

Abstract
BACKGROUND
Type 1 diabetes is a chronic autoimmune disease that leads to destruction of insulin-producing beta cells and dependence on exogenous insulin for survival. Some interventions have delayed the loss of insulin production in patients with type 1 diabetes, but interventions that might affect clinical progression before diagnosis are needed.
METHODS
We conducted a phase 2, randomized, placebo-controlled, double-blind trial of teplizumab (an Fc receptor–nonbinding anti-CD3 monoclonal antibody) involving relatives of patients with type 1 diabetes who did not have diabetes but were at high risk for development of clinical disease. Patients were randomly assigned to a single 14-day course of teplizumab or placebo, and follow-up for progression to clinical type 1 diabetes was performed with the use of oral glucose-tolerance tests at 6-month intervals.
RESULTS
A total of 76 participants (55 [72%] of whom were ≤18 years of age) underwent randomization — 44 to the teplizumab group and 32 to the placebo group. The median time to the diagnosis of type 1 diabetes was 48.4 months in the teplizumab group and 24.4 months in the placebo group; the disease was diagnosed in 19 (43%) of the participants who received teplizumab and in 23 (72%) of those who received placebo. The hazard ratio for the diagnosis of type 1 diabetes (teplizumab vs. placebo) was 0.41 (95% confidence interval, 0.22 to 0.78; P= 0.006 by adjusted Cox proportional-hazards model). The annualized rates of diagnosis of diabetes were 14.9% per year in the teplizumab group and 35.9% per year in the placebo group. There were expected adverse events of rash and transient lymphopenia. KLRG1+TIGIT+CD8+ T cells were more common in the teplizumab group than in the placebo group. Among the participants who were HLA-DR3–negative, HLA-DR4–positive, or anti–zinc transporter 8 antibody–negative, fewer participants in the teplizumab group than in the placebo group had diabetes diagnosed.
CONCLUSIONS
Teplizumab delayed progression to clinical type 1 diabetes in high-risk participants. (Funded by the National Institutes of Health and others; ClinicalTrials.gov number, .)
TYPE 1 DIABETES IS CAUSED BY THE AUTO-immune destruction of insulin-producing beta cells in the islets of Langerhans, which leads to dependence on exogenous insulin for survival. Approximately 1 million to 1.5 million Americans have type 1 diabetes, which is one of the most common diseases of childhood (second-most-common after asthma).1 Despite improvements in care, the desired glycemic targets are not achieved in most patients with type 1 diabetes,2 and an increased risk of complications and death persists. Two studies involving Scottish men and women noted the loss of 14.2 and 17.7 life-years, respectively, among those in whom the condition was diagnosed before the age of 10 years and of 11 and 13 life-years, respectively, among those in whom it was diagnosed before the age of 20 years.3,4
In genetically susceptible persons, type 1 diabetes progresses through asymptomatic stages before the development of overt hyperglycemia. These stages are characterized by the appearance of autoantibodies (stage 1) and then dysglycemia (stage 2). In stage 2, metabolic responses to a glucose load are impaired, but other metabolic indexes — for example, the level of glycosylated hemoglobin — remain normal, and insulin treatment is not needed.5 These immunologic and metabolic features can identify persons at high risk for development of clinical disease; overt hyperglycemia, once it develops, requires insulin treatment.
Several immune interventions, when studied in patients with recent-onset clinical type 1 diabetes, have been reported to delay the decline in beta-cell function.6 One promising type of therapy appears to be Fc receptor–nonbinding anti-CD3 monoclonal antibodies, such as teplizumab; multiple studies involving patients with type 1 diabetes have shown that teplizumab treatment reduces the loss of beta-cell function, even as long as 7 years after diagnosis.7–11 The drug modifies CD8+ T lymphocytes, which are thought to be the important effector cells that kill beta cells.12,13
Whether interventions at stage 1 or 2 might alter the progression to clinical type 1 diabetes has been unclear. We therefore tested whether teplizumab treatment would prevent or delay the onset of clinical type 1 diabetes in high-risk persons.
METHODS
TRIAL PARTICIPANTS
Participants were identified through the TrialNet Natural History Study.14 The trial was conducted from July 2011 through November 2018 at sites in the United States, Canada, Australia, and Germany (Fig. S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org). The full protocol is available at NEJM.org. Institutional-review-board approval was obtained at each participating site (see the Supplementary Appendix for a full listing). The participants, their parents, or both provided written informed consent or assent before trial entry.
Eligible participants were nondiabetic relatives of patients with type 1 diabetes and were at least 8 years of age at the time of randomization and at high risk for development of clinical diabetes. Participants also had to have had two or more diabetes-related autoantibodies detected in two samples obtained within 6 months before randomization. In addition, participants had to have had evidence of dysglycemia during an oral glucose-tolerance test, with dysglycemia defined as a fasting glucose level of 110 to 125 mg per deciliter (6.1 to 6.9 mmol per liter), a 2-hour postprandial plasma glucose level of at least 140 mg per deciliter (7.8 mmol per liter) and less than 200 mg per deciliter (11.1 mmol per liter), or an intervening postprandial glucose level at 30, 60, or 90 minutes of greater than 200 mg per deciliter on two occasions, within 52 days before enrollment. The protocol was amended in 2014 to allow enrollment of participants younger than 18 years of age who had a single abnormal oral glucose-tolerance test result, because the rates of type 1 diabetes progression were similar with or without a confirmatory oral glucose-tolerance test in this age group. In eight participants (five in the teplizumab group and three in the placebo group), the second pretreatment oral glucose-tolerance test was performed on the first day of administration of teplizumab or placebo. Persons with other clinically important medical histories, abnormal laboratory chemical values, or abnormal blood counts were excluded.
TRIAL DESIGN AND INTERVENTION
Participants were randomly assigned in a 1:1 ratio to receive either teplizumab or placebo. Randomization was stratified according to TrialNet site, age (<18 years or ≥18 years), and second oral glucose-tolerance test result before treatment (impaired tolerance, normal tolerance, or diabetes). The treatment-group assignments were double-masked. Participants received a 14-day outpatient course of teplizumab or saline to be administered intravenously in a clinical research center. Teplizumab was given at a dose of 51 μg per square meter of body-surface area on day 0, a dose of 103 μg per square meter on day 1, a dose of 207 μg per square meter on day 2, and a dose of 413 μg per square meter on day 3, followed by a dose of 826 μg per square meter on each of days 4 through 13, as described previously.7,10
END POINTS AND ASSESSMENTS
The primary end point was the elapsed time from randomization to the clinical diagnosis of diabetes, determined with the use of criteria from the American Diabetes Association.15 Scheduled oral glucose-tolerance tests were performed 3 months and 6 months after the infusions and every 6 months thereafter. Random screening glucose levels were evaluated at 3-month intervals, and an oral glucose-tolerance test was performed if the random glucose level was higher than 200 mg per deciliter (11.1 mmol per liter) in association with standardized symptoms of diabetes.
Oral glucose-tolerance test results that indicated diabetes were then sequentially confirmed, and the date of diagnosis was identified as the time of the first of the two diagnostic tests.16 Outcomes were reviewed by the TrialNet Eligibility and Events Committee, the members of which were unaware of the treatment-group assignments.
TRIAL OVERSIGHT
The trial was developed and conducted by Type 1 Diabetes TrialNet, which is funded by the National Institutes of Health and the Juvenile Diabetes Research Foundation. MacroGenics was the holder of the investigational new drug application at the start of the trial. Currently, Provention Bio holds the application, and employees of Provention Bio reviewed the manuscript before submission.
The trial coordination, laboratory tests, and data management were conducted centrally, with the exception of complete blood count and differential and routine chemical analyses, which were performed at the infusion sites. Flow cytometry was performed centrally (Table S1 in the Supplementary Appendix). TrialNet investigators designed the trial. Members of the TrialNet Coordinating Center, including two of the authors, gathered and analyzed the data and vouch for the accuracy and completeness of the data and for the fidelity of the trial to the protocol. An independent medical monitor (who was unaware of the treatment-group assignments) reviewed all accruing safety data. MacroGenics provided teplizumab and matching placebo but was not involved in the conduct of the trial or in data analysis. Representatives from the sponsoring institute of the National Institutes of Health (National Institute of Diabetes and Digestive and Kidney Diseases) participated in the design and conduct of the trial; interpretation of the data; preparation, review, and approval of the manuscript for submission; and the decision to submit the manuscript for publication. The sponsor did not have the right or ability to veto submission for publication.
STATISTICAL ANALYSIS
The cumulative incidence of diabetes diagnosis within each group over time after randomization was estimated in a Kaplan–Meier analysis with the “diabetes-free” survival function.17 The difference between the treatment groups in the 6-month-interval cumulative-incidence functions was estimated as the hazard ratio, and hypotheses were evaluated with the use of a likelihood-ratio test; both analyses were based on the Cox proportional-hazards model.18
Because of slower-than-expected rates of enrollment, the original protocol (which called for the enrollment of 144 participants) was revised to detect a 60% (previously 50%) lower risk in the teplizumab group than in the placebo group (i.e., a hazard ratio of 0.4) with 80% statistical power at an alpha level of 0.025 (one-sided). This update set the goal of enrolling at least 71 participants and following them until 40 participants had received a diagnosis of type 1 diabetes.19
Data on safety and efficacy were evaluated twice yearly by an independent data and safety monitoring board. An interim analysis was conducted when 18 (of 40) cases of type 1 diabetes had been observed, and a formal comparison was presented to the data and safety monitoring board. Lan–DeMets stopping rules were used.20 Data were analyzed according to the intention-to-treat principle. Tests of significance reported herein are two-sided, with a threshold of significance of 0.05. The interim assessment had a negligible effect on the threshold of significance for the final analysis (one-sided P = 0.0247), and therefore fixed-sample significance levels are reported. All confidence intervals reported are 95% confidence intervals. Subgroup analyses were prespecified but were not adjusted for multiple testing. Flow-cytometry data were analyzed by means of analysis of variance at four time points. Statistical analyses were performed with either TIBCO Spotfire S+ Workbench, version 8.2 (TIBCO), or SAS software, version 9.4 (SAS Institute).
RESULTS
PARTICIPANTS
Of the 112 potential participants who were screened for eligibility, 76 underwent randomization — 44 to the teplizumab group and 32 to the placebo group (Fig. S2 in the Supplementary Appendix). The randomization process resulted in unequal numbers of participants in the treatment groups, perhaps because of the small number of enrolled participants (<4) at some study sites, randomization stratification, or other, unclear factors. Before enrollment, all participants were positive for at least two autoantibodies, and 71% were positive for three or more autoantibodies. The treatment groups were generally well balanced with regard to baseline characteristics (Table 1, and Table S2 in the Supplementary Appendix). The majority of participants (55 [72%]) were children (<18 years), most were white, and more than half were siblings of patients with type 1 diabetes. Of the 55 participants who were younger than 18 years of age, 47 had a confirmed dysglycemic oral glucose-tolerance test result before randomization. Of the participants who underwent randomization after a single dysglycemic test result, 2 had diabetic and 6 had normal oral glucose-tolerance test results on the day of randomization. These participants were included in the intention-to-treat analysis, which was adjusted for the results of the blinded oral glucose-tolerance test before randomization.
Table 1.
Baseline Characteristics of the Participants.*
| Characteristic | Teplizumab (N = 44) | Placebo (N = 32) |
|---|---|---|
| Age — yr | ||
| Median (IQR) | 14 (12–22) | 13 (11–16) |
| Range | 8.5–49.5 | 8.6–45.0 |
| Age <18 yr — no. (%) | 29 (66) | 26 (81) |
| Male sex — % | 57 | 53 |
| Relationship to person with type 1 diabetes — no. (%) | ||
| Sibling† | 28 (64) | 16 (50) |
| Offspring | 6 (14) | 6 (19) |
| Parent | 6 (14) | 3 (9) |
| Sibling and another first-degree relative | 2 (5) | 3 (9) |
| Second-degree relative | 2 (5) | 3 (9) |
| Third-degree relative or further removed | 0 | 1 (3) |
| Autoantibodies — no. of participants positive (%)‡ | ||
| Anti-GAD65, harmonized | 40 (91) | 28 (88) |
| Micro insulin | 20 (45) | 11 (34) |
| Anti–IA-2, harmonized | 27 (61) | 24 (75) |
| ICA | 29 (66) | 28 (88) |
| Anti-ZnT8 | 32 (73) | 24 (75) |
| Median glycated hemoglobin level (IQR) — % | 5.2 (4.9–5.4) | 5.3 (5.1–5.4) |
Percentages may not total 100 because of rounding. GAD65 denotes glutamic acid decarboxylase 65, IA-2 islet antigen 2, ICA islet-cell autoantibody, IQR interquartile range, and ZnT8 zinc transporter 8.
Participants in this category may have had more than one sibling with type 1 diabetes.
Shown are the autoantibodies for which participants were positive at the time of randomization. All participants were positive for at least two autoantibodies before randomization.
In total, 93% of participants in the teplizumab group (41 of 44) and 88% of participants in the placebo group (28 of 32) completed the 14-day course of the assigned trial agent. The median total dose of teplizumab administered was 9.14 mg per square meter (interquartile range, 9.01 to 9.37). Three participants in the teplizumab group and 4 participants in the placebo group did not complete the trial regimen; the reasons were laboratory abnormalities (4 participants), an inability to have intravenous access established (2), or rash (1). The median follow-up duration was 745 days (range, 74 to 2683). The duration of follow-up was more than 3 years in 57 participants (75%). Type 1 diabetes was diagnosed in 42 participants (55%).
EFFICACY
Treatment with a single course of teplizumab delayed the time to diagnosis of type 1 diabetes (Fig. 1): 19 (43%) of the 44 participants who received teplizumab and 23 (72%) of the 32 participants who received placebo had type 1 diabetes diagnosed. The annualized rates of diagnosis of type 1 diabetes were 14.9% per year in the teplizumab group and 35.9% per year in the placebo group. The median time to diagnosis was 48.4 months in the teplizumab group and 24.4 months in the placebo group (hazard ratio, 0.41; 95% confidence interval [CI], 0.22 to 0.78; two-sided P = 0.006). The hazard ratio remained significant when adjusted for prespecified covariates of age, the results of the second oral glucose-tolerance test before randomization, or the presence of anti-GAD65 antibody.
Figure 1. Effects of Teplizumab on Development of Type 1 Diabetes.
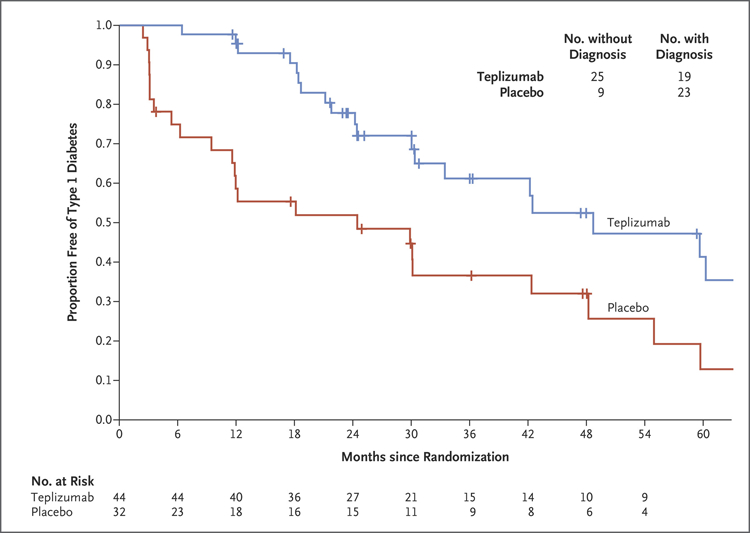
Shown are Kaplan–Meier estimates of the proportions of participants in whom clinical diabetes was not diagnosed. The overall hazard ratio was 0.41 (95% confidence interval [CI], 0.22 to 0.78; two-sided P = 0.006 by adjusted Cox proportional-hazards model). The median time to diagnosis of type 1 diabetes was 48.4 months in the teplizumab group and 24.4 months in the placebo group. The numbers of participants with or without a diagnosis of clinical type 1 diabetes (upper right) represent data at the conclusion of the trial. Tick marks indicate censored data.
The percentage of participants with progression to clinical type 1 diabetes in the overall trial population was greater in the first year after trial entry (17 of the 42 participants with progression, 40%) than in year 2 (10 participants, 24%), year 3 (6 participants, 14%), or year 4 (5 participants, 12%) (Table S3 in the Supplementary Appendix). The largest effect of teplizumab treatment was found in the first year: diabetes was diagnosed in only 3 of 44 participants (7%) in the teplizumab group, in contrast to 14 of 32 participants (44%) in the placebo group (unadjusted hazard ratio, 0.13; 95% CI, 0.05 to 0.34).
SAFETY
Teplizumab treatment was associated with adverse events, which are listed in Table 2. Similar to findings in previous trials of teplizumab in patients with new-onset type 1 diabetes, the lymphocyte count decreased to a nadir on day 5 (total decrease, 72.3%; interquartile range, 82.1 to 68.4; P<0.001) (Fig. 2A).7,8 A total of 15 (75%) of the 20 grade 3 events in the teplizumab group involved lymphopenia during the first 30 days after administration. Lymphopenia resolved by day 45 in all participants except one; in that participant, the lymphocyte counts returned to the normal range on day 105. A spontaneously resolving rash, as previously noted, occurred in 16 (36%) of participants who received teplizumab.8 The rates of infection were similar in the two treatment groups.
Table 2.
Adverse Events during Active Follow-up.*
| Adverse Event Category | Teplizumab | Placebo | ||
|---|---|---|---|---|
| Events (N = 112) |
Participants (N = 44) |
Events (N = 23) |
Participants (N = 32) |
|
| no. | no. (%) | no. | no. (%) | |
| Blood or bone marrow† | 45 | 33 (75) | 2 | 2 (6) |
| Dermatologic or skin† | 17 | 16 (36) | 1 | 1 (3) |
| Pain | 11 | 5 (11) | 5 | 3 (9) |
| Infection | 8 | 5 (11) | 5 | 3 (9) |
| Gastrointestinal | 5 | 4 (9) | 3 | 3 (9) |
| Metabolic or laboratory | 7 | 4 (9) | 2 | 2 (6) |
| Pulmonary or upper respiratory | 6 | 4 (9) | 0 | 0 |
| Constitutional symptoms | 3 | 2 (5) | 0 | 0 |
| Allergy or immunologic | 2 | 2 (5) | 0 | 0 |
| Cardiac, general | 1 | 1 (2) | 1 | 1 (3) |
| Endocrine | 0 | 0 | 2 | 2 (6) |
| Vascular | 1 | 1 (2) | 1 | 1 (3) |
| Neurologic | 1 | 1 (2) | 0 | 0 |
| Ocular or visual | 1 | 1 (2) | 0 | 0 |
| Musculoskeletal or soft tissue | 2 | 1 (2) | 0 | 0 |
| Hepatobiliary or pancreatic | 0 | 0 | 1 | 1 (3) |
| Syndrome | 1 | 1 (2) | 0 | 0 |
| Hemorrhage or bleeding | 1 | 1 (2) | 0 | 0 |
Events listed were attributed as possibly, probably, or definitely related to the trial agent by the trial-site investigator.
The frequency of this type of event differed significantly between the two groups (P<0.001).
Figure 2. Changes in T-Cell Subsets in the Treatment Groups.
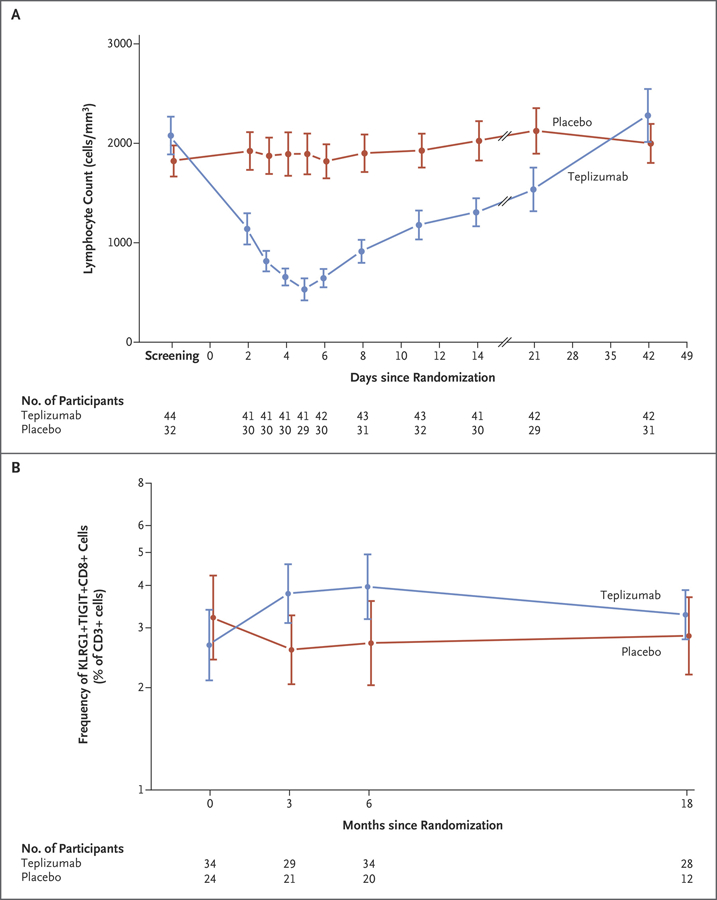
Panel A shows the absolute lymphocyte counts in the treatment groups over the first 7 weeks after enrollment. Panel B shows the frequency of KLRG1+TIGIT+CD8+ T cells as a percentage of total CD3+ T cells in the teplizumab and placebo groups. The estimates of the percentage differences between the teplizumab group and the placebo group are 46.5% at 3 months (95% CI, 8.23 to 98.4), 49% at 6 months (95% CI, 4.13 to 113), and 15.9% at 18 months (95% CI, −14.2 to 56.4). The analysis was performed with log-transformed values by analysis of covariance and corrected for the baseline values. In both panels, means and 95% confidence intervals are shown.
Anti-CD3 monoclonal antibody treatment has been associated with Epstein–Barr virus (EBV) reactivation.21,22 At trial entry, 30 participants (39%; 16 in the teplizumab group and 14 in the placebo group) had antibodies against EBV. At weeks 3 through 6 after receipt of the trial regimen, there was quantifiable EBV DNA in whole blood in 8 of the seropositive participants — all in the teplizumab group — one of whom had symptoms of pharyngitis, rhinorrhea, and cough on day 38. In these participants, the EBV DNA levels decreased to below the level of quantification between day 43 and day 134 (mean day 77). At trial entry, 17 participants (10 in the teplizumab group and 7 in the placebo group) had antibodies against cytomegalovirus (CMV). One participant in the teplizumab group who was CMV-seropositive had detectable levels of CMV DNA at day 20, but CMV DNA was undetectable by day 42.
CHANGES IN IMMUNE-CELL SUBSETS
An increased frequency of KLRG1+TIGIT+EOMES+ CD8+ T cells, associated with T-cell unresponsiveness, has previously been reported among patients with new-onset diabetes who had a response to teplizumab.12,13 To determine whether treatment with teplizumab in the current prevention trial was associated with similar changes, we analyzed the frequency of KLRG1+TIGIT+CD8+ T cells among the total CD3+ T cells in the two treatment groups. These cells were more common at months 3 and 6 than at baseline in participants who received teplizumab (mean, 3.79% [95% CI, 3.1 to 4.62] at month 3 and 3.97% [95% CI, 3.18 to 4.94] at month 6, vs. 2.67% [95% CI, 2.1 to 3.39] at baseline), and the levels at months 3 and 6 were higher than those in participants who received placebo (mean, 2.59% [95% CI, 2.05 to 3.27] at month 3 and 2.71% [95% CI, 2.03 to 3.6] at month 6) (Fig. 2B, and Figs. S3 and S4 in the Supplementary Appendix). In contrast, the frequency of CD4+ regulatory T cells or KLRG1− TIGIT−CD8+ T cells did not differ significantly between the two treatment groups, which suggested that there was selectivity in the effect of teplizumab23,24 (Fig. S3 in the Supplementary Appendix).
SUBGROUP ANALYSIS
In prespecified analyses, we compared the effects of teplizumab in subgroups based on age, HLA type, pretreatment C-peptide and glucose levels during the oral glucose-tolerance tests, and autoantibodies (Fig. 3). Among the 43 participants in the teplizumab group for whom data were available, 21 (49%) had HLA-DR3 and 28 (65%) had HLA-DR4 major histocompatibility complex (MHC) molecules. The presence of HLA-DR4 and absence of HLA-DR3 were associated with more robust responses to teplizumab (hazard ratio, 0.20 [95% CI, 0.09 to 0.45] and 0.18 [95% CI, 0.07 to 0.45], respectively, without adjustment for multiplicity) (Figs. S5 and S6 in the Supplementary Appendix). The response to teplizumab as compared with placebo was greater among participants without anti–zinc transporter 8 (ZnT8) antibodies than among those with these antibodies (hazard ratio, 0.07; 95% CI, 0.02 to 0.26) (Fig. S7 in the Supplementary Appendix). The presence or absence of other autoantibodies was not associated with clinical response. The response to teplizumab was also greater among participants whose C-peptide responses to the oral glucose-tolerance test at baseline were below the median (1.75 nmol per liter) than among those whose responses were above the median (hazard ratio, 0.19; 95% CI, 0.08 to 0.47) (Fig. S8 in the Supplementary Appendix).
Figure 3. Subgroup Analysis of Responses to Teplizumab.
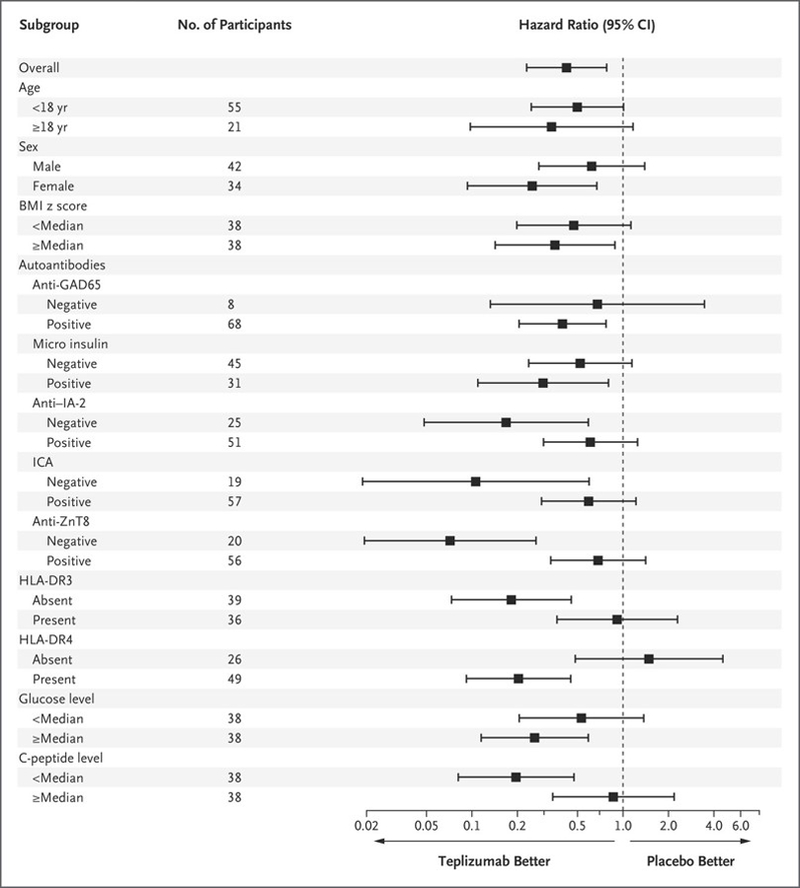
The forest plot shows the hazard ratios and 95% confidence intervals for a diagnosis of type 1 diabetes in the teplizumab group as compared with the placebo group for the two categories of each baseline feature. The Cox model was adjusted for age, with the exception of the interaction test for age (<18 years vs. ≥18 years), but was not adjusted for multiple testing. BMI denotes body-mass index, GAD65 glutamic acid decarboxylase 65, IA-2 islet antigen 2, ICA islet-cell autoantibody, and ZnT8 zinc transporter 8.
DISCUSSION
In this phase 2 trial, a single course of teplizumab significantly slowed progression to clinical type 1 diabetes in high-risk, nondiabetic relativestives of patients with diabetes who had at least two autoantibodies and abnormal results of an oral glucose-tolerance test at trial entry. The median delay in the diagnosis of diabetes was 2 years; at the conclusion of the trial, the percentage of diabetes-free persons in the teplizumab group (57%) was double that in the placebo group (28%). The safety analysis revealed expected adverse events of rash and transient lymphopenia among both children and adults. The delay of progression to diabetes is of clinical importance, particularly for children, in whom the diagnosis is associated with adverse outcomes, and given the challenges of daily management of the condition.2,4 Our findings support the notion that type 1 diabetes is a chronic T-cell–mediated disease and suggest that immunomodulation before the development of clinical disease can be useful.6,25
The effects of teplizumab were greatest in the first 3 years after administration. Among the participants in whom diabetes was diagnosed, 41% had the disease within the first year after randomization, and the risk was lowest at that time for those exposed to teplizumab. The relatively rapid rate of progression to clinical diabetes in the placebo group reflects the very high risk among children with autoantibodies.5,26,27 Indeed, our observations among young persons who did not yet have clinical disease reflect the likely progression when two or more autoantibodies and dysglycemia are found and are consistent with our report of high rates of beta-cell death in these persons.26–28 Preclinical studies suggested that an active autoimmune response is needed for the actions of an anti-CD3 monoclonal antibody29,30; thus, earlier interventions (i.e., during stage 1) may be less efficacious. Consistent with such observations, the response to teplizumab was greatest among participants with C-peptide responses that were below the median. We speculate that the efficacy during the period before diagnosis supports the development of an active screening program to identify persons who are at extremely high risk for disease progression.
Our data suggest that responses to teplizumab differ on the basis of characteristics of the participants. The absence of one type 1 diabetes–associated MHC allele, HLA-DR3, but the presence of HLA-DR4, as well as the absence of anti-ZnT8 antibodies identified the persons most likely to have a response. The MHC may modulate responsiveness to teplizumab through its effect on the T-cell repertoire, perhaps altering T-cell activation status and susceptibility to the effects of the drug. We speculate that anti-ZnT8 antibodies may identify persons with a more fulminant immune response or other features that make their T cells less susceptible to teplizumab.
The transient decline in lymphocyte counts with teplizumab treatment most likely reflects egress from the peripheral blood.31,32 Our flow-cytometry studies may suggest that teplizumab treatment causes changes in the phenotype of CD8+ T cells; we have previously associated these changes with a nonresponsive or “exhausted” phenotype.13 These CD8+ T cells are not, however, inactive, since the few participants with detectable EBV and CMV DNA had rapid clearance of these DNA loads.33–35 The resolution of EBV and CMV activation and the absence of an increased rate of infectious adverse events lead us to hypothesize that the duration of the functional effects of teplizumab on T cells may be affected by their avidity for autoantigens, viral antigens, or other antigens. The effects may be short-lived in T cells that have high avidity for viral antigens such as those associated with EBV but longer-lived in autoreactive T cells, which have lower avidity. Future studies with antigen-reactive T cells will be needed to address this hypothesis.
Our trial had certain limitations. The cohort was relatively small, and the estimated power was limited. The participants were relatives of patients with type 1 diabetes, and we do not know whether these findings will be generally applicable to persons who do not have first-degree relatives with diabetes and who appear to be at risk for type 1 diabetes. Although it reflected the known incidence of disease, our trial population was overwhelmingly made up of non-Hispanic white participants. The drug was given for only one course, and although repeated dosing may provide additional benefits and capture more persons with active disease or prolong the therapeutic effect, this strategy was not tested in this trial.7,11 We have not fully assessed the potential development of antibodies to teplizumab, which would be a concern. An assay to detect such autoantibodies has not yet been fully vetted and validated. In previous trials, antidrug antibodies have been found in approximately 20% to 55% of teplizumab-treated participants after the first course, but the effects on the immunologic or clinical outcomes are not clear.10,36
In conclusion, in our trial, a 2-week course of treatment with teplizumab delayed the diagnosis of clinical type 1 diabetes in high-risk participants.
Supplementary Material
Acknowledgments
A data sharing statement provided by the authors is available with the full text of this article at NEJM.org.
Supported by the National Institutes of Health through the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), the National Institute of Allergy and Infectious Diseases, and the Eunice Kennedy Shriver National Institute of Child Health and Human Development, through cooperative agreements U01 DK061010, U01 DK061034, U01 DK061042, U01 DK061058, U01 DK085453, U01 DK085461, U01 DK085465, U01 DK085466, U01 DK085476, U01 DK085499, U01 DK085504, U01 DK085509, U01 DK103153, U01 DK103180, U01 DK103266, U01 DK103282, U01 DK106984, U01 DK106994, U01 DK107013, U01 DK107014, UC4 DK097835, and UC4 DK106993; the Juvenile Diabetes Research Foundation; and the American Diabetes Association. Additional support for clinical studies was provided by the National Center for Research Resources through Clinical Translational Science Awards UL1TR000142, UL1TR002366, UL1TR000445, UL1TR000064, UL1TR002537, UL1TR001082, UL1TR000114, UL1TR001857, UL1TR002529, UL1TR001872 and by the Immune Tolerance Network (UM1 AI09565). MacroGenics donated the study agents and provided funds for additional site monitoring.
Dr. Herold reports receiving consulting fees from Provention Bio, Bristol-Myers Squibb, Eli Lilly, and Semma Bio, advisory board fees from Tiziana Life Sciences, Forkhead Bio, and Toleron, and grant support and consulting fees from Merck, and holding patent 10,125,394, on beta cell death assay, licensed to Yale University; Dr. Bluestone, receiving fees for serving as a member on the board of directors and holding stock in Rheos Medicines, receiving fees for serving as founder and holding stock in Celsius, receiving fees for serving as a science advisory board member and holding stock in Quentis, Vir, and Arcus, receiving grant support and fees for serving as a science advisory board member from Pfizer, receiving grant support and advisory fees and holding stock in Celgene, holding stock in Kadmon and Viacyte, receiving fees for serving on the board of directors, holding stock, and receiving fees from a licensing asset, from Provention Bio, receiving income and holding stock in Macrogenics, and holding patent 5,885,573 and patent 6,491,916, on methods and materials for modulation of the immunosuppressive activity and toxicity of monoclonal antibodies, licensed to Provention Bio; Dr. DiMeglio, receiving grant support from Caladrius and Janssen; Dr. Gitelman, receiving fees for serving as site principal investigator (PI) from Janssen and Pfizer, advisory fees and fees for serving as study PI from Caladrius, advisory board fees from Eli Lilly, Genentech, Roche, and ImmunoMolecular Therapeutics, and advisory board fees and serving as site PI for Tolerion; Dr. Gottlieb, receiving grant support from Pfizer, Novo Nordisk, and Caladrius, advisory board fees from GlaxoSmithKline, fees for serving on a data and safety monitoring board (DSMB) from Viacyte, consulting fees from Kamada, and serving as cofounder and holding shares of IM Therapeutics; Dr. Rodriguez, receiving grant support from Boehringer Ingelheim, Bristol-Myers Squibb, Daiichi, Mannkind, Takeda, Intrexon, and Tolerion, fees for serving on a DSMB from Merck and Novartis, grant support and fees for serving on a DSMB from Novo Nordisk, and travel support from Roche Diagnostics; Dr. Skyler, receiving advisory board fees from Adocia, Dance Biopharm, and Orgenesis, consulting fees from AstraZeneca, BD Technologies, Boehringer Ingelheim, Debiopharm, Elcelyx Therapeutics, Eli Lilly, Genentech, Immunomolecular Therapeutics, Intrexon/ActoBio, Kamada, Merck, Novo Nordisk, Sanofi, Servier, Tolerion, vTv Therapeutics, Valeritas, Duologics, Zafgen, Applied Therapeutics, Dalcor, Esperion, Geneuro, and Ideal Life, fees for serving on the board of directors for Dexcom, Intarcia Therapeutics, and Moerae Matrix, and fees for serving as chair and fees for serving on an advisory committee from INNODIA; and Dr. Greenbaum, receiving grant support and consulting fees from Novo Nordisk, consulting fees and travel support from Bristol-Myers Squibb, grant support from Janssen Pharmaceuticals, and consulting fees from Eli Lilly. No other potential conflict of interest relevant to this article was reported.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
We thank Lisa Spain, Ph.D., Ellen Leschek, M.D., and Judy Fradkin, M.D., of the NIDDK for their guidance and support; Noha Lim, Ph.D., Elisavet Serti, Ph.D., Sarah Muller, Adriana Weinberg, M.D., Michael Green, M.D., and Brett Loechelt, M.D., for analysis of clinical and laboratory data; Courtney Henderson, Darlene Amado, and Sarah Muller for data collection and assistance with preparation of an earlier version of the manuscript, and Elisavet Serti, Ph.D., of the Immune Tolerance Network for assistance with data analysis.
Footnotes
A complete list of investigators in the Type 1 Diabetes TrialNet Study Group is provided in the Supplementary Appendix, available at NEJM.org.
A Quick Take is available at NEJM.org
Contributor Information
Kevan C. Herold, Departments of Immunobiology and Internal Medicine, Yale University, New Haven, CT, Florida
Brian N. Bundy, Departments of Epidemiology and Pediatrics, University of South Florida, Tampa, Florida
S. Alice Long, Benaroya Research Institute, Seattle
Jeffrey A. Bluestone, Diabetes Center, University of California at San Francisco, San Francisco
Linda A. DiMeglio, Department of Pediatrics, Indiana University, Indianapolis
Matthew J. Dufort, Benaroya Research Institute, Seattle
Stephen E. Gitelman, Diabetes Center, University of California at San Francisco, San Francisco
Peter A. Gottlieb, Barbara Davis Diabetes Center, University of Colorado, Anschultz
Jeffrey P. Krischer, Departments of Epidemiology and Pediatrics, University of South Florida, Tampa, Florida
Peter S. Linsley, Benaroya Research Institute, Seattle
Jennifer B. Marks, Department of Medicine, University of Miami, Miami, Florida
Wayne Moore, Children’s Mercy Hospital, Kansas City, MO
Antoinette Moran, Department of Pediatrics, University of Minnesota, Minneapolis
Henry Rodriguez, Departments of Epidemiology and Pediatrics, University of South Florida, Tampa, Florida
William E. Russell, Department of Pediatrics and Cell and Developmental Biology, Vanderbilt University, Nashville
Desmond Schatz, Department of Pediatrics, University of Florida, Gainesville, Florida
Jay S. Skyler, Department of Medicine, University of Miami, Miami, Florida
Eva Tsalikian, Department of Pediatrics, University of Iowa, Iowa City
Diane K. Wherrett, Hospital for Sick Children, University of Toronto, Toronto
Anette-Gabriele Ziegler, Forschergruppe Diabetes, Technical University Munich, at Klinikum rechts der Isar, Munich, Germany
Carla J. Greenbaum, Benaroya Research Institute, Seattle
References
- 1.Menke A, Orchard TJ, Imperatore G, Bullard KM, Mayer-Davis E, Cowie CC. The prevalence of type 1 diabetes in the United States. Epidemiology 2013;24: 773–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller KM, Foster NC, Beck RW, et al. Current state of type 1 diabetes treatment in the U.S.: updated data from the T1D Exchange clinic registry. Diabetes Care 2015;38:971–8. [DOI] [PubMed] [Google Scholar]
- 3.Livingstone SJ, Levin D, Looker HC, et al. Estimated life expectancy in a Scottish cohort with type 1 diabetes, 2008–2010. JAMA 2015;313:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rawshani A, Sattar N, Franzén S, et al. Excess mortality and cardiovascular disease in young adults with type 1 diabetes in relation to age at onset: a nationwide, register-based cohort study. Lancet 2018; 392:477–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Insel RA, Dunne JL, Atkinson MA, et al. Staging presymptomatic type 1 diabetes: a scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015;38:1964–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Atkinson MA, Roep BO, Posgai A, Wheeler DCS, Peakman M. The challenge of modulating β-cell autoimmunity in type 1 diabetes. Lancet Diabetes Endocrinol 2019;7:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herold KC, Gitelman SE, Ehlers MR, et al. Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: metabolic and immunologic features at base-line identify a subgroup of responders. Diabetes 2013;62:3766–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herold KC, Hagopian W, Auger JA, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med 2002;346:1692–8. [DOI] [PubMed] [Google Scholar]
- 9.Keymeulen B, Vandemeulebroucke E, Ziegler AG, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med 2005;352:2598–608. [DOI] [PubMed] [Google Scholar]
- 10.Hagopian W, Ferry RJ Jr, Sherry N, et al. Teplizumab preserves C-peptide in recent-onset type 1 diabetes: two-year results from the randomized, placebo-controlled Protégé trial. Diabetes 2013;62:3901–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sherry N, Hagopian W, Ludvigsson J, et al. Teplizumab for treatment of type 1 diabetes (Protégé study): 1-year results from a randomised, placebo-controlled trial. Lancet 2011;378:487–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tooley JE, Vudattu N, Choi J, et al. Changes in T-cell subsets identify responders to FcR-nonbinding anti-CD3 mAb (teplizumab) in patients with type 1 diabetes. Eur J Immunol 2016;46:230–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Long SA, Thorpe J, DeBerg HA, et al. Partial exhaustion of CD8 T cells and clinical response to teplizumab in new-onset type 1 diabetes. Sci Immunol 2016; 1(5):eaai7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bingley PJ, Wherrett DK, Shultz A, Rafkin LE, Atkinson MA, Greenbaum CJ. Type 1 Diabetes TrialNet: a multifaceted approach to bringing disease-modifying therapy to clinical use in type 1 diabetes. Diabetes Care 2018;41:653–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.American Diabetes Association. Classification and diagnosis of diabetes: Standards of Medical Care in Diabetes-2019. Diabetes Care 2019;42:Suppl 1:S13–S28. [DOI] [PubMed] [Google Scholar]
- 16.Diabetes Prevention Trial–Type 1 Diabetes Study Group. Effects of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J Med 2002;346:1685–91. [DOI] [PubMed] [Google Scholar]
- 17.Therneau TM, Grambsch PM. Modeling survival data: extending the Cox Model. New York: Springer-Verlag, 2000. [Google Scholar]
- 18.Cox DR. Regression model and life-tables. J R Stat Soc [B] 1972;34:187–220. [Google Scholar]
- 19.Schoenfeld DA. Sample-size formula for the proportional-hazards regression model. Biometrics 1983;39:499–503. [PubMed] [Google Scholar]
- 20.Lan KKG, DeMets DL. Discrete sequential boundaries for clinical trials. Biometrika 1983;70:659–63. [Google Scholar]
- 21.Junker AK, Chan KW, Lirenman DS. Epstein-Barr virus infections following OKT3 treatment. Transplantation 1989; 47:574–5. [PubMed] [Google Scholar]
- 22.Keymeulen B, Candon S, Fafi-Kremer S, et al. Transient Epstein-Barr virus reactivation in CD3 monoclonal antibodytreated patients. Blood 2010;115:1145–55. [DOI] [PubMed] [Google Scholar]
- 23.Perdigoto AL, Preston-Hurlburt P, Clark P, et al. Treatment of type 1 diabetes with teplizumab: clinical and immunological follow-up after 7 years from diagnosis. Diabetologia 2019;62:655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herold KC, Burton JB, Francois F, Poumian-Ruiz E, Glandt M, Bluestone JA. Activation of human T cells by FcR nonbinding anti-CD3 mAb, hOKT3gamma1(Ala-Ala). J Clin Invest 2003;111:409–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herold KC, Vignali DA, Cooke A, Bluestone JA. Type 1 diabetes: translating mechanistic observations into effective clinical outcomes. Nat Rev Immunol 2013; 13:243–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greenbaum CJ, Beam CA, Boulware D, et al. Fall in C-peptide during first 2 years from diagnosis: evidence of at least two distinct phases from composite Type 1 Diabetes TrialNet data. Diabetes 2012;61: 2066–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wherrett DK, Chiang JL, Delamater AM, et al. Defining pathways for development of disease-modifying therapies in children with type 1 diabetes: a consensus report. Diabetes Care 2015;38:1975–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herold KC, Usmani-Brown S, Ghazi T, et al. β Cell death and dysfunction during type 1 diabetes development in at-risk individuals. J Clin Invest 2015;125:1163–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chatenoud L, Thervet E, Primo J, Bach JF. Anti-CD3 antibody induces long-term remission of overt autoimmunity in non-obese diabetic mice. Proc Natl Acad Sci U S A 1994;91:123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chatenoud L, Primo J, Bach JF. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J Immunol 1997;158:2947–54. [PubMed] [Google Scholar]
- 31.Esplugues E, Huber S, Gagliani N, et al. Control of TH17 cells occurs in the small intestine. Nature 2011;475:514–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waldron-Lynch F, Henegariu O, Deng S, et al. Teplizumab induces human guttropic regulatory cells in humanized mice and patients. Sci Transl Med 2012;4: 118ra12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wherry EJ. T cell exhaustion. Nat Immunol 2011;12:492–9. [DOI] [PubMed] [Google Scholar]
- 34.Wherry EJ, Ha SJ, Kaech SM, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007;27:670–84. [DOI] [PubMed] [Google Scholar]
- 35.McKinney EF, Lee JC, Jayne DR, Lyons PA, Smith KG. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 2015;523:612–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Long SA, Thorpe J, Herold KC, et al. Remodeling T cell compartments during anti-CD3 immunotherapy of type 1 diabetes. Cell Immunol 2017;319:3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.