 subcomplex of the hKGDHc (when E3 is scarce) [20], principally in the course of acidosis [30].
ROS Generation by hE3, Pathogenic hE3 Mutants, and hE3-Harboring Multienzyme Complexes
Among all the mitochondrial alpha-keto (or 2-oxo) acid dehydrogenase complexes (OADHc), the KGDHc exhibits the most dominant ROS generation under pathologically relevant conditions [20–22, 31–42]. ROS generation by the KGDHc can occur in the forward catalytic direction in case the physiological electron acceptor NAD+ is scarce or absent (in vitro), or alternatively in the reverse reaction driven by a high NADH/NAD+ ratio [20, 30, 33, 34]; superoxide (as a primary ROS) is generated at the flavoenzyme E3 component [20, 33, 34] (see Scheme 1). The isolated E3 component [43, 44] is also capable of generating ROS, in an oxidase reaction, in either direction of the catalytic reaction [20, 24, 30, 45–51] (Scheme 1). The E3 subunit forms a functional (obligate), non-covalent homodimer that uses residues from both monomers for the physiological LADH activity [45, 52–55], but not for the ROS-generating activity [56–58]; hE3 comprises four domains: a FAD-binding (1–149), a NAD+/NADH-binding (150–282), a central (283–350), and an interface domain (351–474). ROS generation in the reverse reaction of the isolated E3 component is stimulated in acidosis; this same pathological condition also enhances the ROS production in the reverse, but not in the forward reaction of KGDHc [30]. Sensitivity to a decreasing pH of ROS-generation by isolated disease-causing hE3 mutants that display increased ROS-generating capacities in the reverse reaction was reported to be even more pronounced [24]; pathogenic substitutions which stimulated ROS production took place at the disulfide-exchange site (P453L), the dimerization surface (E340K, D444V), or the cofactor-binding site (G194C) [24]. Calibrated gel filtration, molecular dynamics (MD) simulation, hydrogen–deuterium exchange mass spectrometry (HDX-MS), diffusion-ordered (DOSY) NMR, (soft-ionizing) nano-LC MS, and X-ray crystallography (see below) all confirmed that neither (mild) acidosis nor the hitherto investigated disease-causing homodimerization surface mutations led to monomerization of the E3 dimer [24, 25, 30, 59–62]. Importantly, ROS production could also be stimulated by a relevant disease-causing hE3 mutant (G194C-hE3) when complexed to a multienzyme complex (hKGDHc) [20]. The D444V-, G194C-, E340K-, R460G-, and R447G-hE3 pathogenic mutants were reported to oxidatively deteriorate the lipoic acid (LA) cofactors of the PDHc and KGDHc in a yeast model, and in case of D444V-hE3, in human homozygous fibroblasts [23]. In mutants exhibiting stimulated ROS production, LADH activity was generally impaired in both catalytic directions. In P453L-hE3 the physiological activity was almost entirely lost while the ROS-generating activity became predominant, whereas in G194C-hE3 the LADH activity was not altered, but the ROS-producing capacity increased [24]. For P453L-hE3 the clinical phenotype was very severe [16, 63] and the excessive ROS production was proposed to be a contributing factor to this [24]. G194C-hE3 leads often to adult-onset manifestations, which is in accord with the retained LADH activity and the moderately enhanced ROS generation [24]. FAD contents were almost entirely retained in D444V-hE3 and E340K-hE3, while G194C-hE3 and P453L-hE3 exhibited ~ 30% loss of FAD [24]. Circular dichroism (CD) spectroscopy represented no significant overall structural alterations in the above four mutants [24]. HDX-MS however detected significant changes in flexibility/exposure in the lipoic acid (LA) binding channel of P453L-hE3 and G194C-hE3, which could potentially result in stimulation of ROS production. HDX-MS data for D444V-hE3 and E340K-hE3 were rather inconclusive in terms of the mechanism of action of increased ROS production [25]. MD simulation of 13 disease-causing hE3 mutants was also carried out [59, 60]; good correlation with HDX-MS results was reported for the P453L, K37E, G194C, I445M, and R460G substitutions [4].
Scheme 1.

Forward, reverse, and ROS-generating reactions of LADH
Conclusions of the First Disease-Causing Mutant Structure (D444V-hE3) Relevant to Compromised Enzymatic Activity and ROS Generation
Crystal structures have been published for hE3 [28, 29, 52, 61, 64] and now also for the D444V-hE3 disease-causing mutant from our laboratory [61]. High-resolution crystal structures have very recently been determined in our laboratory also for the P453L- (PDB ID: 6I4Z), G194C- (PDB ID: 6I4P), R460G- (PDB IDs: 6I4R and 6HG8), R447G- (PDB ID: 6I4S), I445M- (PDB ID: 6I4T), and G426E-hE3 (PDB ID: 6I4U) disease-causing variants and for hE3 at the hitherto highest 1.75 Å resolution (PDB ID: 6I4Q), however, the thorough and comparative analysis of these structures is still in progress. The already published and analyzed D444V-hE3 crystal structure demonstrated a shorter and wider H+/H2O-releasing channel when compared to the wild type structure. This channel is solvent accessible, leads to the active site and it is the continuation of the LA-binding substrate channel (Fig. 1); the H+/H2O channel appears to have catalytic roles in LADH function and perhaps ROS generation by hE3 [61]. A drop in surface potential around the exit of this channel was also found when compared to the hE3 structure. Several helices and random coils form the above channel, the helices all pointing with the positive ends of their dipoles towards the active site; structural alterations in the channel-forming helices likely affect the active site via relayed helix dipole moment contributions. All the above mentioned structural alterations in D444V-hE3 hence indeed may lead to a drop in enzyme activity and the positive shift in ROS-generating capacity [61]. Another indirect effect that might also have contribution to the pathological behaviors is an overall change in penetration through the channel upon structural changes, which was suggested altering the apparent redox potential of the FAD moiety [65, 66]; the redox status of FAD has direct influence on both the regular catalytic and ROS-generating activities. The C-terminus in D444V-hE3 was shown by HDX-MS to possess higher flexibility as compared to hE3 [25]; this effect could not be detected by crystallography, perhaps due to the cryogenic conditions. Since the C-terminus separates and hence forms connection between the LA-binding and the H+/H2O channels, any change in this region might also affect the LA-binding substrate channel, which is also implicated in both the normal catalytic action as well as superoxide generation [25, 61]. Since the disease-causing dimer interface substitutions (D444V, E340K, R447G, R460G, I445M) all take place in the vicinity of the H+/H2O channel (Fig. 2), considerably far from the LA-binding channel, the cofactor-binding sites, and the active site, this channel was connected to the potential existence of a generalized pathomechanism of human E3-deficiency for the disease-causing dimerization interface mutations [61].
Fig. 1.

The LA(/DHLA)-binding and H+/H2O channels in the A-B dimers of hE3 (A, PDB ID: 5NHG) and D444V-hE3 (B, PDB ID: 5J5Z). Monomer A is labeled in both proteins. The redox-active C45-C50 pair and FAD are represented as yellow and red sticks, respectively. (Color figure online)
Fig. 2.
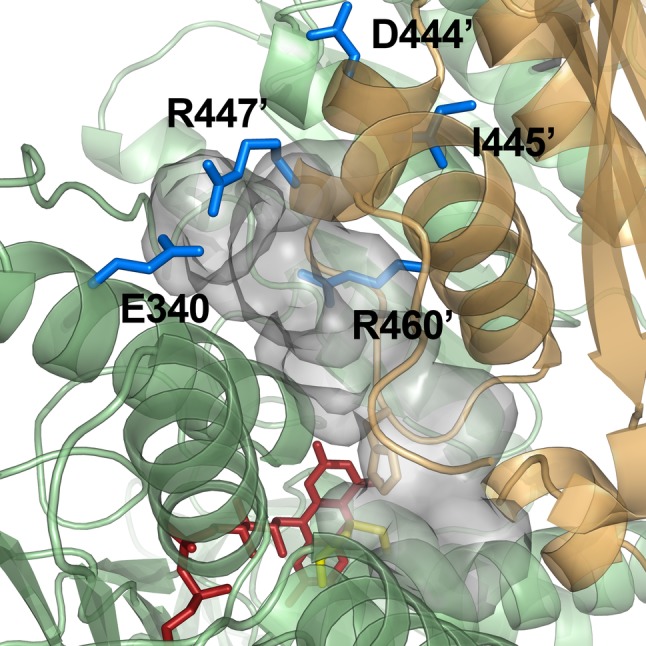
Pathogenic amino acid substitution sites near the H+/H2O channel in the hE3 crystal structure (PDB ID: 5NHG). The inner surface of the channel is displayed (A-B dimer). The C45-C50 pair and FAD are colored as for Fig. 1. (Color figure online)
Affinity Loss for Multienzyme Complexes and Respective Structural Conclusions for D444V-hE3
The affinity of E3 for the KGDHc proved to be low [67–69] and even lower in acidosis [30]; E3 binds ~ 30 times stronger to the PDHc [68, 70, 71]. Several experimental evidence suggest that E1, and not E2, would directly bind E3 in the KGDHc [72–74]. LADH can also exist as a liberated protein in vivo [30, 68, 75–78]; it is the most abundant flavoprotein in brain and muscle mitochondria [79]. Several disease-causing hE3 variants (R447G-, D444V-, R460G-, and E340K-hE3) exhibit significantly impaired affinity for the hPDHc leading to greatly compromised overall hPDHc activities [25–27, 29]. The D444V-hE3 crystal structure demonstrated a drop in surface potential over the entire protein molecule, while HDX-MS showed an enhanced flexibility on the surface where the E3-binding protein (E3BP) of hPDHc is tethered [25, 61]; both effects likely contribute to the compromised affinity for hPDHc. The D444V-hE3 crystal structure also confirmed previous experimental data on the lack of monomerization and FAD loss in this mutant [61].
ROS Generation by the E1–E2 Subcomplex of the hKGDHc
In case hE3 is untethered from the hKGDHc, as is likely the case for several pathogenic variants and in acidosis, the E1-E2 subcomplex is potentially also capable of generating ROS at a very considerable rate in the forward catalytic direction [20] (Scheme 1). Thus, under such conditions, ROS production might proceed simultaneously from E3 (principally in the reverse catalytic direction) as well as from the E1-E2 subcomplex (in the forward catalytic direction), provided that substrate provision is sufficient [4, 20]; an intact population of KGDHc may still retain some overall activity [6], unless the LA prosthetic group suffers oxidative damage [23]. Since the E2 (dihydrolipoamide succinyltransferase) component could potentially also be targeted against ROS generation, we very recently determined its cryo-electronmicroscopic structure; we were able to detect residues 218–453 at 3.31 Å resolution (PDB ID: 6H05), which in a further and still progressing stage of structure determination appears to develop to be 2.9 Å resolution (unpublished result).
Conclusions
In conclusion, human E3-deficiency is still an incurable and hardly manageable disease, which might be taken under better control by dietary restrictions, nutritional support, correction of metabolic acidosis and coagulopathy, administration of flavins, lipoic acid, thiamine, trial of dichloroacetate, for selected and respective cases [1, 4]. Since in many cases with impaired affinity for multienzyme complexes the residual hE3 activity is much higher than the residual overall multienzyme complex activity, with the high-resolution crystal structures in hand, adaptor molecules could potentially be developed to retether the respective pathogenic hE3 mutants to their harboring multienzyme complexes; although this might only be a limited therapeutic solution, the approach can potentially be advantageous for selected patients. Development of specific ROS generation inhibitors against selected hE3 mutants and the E2 subunit of the hKGDHc, besides a general antioxidant therapy, might also be a valid approach towards possible therapeutic solutions. Besides gene therapy, which still requires substantial development to be efficient and completely safe, enzyme replacement therapy might gain potentials in the treatment of human E3 deficiency in the near future [80, 81].
Acknowledgements
Open access funding provided by Semmelweis University (SE). Financial support is acknowledged from the Hungarian Academy of Sciences (MTA Grant 02001 to Vera Adam-Vizi), the Hungarian Scientific Research Fund (OTKA Grant 112230 to V.A.-V.), and the Hungarian Brain Research Program (Grant 2017-1.2.1-NKP-2017-00002 to V.A.-V.). The author thanks Eszter Szabo for the figures and her valuable comments and Prof. Vera Adam-Vizi for her long time support.
Compliance with Ethical Standards
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Special Issue: In honor of Prof. Vera Adam-Vizi.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Quinonez SC, Thoene JG, et al. Dihydrolipoamide Dehydrogenase Deficiency. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet] 1993–2016. Seattle (WA): University of Washington, Seattle; 2014. pp. 1–37. [PubMed] [Google Scholar]
- 2.Scott SA, Edelmann L, Liu L, Luo MJ, Desnick RJ, Kornreich R. Experience with carrier screening and prenatal diagnosis for 16 Ashkenazi Jewish genetic diseases. Hum Mutat. 2010;31:1240–1250. doi: 10.1002/humu.21327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shaag A, Saada A, Berger I, Mandel H, Joseph A, Feigenbaum A, Elpeleg ON. Molecular basis of lipoamide dehydrogenase deficiency in Ashkenazi Jews. Am J Med Genet. 1999;82:177–182. [PubMed] [Google Scholar]
- 4.Ambrus A, Adam-Vizi V. Human dihydrolipoamide dehydrogenase (E3) deficiency: novel insights into the structural basis and molecular pathomechanism. Neurochem Int. 2018;117:5–14. doi: 10.1016/j.neuint.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 5.Carrozzo R, Torraco A, Fiermonte G, Martinelli D, Di Nottia M, Rizza T, Vozza A, Verrigni D, Diodato D, Parisi G, Maiorana A, Rizzo C, Pierri CL, Zucano S, Piemonte F, Bertini E, Dionisi-Vici C. Riboflavin responsive mitochondrial myopathy is a new phenotype of dihydrolipoamide dehydrogenase deficiency. The chaperon-like effect of vitamin B2. Mitochondrion. 2014;18:49–57. doi: 10.1016/j.mito.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 6.Quinonez SC, Leber SM, Martin DM, Thoene JG, Bedoyan JK. Leigh syndrome in a girl with a novel DLD mutation causing E3 deficiency. Pediatr Neurol. 2013;48:67–72. doi: 10.1016/j.pediatrneurol.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quintana E, Pineda M, Font A, Vilaseca MA, Tort F, Ribes A, Briones P. Dihydrolipoamide dehydrogenase (DLD) deficiency in a Spanish patient with myopathic presentation due to a new mutation in the interface domain. J Inherit Metab Dis. 2010;33:S315–S319. doi: 10.1007/s10545-010-9169-4. [DOI] [PubMed] [Google Scholar]
- 8.Cameron JM, Levandovskiy V, MacKay N, Raiman J, Renaud DL, Clarke JTR, Feigenbaum A, Elpeleg O, Robinson BH. Novel mutations in dihydrolipoamide dehydrogenase deficiency in two cousins with borderline-normal PDH complex activity. Am J Med Genet A. 2006;140A:1542–1552. doi: 10.1002/ajmg.a.31313. [DOI] [PubMed] [Google Scholar]
- 9.Odievre MH, Chretien D, Munnich A, Robinson BH, Dumoulin R, Masmoudi S, Kadhom N, Rötig A, Rustin P, Bonnefont JP. A novel mutation in the dihydrolipoamide dehydrogenase E3 subunit gene (DLD) resulting in an atypical form of alpha-ketoglutarate dehydrogenase deficiency. Hum Mutat. 2005;25:323–324. doi: 10.1002/humu.9319. [DOI] [PubMed] [Google Scholar]
- 10.Hong YS, Korman SH, Lee J, Ghoshal P, Qu Q, Barash V, Kang S, Oh S, Kwon M, Gutman A, Rachmel A, Patel MS. Identification of a common mutation (Gly194Cys) in both Arab Moslem and Ashkenazi Jewish patients with dihydrolipoamide dehydrogenase (E3) deficiency: possible beneficial effect of vitamin therapy. J Inherit Metab Dis. 2003;26:816–818. doi: 10.1023/b:boli.0000010004.12053.5b. [DOI] [PubMed] [Google Scholar]
- 11.Grafakou O, Oexle K, van den Heuvel L, Smeets R, Trijbels F, Goebel HH, Bosshard N, Superti-Furga A, Steinmann B, Smeitink J. Leigh syndrome due to compound heterozygosity of dihydrolipoamide dehydrogenase gene mutations. Description of the first E3 splice site mutation. Eur J Pediatr. 2003;162:714–718. doi: 10.1007/s00431-003-1282-z. [DOI] [PubMed] [Google Scholar]
- 12.Cerna L, Wenchich L, Hansiková H, Kmoch S, Peskova K, Chrastina P, Brynda J, Zeman J. Novel mutations in a boy with dihydrolipoamide dehydrogenase deficiency. Med Sci Monitor. 2001;7:1319–1325. [PubMed] [Google Scholar]
- 13.Shany E, Saada A, Landau D, Shaag A, Hershkovitz E, Elpeleg ON. Lipoamide dehydrogenase deficiency due to a novel mutation in the interface domain. Biochem Biophys Res Commun. 1999;262:163–166. doi: 10.1006/bbrc.1999.1133. [DOI] [PubMed] [Google Scholar]
- 14.Hong YS, Kerr DS, Liu TC, Lusk M, Powell BR, Patel MS. Deficiency of dihydrolipoamide dehydrogenase due to two mutant alleles (E340K and G101del)—analysis of a family and prenatal testing. Biochim Biophys Acta-Mol Basis Dis. 1997;1362:160–168. doi: 10.1016/s0925-4439(97)00073-2. [DOI] [PubMed] [Google Scholar]
- 15.Hong YS, Kerr DS, Craigen WJ, Tan J, Pan YZ, Lusk M, Patel MS. Identification of two mutations in a compound heterozygous child with dihydrolipoamide dehydrogenase deficiency. Hum Mol Genet. 1996;5:1925–1930. doi: 10.1093/hmg/5.12.1925. [DOI] [PubMed] [Google Scholar]
- 16.Liu TC, Kim H, Arizmendi C, Kitano A, Patel MS. Identification of two missense mutations in a dihydrolipoamide dehydrogenase-deficient patient. Proc Natl Acad Sci USA. 1993;90:5186–5190. doi: 10.1073/pnas.90.11.5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brassier A, Ottolenghi C, Boutron A, Bertrand AM, Valmary-Degano S, Cervoni JP, Chretien D, Arnoux JB, Hubert L, Rabier D, Lacaille F, de Keyzer Y, Di Martino V, de Lonlay P. Dihydrolipoamide dehydrogenase deficiency: a still overlooked cause of recurrent acute liver failure and Reye-like syndrome. Mol Genet Metab. 2013;109:28–32. doi: 10.1016/j.ymgme.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 18.Barak N, Huminer D, Segal T, Ben Ari Z, Halevy J, Kaspa RT. Lipoamide dehydrogenase deficiency: a newly discovered cause of acute hepatitis in adults. J Hepatol. 1998;29:482–484. doi: 10.1016/s0168-8278(98)80069-x. [DOI] [PubMed] [Google Scholar]
- 19.Elpeleg ON, Saada AB, Shaag A, Glustein JZ, Ruitenbeek W, Tein I, Halevy J. Lipoamide dehydrogenase deficiency: a new cause for recurrent myoglobinuria. Muscle Nerve. 1997;20:238–240. doi: 10.1002/(sici)1097-4598(199702)20:2<238::aid-mus18>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 20.Ambrus A, Nemeria NS, Torocsik B, Tretter L, Nilsson M, Jordan F, Adam-Vizi V. Formation of reactive oxygen species by human and bacterial pyruvate and 2-oxoglutarate dehydrogenase multienzyme complexes reconstituted from recombinant components. Free Radic Biol Med. 2015;89:642–650. doi: 10.1016/j.freeradbiomed.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nemeria NS, Gerfen G, Yang LY, Zhang X, Jordan F. Evidence for functional and regulatory cross-talk between the tricarboxylic acid cycle 2-oxoglutarate dehydrogenase complex and 2-oxoadipate dehydrogenase on the l-lysine, l-hydroxylysine and l-tryptophan degradation pathways from studies in vitro. Biochim Biophys Acta-Bioenerg. 2018;1859:932–939. doi: 10.1016/j.bbabio.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 22.Nemeria NS, Gerfen G, Nareddy PR, Yang LY, Zhang X, Szostak M, Jordan F. The mitochondrial 2-oxoadipate and 2-oxoglutarate dehydrogenase complexes share their E2 and E3 components for their function and both generate reactive oxygen species. Free Radic Biol Med. 2018;115:136–145. doi: 10.1016/j.freeradbiomed.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 23.Vaubel RA, Rustin P, Isaya G. Mutations in the dimer interface of dihydrolipoamide dehydrogenase promote site-specific oxidative damages in yeast and human cells. J Biol Chem. 2011;286:40232–40245. doi: 10.1074/jbc.M111.274415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ambrus A, Torocsik B, Tretter L, Ozohanics O, Adam-Vizi V. Stimulation of reactive oxygen species generation by disease-causing mutations of lipoamide dehydrogenase. Hum Mol Genet. 2011;20:2984–2995. doi: 10.1093/hmg/ddr202. [DOI] [PubMed] [Google Scholar]
- 25.Ambrus A, Wang JJ, Mizsei R, Zambo Z, Torocsik B, Jordan F, Adam-Vizi V. Structural alterations induced by ten disease-causing mutations of human dihydrolipoamide dehydrogenase analyzed by hydrogen/deuterium-exchange mass spectrometry: implications for the structural basis of E3 deficiency. Biochim Biophys Acta-Mol Basis Dis. 2016;1862:2098–2109. doi: 10.1016/j.bbadis.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park Y-H, Patel MS. Characterization of interactions of dihydrolipoamide dehydrogenase with its binding protein in the human pyruvate dehydrogenase complex. Biochem Biophys Res Commun. 2010;395:416–419. doi: 10.1016/j.bbrc.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patel MS, Korotchkina LG, Sidhu S. Interaction of E1 and E3 components with the core proteins of the human pyruvate dehydrogenase complex. J Mol Catal B-Enzym. 2009;61:2–6. doi: 10.1016/j.molcatb.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ciszak EM, Makal A, Hong YS, Vettaikkorumakankauv AK, Korotchkina LG, Patel MS. How dihydrolipoamide dehydrogenase-binding protein binds dihydrolipoamide dehydrogenase in the human pyruvate dehydrogenase complex. J Biol Chem. 2006;281:648–655. doi: 10.1074/jbc.M507850200. [DOI] [PubMed] [Google Scholar]
- 29.Brautigam CA, Wynn RM, Chuang JL, Machius M, Tomchick DR, Chuang DT. Structural insight into interactions between dihydrolipoamide dehydrogenase (E3) and E3 binding protein of human pyruvate dehydrogenase complex. Structure. 2006;14:611–621. doi: 10.1016/j.str.2006.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ambrus A, Tretter L, Adam-Vizi V. Inhibition of the alpha-ketoglutarate dehydrogenase-mediated reactive oxygen species generation by lipoic acid. J Neurochem. 2009;109:222–229. doi: 10.1111/j.1471-4159.2009.05942.x. [DOI] [PubMed] [Google Scholar]
- 31.Mailloux RJ, Gardiner D, O’Brien M. 2-Oxoglutarate dehydrogenase is a more significant source of O2·/H2O2 than pyruvate dehydrogenase in cardiac and liver tissue. Free Radic Biol Med. 2016;97:501–512. doi: 10.1016/j.freeradbiomed.2016.06.014. [DOI] [PubMed] [Google Scholar]
- 32.Quinlan CL, Goncalves RL, Hey-Mogensen M, Yadava N, Bunik VI, Brand MD. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J Biol Chem. 2014;289:8312–8325. doi: 10.1074/jbc.M113.545301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tretter L, Adam-Vizi V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J Neurosci. 2004;24:7771–7778. doi: 10.1523/JNEUROSCI.1842-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi QL, Xu H, Kleinman WA, Gibson GE. Novel functions of the alpha-ketoglutarate dehydrogenase complex may mediate diverse oxidant-induced changes in mitochondrial enzymes associated with Alzheimer’s disease. Biochim Biophys Acta Mol Basis Dis. 2008;1782:229–238. doi: 10.1016/j.bbadis.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Starkov AA. An update on the role of mitochondrial alpha-ketoglutarate dehydrogenase in oxidative stress. Mol Cell Neurosci. 2013;55:13–16. doi: 10.1016/j.mcn.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adam-Vizi V, Tretter L. The role of mitochondrial dehydrogenases in the generation of oxidative stress. Neurochem Int. 2013;62:757–763. doi: 10.1016/j.neuint.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 38.Andreyev AY, Kushnareva YE, Murphy AN, Starkov AA. Mitochondrial ROS metabolism: 10years later. Biochemistry (Moscow) 2015;80:517–531. doi: 10.1134/S0006297915050028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andreyev AI, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Moscow) 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 40.Rimessi A, Previati M, Nigro F, Wieckowskic MR, Pinton P. Mitochondrial reactive oxygen species and inflammation: Molecular mechanisms, diseases and promising therapies. Int J Biochem Cell Biol. 2016;81:281–293. doi: 10.1016/j.biocel.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 41.Nemeria NS, Gerfen G, Guevara E, Nareddy PR, Szostak M, Jordan F. The human Krebs cycle 2-oxoglutarate dehydrogenase complex creates an additional source of superoxide/hydrogen peroxide from 2-oxoadipate as alternative substrate. Free Radic Biol Med. 2017;108:644–654. doi: 10.1016/j.freeradbiomed.2017.04.017. [DOI] [PubMed] [Google Scholar]
- 42.Fisher-Wellman KH, Gilliam LAA, Lin CT, Cathey BL, Lark DS, Neufer PD. Mitochondrial glutathione depletion reveals a novel role for the pyruvate dehydrogenase complex as a key H2O2-emitting source under conditions of nutrient overload. Free Radic Biol Med. 2013;65:1201–1208. doi: 10.1016/j.freeradbiomed.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ambrus A, Torocsik B, Adam-Vizi V. Refolding of the human dihydrolipoamide dehydrogenase. Biochem Eng J. 2009;45:120–125. doi: 10.1016/j.pep.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 44.Ambrus A, Torocsik B, Adam-Vizi V. Periplasmic cold expression and one-step purification of human dihydrolipoamide dehydrogenase. Protein Expr Purif. 2009;63:50–57. doi: 10.1016/j.pep.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 45.Gazaryan IG, Krasnikov BF, Ashby GA, Thorneley RNF, Kristal BS, Brown AM. Zinc is a potent inhibitor of thiol oxidoreductase activity and stimulates reactive oxygen species production by lipoamide dehydrogenase. J Biol Chem. 2002;277:10064–10072. doi: 10.1074/jbc.M108264200. [DOI] [PubMed] [Google Scholar]
- 46.Bando Y, Aki K. Mechanisms of generation of oxygen radicals and reductive mobilization of ferritin iron by lipoamide dehydrogenase. J Biochem. 1991;109:450–454. doi: 10.1093/oxfordjournals.jbchem.a123402. [DOI] [PubMed] [Google Scholar]
- 47.Massey V, Strickland S, Mayhew SG, Howell LG, Engel PC, Matthews RG, Schuman M, Sullivan PA. The production of superoxide anion radicals in the reaction of reduced flavins and flavoproteins with molecular oxygen. Biochem Biophys Res Commun. 1969;36:891–897. doi: 10.1016/0006-291x(69)90287-3. [DOI] [PubMed] [Google Scholar]
- 48.Huennekens FM, Basford RE, Gabrio BW. An oxidase for reduced diphosphopyridine nucleotide. J Biol Chem. 1955;213:951–967. [PubMed] [Google Scholar]
- 49.Tahara EB, Barros MH, Oliveira GA, Netto LES, Kowaltowski AJ. Dihydrolipoyl dehydrogenase as a source of reactive oxygen species inhibited by caloric restriction and involved in Saccharomyces cerevisiae aging. Faseb J. 2007;21:274–283. doi: 10.1096/fj.06-6686com. [DOI] [PubMed] [Google Scholar]
- 50.Kareyeva AV, Grivennikova VG, Vinogradov AD. Mitochondrial hydrogen peroxide production as determined by the pyridine nucleotide pool and its redox state. Biochim Biophys Acta Bioenerg. 2012;1817:1879–1885. doi: 10.1016/j.bbabio.2012.03.033. [DOI] [PubMed] [Google Scholar]
- 51.Tretter L, Ambrus A. Measurement of ROS homeostasis in isolated mitochondria. Methods Enzymol. 2014;547:199–223. doi: 10.1016/B978-0-12-801415-8.00012-6. [DOI] [PubMed] [Google Scholar]
- 52.Brautigam CA, Chuang JL, Tomchick DR, Machius M, Chuang DT. Crystal structure of human dihydrolipoamide dehydrogenase: NAD(+)/NADH binding and the structural basis of disease-causing mutations. J Mol Biol. 2005;350:543–552. doi: 10.1016/j.jmb.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 53.Qi F, Pradhan RK, Dash RK, Beard DA. Detailed kinetics and regulation of mammalian 2-oxoglutarate dehydrogenase. BMC Biochem. 2011;12:53. doi: 10.1186/1471-2091-12-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu TC, Korotchkina LG, Hyatt SL, Vettakkorumakankav NN, Patel MS. Spectroscopic studies of the characterization of recombinant human dihydrolipoamide dehydrogenase and its side-directed mutants. J Biol Chem. 1995;270:15545–15550. doi: 10.1074/jbc.270.26.15545. [DOI] [PubMed] [Google Scholar]
- 55.Kim H, Patel MS. Characterization of 2 site specifically mutated human dihydrolipoamide dehydrogenases (His-452-]Gln and Glu-457-]Gln) J Biol Chem. 1992;267:5128–5132. [PubMed] [Google Scholar]
- 56.Klyachko NL, Shchedrina VA, Efimov AV, Kazakov SV, Gazaryan IG, Kristal BS, Brown AM. pH-dependent substrate preference of pig heart lipoamide dehydrogenase varies with oligomeric state—response to mitochondrial matrix acidification. J Biol Chem. 2005;280:16106–16114. doi: 10.1074/jbc.M414285200. [DOI] [PubMed] [Google Scholar]
- 57.Visser J, Veeger C. Relations between conformations and activities of lipoamide dehydrogenase. 3. Protein association-dissociation and the influence on catalytic properties. Biochim Biophys Acta. 1968;159:265–275. doi: 10.1016/0005-2744(68)90075-2. [DOI] [PubMed] [Google Scholar]
- 58.Tsai CS, Templeton DM, Wand AJ. Multifunctionality of lipoamide dehydrogenase: activities of chemically trapped monomeric and dimeric enzymes. Arch Biochem Biophys. 1981;206:77–86. doi: 10.1016/0003-9861(81)90068-0. [DOI] [PubMed] [Google Scholar]
- 59.Ambrus A, Mizsei R, Adam-Vizi V. Structural alterations by five disease-causing mutations in the low-pH conformation of human dihydrolipoamide dehydrogenase (hLADH) analyzed by molecular dynamics—implications in functional loss and modulation of reactive oxygen species generation by pathogenic hLADH forms. Biochem Biophys Reports. 2015;2:50–56. doi: 10.1016/j.bbrep.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ambrus A, Adam-Vizi V. Molecular dynamics study of the structural basis of dysfunction and the modulation of reactive oxygen species generation by pathogenic mutants of human dihydrolipoamide dehydrogenase. Arch Biochem Biophys. 2013;538:145–155. doi: 10.1016/j.abb.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 61.Szabo E, Mizsei R, Wilk P, Zambo Z, Torocsik B, Weiss MS, Adam-Vizi V, Ambrus A. Crystal structures of the disease-causing D444V mutant and the relevant wild type human dihydrolipoamide dehydrogenase. Free Radic Biol Med. 2018;124:214–220. doi: 10.1016/j.freeradbiomed.2018.06.008. [DOI] [PubMed] [Google Scholar]
- 62.Ambrus A, Friedrich K, Somogyi A. Oligomerization of nitrophorins. Anal Biochem. 2006;352:286–295. doi: 10.1016/j.ab.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 63.Sakaguchi Y, Yoshino M, Aramaki S, Yoshida I, Yamashita F, Kuhara T, Matsumoto I, Hayashi T. Dihydrolipoyl dehydrogenase deficiency—a therapeutic trial with branched-chain amino acid restriction. Eur J Pediatr. 1986;145:271–274. doi: 10.1007/BF00439399. [DOI] [PubMed] [Google Scholar]
- 64.Brautigam CA, Wynn RM, Chuang JL, Naik MT, Young BB, Huang TH, Chuang DT. Structural and thermodynamic basis for weak interactions between dihydrolipoamide dehydrogenase and subunit-binding domain of the branched-chain alpha-ketoacid dehydrogenase complex. J Biol Chem. 2011;286:23476–23488. doi: 10.1074/jbc.M110.202960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Benen J, Vanberkel W, Dieteren N, Arscott D, Williams C, Veeger C, Dekok A. Lipoamide dehydrogenase from Azotobacter vinelandii-site-directed mutagenesis of the His450-Glu455 diad-kinetics of wild-type and mutated enzymes. Eur J Biochem. 1992;207:487–497. doi: 10.1111/j.1432-1033.1992.tb17075.x. [DOI] [PubMed] [Google Scholar]
- 66.Benen J, Vanberkel W, Veeger C, Dekok A. Lipoamide dehydrogenase from Azotobacter vinelandii—the role of the C-terminus in catalysis and dimer stabilization. Eur J Biochem. 1992;207:499–505. doi: 10.1111/j.1432-1033.1992.tb17076.x. [DOI] [PubMed] [Google Scholar]
- 67.Bunik V, Westphal AH, de Kok A. Kinetic properties of the 2-oxoglutarate dehydrogenase complex from Azotobacter vinelandii—evidence for the formation of a precatalytic complex with 2-oxoglutarate. Eur J Biochem. 2000;267:3583–3591. doi: 10.1046/j.1432-1327.2000.01387.x. [DOI] [PubMed] [Google Scholar]
- 68.Reed LJ, Oliver RM. The multienzyme alpha-keto acid dehydrogenase complexes. Brookhaven Symp Biol. 1968;21:397–412. [PubMed] [Google Scholar]
- 69.Massey V. The composition of the ketoglutarate dehydrogenase complex. Biochim Biophys Acta. 1960;38:447–460. doi: 10.1016/0006-3002(60)91280-4. [DOI] [PubMed] [Google Scholar]
- 70.Poulsen LL, Wedding RT. Purification and properties of the a-ketoglutarate dehydrogenase complex of cauliflower mitochondria. J Biol Chem. 1970;245:5709–5717. [PubMed] [Google Scholar]
- 71.Erfle JD, Sauer F. The inhibitory effects of acyl-coenzyme A esters on the pyruvate and a-oxoglutarate dehydrogenase complexes. Biochim Biophys Acta. 1969;178:441–452. doi: 10.1016/0005-2744(69)90213-7. [DOI] [PubMed] [Google Scholar]
- 72.Zhou J, Yang L, Ozohanics O, Zhang X, Wang J, Ambrus A, Arjunan P, Brukh R, Nemeria NS, Furey W, Jordan F. A multipronged approach unravels unprecedented protein-protein interactions in the human 2-oxoglutarate dehydrogenase multienzyme complex. J Biol Chem. 2018;293:19213–19227. doi: 10.1074/jbc.RA118.005432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rice JE, Dunbar B, Lindsay JG. Sequences directing dihydrolipoamide dehydrogenase (E3) binding are located on the 2-oxoglutarate dehydrogenase (E1) component of the mammalian 2-oxoglutarate dehydrogenase multienzyme complex. EMBO J. 1992;11:3229–3235. doi: 10.1002/j.1460-2075.1992.tb05400.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McCartney RG, Rice JE, Sanderson SJ, Bunik V, Lindsay H, Lindsay JG. Subunit interactions in the mammalian alpha-ketoglutarate dehydrogenase complex - Evidence for direct association of the alpha-ketoglutarate dehydrogenase and dihydrolipoamide dehydrogenase components. J Biol Chem. 1998;273:24158–24164. doi: 10.1074/jbc.273.37.24158. [DOI] [PubMed] [Google Scholar]
- 75.Yan LJ, Thangthaeng N, Sumien N, Forster MJ. Serum dihydrolipoamide dehydrogenase is a labile enzyme. J Biochem Pharmacol Res. 2013;1:30–42. [PMC free article] [PubMed] [Google Scholar]
- 76.Constantinescu A, Pick U, Handelman GJ, Haramaki N, Han D, Podda M, Tritschler HJ, Packer L. Reduction and transport of lipoic acid by human erythrocytes. Biochem Pharmacol. 1995;50:253–261. doi: 10.1016/0006-2952(95)00084-d. [DOI] [PubMed] [Google Scholar]
- 77.Reed LJ, Hackert ML. Structure-function relationships in dihydrolipoamide acyltransferases. J Biol Chem. 1990;265:8971–8974. [PubMed] [Google Scholar]
- 78.Jiang ZH, Chen QY, Harrison TJ, Li GJ, Wang XY, Li H, Hu LP, Li KW, Yang QL, Tan C, Fang ZL. Hepatitis B virus core promoter double mutations (A1762T, G1764A) are associated with lower levels of serum dihydrolipoyl dehydrogenase. Intervirology. 2016;59:1–7. doi: 10.1159/000445319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kunz WS, Gellerich FN. Quantification of the content of fluorescent flavoproteins in mitochondria from liver, kidney cortex, skeletal muscle, and brain. Biochem Med Metab Biol. 1993;50:103–110. doi: 10.1006/bmmb.1993.1051. [DOI] [PubMed] [Google Scholar]
- 80.Rapoport M, Saada A, Elpeleg O, Lorberboum-Galski H. TAT-mediated delivery of LAD restores pyruvate dehydrogenase complex activity in the mitochondria of patients with LAD deficiency. Mol Ther. 2008;16:691–697. doi: 10.1038/mt.2008.4. [DOI] [PubMed] [Google Scholar]
- 81.Rapoport M, Salman L, Sabag O, Patel MS, Lorberboum-Galski H. Successful TAT-mediated enzyme replacement therapy in a mouse model of mitochondrial E3 deficiency. J Mol Med. 2011;89:161–170. doi: 10.1007/s00109-010-0693-3. [DOI] [PubMed] [Google Scholar]