

Abstract
Extracellular HMGB1 triggers inflammation following infection or injury and supports tumorigenesis in inflammation‐related malignancies. HMGB1 has several redox states: reduced HMGB1 recruits inflammatory cells to injured tissues forming a heterocomplex with CXCL12 and signaling via its receptor CXCR4; disulfide‐containing HMGB1 binds to TLR4 and promotes inflammatory responses. Here we show that diflunisal, an aspirin‐like nonsteroidal anti‐inflammatory drug (NSAID) that has been in clinical use for decades, specifically inhibits in vitro and in vivo the chemotactic activity of HMGB1 at nanomolar concentrations, at least in part by binding directly to both HMGB1 and CXCL12 and disrupting their heterocomplex. Importantly, diflunisal does not inhibit TLR4‐dependent responses. Our findings clarify the mode of action of diflunisal and open the way to the rational design of functionally specific anti‐inflammatory drugs.
Keywords: CXCL12, diflunisal, HMGB1, inflammation, NMR
Subject Categories: Chemical Biology, Immunology
Introduction
High‐mobility group Box‐1 (HMGB1), a highly conserved and abundant nuclear protein expressed in all eukaryotic cells, is a key trigger of inflammation 1, 2. HMGB1 consists of two structurally independent L‐shaped tandem HMG‐box domains (~80 amino acids each), referred to as Box A and Box B, connected by a flexible linker and followed by a long acidic C‐terminal tail of 30 amino acids 3. Within the nucleus, HMGB1 is a DNA chaperone that regulates nucleosome assembly and chromatin structure, promotes interactions between several transcription factors, and facilitates their binding to DNA 4. HMGB1 is passively released in the extracellular space by dead cells and is actively secreted by severely stressed cells (reviewed in 2). Extracellular HMGB1 alerts the host to unscheduled cell death, to stress, and to microbial invasion, thus playing a key role in inflammation and immune responses 2. In particular, it triggers inflammation following injury or infection by first recruiting inflammatory cells 5 and then activating them 6.
HMGB1‐induced recruitment of inflammatory cells depends on the formation of a heterocomplex between its fully reduced form and the chemokine CXCL12, which in turn activates CXCR4, a G protein‐coupled receptor (GPCR) 6. The HMGB1/CXCL12 heterocomplex triggers specific CXCR4 homodimer rearrangements 5 and promotes CXCR4‐mediated signaling, resulting in increased ERK activation and calcium rise 7. The HMGB1/CXCL12 heterocomplex has enhanced efficiency compared to CXCL12 alone in promoting human monocyte migration 5, 6 and tissue regeneration 2, 8. Heterophilic interactions between chemokines are known to modulate human leukocyte migration 9, 10. In this context, functional synergism through formation of heterocomplexes between distinct chemokines and inflammatory mediators appears to be a common strategy to fine‐tune and trigger diverse signaling pathways, and increases the multi‐tasking potential of the molecules involved in heterodimerization (reviewed in 7, 11, 12).
As a prototype of multi‐tasking alarmin 13, HMGB1 modulates its function and its interaction partners according to its oxidation state. The formation of a disulfide bond between cysteines 22 and 44 makes HMGB1 unable to form the heterocomplex with CXCL12, but makes it a ligand of TLR4, a receptor that also recognizes LPS 6. Since the two redox states of HMGB1 have alternative activities, it would be desirable to inhibit them selectively.
Interfering with the formation of the HMGB1‐CXCL12 heterocomplex with small molecules might offer new pharmacological opportunities against inflammation‐related diseases 14, 15. This is an attractive but challenging route, mainly because the large and flat interfaces typically associated with protein–protein interactions have reduced druggability 16, 17. Here, we show that diflunisal (DFL; Fig 1A), an aspirin‐like nonsteroidal anti‐inflammatory drug that was identified in the 1970s through phenotypic screens 18, exerts its anti‐inflammatory action at least in part by disrupting the interaction between HMGB1 and CXCL12.
Figure 1. Diflunisal binds HMGB1.
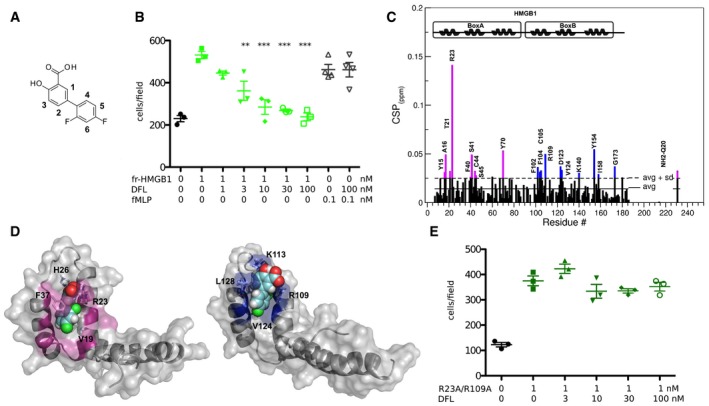
- Chemical structure of DFL.
- DFL inhibits HMGB1‐induced, but not fMLP‐induced cell migration. Mouse 3T3 fibroblasts were subjected to chemotaxis assays in Boyden chambers, 1 nM HMGB1, or no chemoattractant was added in the lower chamber, together with the indicated concentrations of DFL. Data points with average ± standard deviation (avg ± SD; n = 3, each point represents a biological replicate) in a representative experiment. The reproducibility of this experiment is shown in Appendix Fig S1. Statistics: one‐way ANOVA (P < 0.0001), followed by Dunnett's post‐tests. **P < 0.01, ***P < 0.001 relative to no DFL addition.
- Histogram showing residue‐specific CSPs of 15N‐labeled HMGB1 (˜0.1 mM) upon addition of 10‐fold excess of DFL (helices are schematically represented on the top). Missing residues are prolines or are absent because of exchange with the solvent. Box A and Box B residues with CSP > avg + SD are represented in magenta and blue, respectively.
- HADDOCK models of interaction of DFL (CPK representation) with Box A (left) and Box B (right) (color, residues with CSP > avg + SD). HMGB1 residues (sticks) involved in hydrophobic and electrostatic interactions with DFL are explicitly labeled.
- The R23A/R109A HMGB1 double mutant has chemotactic activity but is not inhibited by DFL (the experiment was carried out as described in panel B). The data points represent three biological replicates in one experiment (of two performed in different days); the avg ± SD is shown. The difference in migration toward R23A/R109A HMGB1 in the presence of increasing concentrations of DFL is statistically non‐significant (one‐way ANOVA plus post‐tests).
Source data are available online for this figure.
Results
DFL is a potent inhibitor of HMGB1
We recently found that salicylic acid (SA) suppresses HMGB1's pro‐inflammatory activity and inhibits HMGB1‐induced motility, migration, invasion, and anchorage‐independent colony formation of mesothelioma cells via a cyclooxygenase‐2‐independent mechanism 19, 20. We also found that adding to the SA core an aliphatic extension (e.g. acetyl‐3‐aminoethyl‐SA, ac3AESA) improved the inhibition of HMGB1 activities 19. Although ac3AESA is not metabolically stable, its improved activity relative to SA suggested that other FDA‐approved salicylates might be more potent HMGB1 inhibitors. We focused our attention on DFL, a difluorophenyl that is structurally related to salicylates but is not hydrolyzed in vivo to SA 18. We used migration of mouse 3T3 fibroblasts toward HMGB1 as cellular readout, and we found that DFL halved the number of migrating cells (IC50) already at a concentration below 10 nM, the lowest documented IC50 so far for HMGB1 inhibitors 14. Importantly, DFL did not affect chemotaxis toward fMLP (Fig 1B and Appendix Fig S1), indicating that it does not influence the general motility of fibroblasts. Specifically, DFL appeared much more active than Glycyrrhizin, the most used HMGB1 inhibitor, which has an IC50 of about 150 μM in the fibroblast migration assay 21.
We then asked whether DFL was a direct ligand of HMGB1. We performed NMR saturation transfer difference (STD) 22 and water‐Ligand Observation via Gradient SpectroscopY (waterLOGSY) experiments 23 on DFL (1 mM) in the presence of sub‐stoichiometric concentrations of recombinant HMGB1 (50 μM). We observed STD signals and inversion of the sign in waterLOGSY spectra for all DFL protons to varying extents, thus demonstrating a direct DFL‐HMGB1 interaction (Fig EV1A and B).
Figure EV1. Binding of DFL to HMGB1 assessed by NMR and chemotaxis experiments on R23A and R109A HMGB1 mutants.
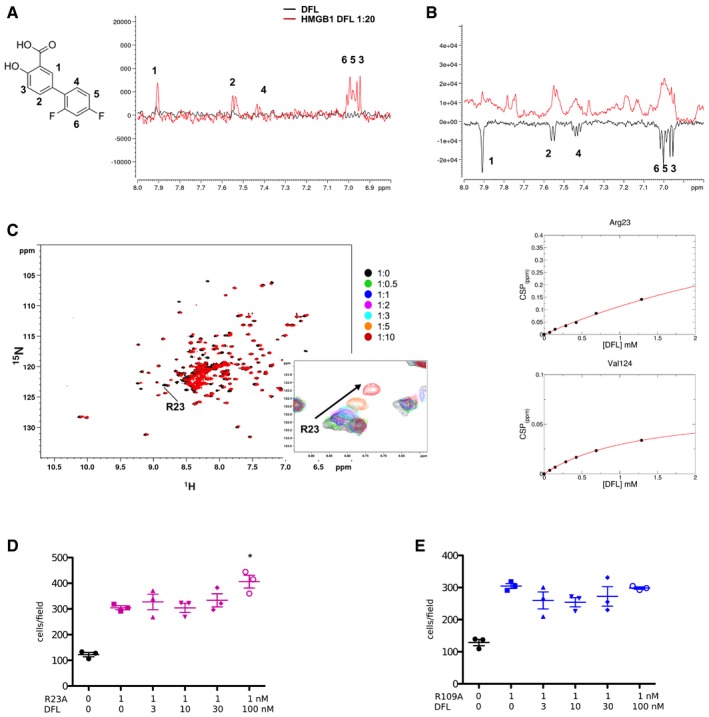
-
ASaturation transfer difference (STD) results obtained for 1 mM DFL alone (black line) and with 0.05 mM HMGB1 (red line) in 20 mM phosphate buffer, 150 mM NaCl, 1 mM DTT pH 7.3 (on resonance: 0 ppm; saturation time: 3 s). STD signals with different intensities are observed for all protons in the presence of HMGB1, indicating that DFL directly binds to HMGB1. The numbered peaks correspond to proton resonance assignments.
-
BWaterLOGSY spectra obtained for 1 mM DFL alone (black line) and in complex with 0.05 mM HMGB1 (red line). All DFL protons display signals inversion, indicating binding to HMGB1.
-
CSuperimposition of the 1H‐15N HSQC spectra of HMGB1 (˜0.1 mM, pH 7.3, phosphate buffer, 1 mM DTT) without (black) and with 10‐fold excess of DFL (red). Inset: selected region of the superimposition of 1H‐15N HSQC spectra of HMGB1 during the titration with DFL (0.5, 1, 2, 3, 5, and 10 equivalents) showing the displacement of the peak associated to R23 along the titration. On the right: weighted average of R23 and V124 amide 1H and 15N chemical shift changes in the presence of increasing concentration of DFL. Nonlinear curve fitting yields K d = 2.8 ± 1.4 mM.
-
D, EThe R23A (D) and R109A (E) HMGB1 mutants have chemotactic activity but are not inhibited by DFL. Data points with average ± standard deviation (avg ± SD; n = 3, each point represents a biological replicate) in one representative experiment (of two experiments performed in different days). Statistics: one‐way ANOVA (P < 0.0001), followed by Dunnett's post‐tests. *P < 0.05 relative to no DFL addition. The difference in migration toward R109A HMGB1 in the presence of increasing concentrations of DFL is statistically non‐significant (one‐way ANOVA plus post‐tests).
Source data are available online for this figure.
To identify the DFL binding site on HMGB1, we monitored perturbations of its 1H‐15N Heteronuclear Single Quantum Coherence (HSQC) spectrum upon DFL addition. Binding occurred in fast regime on the NMR time scale (Fig EV1C). The pattern of chemical shift perturbations (CSPs) indicated that DFL binds to both boxes in full‐length HMGB1 (Fig 1C). Indeed, residues whose amide resonances showed significant CSPs (CSP > avg + SD), i.e., Y15, A16, Q20, T21, R23, F40, S41, C44, S45 in Box A, and F102, F104, C105, R109, D123, V124, K140 in Box B, were similar to the ones mostly affected in DFL titrations into isolated HMG boxes (Appendix Fig S2). When mapped on the structure of the two HMG boxes, residues with significant CSP defined a surface located at the crux of the typical L‐shaped fold, and characterized by a small solvent‐exposed hydrophobic surface suitable for favorable van der Waals (vdW) interactions with the aromatic rings of DFL (Fig 1D). We also observed that some peaks not directly in the binding site (such as E39, W48, F59, A63 and H116, I121, E144) were affected by line broadening effects, likely due to changes in internal motion upon ligand binding.
CSP data were next used to generate HADDOCK 24 data‐driven docking models of DFL in complex with Box A and Box B. The binding modes are highly reminiscent of the one observed for Glycyrrhizin 21. DFL accommodates at the junction of the two arms of both individual HMG boxes, establishing favorable vdW interactions with the hydrophobic side chains of V19 and F37 in Box A and of V124 and L128 in Box B (Fig 1D). The models predict stabilizing electrostatic interactions between the DFL carboxylate and R23 and R109 side chains (Fig 1D). We then mutated R23 and R109 to alanines and also produced the double‐mutant R23A/R109A. The 15N‐labeled R23A/R109A mutant was still able to form a heterocomplex with CXCL12 (Appendix Fig S3A) and maintained the chemoattractant activity. Conversely, DFL did not inhibit cell migration induced by the single and double mutants (Figs 1E, and EV1D and E). Indeed, NMR titration of DFL into the 15N‐labeled R23A/R109A mutant induced significantly lower CSPs as compared to the wild‐type protein (Appendix Fig S3B), indicative of weaker binding.
Microscale thermophoresis (MST) measurements 25, performed by titrating DFL into fluorescently labeled HMGB1, yielded an apparent dissociation constant (Kd) of 1.6 ± 0.8 mM (Appendix Fig S4), in line with the mM affinity (Kd = 2.8 ± 1.4 mM) estimated by fitting the shift of selected NMR peaks upon addition of increasing DFL concentrations (Fig EV1C).
Overall, these experiments indicate that DFL binds to HMGB1. However, the low binding affinity of DFL to HMGB1 is in striking contrast with its high activity in cell migration experiments, thus arguing for a more complex inhibition mechanism than a conventional ligand–protein binary interaction.
DFL binds to CXCL12 but does not inhibit CXCL12‐induced cell migration
We thus hypothesized that DFL might not target HMGB1 alone, but rather the HMGB1/CXCL12 heterocomplex that actually drives cell migration 5. To explore this possibility, we first investigated whether DFL could be a direct ligand of CXCL12. NMR titration experiments of DFL into 0.1 mM 15N‐labeled CXCL12 clearly demonstrated an interaction between CXCL12 and DFL in fast exchange regime, with an apparent Kd of 800 ± 102 μM, in agreement with the millimolar affinity measured by MST (Kd = 2.6 ± 1.2 mM; Fig EV2A–C).
Figure EV2. Binding of DFL to CXCL12 assessed by NMR and MST .
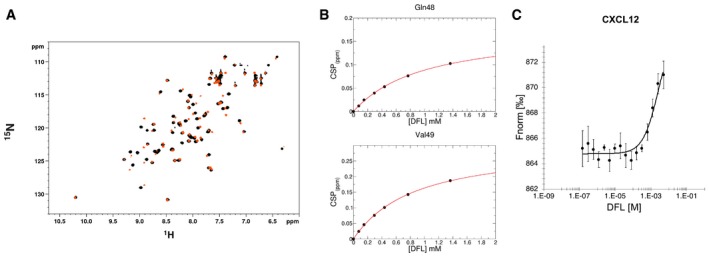
- Superposition of 1H‐15N HSQC spectra of CXCL12 (˜0.1 mM, pH 6, phosphate buffer) without (black) and with 10‐fold excess of DFL (orange).
- Weighted average of Q48 and V49 amide 1H and 15N chemical shift changes in the presence of increasing concentration of DFL. Nonlinear curve fitting yields K d = 802 ± 102 μM.
- Dose–response curve of CXCL12‐DFL interaction followed by MST. DFL concentrations ranged from 5 mM to 153 nM on 50 nM labeled 6His‐CXCL12. The K d is 2.6 ± 1.2 mM (n = 3, error bars correspond to SD). Saturation of the curve could not be reached because of solubility issues for DFL stock solutions > 10 mM. (Fnorm = normalized fluorescence).
Source data are available online for this figure.
The highest resonance shifts involved residues located on the β1 strand (V23, H25, K27) and residues (A40, N45, Q48, V49) around the so‐called sY21 CXCR4 binding site 26, 27, which is recognized by the sulfated CXCR4 extracellular N‐terminus (Fig 2A). Prompted by this observation, we performed STD and waterLOGSY competition experiments between DFL and the CXCR41‐38sY21 peptide that corresponds to the N‐terminal tail of CXCR4 sulfated in position Y21 26, 27. Reduction of DFL signal intensities in STD and waterLOGSY spectra upon addition of CXCR41‐38sY21 indicated that both ligands compete for the same binding site (Fig 2B). Accordingly, data‐driven docking calculations indicated that the carboxylate and the hydroxyl of DFL are, respectively, well suited for polar interactions with the side chains of N45, R47, and E15 located within the sY21 binding pocket (Fig 2C).
Figure 2. Diflunisal binds CXCL12.

- Histogram showing the CSPs of 15N‐labeled CXCL12 amides (˜0.1 mM) upon addition of 10‐fold excess of DFL. Missing residues are prolines. Elements of secondary structure are depicted on top.
- Top: 1H spectrum of DFL, where the numbered peaks correspond to DFL protons (left). Middle: superimposition of STD spectra obtained for 0.5 mM DFL with 0.05 mM CXCL12 (black line) and upon addition of 0.2 mM of CXCR41‐38sY21 (red line). Bottom: waterLOGSY spectra obtained for 0.5 mM DFL with 0.05 mM CXCL12 (black line) upon addition of 0.2 mM of CXCR41‐38sY21 (red line).
- Left: HADDOCK model of interaction of DFL (CPK representation) with CXCL12 (gray surface and cartoon). CXCL12 residues with CSP > avg + SD located around the sY21 binding site and on the β1 strand are in red and orange, respectively. Right: Zoom in the binding site, CXCL12 residues (sticks) involved in hydrophobic and electrostatic interactions with DFL (sticks) are explicitly labeled (right).
- DFL does not inhibit CXCL12‐induced chemotaxis. Mouse 3T3 fibroblasts were subjected to chemotaxis assays in Boyden chambers; 1 nM CXCL12 or no chemoattractant was added in the lower chamber, together with the indicated concentrations of DFL. Data points (n = 3) with avg ± SD in one representative experiment (of two performed in different days); the migration in the presence of increasing concentrations of DFL is not significantly different (one‐way ANOVA plus post‐tests).
- DFL inhibits chemotaxis toward the HMGB1/CXCL12 heterocomplex. Data points (n = 3) with avg ± SD in one representative experiment (of three performed in different days). Migration in the presence or absence of DFL is significantly different (P = 0.0037, one‐way ANOVA plus Dunnett's post‐test; **P < 0.01 relative to no DFL addition).
Source data are available online for this figure.
Ligands targeting the sY21 binding pocket are also known to allosterically trigger CXCL12 dimerization upon binding 28. Thus, we hypothesized that CSPs involving the β1 strand (Fig 2A and C) might be due to protein dimerization. Indeed, the increase in CXCL12 correlation time upon DFL addition (τc of CXCL12free = 5.5 ± 0.15 ns, τc of CXCL12DFL = 6.3 ± 0.4 ns, Appendix Fig S5A), as assessed by heteronuclear NMR relaxation experiments, suggests that DFL favors CXCL12 self‐association. In agreement with this notion, we observed that the chemical shifts of residues K27 moved toward the dimeric form 29 upon addition of DFL (Appendix Fig S5B and C), even though the observed chemical shifts likely arise from a combination of conformational effects upon ligand binding and dimer formation 26, 30. The extent of dimer formation was assessed by monitoring CXCL12 chemical shift changes as a function of CXCL12 concentration, in the absence and in the presence of 1 mM DFL. The dimerization constants were 5.6 ± 0.4 and 2.1 ± 0.8 mM, respectively (Appendix Fig S5D), corresponding to an increase in dimer content from 3 to 8% for a 0.1 mM CXCL12 solution upon the addition of 1 mM DFL. It is known that CXCL12 homodimers are chemotactically inactive 27; however, the amount of CXCL12 dimer induced by DFL is almost negligible and with no expected impact on CXCL12‐induced chemotaxis. In fact, cell migration experiments showed that DFL was unable to inhibit CXCL12‐dependent chemotaxis at concentrations where it inhibited chemotaxis induced by HMGB1 (Fig 2D).
Overall, these experiments show that DFL binds to CXCL12 in addition to HMGB1, although the affinity is also low.
DFL targets the HMGB1/CXCL12 heterocomplex
Since DFL binds both HMGB1 and CXCL12, we expected that DFL would also target the HMGB1/CXCL12 heterocomplex. Indeed, DFL significantly reduced the migration of 3T3 fibroblasts toward the preformed HMGB1/CXCL12 heterocomplex (Fig 2E). To verify biophysically that DFL interferes with the HMGB1/CXCL12 heterocomplex, we first produced the heterocomplex by titrating unlabeled CXCL12 (up to 0.2 mM) into 15N‐labeled HMGB1 (0.1 mM; Fig 3A and B) and then added DFL (Fig 3D and E). As already reported 5, complex formation induced the reduction or disappearance of peak intensities of HMGB1 amide resonances, indicative of micromolar affinities in intermediate exchange regime on the NMR time scale (Fig 3B). These observations were in agreement with Kd = 4 ± 0.4 μM, as measured by MST (Fig 3C).
Figure 3. Effect of DFL on the HMGB1/CXCL12 heterocomplex.
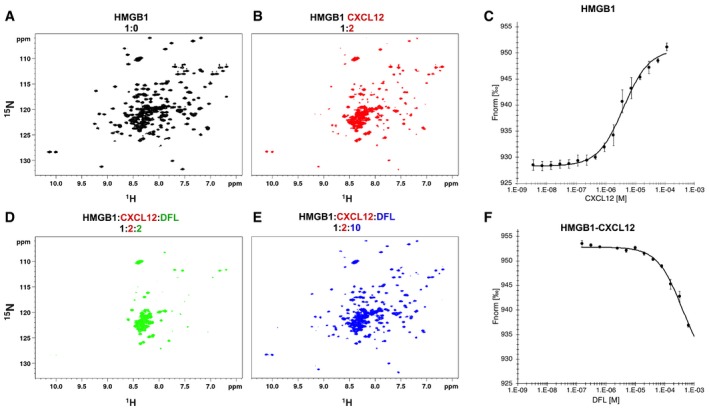
-
A, B1H‐15N HSQC HMGB1 (0.1 mM) spectrum (A) without (black) and (B) with 0.2 mM CXCL12.
-
CMST measurements of CXCL12 titrated into 50 nM fluorescently labeled HMGB1; the signal increases from ˜930 (free) to ˜950 a.u. (bound) yielding an apparent K d of 4 ± 0.4 μM. n = 3; data represent avg ± SD.
-
D, EH‐15N HSQC of HMGB1 (0.1 mM) with CXCL12 (0.2 mM) upon addition of (D) 0.2 mM and (E) 1 mM DFL.
-
FMST measurements of the heterocomplex (preformed using 50 nM HMGB1 and 50 μM CXCL12) in the presence of increasing concentrations of DFL. Decreasing MST signal (from ˜950 to ˜930 a.u.) upon DFL titration indicates that CXCL12 has been displaced from HMGB1. n = 3; data represent avg ± SD.
Source data are available online for this figure.
Intriguingly, HMGB1 residues involved in interactions with CXCL12 and DFL partially coincide (Fig EV3A and B), suggesting that DFL might interfere with the surface of interaction between HMGB1 and CXCL12. Upon addition of 0.2 mM DFL to the preformed 15N‐HMGB1/CXCL12 heterocomplex, we observed a drastic line broadening in the 1H‐15N HSQC spectrum, with the disappearance of the majority of HMGB1 peaks (Fig 3D, green). These line broadening effects were conceivably associated with multiple equilibria involving free HMGB1, HMGB1 bound to DFL, HMGB1 bound to CXCL12, and the ternary complex involving HMGB1, CXCL12, and DFL. The remaining peaks, in the typical random coil region of the C‐terminal HMGB1 acidic tail, were not affected. By ultimately adding a 10‐fold excess of DFL, the 1H‐15N HSQC spectrum of HMGB1 was restored (Figs 3E, blue and EV3C), indicating that CXCL12 was displaced and the heterocomplex disrupted. Notably, some HMGB1 cross‐peaks were shifted with respect to free HMGB1 due to binding of DFL (Fig EV3C). In MST experiments, addition of increasing concentrations of DFL to a preformed HMGB1/CXCL12 complex led to a sharp transition at 380 ± 17 μM, consistent with the disassembly of fluorescently labeled HMGB1 from the heterocomplex (Fig 3F).
Figure EV3. DFL and CXCL12 binding surfaces on HMGB1 partially coincide and details of DFL effects on NMR spectra of the HMGB1/CXCL12 heterocomplex.
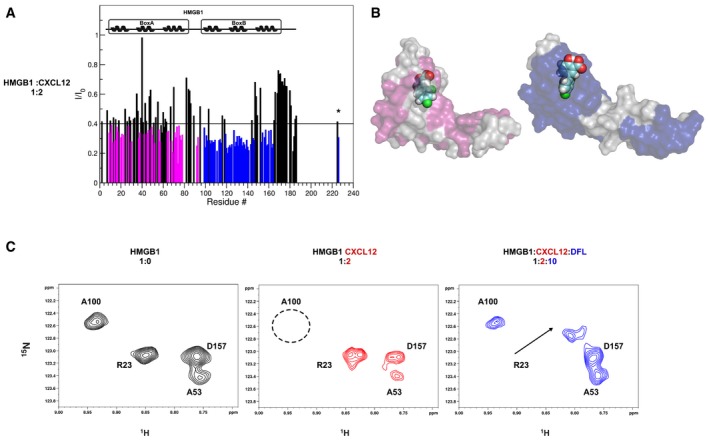
- Histograms showing the I/I0 peak intensity ratio of HMGB1 amides (˜0.1 mM, pH 6, phosphate buffer, 1 mM DTT) with and without twofold excess of CXCL12 (bottom). Io and I are peak intensities in free and bound HMGB1, respectively. Residues showing significant decrease of I/I0 ratio (I/I0 ratio < avg) upon CXCL12 binding are represented with magenta (Box A) and blue (Box B) histograms. α‐helices are schematically represented on the top of the histogram.
- Surface representations of Box A‐DFL and Box B‐DFL molecular models are reported where the residues showing significant decrease of I/I0 ratio (I/I0 ratio < avg) upon CXCL12 binding are mapped in magenta and blue, respectively.
- Selected region of 1H‐15N‐HSQC spectra of HMGB1 (˜0.1 mM, pH 6, phosphate buffer, 1 mM DTT) without (black), with twofold excess of CXCL12 (red) and 10‐fold excess of DFL (blue). In the presence of CXCL12, the HMGB1 spectrum undergoes line broadening and some peaks (e.g., A100 in Box B, black dotted circle) disappear, indicating complex formation. The A100 peak reappears in the 1H‐15N‐HSQC spectrum of HMGB1 upon addition of DFL, whereas the sites involved in binding to DFL are shifted (e.g., R23).
These experiments show that DFL binds to the HMGB1/CXCL12 heterocomplex and is able to disrupt it.
DFL does not inhibit HMGB1‐dependent inflammatory cell activation
In line with the evidence that DFL affects the HMGB1/CXCL12 heterocomplex, we predicted that it would not interfere with the transcription and release of pro‐inflammatory cytokines, which depends on TLR4 activation. Only HMGB1 containing a disulfide bond between cysteines 22 and 44 induces cytokine production by binding to receptor TLR4 5, 31; conversely, disulfide‐containing HMGB1 cannot form a complex with CXCL12 5.
Indeed, addition of DFL did not affect the transcription of TNF in mouse macrophages stimulated with disulfide HMGB1 (Fig 4A); likewise, DFL did not affect cytokine induction by disulfide HMGB1 in human macrophages (Fig EV4A–C). This is at variance with the effect of SA, which inhibits the cytokine‐inducing activities of disulfide HMGB1 19.
Figure 4. Diflunisal affects the HMGB1/CXCL12/CXCR4 axis but not the HMGB1/TLR4 axis.
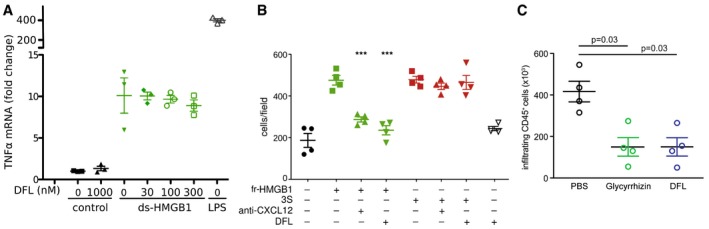
- DFL does not affect the cytokine‐inducing activities of disulfide HMGB1 (ds‐HMGB1). Mouse bone marrow‐derived macrophages were activated or not for 3 h with 3 μg/ml ds‐HMGB1 (˜100 nM) or 10 ng/ml LPS, in the presence of the indicated concentrations of DFL. The data points represent n = 3 biological replicates, with avg ± SD, in one representative experiment (of two performed in different days). The levels of TNFα mRNA are not significantly different in the presence of increasing concentrations of DFL (one‐way ANOVA plus post‐tests).
- DFL does not inhibit chemotaxis toward 3S. Mouse 3T3 fibroblasts were subjected to chemotaxis assays in Boyden chambers; 1 nM fully reduced HMGB1 (fr‐HMGB1; green) or 3S (red), or no chemoattractant (black), were added in the lower chamber, together with 1 μg/ml of anti‐CXCL12 monoclonal antibody or 30 nM DFL. Data points (n = 4) with avg ± SD in one representative experiment (of two performed in different days). Statistics: one‐way ANOVA (P < 0.0001), followed by Dunnett's post‐test. ***P < 0.001 relative to migration toward HMGB1, the migrations toward 3S are not significantly different among each other.
- Quantification of CD45+ cells isolated with immunobeads from injured muscles 6 h after damage. Before damage mice were treated either with phosphate‐buffered saline vehicle, Glycyrrhizin, or DFL. Two independent experiments. Each point represents a different mouse, and avg ± SD are shown (statistics: Kruskal–Wallis plus post‐tests).
Source data are available online for this figure.
Figure EV4. DFL does not affect the cytokine‐inducing activities of disulfide HMGB1 on human macrophages.
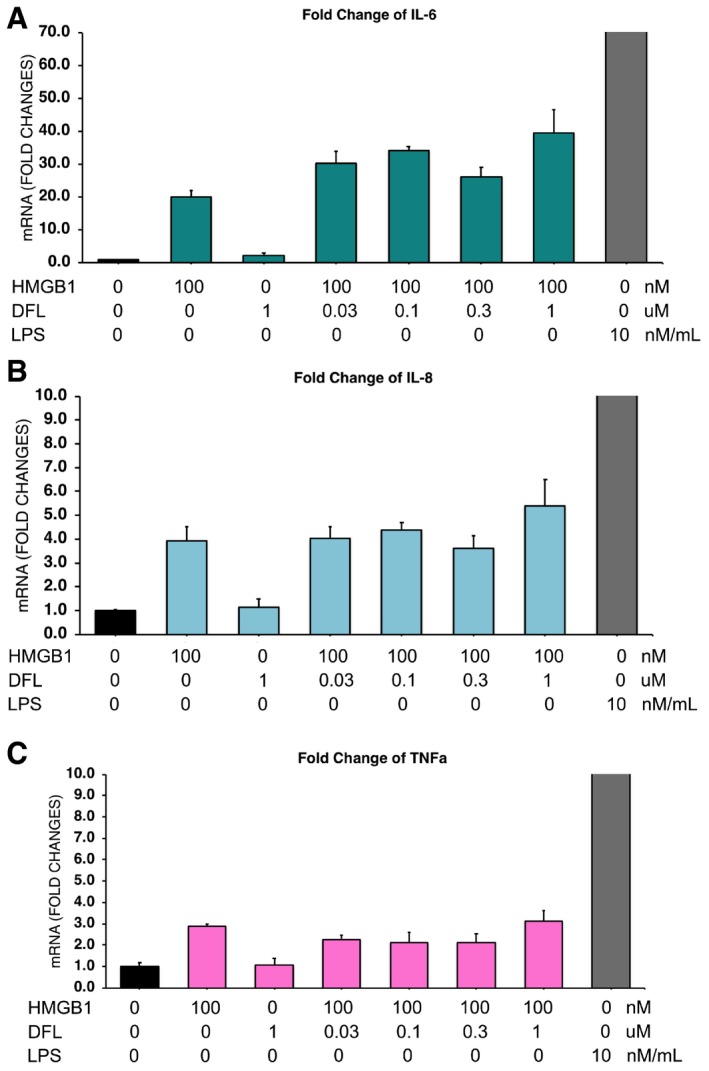
-
A–CHuman macrophages were obtained as described 6 and activated or not for 3 h with 3 μg/ml disulfide HMGB1 (˜100 nM) or 10 ng/ml LPS, in the presence of the indicated concentrations of DFL. The bars represent the avg ± SD (n = 3 biological replicates). In all cases, the mRNA levels (of IL6, IL8, TNF) in cells exposed to disulfide HMGB1 (ds‐HMGB1) are not different in the presence of increasing concentrations of DFL (one‐way ANOVA plus post‐tests). This experiment was performed twice with macrophages generated from different donors.
Source data are available online for this figure.
We also predicted that DFL would not affect the chemoattractant activity of 3S, a mutant of HMGB1 where all cysteines are replaced by serines. 3S does not interact with TLR4, but interacts directly with CXCR4 without the need to form a complex with CXCL12, and has the same chemoattractant properties as fully reduced HMGB1 32. Indeed, both DFL and a monoclonal antibody against CXCL12 inhibited HMGB1‐induced chemotaxis, but neither was effective on chemotaxis induced by 3S (Fig 4B).
Together, these experiments show that DFL only acts on the activities of HMGB1 that depend on the formation of the HMGB1/CXCL12 heterocomplex and its binding to CXCR4, and not on those that depend on TLR4 activation. The two axes are mutually exclusive because they require alternative oxidation states of cysteines 22 and 44 of HMGB1.
DFL inhibits inflammatory cell migration in vivo
Next, we tested in vivo the ability of DFL to inhibit the migration of inflammatory cells into injured muscle, which also depends on the HMGB1/CXCL12 heterocomplex 32. Muscle injury was induced by a single dose of cardiotoxin in the tibialis anterior muscle of C57Bl/6 wild‐type mice, and the infiltration of leukocytes (CD45+ cells) in injured muscle was assessed 6 h later 32. DFL significantly decreased the recruitment of CD45+ cells into the injured muscle compared to vehicle‐treated mice (Fig 4C). As previously reported 5, Glycyrrhizin also inhibited recruitment of leukocytes into injured muscle (Fig 4C); we thus tested whether its inhibitory activity was mechanistically similar to that of DFL. Indeed, NMR titration experiments clearly showed that Glycyrrhizin binds directly to CXCL12, affecting a binding surface similar to that targeted by DFL (Appendix Fig S6). Most importantly, Glycyrrhizin was able to disrupt the HMGB1‐CXCL12 complex, as shown in 15N‐HSQC experiments by the recovery of 15N HMGB1 resonances upon addition to a preformed heterocomplex (Fig EV5A–D). Of note, in strong analogy to DFL, Glycyrrhizin is not an inhibitor of CXCL12‐induced chemotaxis 5.
Figure EV5. Effects of Glycyrrhizin on the HMGB1/CXCL12 heterocomplex.
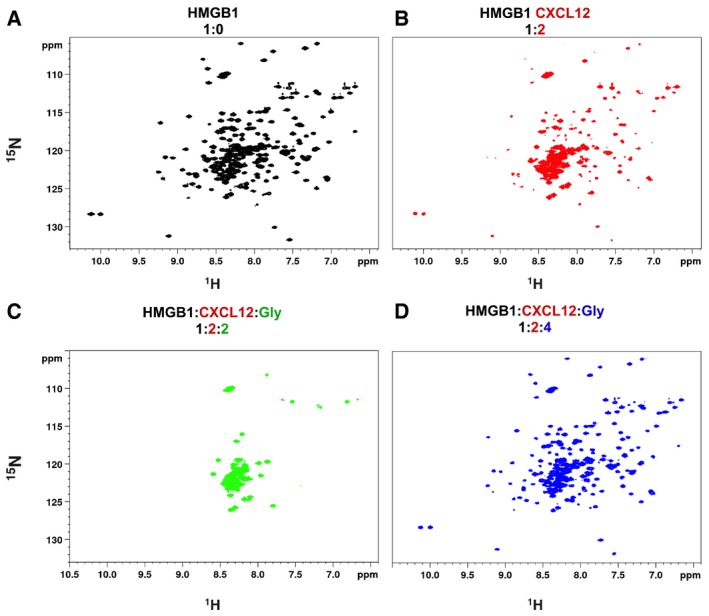
-
A–D1H‐15N HSQC HMGB1 (˜0.1 mM) spectrum (A) without (black), (B) with a twofold excess of CXCL12 (red), (C) upon addition of twofold (green), and (D) fourfold excess of Glycyrrhizin (Glyc) (blue).
Discussion
Salicylates and their derivatives are potent and widely used nonsteroidal anti‐inflammatory drugs with a wide spectrum of modulating activities, comprising inhibition of cyclooxygenases 33 and of NF‐kB 34, allosteric activation of AMP kinase (AMPK) 35, alteration of mTOR signaling 36, perturbation of EGFR internalization 37, and inhibition of CBP/p300 acetyltransferase activity 38. Recently, we showed that salicylates also inhibit signaling by HMGB1 19. Here we show that DFL, a synthetic salicylate that was developed via phenotypic assays, is a potent inhibitor of inflammatory cell recruitment by HMGB1, which is fully consistent with its anti‐inflammatory properties.
Recruitment of inflammatory cells depends at least in part on the formation of a heterocomplex between HMGB1 and the chemokine CXCL12 that activates CXCR4 5. DFL inhibits cell migration induced by the HMGB1/CXCL12 heterocomplex at concentrations as low as 10 nM. Moreover, DFL injected i.p. in mice inhibits the recruitment of monocytes into injured muscle, which is an inflammatory response that also depends on the HMGB1/CXCL12 heterocomplex 5. Importantly, DFL selectively interferes with the HMGB1/CXCL12/CXCR4 inflammatory axis but does not affect signaling by disulfide HMGB1 via the TLR4/MD‐2 axis 6, 39, 40, as it does not inhibit the transcription and the release of pro‐inflammatory cytokines. Thus, DFL selectively modulates only one specific signaling axis in which HMGB1 is involved, and can represent a powerful tool for probing which of the different axes are relevant in a specific biological situation.
The molecular mechanism whereby DFL inhibits signaling by the HMGB1/CXCL12 heterocomplex is rather unusual: DFL directly binds both HMGB1 and CXCL12 individually, the former on each HMG box, and the latter on the cleft that is engaged by the N‐terminal tail of the CXCR4 receptor. Glycyrrhizin, a previously known HMGB1 inhibitor 21, also binds CXCL12 and similarly to DFL inhibits inflammatory cell migration in vivo (this work and 5).
The functional target of DFL appears to be the HMGB1/CXCL12 heterocomplex. HMGB1 cannot induce chemotaxis without CXCL12 5, 32. CXCL12 alone induces CXCR4‐dependent chemotaxis, but DFL does not affect such chemotaxis at the same concentrations where it inhibits chemotaxis induced by the HMGB1/CXCL12 heterocomplex. Finally, CXCL12 is an essential component of the DFL target, since DFL does not inhibit cell migration induced by 3S, a mutated form of HMGB1 that activates CXCR4 without forming a complex with CXCL12 32.
We confirmed the involvement of HMGB1 as a component of the DFL target by mutagenesis. According to our HADDOCK model, DFL interacts electrostatically with both residues R23 in Box A and R109 of Box B in HMGB1, and mutations R23A and R109A preserve the chemotactic activity of HMGB1 and the formation of the heterocomplex with CXCL12, but abrogate inhibition by DFL.
It is clear that our biophysically defined system containing only HMGB1 and CXCL12 can prove that these molecules are targets of DFL, but cannot fully recapitulate the effect of DFL on cell migration, where the heterocomplex is already active at 1 nM and DFL is inhibitory at a concentration of about 10 nM. Discrepancies between in vitro and in vivo efficacy have been already reported for DFL, whose inhibition of CBP/p300 acetyltransferase activity in cells is far more effective than in vitro 38. It must be noted that chemotaxis depends on signaling events at the cell membrane, where cooperative binding phenomena among multiple actors can take place 41. For example, interactions of both HMGB1 and CXCL12 with cell surface glycosaminoglycans (GAGs) 28, 42, 43 might lead to their co‐localization in the membrane proximity and increase their local concentration. Second, the heterocomplex might bind tightly to CXCR4, shifting the equilibrium away from unbound CXCL12 and HMGB1 toward the HMGB1/CXCL12/CXCR4 triple complex. Third, allosteric effects may occur in the conformation of the heterocomplex upon receptor binding 41 increasing its affinity for the DFL binding site(s). Binding of DFL may also modulate the heterocomplex‐induced self‐association of CXCR4 molecules 5, likely translating into an enhanced inhibition of the associated signaling cascade.
Indeed, even the binding of DFL to the HMGB1/CXCL12 heterocomplex in our defined system is remarkably composite. The binding affinities of DFL to HMGB1 alone or to CXCL12 alone are in the millimolar range, but DFL leads to the disassembly of the heterocomplex at lower concentrations. DFL could not cause the disassembly of the complex without physically interacting with it. We thus infer that DFL binds to the heterocomplex preferentially relative to its components, possibly due to allosteric rearrangements in the heterocomplex of the DFL binding sites on CXCL12 and HMGB1. We also speculate that binding of DFL to the heterocomplex and heterocomplex dissociation might be separate events, and that heterocomplex dissociation may require sequential DFL binding to multiple sites. Unfortunately, the details of DFL binding to the HMGB1/CXCL12 heterocomplex cannot be investigated by NMR nor by MST. Addition of high micromolar concentrations of DFL to the heterocomplex causes the near complete collapse of the 15N HSQC spectrum of HMGB1, in line with the presence of multiple equilibria; microscale thermophoresis diffusion rates of the HMGB1/CXCL12 heterocomplex do not change detectably upon DFL addition until the heterocomplex disassembles.
Undoubtedly, DFL represents the prototype of a multitarget drug 44, 45, displaying a low affinity toward its multiple targets but resulting in high efficacy through their simultaneous engagement 46. DFL inhibits COX enzymes, reduces CBP/p300 lysine acetyltransferase activity 38, the activation of NF‐kB 34, and activates AMPK 35. All these molecular interactions, including the ones we have described in this work and most likely others, may synergistically contribute to the complex pharmacological profile of DFL.
In conclusion, the ability of DFL to selectively interfere with the HMGB1/CXCL12/CXCR4 inflammatory axis offers unprecedented structural/functional insights into the anti‐inflammatory activity of DFL as a NSAID. These insights also show that protein–protein interactions within the HMGB1‐CXCL12 heterocomplex are druggable with high specificity and selectivity. Targeting functional chemokine interactions, as shown for example for CXCL4/CCL5 heterocomplex 47, may offer important advantages over direct antagonism of chemokines or their receptors, thus opening new strategies for the rational design of novel inhibitors of this crucial inflammatory axis.
Materials and Methods
Reagents
Recombinant HMGB1 constructs, including Box A (residues 1–89), Box B (residues 90–175), and full‐length HMGB1 (residues 1–214), were produced by expressing constructs in a pETM‐11 vector (EMBL, Heidelberg, DE). The recombinant proteins contained an N‐terminal 6His‐tag, removable by cleavage with TEV protease. After expression and cleavage with TEV protease, the proteins have a residual N‐terminal three‐residue tag (GAM). Box A and Box B proteins were expressed in Escherichia coli BL21 (DE3) cells, whereas HMGB1 was expressed in pLys BL21 (DE3) E. coli cells. Cells were grown at 37°C until the optical density at 600 nm reached 0.8 absorbance units. Gene expression was induced by the addition of isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) to a final concentration of 1 mM. After 18 h of incubation at 25°C with shaking, cells were harvested by centrifugation. The cells were re‐suspended in lysis buffer [20 mM Tris–HCl pH 8.0, 150 mM NaCl, 10 mM imidazole, 2 mM β‐mercaptoethanol, 0.2% NP‐40, Complete EDTA‐free protease inhibitor (Roche), 2 ng/ml DNase, 20 ng/ml RNase)] and lysed by sonication. Cell debris was removed by centrifugation at 11,000 g for 45 min at 4°C. The soluble 6His‐tagged proteins were purified from the supernatant by affinity chromatography using Ni2+‐NTA agarose resin (Qiagen, Hilden, Germany). After several washing steps, proteins were eluted in 20 mM Tris pH 8.0, 150 mM NaCl, 300 mM imidazole, and 2 mM β‐mercaptoethanol. The 6His‐tag was removed by overnight incubation at 4°C with TEV protease. During incubation, the sample was dialyzed against 20 mM Tris pH 8.0, 150 mM NaCl, 2 mM β‐mercaptoethanol for Box A and Box B, whereas for HMGB1 the dialysis buffer was 20 mM Tris pH 8.0, 20 mM NaCl, and 2 mM β‐mercaptoethanol. Uncleaved 6His‐tagged protein and TEV protease were then removed by repassing the sample over Ni2+‐NTA resin. HMGB1 was further purified on a HitrapQ ion‐exchange column using buffer A (20 mM Tris pH 8, 20 mM NaCl, 2 mM β‐mercaptoethanol) and buffer B (20 mM Tris pH 8, 1 M NaCl, 2 mM β‐mercaptoethanol) to create a linear gradient of NaCl. HMGB1 elutes at 500 mM NaCl. Box A and B samples were purified by gel filtration on a Superdex‐75 column (Amersham Biosciences, Milan, Italy) equilibrated in 20 mM phosphate buffer (pH 7.3), 150 mM NaCl, and 1 mM dithiothreitol (DTT). For MST experiments, the 6His‐tag was not removed. Uniformly 15N‐ and 15N/13C‐labeled proteins were prepared using M9 minimal bacterial growth media appropriately supplemented with 15N‐labeled ammonium chloride and 13C‐labeled glucose. R23A, R109A, and R23A/R109A mutants were generated in the pETM11‐HMGB1 vector by PCR‐site directed mutagenesis. The mutant proteins were expressed and purified as described for the wild‐type protein, and their fold was checked by 1H monodimensional NMR spectra. Unlabeled and uniformly 15N‐ and 15N/13C‐labeled CXCL12 were provided by HMGBiotech (Milan, Italy). For biophysical measurements, all HMGB1 constructs, after expression and purification, were dialyzed against NMR buffer, containing 20 mM phosphate buffer pH 7.3, 150 mM NaCl, 1 mM DTT. CXCL12 was dialyzed against a buffer containing 20 mM phosphate buffer pH 6, 20 mM NaCl.
Proteins concentrations were determined measuring the absorbance at 280 nm considering molar extinction coefficients of 9,970, 10,810, 21,430, and 8,700 M−1 cm−1 for Box A, Box B, HMGB1, and CXCL12, respectively.
Proteins used for cell‐based and in vivo assays were provided by HMGBiotech (Milan).
Diflunisal (5‐(2,4‐difluorophenyl)‐2‐hydroxybenzoic acid) was purchased from Sigma‐Aldrich.
The antibody to human/mouse CXCL12 was from R&D Systems (MAB350).
Peptide synthesis and purification
The CXCR41‐38sY21 peptide with sequence MEGIDIYTSDNYTEEMGSGDY(Sulfo)DSMKEPAFREENANFNK was synthesized using the Fmoc method 48 and purified by preparative reversed‐phase chromatography (RP‐HPLC). Details on peptide synthesis and purification are reported in Appendix Supplementary Methods and Appendix Fig S7.
NMR measurements
NMR spectra were recorded at 298 K on a Bruker Avance 600 MHz spectrometer (Karlsruhe, Germany) equipped with a triple‐resonance TCI cryoprobe with an x, y, z‐shielded pulsed‐field gradient coil. Spectra were processed with Topspin™ 3.2 (Bruker) and analyzed with CcpNmr Analysis 2.3. 49 1H‐15N‐HSQC assignments of HMGB1 and its constructs were taken from the BMRB databank (accession numbers: 15148, 15149). The 1H‐15N‐HSQC assignment of CXCL12 was obtained from the BMRB databank (accession number 16143) 50 and confirmed via acquisition of 3D HNCA, CBCA(CO)NH experiments. Side chain assignment of Box A (Box B) in the presence of DFL (stoichiometric ratio 1:2) was obtained through the analysis of 3D HNCA, CBCA(CO)NH, CBCANH, H(CCO)NH, CC(CO)NH, and HCCH‐TOCSY experiments and 2D 1H‐1H TOCSY (mixing time: 60 ms) and NOESY (mixing time: 120 ms) spectra. Intermolecular nuclear Overhauser effect (nOes) between DFL and Box A (Box B) was obtained from 3D 13C‐NOESY‐HSQC with no evolution on 13C dimension (2,048 × 1 × 256 increments) experiments with 15N/13C filter in F1 (mixing time 200 ms); protein and ligand concentration were 0.8 and 1.6 mM, respectively, in D2O.
Titrations
For NMR titrations, at each titration point a 2D water‐flip‐back 1H‐15N‐edited HSQC spectrum was acquired with 2,048 (160) complex points for 1H (15N), respectively, apodized by 90° shifted squared (sine) window functions, and zero filled to 256 points for indirect dimension. Assignment of the labeled proteins in the presence of the ligands (DFL or unlabeled protein) was obtained following individual cross‐peaks through the titration series. For each residue, the weighted average of the 1H and 15N chemical shift perturbation (CSP) was calculated as CSP = [(Δδ2HN + Δδ2N/25)/2]1/2 51. In titrations of 15N HMGB1 with unlabeled CXCL12, because of extensive line broadening, the effect of the binding was monitored measuring the variation of 1H‐15N peak intensities ratios I/Io upon CXCL12 addition, where Io and I are peak intensities in free and bound HMGB1, respectively. Intensity ratio I/Io values were normalized on the highest value.
DFL titrations have been performed on 15N HMGB1 constructs (full‐length protein, Box A, Box B, 20 mM phosphate buffer, pH 7.3, 20 mM NaCl, 1 mM DTT) and on 15N CXCL12 (20 mM phosphate buffer, pH 6, 20 mM NaCl) adding 0.5, 1, 2, 3, 5, 10 equivalents of DFL to the labeled proteins. In order to minimize dilution and NMR signal loss, titrations were carried out by adding small aliquots of concentrated DFL (10 mM in 20 mM phosphate buffer, pH 7.3, 150 mM NaCl) to the 15N‐labeled protein samples (0.1 mM).
CXCL12 (1 mM stock solution, 20 mM phosphate buffer, pH 6, 20 mM NaCl) was titrated adding 0.5, 1, 1.5, 2 equivalents into 0.1 mM 15N‐labeled HMGB1 (20 mM phosphate buffer, pH 6, 20 mM NaCl, 1 mM DTT). DTT is required to maintain HMGB1 in its fully reduced form. We suspected that DTT might break important disulfide bridges within CXCL12; however, control 1H‐15N HSQC spectra acquired in the presence of 1 mM DTT indicated that 15N‐labeled CXCL12 in these conditions was stable for at least 4 h, a time scale which was well within the time required for NMR titrations.
NMR‐based antagonist‐induced dissociation assays 52 were performed titrating DFL on the 15N‐HMGB1/CXCL12 heterocomplex (ratio 1:2) adding 1, 2, 3, 5, 10 equivalents of ligand to the heterocomplex. Similar NMR‐based antagonist‐induced dissociation assays were performed titrating Glycyrrhizin into the 15N‐HMGB1/CXCL12 heterocomplex (ratio 1:2) adding 0.25, 0.5, 0.75, 1, 1.5, 2.5, and 4 equivalents of ligand to the heterocomplex.
Glycyrrhizin (Acros Organics) titration on 15N CXCL12 (20 mM phosphate buffer, pH 6, 20 mM NaCl) has been performed adding 0.25, 0.5, 1, 2 equivalents of ligand to the labeled proteins using a 10 mM stock solution (20 mM phosphate buffer, pH 7.3, 150 mM NaCl).
Saturation transfer difference and water‐ligand observed via gradient spectroscopy
STD and waterLOGSY experiments have been performed on 0.5 mM DFL in the presence of 0.05 mM HMGB1 or CXCL12 in NMR buffer. STD experiments were acquired using a pulse scheme (Bruker pulse sequence: stddiffesgp.3) with excitation sculpting with gradients for water suppression and spin‐lock field to suppress protein signals. The spectra were acquired using 128 scans, a spectral width of 9,600 Hz, 64 K data points for acquisition. For protein saturation, a train of 60 Gaussian‐shaped pulses of 50 ms was applied, for a total saturation time of 3 s. Relaxation delay was set to 3 s. On‐ and off‐resonance irradiations were set at 0 ppm and at 107 ppm, respectively. STD spectra were obtained by internal subtraction of the on‐resonance spectrum from the off‐resonance spectrum.
WaterLOGSY experiments were acquired using a pulse scheme as described 53 with excitation sculpting and flip‐back for water suppression. The spectra were acquired using 128 scans, 32 K data points for acquisition, mixing time was set to 1 s.
Competition experiments have been performed comparing STD and waterLOGSY performed on 0.5 mM DFL with 0.05 mM CXCL12 and upon addition of 0.2 mM of CXCR41‐38sY21 peptide.
Dissociation constant estimation
The apparent dissociation constants of the DFL‐15N‐HMGB1 and DFL‐15N‐CXCL12 interactions were estimated from least‐squares fitting of CSPs as a function of total DFL concentration according to the equation:
with a = (K a/δb) [P t], b = 1 + K a([L ti]+[P t]), and c = δ b K a[L ti], where δ i is the absolute change in chemical shift for each titration point, [L ti] is the total DFL concentration at each titration point, [P t] is the total protein concentration, K a = 1/K d is the association constant, and δ b is the chemical shift of the resonance in the complex. The K d of DFL‐15N‐HMGB1 and DFL‐15N‐CXCL12 interactions was the average of the fitting of 5 and 9 residues, respectively, plus the standard deviation. K d and δ b were used as fitting parameters using the Xmgrace program (http://plasma-gate.weizmann.ac.il/Grace/).
The apparent equilibrium constant for the CXCL12 monomer‐dimer equilibrium in the presence and in the absence of DFL was estimated from nonlinear fitting of CXCL12 chemical shifts as a function of increasing CXCL12 concentrations (0.025, 0.1, 0.18, 0.37, 0.73, 1.47 mM) according to the equation 54:
where δ i is the absolute change in chemical shift, δ d is the change in chemical shift of the dimer, [P] is the protein concentration, and K a = 1/K d is the association constant. K a and δ d were used as fitting parameters using the Xmgrace program (http://plasma-gate.weizmann.ac.il/Grace/).
At a given total CXCL12 concentration [M]T and a given estimated dimerization K d, the molar extent of CXCL12 monomer [M] and dimer [D] is calculated by solving the following quadratic equation 55:
Relaxation experiments
Relaxation experiments were performed on 15N‐labeled CXCL12 (0.1 mM in 20 mM phosphate buffer, 20 mM NaCl, pH 6) in the absence and in the presence of 10‐fold excess DFL at 298 K.
Heteronuclear {1H}15N nuclear Overhauser enhancement, longitudinal and transversal 15N relaxation rates (R1, R2) were measured using standard 2D methods 56. For R1 and R2 experiments, a duty‐cycle heating compensation was used 57; the two decay curves were sampled at 12 (ranging from 50 to 2,000 ms) and 11 (from 12 to 244 ms) different time points, respectively. Both R 1 and R 2 experiments were collected in random order and using a recovery delay of 2.5 s. R 1 and R 2 values were fitted to a 2‐parameter exponential decay from the intensities using the fitting routine implemented in the CcpNmr program; duplicate measurements were used to allow a statistical analysis of the uncertainties 58. HSQC spectra measured in an interleaved fashion with and without 4 s of proton saturation during recovery delay were recorded for the {1H}15N heteronuclear nOe experiments. The corresponding values were obtained from the ratio between saturated and unsaturated peaks intensities. The uncertainty was estimated through the ratio between the standard deviation of the noise in both saturated and unsaturated spectra divided by the intensity of the respective peaks. The correlation time (τ c) was estimated from R 2/R 1 according to the following equation 59:
Only residues corresponding to non‐overlapping peaks, with heteronuclear nOe > 0.65, were used, and residues with anomalous R 2 were excluded 60.
Docking models
Molecular docking of DFL on Box A (residues G3‐Y77), Box B (A93‐G173; coordinates were extracted from the PDB structure with code: 2YRQ), and CXCL12 (K1‐K68, coordinates extracted from the PDB structure with code: 4UAI) was performed using the data‐driven software HADDOCK 2.2 24, 61 following the classical three‐stage procedure which includes: (i) randomization of orientations and rigid body minimization, (ii) simulated annealing in torsion angle space, and (iii) refinement in Cartesian space with explicit water. Ambiguous interaction restraints (AIRs) were defined as follows: Residues with CSP > avg + SD or displaying intramolecular nOes were used to define active residues, whose solvent accessible surface neighbors were set as passive (Appendix Table S1). In the case of CXCL12, only the residues located around the sY21 binding site (red CSP in Fig 2D) were set as active (Appendix Table S1), as STD competition experiments of DFL in the presence of CXCR41‐38sY21 demonstrated that they both compete for the sY21 binding site. In the case of Box A (Box B), intermolecular nOes were included as unambiguous restraints in the calculations only in the semi‐flexible refinement stage, setting the maximum distance of the nOe H pairs to 5 Å (Appendix Table S1).
Optimized parameters for liquid simulation (OPLS) were used for the protein (protein‐allhdg5‐4 and protein‐allhdg5‐4‐caro). The geometric coordinates and parameters for DFL were calculated and optimized using the PRODRG server 62. Calculations generated 1,000, 1,000, 500 structures for the rigid body docking (it0), the semi‐flexible refinement (it1), and the explicit solvent refinement (water), respectively. The final 500 structures obtained after water refinement were scored according to their HADDOCK score. The latter (defined as HADDOCKscore = 1.0 EvdW + 0.2 Eelec + 1.0 Edesolv + 0.1 EAIR) is a weighted combination of van der Waals (vdW) and electrostatic energy terms (Lennard–Jones and Coulomb potentials), empirical desolvation term 63, and ambiguous interaction restraint energy term, which reflects the accordance of the model to the input restraints.
HADDOCK models were clustered 64 based on their interface root mean square deviation (rmsd), setting the cutoff and the minimum number of models in a cluster to 1.8 Å and 10 for the boxes, and 2.5 Å and 10 for CXCL12, respectively. Proteins were aligned and fitted on the backbone of active residues reported in Appendix Table S1. The rmsd of DFL was calculated only on the heavy atoms of the entire scaffold.
To remove any bias of the cluster size on the cluster statistics, the final overall score of each cluster was calculated on the four lowest HADDOCK score models in that cluster. For each protein, the cluster with the best fitting with respect to the experimentally driven restraints (lowest number of violations) and the best HADDOCK score (cluster 1 for Box A, cluster 5 for Box B and CXCL12) was selected (Appendix Figs S8, S9, and S10).
The analysis of the docking calculations was performed applying in‐house python and tcl scripts. Molecular images were generated by PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.
MST experiments
MST experiments were performed at 24°C on a NanoTemper® Monolith NT.115 instrument with red filters, using 40% LED power and 60% MST power. Binding experiments were carried out using 6His‐tagged HMGB1 and 6His‐tagged CXCL12, non‐covalently labeled with the NT647 fluorescence dye 65.
For binding assays, DFL or CXCL12 were titrated (16‐points) on 6His‐tagged HMGB1 (full‐length protein, MST buffer containing 20 mM phosphate buffer pH 7.3, 20 mM NaCl, 0.05% Tween, 1 mM DTT), and DFL was titrated also on 6His‐tagged CXCL12 (MST buffer containing 20 mM phosphate buffer pH 7.3, 20 mM NaCl, 0.05% Tween). The ligand dilutions were generated as a 1:2 dilution of the stock solution using MST buffer; a constant amount of labeled proteins (50 nM) was added to all dilution steps. Maximum concentrations of DFL and CXCL12 in the titrations series were 5 mM and 217 μM, respectively. Complex samples were incubated for 15 min before loading into NanoTemper premium capillaries.
Competition experiments were carried out pre‐forming a complex between labeled 6His‐tagged HMGB1 (50 nM) and unlabeled CXCL12 (10 μM, i.e., 2 times the estimated K d) 66. For 16‐point titration series of DFL, serial 1:2 dilutions of the DFL stock solution were made into MST buffer, and a constant amount of preformed heterocomplex was added to all dilution steps. All samples were incubated for 15 min and centrifuged at 15,000 g for 10 min before measurements. Maximum concentration of DFL in the titrations series was 0.63 mM. Addition of DFL induced the recovery of the MST signal of HMGB1 toward the unbound state value (Fig 3C). Attempts to reach saturation failed, as higher DFL concentrations (up to 5 mM) induced aggregation phenomena, as assessed by the bumpiness of the MST traces (data not shown).
For all MST experiments, data points were the average of three measurements (error bars correspond to standard deviation). All data analyses were carried out using NanoTemper analysis software using the K d model fitting for the binding assays and Hill model for competition experiments. For titrations of HMGB1 with DFL and CXCL12, based on previous data obtained on Glycyrrhizin 21 and CXCL12 5, we assumed that the stoichiometry would be 1:2. The form of the binding isotherm was indistinguishable from a one binding site isotherm, thus supporting the presence of equivalent binding sites with similar apparent affinity (i.e., similar K d values) 67. Fitting of the data with the following equation:
yielded apparent K ds of 1.6 mM and 3.9 μM for DFL and CXCL12, respectively; i.e., values virtually indistinguishable from those obtained using the one binding site model implemented in Nanotemper analysis software.
For the sake of completeness, to exclude any cooperative binding, we also tried to fit the data with the Hill model, yielding an EC50 of 3.8 ± 0.5 μM, perfectly in line with the estimated K d. Notably, the fitting yielded a Hill coefficient of 1.1, thus suggesting no cooperativity.
Cell migration experiments
For fibroblast chemotaxis, modified Boyden chambers were used with filters (pore diameter 8 μm; Neuro Probe) coated with 50 μg/ml fibronectin (Roche). Mouse 3T3 cells (50,000 in 200 μl) were added to the upper chamber. Serum‐free DMEM as negative control, HMGB1, and/or other molecules was added to the lower chamber at the indicated concentration, and then cells were left to migrate for 3 h at 37°C. Cells were fixed with ethanol and stained with Giemsa Stain (Sigma), and then non‐migrating cells were removed with a cotton swab. All assays were done at least in biological triplicate. The migrated cells were acquired with Zeiss Imager M.2 microscope at 10× magnification and then evaluated with an automated counting program. All assays were done at least in biological triplicate and were repeated at least twice.
Assay for cytokine induction on mouse BMDMs
Bone marrow‐derived macrophages (BMDMs) were obtained as described 68 and stimulated as described in the legend to Fig 4. Total RNAs were isolated using the Illustra RNAspin Mini kit (GE Healthcare), and complementary DNAs (cDNAs) were obtained by reverse transcription with oligo(dT) primers (Invitrogen, Carlsbad, CA, USA) and SuperScript II Reverse Transcriptase (Invitrogen) following the manufacturers’ instructions. Quantitative real‐time PCR was performed using a LightCycler480 (Roche Molecular Diagnostics), in triplicates, using SYBR Green I master mix. The ΔC t method was used for quantification, and the β‐actin gene was used for normalization.
The sequence of the primers was
TNF forward: CTTCTCATTCCTGCTTGTGG
TNF reverse: GCAGAGAGGAGGTTGACTTTC
Beta‐actin forward: AGACGGGGTCACCCACACTGTGCCCATCTA
Beta‐actin reverse: CTAGAAGCACTTGCGGTGCACGATGGAGGG
Human macrophages
Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats of donor blood (Hospital of Magenta, Italy) by Ficoll gradient centrifugation (Lymphoprep, AXIS‐SHIELD). CD14+ monocytes were isolated by positive immunoselection (CD14 MicroBeads, Miltenyi Biotec, Germany) according to the manufacturer's instructions and differentiated into macrophages using X‐Vivo medium supplemented with 1% heat‐inactivated human serum, GM‐CSF, and M‐CSF.
In vivo injury model
Eight‐week‐old wild‐type male C57Bl6 mice were purchased from Charles River, Calco, Italy, and housed in the San Raffaele animal house for 3 days before experimentation. To assess the initial leukocyte recruitment after muscle injury, mice were injected intravenously with 200 μg of DFL (Sigma‐Aldrich) or Glycyrrhizin (Acros Organics) 3 h before muscle injection with 50 μl of 15 μM cardiotoxin (Latoxan). After 6 h, the injured tibialis anterior muscles were collected and dissociated in RPMI 1640 containing 0.2% collagenase B (Roche Diagnostics) at 37°C for 1 h. CD45+ cells were purified by magnetic cell sorting by using anti‐CD45 beads (Miltenyi Biotec) according to the manufacturer's instructions and quantified by Countess (Invitrogen).
Protocol 838 was approved by the San Raffaele IACUC and by the Italian Istituto Superiore di Sanità.
Author contributions
FDL performed the NMR experiments, computational studies, analyzed the data, and prepared the manuscript. GQ performed the NMR experiments, and MT performed the in vivo experiments and analyzed the data. VM, CZ, AP, and MC expressed and purified recombinant proteins. FDM performed the cell migration experiments. AG synthesized and purified the peptide. RM performed the cytokine induction experiments. GM and MEB directed the study and were involved in all aspects of the experimental design, data analysis, and manuscript preparation. All authors critically reviewed the text and figures.
Conflict of interest
The authors declare that they have no conflict of interest. However, M.E.B. is founder and part‐owner of HMGBiotech, a company that provides goods and services related to HMGB proteins. A.P. and M.C. are employees of HMGBiotech.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
This work was supported by an iCARE Fellowship funded by AIRC and Marie Curie Actions—People—COFUND (Project code 16258), by AIRC (IG18623; IG21440), and by Fellowship Fondazione Umberto Veronesi 2019. We gratefully acknowledge Dr. Francesca Viganò and Dr. Katarzyna Walkiewicz (NanoTemper Technologies GmbH, Munich), and Dr. Delia Tarantino (University of Milan), for technical assistance and discussion in MST experiments, and Dr. Samuel Zambrano for mathematical modeling and useful discussions.
EMBO Reports (2019) 20: e47788
Contributor Information
Marco E Bianchi, Email: bianchi.marco@hsr.it.
Giovanna Musco, Email: musco.giovanna@hsr.it.
References
- 1. Andersson U, Tracey KJ (2011) HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol 29: 139–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bianchi ME, Crippa MP, Manfredi AA, Mezzapelle R, Rovere Querini P, Venereau E (2017) High‐mobility group box 1 protein orchestrates responses to tissue damage via inflammation, innate and adaptive immunity, and tissue repair. Immunol Rev 280: 74–82 [DOI] [PubMed] [Google Scholar]
- 3. Thomas JO (2001) HMG1 and 2: architectural DNA‐binding proteins. Biochem Soc Trans 4: 395–401 [DOI] [PubMed] [Google Scholar]
- 4. Štros M (2010) HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta Gene Regul Mech 1799: 101–113 [DOI] [PubMed] [Google Scholar]
- 5. Schiraldi M, Raucci A, Muñoz LM, Livoti E, Celona B, Venereau E, Apuzzo T, De Marchis F, Pedotti M, Bachi A et al (2012) HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR5. J Exp Med 209: 551–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, Liu J, Antonelli A, Preti A, Raeli L et al (2012) Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med 209: 1519–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cecchinato V, D'Agostino G, Raeli L, Uguccioni M (2016) Chemokine interaction with synergy‐inducing molecules: fine tuning modulation of cell trafficking. J Leukoc Biol 99: 851–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee G, Espirito Santo AI, Zwingenberger S, Cai L, Vogl T, Feldmann M, Horwood NJ, Chan JK, Nanchahal J (2018) Fully reduced HMGB1 accelerates the regeneration of multiple tissues by transitioning stem cells to G Alert. Proc Natl Acad Sci USA 115: E4463–E4472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Paoletti S (2005) A rich chemokine environment strongly enhances leukocyte migration and activities. Blood 105: 3405–3412 [DOI] [PubMed] [Google Scholar]
- 10. Collins PJ, McCully ML, Martínez‐Muñoz L, Santiago C, Wheeldon J, Caucheteux S, Thelen S, Cecchinato V, Laufer JM, Purvanov V et al (2017) Epithelial chemokine CXCL14 synergizes with CXCL12 via allosteric modulation of CXCR10. FASEB J 31: 3084–3097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Proudfoot AEI, Uguccioni M (2016) Modulation of chemokine responses: synergy and cooperativity. Front Immunol 7: 183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gouwy M, Schiraldi M, Struyf S, Van Damme J, Uguccioni M (2012) Possible mechanisms involved in chemokine synergy fine tuning the inflammatory response. Immunol Lett 145: 10–14 [DOI] [PubMed] [Google Scholar]
- 13. Kapurniotu A, Gokce O, Bernhagen J (2019) The multitasking potential of alarmins and atypical chemokines. Front Med 6: 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Venereau E, De Leo F, Mezzapelle R, Careccia G, Musco G, Bianchi ME (2016) HMGB1 as biomarker and drug target. Pharmacol Res 111: 534–544 [DOI] [PubMed] [Google Scholar]
- 15. VanPatten S, Al‐Abed Y (2018) High Mobility Group Box‐1 (HMGb1): current wisdom and advancement as a potential drug target. J Med Chem 61: 5093–5107 [DOI] [PubMed] [Google Scholar]
- 16. Laraia L, McKenzie G, Spring DR, Venkitaraman AR, Huggins DJ (2015) Overcoming chemical, biological, and computational challenges in the development of inhibitors targeting protein‐protein interactions. Chem Biol 22: 689–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scott DE, Bayly AR, Abell C, Skidmore J (2016) Small molecules, big targets: drug discovery faces the protein–protein interaction challenge. Nat Rev Drug Discov 15: 533–550 [DOI] [PubMed] [Google Scholar]
- 18. Steelman S, Cirillo V, Tempero K (1978) The chemistry, pharmacology and clinical pharmacology of diflunisal. Curr Med Res Opin 5: 506–514 [DOI] [PubMed] [Google Scholar]
- 19. Choi HW, Tian M, Song F, Venereau E, Preti A, Park S‐W, Hamilton K, Swapna GVT, Manohar M, Moreau M et al (2015) Aspirin's active metabolite salicylic acid targets high mobility group box 1 to modulate inflammatory responses. Mol Med 21: 526–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang H, Pellegrini L, Napolitano A, Giorgi C, Jube S, Preti A, Jennings CJ, De Marchis F, Flores EG, Larson D et al (2015) Aspirin delays mesothelioma growth by inhibiting HMGB1‐mediated tumor progression. Cell Death Dis 6: e1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mollica L, De Marchis F, Spitaleri A, Dallacosta C, Pennacchini D, Zamai M, Agresti A, Trisciuoglio L, Musco G, Bianchi ME (2007) Glycyrrhizin binds to high‐mobility group box 1 protein and inhibits its cytokine activities. Chem Biol 14: 431–441 [DOI] [PubMed] [Google Scholar]
- 22. Mayer M, Meyer B (2001) Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J Am Chem Soc 123: 6108–6117 [DOI] [PubMed] [Google Scholar]
- 23. Dalvit C, Fogliatto GP, Stewart A, Veronesi M, Stockman B (2001) WaterLOGSY as a method for primary NMR screening: practical aspects and range of applicability. J Biomol NMR 21: 349–359 [DOI] [PubMed] [Google Scholar]
- 24. Dominguez C, Boelens R, Bonvin AMJJ (2003) HADDOCK: a protein‐protein docking approach based on biochemical or biophysical information. J Am Chem Soc 125: 1731–1737 [DOI] [PubMed] [Google Scholar]
- 25. Wienken CJ, Baaske P, Rothbauer U, Braun D, Duhr S (2010) Protein‐binding assays in biological liquids using microscale thermophoresis. Nat Commun 1: 100 [DOI] [PubMed] [Google Scholar]
- 26. Veldkamp CT, Seibert C, Peterson FC, Sakmar TP, Volkman BF (2006) Recognition of a CXCR26 sulfotyrosine by the chemokine stromal cell‐derived factor‐1 α. J Mol Biol 4: 1400–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Veldkamp CT, Seibert C, Peterson FC, De la Cruz NB, Haugner JC, Basnet H, Sakmar TP, Volkman BF (2008) Structural basis of CXCR27 sulfotyrosine recognition by the chemokine SDF‐1/CXCL12. Sci Signal 1: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ziarek JJ, Getschman AE, Butler SJ, Taleski D, Stephens B, Kufareva I, Handel TM, Payne RJ, Volkman BF (2013) Sulfopeptide probes of the CXCR28/CXCL12 interface reveal oligomer‐specific contacts and chemokine allostery. ACS Chem Biol 8: 1955–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Veldkamp CT, Peterson FC, Pelzek AJ, Volkman BF (2005) The monomer – dimer equilibrium of stromal cell‐derived factor‐1 (CXCL 12) is altered by pH, phosphate, sulfate, and heparin. Protein Sci 4: 1071–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baryshnikova OK, Sykes BD (2006) Backbone dynamics of SDF‐1alpha determined by NMR: interpretation in the presence of monomer‐dimer equilibrium. Protein Sci 15: 2568–2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. He M, Bianchi ME, Coleman TR, Tracey KJ, Al‐Abed Y (2018) Exploring the biological functional mechanism of the HMGB1/TLR4/MD‐2 complex by surface plasmon resonance. Mol Med 24: 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tirone M, Tran NL, Ceriotti C, Gorzanelli A, Canepari M, Bottinelli R, Raucci A, Di Maggio S, Santiago C, Mellado M et al (2018) High mobility group box 1 orchestrates tissue regeneration via CXCR32. J Exp Med 215: 303–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Warner TD, Giuliano F, Vojnovic I, Bukasa A, Mitchell JA, Vane JR (1999) Nonsteroid drug selectivities for cyclo‐oxygenase‐1 rather than cyclo‐oxygenase‐2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc Natl Acad Sci USA 96: 7563–7568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kopp E, Ghosh S (1994) Inhibition of NF‐kappa B by sodium salicylate and aspirin. Science 265: 956–959 [DOI] [PubMed] [Google Scholar]
- 35. Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green KA, Mustard KJ et al (2012) The ancient drug salicylate directly activates AMP‐activated protein kinase. Science 336: 918–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Din FVN, Valanciute A, Houde VP, Zibrova D, Green KA, Sakamoto K, Alessi DR, Dunlop MG (2012) Aspirin inhibits mTOR signaling, activates AMP‐activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology 142: 1504–1515 e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bashir A, Kankipati C, Jones S, Newman R, Safrany S, Perry C, Nicholl I (2019) A novel mechanism for the anticancer activity of aspirin and salicylates. Int J Oncol 54: 1256–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shirakawa K, Wang L, Man N, Maksimoska J, Sorum AW, Lim HW, Lee IS, Shimazu T, Newman JC, Schröder S et al (2016) Salicylate, diflunisal and their metabolites inhibit CBP/p300 and exhibit anticancer activity. Elife 5: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, Lu B, Chavan S, Rosas‐Ballina M, Al‐Abed Y et al (2010) A critical cysteine is required for HMGB1 binding to Toll‐like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci USA 107: 11942–11947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang H, Lundbäck P, Ottosson L, Erlandsson‐Harris H, Venereau E, Bianchi M, Al‐Abed Y, Andersson U, Tracey K, Antoine D (2012) Redox modification of cysteine residues regulates the cytokine activity of high mobility group box‐1 (HMGB1). Mol Med 18: 250–259 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41. Whitty A (2008) Cooperativity and biological complexity. Nat Chem Biol 4: 435–439 [DOI] [PubMed] [Google Scholar]
- 42. Merenmies J, Pihlaskari R, Laitinen J, Wartiovaara J, Rauvala H (1991) 30‐kDa heparin‐binding protein of brain (amphoterin) involved in neurite outgrowth: amino acid sequence and localization in the filopodia of the advancing plasma membrane. J Biol Chem 266: 16722–16729 [PubMed] [Google Scholar]
- 43. Rueda P, Richart A, Récalde A, Gasse P, Vilar J, Guérin C, Lortat‐Jacob H, Vieira P, Baleux F, Chretien F et al (2012) Homeostatic and tissue reparation defaults in mice carrying selective genetic invalidation of CXCL12/proteoglycan interactions. Circulation 126: 1882–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bolognesi ML, Cavalli A (2016) Multitarget drug discovery and polypharmacology. ChemMedChem 11: 1190–1192 [DOI] [PubMed] [Google Scholar]
- 45. Anighoro A, Bajorath J, Rastelli G (2014) Polypharmacology: challenges and opportunities in drug discovery miniperspective. J Med Chem 57: 7874–7887 [DOI] [PubMed] [Google Scholar]
- 46. Korcsmáros T, Szalay MS, Böde C, Kovács IA, Csermely P (2007) How to design multi‐target drugs: target search options in cellular networks. Expert Opin Drug Discov 2: 799–808 [DOI] [PubMed] [Google Scholar]
- 47. Koenen RR, von Hundelshausen P, Nesmelova IV, Zernecke A, Liehn EA, Sarabi A, Kramp BK, Piccinini AM, Paludan SR, Kowalska MA et al (2009) Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med 15: 97–103 [DOI] [PubMed] [Google Scholar]
- 48. Fields GB, Noble RL (1990) Solid phase peptide synthesis utilizing 9‐fluorenylmethoxycarbonyl amino acids. Chem Biol Drug Des 35: 161–214 [DOI] [PubMed] [Google Scholar]
- 49. Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins Struct Funct Genet 59: 687–696 [DOI] [PubMed] [Google Scholar]
- 50. Veldkamp CT, Ziarek JJ, Su J, Basnet H, Lennertz R, Weiner JJ, Peterson FC, Baker JE, Volkman BF (2009) Monomeric structure of the cardioprotective chemokine SDF‐1/CXCL12. Protein Sci 18: 1359–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Grzesiek S, Stahl SJ, Wingfield PT, Bax A (1996) The CD4 determinant for downregulation by HIV‐1 Nef directly binds to Nef. Mapping of the Nef binding surface by NMR. Biochemistry 35: 10256–10261 [DOI] [PubMed] [Google Scholar]
- 52. Krajewski M, Rothweiler U, D'Silva L, Majumdar S, Klein C, Holak TA (2007) An NMR‐based antagonist induced dissociation assay for targeting the ligand‐protein and protein‐protein interactions in competition binding experiments. J Med Chem 50: 4382–4387 [DOI] [PubMed] [Google Scholar]
- 53. Dalvit C, Pevarello P, Tatò M, Veronesi M, Vulpetti A, Sundström M (2000) Identification of compounds with binding affinity to proteins via magnetization transfer from bulk water. J Biomol NMR 18: 65–68 [DOI] [PubMed] [Google Scholar]
- 54. Tan HKS (1994) Determination of the NMR monomer shift and dimerization constant in a self‐associating system by direct application of the least‐squares method. J Chem Soc, Faraday Trans 90: 3521 [Google Scholar]
- 55. Benfield CTO, Mansur DS, McCoy LE, Ferguson BJ, Bahar MW, Oldring AP, Grimes JM, Stuart DI, Graham SC, Smith GL (2011) Mapping the IκB Kinase β (IKKβ)‐binding interface of the B14 protein, a Vaccinia virus inhibitor of IKKβ‐mediated activation of nuclear factor κB. J Biol Chem 286: 20727–20735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman‐Kay JD, Kay LE (1994) Backbone dynamics of a free and phosphopeptide‐complexed Src‐ homology 2 domain studied by 15N relaxation. Biochemistry 33: 5984–6003 [DOI] [PubMed] [Google Scholar]
- 57. Yip GNB, Zuiderweg ERP (2005) Improvement of duty‐cycle heating compensation in NMR spin relaxation experiments. J Magn Reson 176: 171–178 [DOI] [PubMed] [Google Scholar]
- 58. Skelton NJ, Palmer AG, Akke M, Kordel J, Rance M, Chazin WJ (1993) Practical aspects of two‐dimensional proton‐detected 15N spin relaxation measurements. J Magn Reson, Ser B 102: 253–264 [Google Scholar]
- 59. Bloembergen N, Purcell EM, Pound RV (1948) Relaxation effects in nuclear magnetic resonance absorption. Phys Rev 73: 679–712 [Google Scholar]
- 60. Barbato G, Ikura M, Kay LE, Bax A, Pastor RW (1992) Backbone dynamics of calmodulin studied by 15N relaxation using inverse detected two‐dimensional NMR spectroscopy: the central helix is flexible. Biochemistry 31: 5269–5278 [DOI] [PubMed] [Google Scholar]
- 61. van Zundert GCP, Rodrigues JPGLM, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond ASJ, van Dijk M, de Vries SJ, Bonvin AMJJ (2016) The HADDOCK2.2 Web server: user‐friendly integrative modeling of biomolecular complexes. J Mol Biol 428: 720–725 [DOI] [PubMed] [Google Scholar]
- 62. Schüttelkopf AW, van Aalten DMF (2004) PRODRG: a tool for high‐throughput crystallography of protein–ligand complexes. Acta Crystallogr D Biol Crystallogr 60: 1355–1363 [DOI] [PubMed] [Google Scholar]
- 63. Fernández‐Recio J, Totrov M, Abagyan R (2004) Identification of protein‐protein interaction sites from docking energy landscapes. J Mol Biol 335: 843–865 [DOI] [PubMed] [Google Scholar]
- 64. Daura X, Antes I, van Gunsteren WF, Thiel W, Mark AE (1999) The effect of motional averaging on the calculation of NMR‐derived structural properties. Proteins 36: 542–555 [PubMed] [Google Scholar]
- 65. Bartoschik T, Galinec S, Kleusch C, Walkiewicz K, Breitsprecher D, Weigert S, Muller YA, You C, Piehler J, Vercruysse T et al (2018) Near‐native, site‐specific and purification‐free protein labeling for quantitative protein interaction analysis by MicroScale Thermophoresis. Sci Rep 8: 4977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jerabek‐Willemsen M, Wienken CJ, Braun D, Baaske P, Duhr S (2011) Molecular interaction studies using microscale thermophoresis. Assay Drug Dev Technol 9: 342–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Copeland RA (2000) Enzymes: a practical introduction to structure, mechanism, and data analysis, pp 76–95. New York, NY: Wileyy‐VCH; [Google Scholar]
- 68. Zhang X, Goncalves R, Mosser DM (2008) The isolation and characterization of murine macrophages In Current protocols in immunology, Coligan JE. (ed.), p 14.1 Hoboken, NJ: John Wiley & Sons, Inc. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4