

Abstract
During mitosis, sister chromatids attach to microtubules which generate ~ 700 pN pulling force focused on the centromere. We report that chromatin‐localized signals generated by Polo‐like kinase 1 (Plk1) maintain the integrity of the kinetochore and centromere against this force. Without sufficient Plk1 activity, chromosomes become misaligned after normal condensation and congression. These chromosomes are silent to the mitotic checkpoint, and many lag and mis‐segregate in anaphase. Their centromeres and kinetochores lack CENP‐A, CENP‐C, CENP‐T, Hec1, Nuf2, and Knl1; however, CENP‐B is retained. CENP‐A loss occurs coincident with secondary misalignment and anaphase onset. This disruption occurs asymmetrically prior to anaphase and requires tension generated by microtubules. Mechanistically, centromeres highly recruit PICH DNA helicase and PICH depletion restores kinetochore disruption in pre‐anaphase cells. Furthermore, anaphase defects are significantly reduced by tethering Plk1 to chromatin, including H2B, and INCENP, but not to CENP‐A. Taken as a whole, this demonstrates that Plk1 signals are crucial for stabilizing centromeric architecture against tension.
Keywords: CENP‐A, chromatin, kinase, mitosis, phosphorylation
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Cell Cycle
Introduction
The human kinetochore is a multi‐protein complex essential for accurate segregation of sister chromosomes during mitosis. Within this structure, microtubule‐binding proteins of the KMN complex (Knl1, Mis12 complex, and Ndc80 complex) assemble on a constitutive layer of chromatin‐embedded proteins, termed the constitutive centromere‐associated network (CCAN). During each cell cycle, the CCAN is replenished and redistributed to the new sister chromosome generated by DNA replication. CENP‐A, a histone H3 variant, epigenetically establishes and maintains the CCAN to ensure genomic integrity and cell viability 1, 2, 3, 4, 5. Immediately following mitosis, new CENP‐A is rapidly integrated into centromeric chromatin as a direct consequence of loss of Cdk1 activity 6, 7, 8, a process that requires HJURP, Mis18, and localized kinase signals from Plk1 9, 10, 11, 12. In this manner, the CCAN and centromere specification is maintained in proliferating cells.
Signals within the kinetochore are crucial to generate stable microtubule attachment and biorientation, in which replicated sister chromatids are linked to opposite spindle poles. Multiple protein kinases regulate kinetochore functions, including Mps1 to initiate the mitotic checkpoint 13, 14, BubR1 to control microtubule attachment and checkpoint signaling 15, 16, Aurora B to correct erroneous microtubule attachments 17, 18, 19, 20, Haspin kinase to align chromosomes 21, and Plk1 to stabilize end‐on microtubule attachments 22, 23, 24 and promote CENP‐A assembly following mitosis 12. Although these kinases all localize within the kinetochore, CCAN, or inner centromere, their distributions within these structures are distinct, reflecting their disparate roles.
Plk1 phosphorylates substrates throughout the kinetochore–CCAN even though the ~ 5 nm size of a kinase domain is dwarfed by the ~ 100‐nm scale of a kinetochore. Plk1 reaches substrates either by binding them directly or by binding adjacent proteins. Indeed, a number of binding partners within the kinetochore are known, including Bub1 25, BubR1 26, and CENP‐U/PBIP 27. Multiple partners are important, as Plk1 tethered to distinct partners within the kinetochore can phosphorylate only regionally within this structure 28. Although most of these binding partners are at the outer kinetochore, Plk1 signals arising from the inner centromere and chromatin are crucial for proper chromosome alignment and accurate anaphase segregation 28.
We previously found that partial loss of Plk1 activity leads to an anaphase segregation defect 29 that did not trigger the mitotic checkpoint. In that study, the ~ 15% of lagging chromosomes had a stretched appearance suggesting merotelic attachments, though these aberrant microtubule attachments were not directly observable. Here, we considered the alternative that partial Plk1 inhibition causes chromosome mis‐segregation via a novel mechanism also silent to the mitotic checkpoint. Indeed, we find that Plk1 activity is required at the centromere to stabilize the CCAN and maintain genomic integrity against spindle tension. The mitotic spindle exerts ~ 700 pN force across the mitotic kinetochore 30, causing stretch between sister kinetochores and within each kinetochore 31, 32, which necessarily is transmitted across centromeric chromatin. In the absence of full Plk1 activity, chromosomes align on the metaphase plate initially, followed by loss of multiple kinetochore and CCAN components, including CENP‐A. As cells progress into anaphase, the chromosomes lacking CENP‐A lag behind the segregating masses, yielding cells with chromatin devoid of either ancestral or nascent CENP‐A. Kinetochore disruption is restored, in part, by depletion of the DNA helicase PICH or enforcing Plk1 signals at chromatin or the inner centromere. Thus, “kinetochore rupture” is a new effect of Plk1 inhibition that is not detected by the mitotic checkpoint.
Results
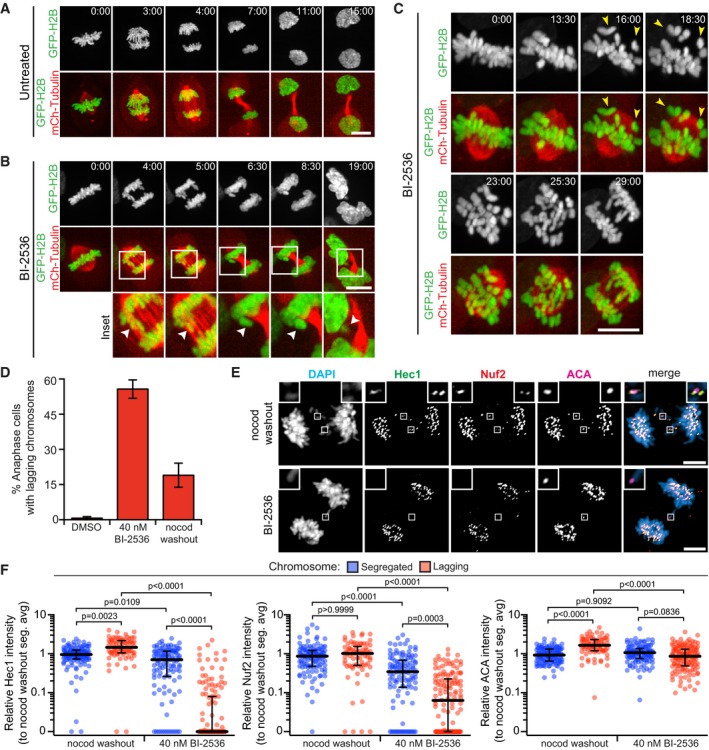
To confirm previous findings and to identify the specific roles of Plk1 at the kinetochore, we performed time‐lapse videomicroscopy with histone H2B‐GFP‐ and mCherry‐tubulin‐labeled RPE1 cells (Fig 1A–C and Movies EV1, EV2 and EV3). As expected, untreated cells efficiently aligned and segregated chromosomes. Next, these cells were challenged with low nanomolar concentrations of BI‐2536, a specific inhibitor of Plk1 23. With treatment, chromosomes aligned in metaphase followed by a secondary misalignment (yellow arrowheads). Cells progressing to anaphase commonly exhibited lagging chromosomes, which were later ensconced into micronuclei in daughter cells (white arrowheads). These findings confirm previous observations that high Plk1 activity is required to maintain metaphase chromosome alignment and to ensure accurate chromosome segregation in anaphase, but not required for mitotic progression.
Figure 1. Plk1 activity is important for outer kinetochore integrity during mitosis in RPE1 cells.
-
A, BMaximum‐intensity projection frames from live‐cell fluorescence microscopy of RPE1 cells undergoing mitosis untreated (A) and exhibiting lagging chromosomes after Plk1 inhibition with 40 nM BI‐2536 (B). Arrowheads in insets highlight micronucleus formation. Time, in min:s from metaphase onset. Scale bars, 5 μm.
-
CMaximum‐intensity projection frames from live‐cell fluorescence microscopy of RPE1 cell exhibiting misaligned chromosomes after Plk1 inhibition with 40 nM BI‐2536. Yellow arrowheads highlight misaligned chromosomes. Time, in min:s from metaphase onset. Scale bars, 5 μm.
-
DGraph shows average percentage (± SEM) of anaphase cells exhibiting lagging chromosomes after Plk1 inhibition (BI‐2536) or nocodazole washout (n > 25 cells/experiment; three independent experiments).
-
ERepresentative maximum‐intensity projection micrographs of kinetochore protein localization in anaphase cells after Plk1 inhibition (BI‐2536) or nocodazole washout. Insets highlight lagging kinetochores, marked by ACA. Scale bars, 5 μm.
-
FGraphs show relative volume intensities of indicated proteins from (E) at the kinetochores of segregated (blue) and lagging (red) chromosomes. Each circle represents a single segregated (n = 4/cell) or lagging (n = 1–7/cell) kinetochore from the same cell (10 cells/experiment; three independent experiments). Bars indicate median kinetochore intensity and interquartile range. Significance determined by Kruskal–Wallis test with Dunn's correction for multiple comparisons.
Lagging chromosomes can arise by distinct mechanisms. One common mechanism is merotelic attachments, the link of a single kinetochore to both mitotic poles 33. Such attachments of single kinetochores to both poles are generated by nocodazole washout and impair the ability of a lagging chromosome to segregate in either direction. To compare this common mechanism with Plk1 inhibition, we generated lagging chromosomes with BI‐2536 treatment or nocodazole treatment/washout (Fig 1D–F) and evaluated two components of the heterotetrameric Ndc80 complex that directly bind microtubules at the outer kinetochore. Strikingly, both Hec1/Ndc80 and Nuf2 intensities are largely reduced on lagging chromosomes with Plk1 inhibition compared to nocodazole washout segregants. By contrast, these Ndc80 components are retained in properly segregated Plk1‐inhibited chromosomes, and in merotelic lagging chromosomes generated with nocodazole washout. To ensure the observations are a direct effect of Plk1 inhibition, we evaluated lagging chromosomes in Plk1as RPE1 cells, which express a modified Plk1 allele sensitive to the bulky ATP analog, 3‐MB‐PP1 29, 34. Indeed, partial inhibition of Plk1 yielded lagging chromosomes with marked reduction of Hec1 (Fig EV1A–C), consistent with our findings in Plk1‐wild‐type RPE1 cells. These results demonstrate a novel mechanism by which Plk1 regulates kinetochore integrity and chromosome segregation during human mitosis.
Figure EV1. Defects in outer kinetochore integrity are specific to loss of Plk1 activity (related to Fig 1).
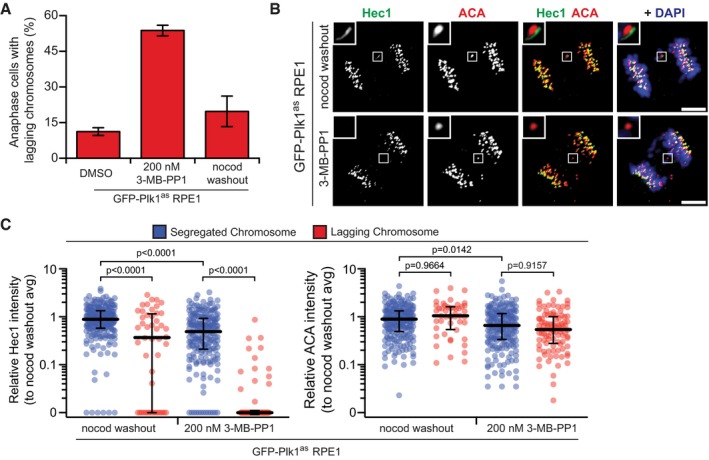
- Graph shows average percentage (± SEM) of EGFP‐Plk1as‐expressing RPE1 cells exhibiting lagging chromosomes in anaphase after Plk1as inhibition (200 nM 3‐MB‐PP1) or nocodazole washout (n = 30 cells/experiment; four independent experiments).
- Representative maximum‐intensity micrographs of anaphase cells from (A). Insets highlight presence/absence of Hec1 at lagging kinetochores, marked by ACA. Scale bars, 5 μm.
- Graph shows relative volume intensities of Hec1 (left) and ACA (right) at segregated (blue) and lagging (red) kinetochores from (A, B). Each circle represents a single kinetochore (n = 10 segregated kinetochores/cell, 1–8 lagging kinetochores/cell; eight cells/experiment; three independent experiments). Bars indicate median kinetochore intensity and interquartile range. Significance determined by Kruskal–Wallis test with Dunn's correction for multiple comparisons.
Mechanistically, the lack of the Ndc80 complex on lagging chromosome could be explained by failed recruitment or by secondary removal. To test if Plk1 is required for Hec1/Ndc80 recruitment, cells were arrested in mitosis using monastrol or nocodazole and exposed to low (40 nM) or high (200 nM) concentrations of BI‐2536. Neither concentration of BI‐2536 impaired kinetochore recruitment of Hec1 (Fig 2A and B), which is consistent with prior findings 35. To test for secondary removal, cells were challenged with MG‐132 to prevent anaphase entry along with BI‐2536 or microtubule poisons (nocodazole or paclitaxel) to generate misaligned chromosomes (Fig 2C). As expected, nocodazole‐ and paclitaxel‐challenged cells exhibited equal Hec1 kinetochore intensity in the majority (> 90%) of misaligned chromosome pairs (Fig 2D and E). Strikingly, misaligned chromosome pairs in the Plk1‐inhibited group exhibited unequal Hec1 distribution. The loss was restricted to the “pole‐distal” kinetochore, consistent with a model where the chromosome pair misaligns through pulling toward the retained kinetochore. These findings demonstrate that Plk1 mediates Hec1 maintenance at, but not recruitment to, the kinetochore.
Figure 2. Plk1 activity is not required for Hec1 kinetochore recruitment, but maintains it against microtubule pulling forces.
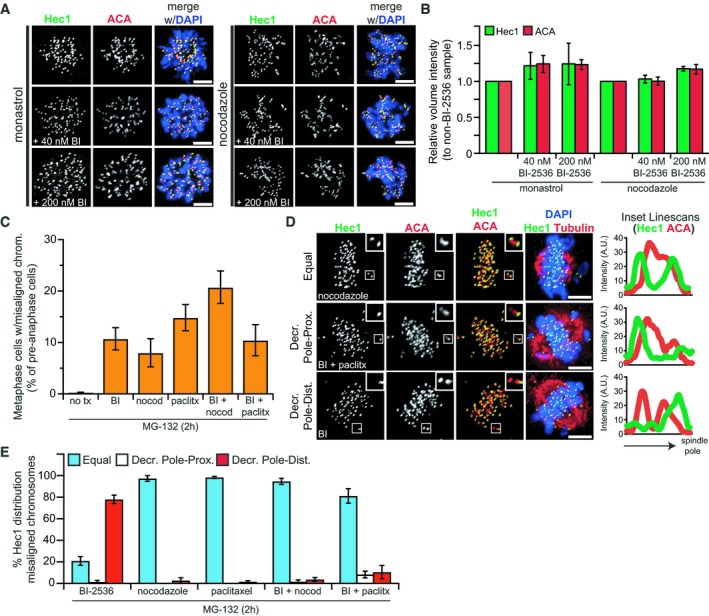
- Representative maximum‐intensity projection micrographs of Hec1 kinetochore localization in prometaphase cells arrested with monastrol (left) or nocodazole (right) with or without Plk1 inhibition (BI‐2536). Scale bars, 5 μm.
- Graph shows average relative volume intensity (± SEM) of total Hec1 and ACA from (A) (n = 10 cells/experiment; three independent experiments).
- Graph shows average percentage (± SEM) of metaphase cells with misaligned chromosomes after 2‐h Plk1 inhibition (BI‐2536) or nocodazole, paclitaxel, or combination challenge. MG‐132 used to prevent mitotic cells from entering anaphase (n = 100 cells/experiment; ≥ 4 independent experiments).
- Representative maximum‐intensity projection micrographs of cells from (C). Insets and linescans highlight 3 types of Hec1 intensity distribution observed between misaligned chromosome pairs. Tubulin indicates approximate position of spindle poles. Scale bars, 5 μm.
- Graph shows average percentage (± SEM) of misaligned chromosome pairs exhibiting each of the distribution types from (D) (n = 1–3 chromosomes/cell; 10 cells/experiment; ≥ 4 independent experiments). “Decreased” intensity indicates ≤ 50% intensity of sister kinetochore.
To determine when Plk1 activity is required to retain Hec1 at kinetochores, we performed time‐lapse imaging of Plk1as cells challenged with 3‐MB‐PP1 at distinct time points following release from an S‐phase thymidine block (Appendix Fig S1A). This demonstrated similar rates of lagging chromosomes whether inhibition was initiated at imaging onset, or for the entire 8.5 h prior to imaging. We conclude that Plk1 inhibition in late G2 and mitosis is sufficient to yield the Hec1 mislocalization.
Because the observed defects occur following initial chromosome alignment and are limited to one kinetochore of a sister pair, we hypothesized that impaired Plk1 activity weakens the kinetochore assembly on centromeric chromatin against spindle microtubule pulling forces, leading to stochastic rupture. Once a kinetochore ruptures, the tension is relieved across the kinetochore of the remaining sister chromosome, leading to poleward migration of the pair. Finally, the sister chromosome lacking a kinetochore would be predicted to fail to segregate in anaphase. To test this model, we employed low‐concentration nocodazole or paclitaxel to relieve tension. As expected, these chemicals can generate misaligned chromosomes individually or with BI‐2536 (Fig 2C). Notably, the misaligned chromosomes co‐challenged with either spindle poison or BI‐2536 retained Hec1 (Fig 2E), indicating that a reduction in microtubule pulling forces at kinetochores is sufficient to prevent kinetochore rupture. In a second experiment, we challenged cells with two distinct inhibitors of Mps1 to abrogate the spindle checkpoint. Inhibiting Mps1 markedly shortens the duration of mitosis, reducing the time for correct microtubule attachments and generation of tension across the kinetochore 36, 37, 38. Consistent with our hypothesis, the kinetochores of lagging chromosomes remained intact when both Plk1 and Mps1 were inhibited (Appendix Fig S1B–D). Taken together, these data support a model where microtubule attachment and tension drive kinetochore loss in the setting of reduced Plk1 activity.
Plk1 activity is required for integrity of the entire kinetochore
To further evaluate the kinetochore components affected by Plk1 inhibition, we probed for the scaffolding protein Knl1, which facilitates kinetochore recruitment of BubR1 and Mad1 (Fig EV2A and C). Like Hec1, Knl1 intensity is markedly reduced in lagging chromosomes (Fig EV2B) or in pole‐distal misaligned chromosomes prior to anaphase (Fig EV2D). Concordant with the loss of Knl1, we observed diminished BubR1 and Mad1 in pole‐distal misaligned chromosomes also lacking Hec1 (Fig EV2E–H). Next, we evaluated two CCAN components that directly link the kinetochore to centromeric chromatin: CENP‐C and CENP‐T (Fig EV2A) 39, 40, 41, 42. Consistent with our other findings with Plk1 inhibition, both CENP‐C and CENP‐T intensities were significantly reduced from kinetochores of lagging chromosomes (Fig 3A–D and Appendix Fig S2A) and disproportionately lost from pole‐distal kinetochores of misaligned chromosomes in metaphase cells (Appendix Fig S2B and C).
Figure EV2. Kinetochore disruption after Plk1 inhibition extends to Knl1, impairing checkpoint protein recruitment.
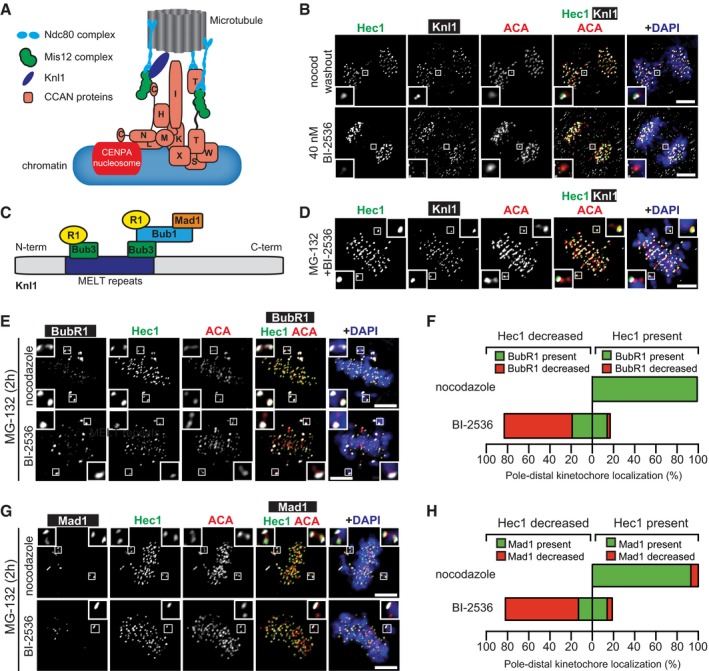
- Illustrative map of the kinetochore highlighting positions of KMN (Knl1, Mis12, Ndc80) and CCAN complexes relative to microtubules and centromeric chromatin.
- Representative maximum‐intensity projection micrographs of kinetochore protein localization in anaphase cells after Plk1 inhibition (BI‐2536) or nocodazole washout. Insets highlight lagging kinetochores, marked by ACA. Scale bars, 5 μm.
- Illustrative diagram of Knl1 indicating method of BubR1 and Mad1 recruitment.
- Representative maximum‐intensity projection micrographs of metaphase cell with misaligned chromosome pairs after 2‐h Plk1 inhibition (BI‐2536). Inset highlights decreased localization of Knl1 and Hec1 to the pole‐distal kinetochores, indicated by ACA. MG‐132 was used to prevent mitotic cells from entering anaphase. Scale bar, 5 μm.
- Representative maximum‐intensity projection micrographs of metaphase cells with misaligned chromosome pair after 2‐h Plk1 inhibition (BI‐2536) or nocodazole challenge. Insets highlight distribution of BubR1 and Hec1 at misaligned kinetochore pairs, indicated by ACA. MG‐132 was used to prevent mitotic cells from entering anaphase. Scale bars, 5 μm.
- Graphs show average percentage of misaligned pole‐distal kinetochores with localized (green) or decreased (red) BubR1 and Hec1 after 2‐h Plk1 inhibition (BI‐2536) or nocodazole challenge (n = 1–10 kinetochore pairs/cell; 10 cells/experiment; two independent experiments). “Decreased” intensity indicates ≤ 50% intensity of sister kinetochore.
- Representative maximum‐intensity projection micrographs of metaphase cells with misaligned chromosome pair after 2‐h Plk1 inhibition (BI‐2536) or nocodazole challenge. Insets highlight distribution of Mad1 and Hec1 at misaligned kinetochore pairs, indicated by ACA. MG‐132 was used to prevent mitotic cells from entering anaphase. Scale bars, 5 μm.
- Graphs show average percentage of misaligned pole‐distal kinetochores with localized (green) or decreased (red) Mad1 and Hec1 after 2‐h Plk1 inhibition (BI‐2536) or nocodazole challenge (n = 1–10 kinetochore pairs/cell; 10 cells/experiment; two independent experiments). “Decreased” intensity indicates ≤ 50% intensity of sister kinetochore.
Figure 3. The integrity defect extends throughout the kinetochore, including CENP‐A.
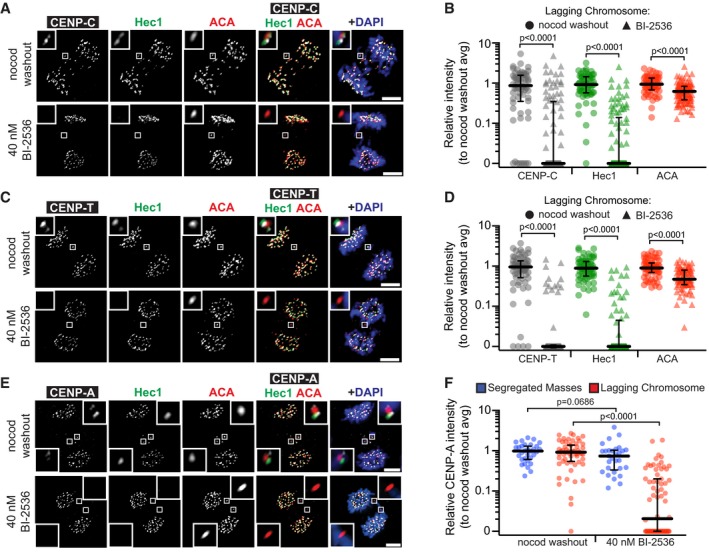
- Representative maximum‐intensity projection micrographs of kinetochore protein localization in anaphase cells after Plk1 inhibition (BI‐2536) or nocodazole washout. Insets highlight lagging kinetochores, marked by ACA. Scale bars, 5 μm.
- Graphs show relative volume intensities of CENP‐C, Hec1, and ACA at lagging chromosomes. Each circle represents a single kinetochore (n = 1–7 kinetochores/cell; 10 cells/experiment; three independent experiments). Bars indicate median kinetochore intensity and interquartile range. Significance determined by two‐tailed Mann–Whitney test.
- Representative maximum‐intensity projection micrographs of kinetochore protein localization in anaphase cells after Plk1 inhibition (BI‐2536) or nocodazole washout. Insets highlight lagging kinetochores, marked by ACA. Scale bars, 5 μm.
- Graph shows relative volume intensities of CENP‐T, Hec1, and ACA at lagging chromosomes. Each circle represents a single kinetochore (n = 1–7 kinetochores/cell; 10 cells/experiment; three independent experiments). Bars indicate median kinetochore intensity and interquartile range. Significance determined by two‐tailed Mann–Whitney test.
- Representative maximum‐intensity projection micrographs of kinetochore protein localization in anaphase cells after Plk1 inhibition (BI‐2536) or nocodazole washout. Insets highlight lagging kinetochores, marked by ACA. Scale bars, 5 μm.
- Graph shows relative volume intensity of CENP‐A at segregated (blue) and lagging kinetochores (red) from (E). Each blue circle represents all segregated kinetochores/cell (n = 10 cells/experiment; three independent experiments), whereas each red circle represents a single kinetochore (n = 1–6 kinetochores/cell; 10 cells/experiment; three independent experiments). Bars indicate median kinetochore intensity and interquartile range. Significance determined by two‐tailed Mann–Whitney test.
Finally, we probed for CENP‐A, the centromere‐specific histone variant that provides the epigenetic mark for the CCAN, and thereby the kinetochore assembly. Strikingly, CENP‐A intensity is significantly diminished in lagging chromosomes with Plk1 inhibition (Figs 3E and F, and EV3A) and, prior to anaphase, CENP‐A is disproportionately lost from the pole‐distal kinetochore of misaligned chromosomes (Fig EV3B and C). Intriguingly, we found only modest loss of ACA signal with Plk1 inhibition. ACA is known to recognize multiple centromere epitopes, including CENP‐B, which binds DNA independently of CENP‐A 43. Consistent with our ACA findings, CENP‐B removal after Plk1 inhibition is less severe than the other examined proteins (Fig EV3D and E). Taken together, these findings indicate that Plk1 activity is required to maintain integrity of the mitotic kinetochore and CCAN, but not for CENP‐B.
Figure EV3. Kinetochore disruption after Plk1 inhibition includes CENP‐A, but not CENP‐B (related to Fig 3).
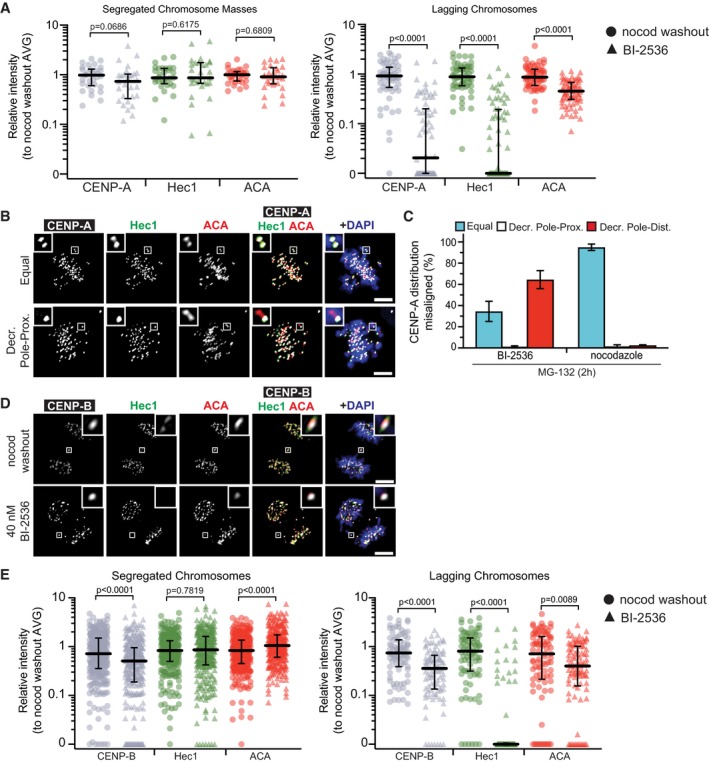
- Graph shows relative volume intensities of CENP‐A (gray), Hec1 (green), and ACA (red) at segregated (left) and lagging (right) kinetochores after Plk1 inhibition or nocodazole washout (Fig 3E and F). Note CENP‐A data are identical to data presented in Fig 3F. Each symbol in the left graph represents all segregated kinetochores/cell (n = 10 cells/experiment; three independent experiments), whereas each symbol in the right graph represents a single kinetochore (n = 1–6 kinetochores/cell; 10 cells/experiment; three independent experiments). Bars indicate median kinetochore intensity and interquartile range. Significance determined by two‐tailed Mann–Whitney test.
- Representative maximum‐intensity projection micrographs of metaphase cells with misaligned chromosomes. Insets highlight distribution of CENP‐A and Hec1 at misaligned kinetochore pair, indicated by ACA. Scale bars, 5 μm.
- Graph shows average percentage (± SEM) of misaligned chromosome pairs exhibiting each of the three distribution types after 2‐h Plk1 inhibition (BI‐2536) or nocodazole challenge (n = 1–8 chromosomes/cell; 10 cells/experiment; two independent experiments). “Decreased” intensity indicates ≤ 50% intensity of sister kinetochore.
- Representative maximum‐intensity micrographs of anaphase cells after Plk1 inhibition (BI‐2536) or nocodazole washout. Insets highlight the presence of CENP‐B and Hec1 at lagging kinetochores, marked by ACA. Scale bars, 5 μm.
- Graph shows relative volume intensities of CENP‐B (gray), Hec1 (green), and ACA (red) at segregated (left) and lagging (right) kinetochores from (B). Each symbol represents a single kinetochore (n = 10 segregated kinetochores/cell, 1–8 lagging kinetochores/cell; 10 cells/experiment; three independent experiments). Bars indicate median kinetochore intensity and interquartile range. Significance determined by two‐tailed Mann–Whitney test.
We next evaluated whether the observed effects altered CENP‐A turnover at the centromere or were specific for its nascent or ancestral pools. Centromere loading of nascent CENP‐A is largely restricted to early G1 6, and turnover of loaded CENP‐A is not known to occur in mitosis 44. Nevertheless, it is possible that Plk1 activity restrains the CENP‐A turnover during late G2 or mitosis. To test this idea, we performed fluorescence recovery after photobleaching (FRAP) on CENP‐A in mitotic cells with and without Plk1 inhibition (Fig EV4A). Consistent with earlier findings 44, there was no observed turnover of CENP‐A during mitosis, and this was unaffected by inhibiting Plk1. Second, we employed quench‐pulse‐chase CENP‐A‐SNAP labeling to differentiate nascent vs. ancestral CENP‐A 6 (Appendix Fig S3). As expected, there was no nascent loading of CENP‐A at the centromere during G2 and this was not altered with Plk1 inhibition, as previously shown 12. Finally, we considered that nascent CENP‐A, loaded during the prior G1, may be more readily extracted from centromeric chromatin than the ancestral pool. To evaluate this, we generated cell lines expressing CENP‐A‐SNAP to differentially label nascent and ancestral CENP‐A nucleosomes (Fig EV4B). We found that both ancestral and nascent CENP‐A pools are extracted with Plk1 inhibition (Fig EV4C and D). We conclude that Plk1 signaling does not modulate CENP‐A loading and turnover, nor is it required for specific stabilization of nascent or ancestral CENP‐A.
Figure EV4. CENP‐A loss is not the result of dysregulated turnover; both ancestral and nascent CENP‐A pools are equally affected.
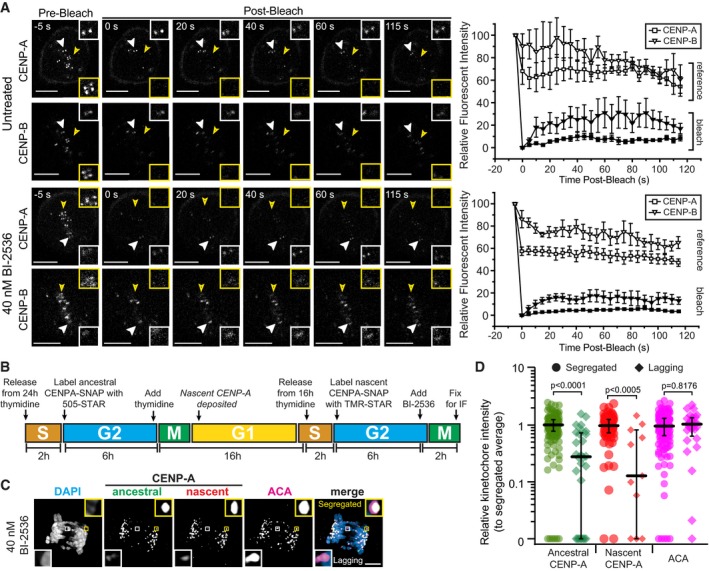
- Representative single‐plane images of untreated and 40 nM BI‐2536‐treated RPE1 cell in mitosis before and after photobleaching CENP‐A and CENPB. The recovery of the CENP‐A and CENPB loci was recorded after photobleaching. White arrowheads mark the kinetochore(s) which was not photobleached (reference), and the yellow arrowhead marks the photobleached kinetochore. The inset on the top right on each image highlights the reference kinetochore(s), and the inset on the bottom right highlights the photobleached kinetochore. Average intensities (± SD) of the CENP‐A and CENPB loci are plotted as a function of time on the right for both untreated (n = 3 kinetochores, two independent experiments) and 40 nM BI‐2536‐treated (n = 10 kinetochores, two independent experiments) conditions. Scale bars, 5 μm.
- Illustrative strategy to differentially label ancestral and nascent CENP‐A pools in CENPA‐SNAP‐expressing RPE1 cells and then determine localization at anaphase kinetochores after Plk1 inhibition (BI‐2536).
- Representative maximum‐intensity projection micrographs of anaphase cell as described in (A). Insets highlight intensities of ancestral (green) and nascent (red) CENP‐A at segregated (yellow box) and lagging (white box) kinetochores, indicated by ACA. Scale bar, 5 μm.
- Graph shows relative volume intensities of ancestral and nascent CENP‐A at segregated (circles) and lagging (diamonds) kinetochores from (A, B). Each symbol represents a single kinetochore (n = 20 segregated kinetochores/cell, 1–8 lagging kinetochores/cell; eight cells from two independent experiments). Bars indicate median kinetochore intensity and interquartile range. Significance determined by two‐tailed Mann–Whitney test.
Kinetochore disruption occurs after chromosome alignment and is mediated in part by PICH
To understand how loss of these kinetochore proteins occurs, we turned to live‐cell imaging of RPE1 cells constitutively expressing YFP‐CENP‐A and mCh‐CENP‐B. Using spinning‐disk confocal microscopy, we monitored CENP‐A and CENP‐B in 250‐nm optical sections of mitotic cells with/without Plk1 inhibition. Consistent with fixed cell analyses, YFP‐CENP‐A was lost while mCh‐CENPB was retained, occurring either at anaphase onset (orange arrowhead/inset, Fig 4A, 0 min, Movie EV4) or after metaphase alignment (orange inset, Fig 4B, 3 min, Movie EV5). These data are consistent with a model where Plk1 inhibition stochastically disrupts kinetochore integrity in early mitosis, yet its effects are not observed until metaphase or anaphase onset (when tension across sister kinetochores is highest), leading to two outcomes: loss of alignment or lagging chromosomes. We are confident that variation in CENP‐A is not due to position in the z‐plane with the 250‐nm optical sections. First, CENP‐A intensity is uniformly equal across sister pairs in early mitosis (Appendix Fig S4A). Second, CENP‐B levels remain uniform before–after CENP‐A loss, as further evidenced by quantitation and single planes (Fig 4A and B right, Appendix Fig S5). Thus, live‐cell imaging confirms disruption of CENP‐A at the inner centromere upon inhibiting Plk1.
Figure 4. CENP‐A loss occurs after midline chromosome alignment.
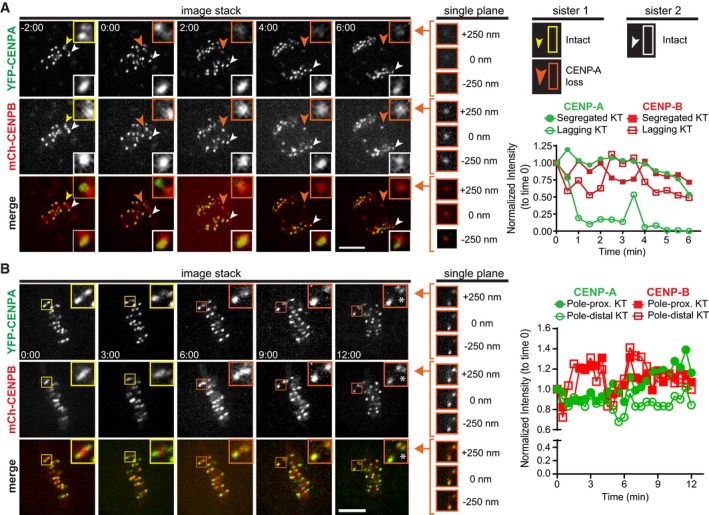
- Maximum‐intensity projection frames from live‐cell fluorescence microscopy of RPE1 cells challenged with 40 nM BI‐2536 to generate lagging chromosomes. White arrowhead follows kinetochore retaining CENP‐A, and yellow/orange arrowheads follow kinetochore losing CENP‐A (yellow—prior to loss; orange—after loss). Insets highlight the kinetochore pair with normalized intensities plotted over time on graph to right. Single‐plane insets of lagging kinetochore at last time point. 0 nm indicates plane with highest CENP‐B intensity. Time, in min:s after anaphase onset. Scale bar, 5 μm.
- Maximum‐intensity projection frames from live‐cell fluorescence microscopy of RPE1 cells challenged with 40 nM BI‐2536 to generate misaligned chromosomes. Insets highlight kinetochore pair with pole‐distal sister losing CENP‐A over time (yellow box—prior to loss; orange box—after loss). Normalized intensities of the pole‐proximal and pole‐distal kinetochores are plotted over time on graph to right. Asterisk marks a kinetochore from an adjacent plane. Single‐plane insets of last time point. 0 nm indicates plane with highest CENP‐B intensity. Time, in min:s, at metaphase onset. Scale bar, 5 μm.
Some chromosomes retain CENP‐A, kinetochore components and maintain metaphase alignment. A likely explanation is a stochastic process of tension‐mediated kinetochore disruption. However, it is possible that the effects are systematic for specific chromosomes at risk. Indeed, CENP‐A levels vary on centromeres of distinct chromosomes 45. We tested if intrinsic factors, such as initial CENP‐A levels, chromosome size, or centromere position, increased the risk of kinetochore disruption. Chromosome pairs with low or moderate initial levels of CENP‐A did not appear to have a higher risk of disruption, nor did asymmetry in initial CENP‐A among chromatid pairs explain which kinetochore was disrupted (Appendix Fig S4A). We did observe a trend toward larger chromosomes more likely to be affected (Appendix Fig S4B). Conversely, acrocentric chromosomes were less likely to experience disruption. These findings are concordant with recent findings 46. Thus, chromosome size and centromere position, but not initial CENP‐A levels, may drive the stochastic nature of kinetochore disruption.
We next considered that stochastic removal of the kinetochore and CENP‐A from the chromosome could result from impaired functions of condensin II or Aurora B. Condensin II operates primarily in the region of centromeric chromatin and is regulated by Plk1 47, 48, 49, 50. However, loss of condensin II is known to increase inter‐kinetochore distance, whereas we observed decreased distance with Plk1 inhibition (Appendix Fig S6A). Additionally, depletion of condensin II is reported to generate microtubule attachment errors with retained kinetochores in human cells 47, 51, both inconsistent with Plk1 loss. Finally, impaired Aurora B kinase activity is associated with condensin depletion 47 and impaired localization of outer kinetochore components 52, 53. However, lagging and misaligned chromosomes generated by the Aurora B inhibitor, ZM447439, retained CENP‐A (Appendix Fig S6B–F). We conclude that the effects of Plk1 inhibition are distinct from those observed by condensin depletion and Aurora B inhibition.
One time‐lapse videomicroscopy movie demonstrated an elastic stretching of CENP‐A and a lengthening gap from CENP‐B, followed by gap closure, with a restored difference (Appendix Fig S7A and Movie EV6). Others showed only partial loss of CENP‐A, sometimes with two apparent foci. These observations suggested a partial unwinding of centromeric chromatin, or ultra‐fine bridge (UFB) of chromatin/DNA linking the residual kinetochore to the extracted CENP‐A and kinetochore. Chromosome spread analysis identified elongated or fragmented CENP‐C signals at an average of 15% (up to 50%) of Plk1‐inhibited kinetochores, compared to 0% in controls (Fig 5A and B), confirming our live‐cell imaging observations. Importantly, CENP‐C signal disruption coincided with similarly stretched or fragmented centromere FISH signals (Fig 5C and D), indicating involvement of centromeric chromatin.
Figure 5. PICH promotes kinetochore and centromeric DNA fragmentation following Plk1 inhibition.
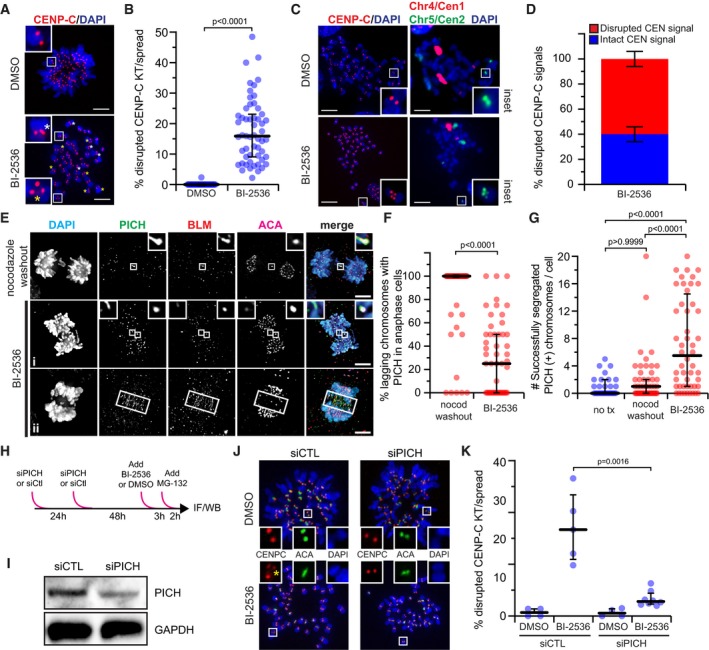
- Representative maximum‐intensity projection micrographs of metaphase spreads after Plk1 inhibition (25 nM BI‐2536) or control (DMSO). Insets highlight kinetochore pairs that are intact (top), elongated (white asterisk), or fragmented (yellow asterisk). Scale bar, 5 μm.
- Graph shows percentage of kinetochore pairs/spread exhibiting disrupted CENP‐C from (A). Each circle represents a spread (n ≥ 30 spreads from five independent experiments). Bars indicate median kinetochore intensity and interquartile range. Significance determined by two‐tailed Mann–Whitney test.
- Representative maximum‐intensity projection micrographs of metaphase spreads after Plk1 inhibition (25 nM BI‐2536) or control (DMSO). Insets highlight integrity of the kinetochore (CENP‐C) and underlying centromeric chromatin (Chromosome 5, Cen2). Note multiple centromere probes used for analysis. Here, centromere probe (Cen1) of Chromosome 4 is indicated in red. Scale bar, 5 μm.
- Graph shows frequency of intact (blue) or disrupted (red) CEN signals associated with disrupted CENP‐C from (C). Bars represent the average percentage (± SEM) of a minimum of 200 centromeres from two independent experiments. Impaired CENP‐C was rarely observed in DMSO control, hence its exclusion from analysis.
- Maximum‐intensity projection micrographs of anaphase cells after nocodazole washout or Plk1 inhibition with 40 nM BI‐2536. Insets highlight localization of PICH and BLM to lagging kinetochores. Elongated, non‐chromatin‐associated midzone signals (ii) were rarely observed and excluded from analysis. Scale bars, 5 μm.
- Graph shows percentage of cells exhibiting lagging chromosomes with PICH(+) kinetochore signals in (E). Each circle represents a cell (n = 50 cells from four independent experiments; average laggards/cell: two nocodazole washout, four BI‐2536). Bars indicate median percentage and interquartile range. Significance determined by two‐tailed Mann–Whitney test.
- Graph shows number of segregated chromosomes with PICH(+) kinetochore signals observed per cell in (E). Each circle represents a cell (n = 30 cells from two independent experiments for no treatment; 50 cells from four independent experiments for nocodazole washout and BI‐2536). Bars indicate median number and interquartile range. Significance determined by Kruskal–Wallis test with Dunn's correction for multiple comparisons.
- Illustrative schematic of the PICH knockdown strategy.
- Immunoblot of protein extracts from RPE1 cells 72 h after PICH (25 nM) or control knockdown. Membranes probed for PICH or GAPDH (loading control).
- Representative maximum‐intensity micrographs of metaphase spreads after control or PICH knockdown with (25 nM BI‐2536) or without (DMSO) Plk1 inhibition. Insets highlight integrity of a kinetochore pair. Yellow asterisk denotes fragmented CENP‐C.
- Graph shows frequency of kinetochore pairs/spread exhibiting disrupted CENP‐C from (J) in the indicated conditions (siPICH 25 and 40 nM). Each circle represents the average frequency observed within a given experiment (n ≥ 30 spreads from 4 to 5 independent experiments). Bars indicate median kinetochore intensity and interquartile range. Significance determined by two‐tailed Mann–Whitney test.
To evaluate for chromatin unwinding, we probed for PICH, a DNA helicase that coats UFBs 54. In untreated metaphase cells, PICH localized to the centromeric region of a small number of chromosomes (Appendix Fig S7B and C), consistent with earlier reports 54, 55. In nocodazole‐challenged cells, PICH localization was more notable at misaligned chromosomes. Strikingly, Plk1 inhibition markedly increased the number of chromosomes with centromeric PICH, particularly in the aligned chromosomes (Appendix Fig S7B and C). During anaphase, PICH localized uniformly to lagging chromosomes generated by nocodazole washout, consistent with earlier findings that PICH DNA binding increases with tension‐induced DNA stretching 56. In contrast, PICH was frequently undetectable (~ 80%) from lagging chromosomes generated by Plk1 inhibition (Fig 5E and F). Moreover, we observed PICH kinetochore localization in segregated chromosomes more frequently with Plk1 inhibition than with nocodazole washout or no treatment (Fig 5G). We rarely observed PICH(+) decorated UFBs (Fig 5E,ii), consistent with our earlier findings 29. We conclude that Plk1 activity prevents access of the PICH helicase to centromeric chromatin.
To evaluate the role of PICH in the previously described kinetochore defect, we used siRNA depletion of PICH in the setting of Plk1 inhibition (Fig 5H–K). Importantly, we found that PICH knockdown largely reversed disrupted CENP‐C signals from Plk1 inhibition in chromosome spreads (Fig 5J and K). Thus, Plk1 activity protects kinetochore and centromere integrity, in part, by preventing PICH accumulation and/or activity at centromeric DNA.
Plk1 signals at chromatin restore chromatin integrity, and mis‐segregated chromosomes fail to re‐establish centromeric CENP‐A
Anaphase defects from Plk1 inhibition are partly rescued by restoring its activity at chromatin or the inner centromere, but not at the outer kinetochore 28. Because loss of inner kinetochore proteins is a salient feature of the anaphase segregation defect (Figs 3 and EV2), we tested if localizing Plk1 activity to the inner kinetochore is sufficient to restore the defect. To this end, we generated cell lines that stably express both a Plk1as allele and a Plk1 chimera, where wild‐type Plk1 kinase domain is fused to the N‐terminus of CENP‐A, CENP‐B, H2B (chromatin localized), or INCENP (inner centromere localized) (Fig 6A and Appendix Fig S8), similar to that reported previously 28. Surprisingly, tethering Plk1 activity to either CENP‐A or CENP‐B failed to restore anaphase chromosome segregation following treatment with BI‐2536 (Figs 6A and EV5A), although CENP‐B‐tethered Plk1 trended toward restoring chromosome segregation. Consistent with our previous experience, tethering Plk1 to chromatin (H2B) or the inner centromere (INCENP) partially restored chromosome segregation (Figs 6A and EV5A). Curiously, we do not observe increased PICH localization to segregated chromosomes after inhibition of Plk1as with 3‐MB‐PP1, which is inconsistent with findings with BI‐2536 (Fig 5G), possibly due to subtle differences in residual Plk1 activity in the two systems. Nevertheless, centromere‐localized Plk1 significantly enhances PICH recruitment (Fig 6C). Improved segregation fidelity suggests, although does not prove, that centromeric Plk1 activity is important for kinetochore integrity. However, the effect of PICH localization during late anaphase remains inconclusive.
Figure 6. Centromere‐localized Plk1 activity promotes kinetochore integrity, which, when lost, is not restored in subsequent G1.
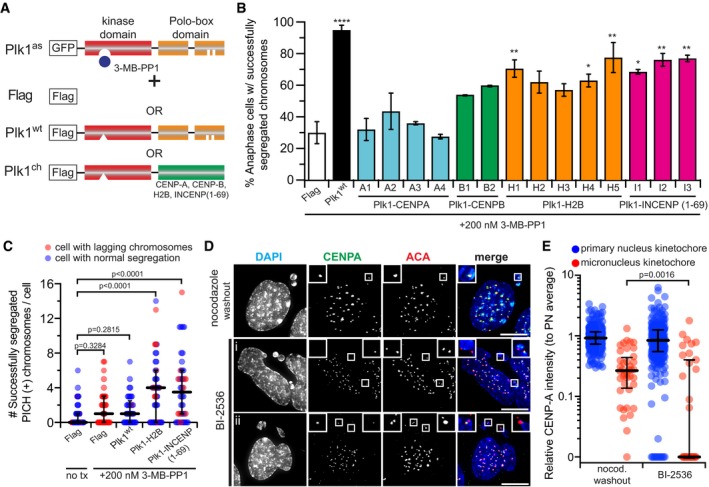
- Illustrative schematic. EGFP‐Plk1as RPE1 cells were transduced with an empty vector (Flag), wild‐type Plk1 (Plk1wt), or wild‐type Plk1 kinase domain tethered to CENP‐A, CENP‐B, H2B, or INCENP(aa 1–69). Cells were challenged with 200 nM 3‐MB‐PP1, which inhibits Plk1as, but not Plk1wt or tethered constructs.
- Graph shows average percentage (± SEM) of anaphase cells with fully segregated chromosomes for each cell line (clones identified alphanumerically) (n = 50 cells/experiment; two independent experiments). Significance of cell lines against Flag control determined by one‐way ANOVA with Holm–Sidak's multiple comparisons test (*P < 0.05, **P < 0.01, ****P < 0.0001).
- Graph shows number of segregated chromosomes with PICH(+) kinetochore signals observed per cell. Each circle represents a cell (n = 45 cells from three independent experiments). Bars indicate median number and interquartile range. Significance determined by Kruskal–Wallis test with Dunn's correction for multiple comparisons.
- Representative maximum‐intensity projection micrographs of interphase cells with micronuclei after BI‐2536 challenge or nocodazole washout. Insets highlight contents of micronuclei. Scale bars, 10 μm.
- Graph shows relative volume intensity of CENP‐A at kinetochores in the primary nucleus (blue) or micronuclei (red) from (E) (n = 368 primary/40 micronuclei kinetochores, nocodazole washout; 308 primary/35 micronuclei kinetochores, BI‐2536; three independent experiments). Bars indicate median intensity and interquartile range. Significance determined by two‐tailed Mann–Whitney test.
Figure EV5. Centromere‐localized Plk1 activity promotes kinetochore integrity, which, when lost, is not restored in subsequent G1 (related to Fig 6).
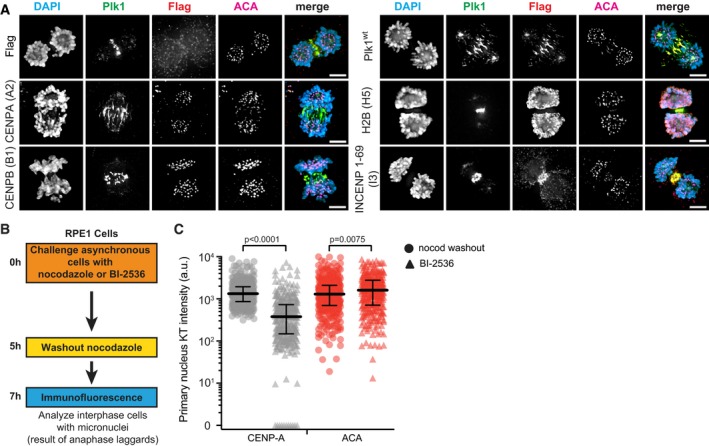
- Representative maximum‐intensity micrographs of anaphase cells indicating localization of the Flag tag, Flag‐Plk1wt, or Flag‐tagged chromatin‐tethered Plk1 constructs. Parentheses indicate clone number. Scale bars, 5 μm.
- Illustrative strategy to analyze the presence of CENP‐A in micronuclei after mitosis compromised by Plk1 inhibition (BI‐2536) or nocodazole washout.
- Graph shows raw kinetochore protein intensities in the primary nuclei of G1 cells following anaphase exposure to nocodazole washout (circle) or 40 nM BI‐2536 (triangle). Bars indicate median protein intensity and interquartile range. Significance determined by two‐tailed Mann–Whitney test.
To evaluate the fate of chromosomes after kinetochore disruption, we challenged cells with BI‐2536 and compared with nocodazole washout control (Fig EV5B). As expected, micronuclei are generated in both experiments, consistent with the finding of lagging chromosomes (Fig 6D). We also find the CENP‐A, but not ACA, signals to be reduced in the primary nucleus of Plk1‐inhibited cells (Fig EV5C), consistent with the role of Plk1 in CENP‐A loading during G1 12. Notably, we observe CENP‐A intensity reduction in micronuclei generated by either Plk1 inhibition or nocodazole washout (Fig 6E), consistent with earlier observations that centromere protein recruitment is impaired in micronuclei 57. Importantly, micronucleus CENP‐A localization after Plk1 inhibition is significantly more impaired compared to nocodazole washout, suggesting the defect is not exclusively generated by impaired nuclear import. We conclude that subtle loss of Plk1 activity at chromatin during mitosis yields centromeres that fail to re‐establish CENP‐A.
Discussion
Herein, we discover that Plk1 activity at chromatin, but not at kinetochores, is crucial to maintain integrity of the entire kinetochore during mitosis. These findings are consistent with our previous report that Plk1 signals at centromeric chromatin maintain genomic integrity 28 and a recent observation that its signals maintain structural integrity of centromeric chromatin 46. As a whole, our data support a model (Fig 7) where Plk1 signals at chromatin secure the integrity of the kinetochore within centromeric chromatin against microtubule‐induced tension at kinetochores. When this activity is lost, centromeric chromatin becomes disrupted, stochastically recruiting PICH. Affected chromosomes align on the metaphase plate, followed by transient elastic stretching (as seen in Appendix Fig S7A and Movie EV6) or a more severe “kinetochore rupture”. The disrupted kinetochore lacks all surveyed components including CENP‐C, CENP‐T, and CENP‐A. However, CENP‐B remains on centromeric chromatin, indicating tight binding to DNA that even chromatin unwinding, sufficient to unwrap histones (as CENP‐A), is not able to remove. Once a kinetochore is ruptured, the chromosome pair migrates toward the pole with a retained kinetochore, as the force across the chromosomes is reduced. We were unable to visualize these ruptured kinetochores pulled toward spindle poles, suggesting that they are rapidly disassembled. These chromatids lacking kinetochores fail to recruit mitotic checkpoint components, permitting progression to anaphase. The result is daughter cells with micronuclei enclosing chromosomes that lack CENP‐A.
Figure 7. Model of kinetochore–centromere dysfunction with loss of Plk1 activity.
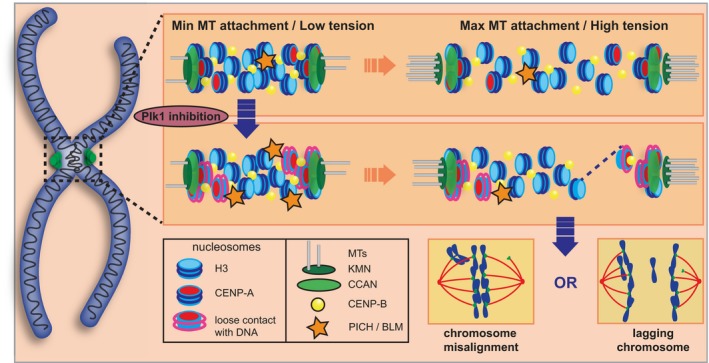
In an unperturbed mitosis, centromeric chromatin is initially compact with DNA tightly wrapped around nucleosomes (Min MT attachment/Low tension). With increasing MT attachment and tension, the chromatin stretches evenly, and DNA remains tightly wrapped around nucleosomes. When Plk1 activity is compromised, PICH recruitment to centromeric chromatin increases and DNA contact around nucleosomes is relaxed (Min MT attachment/Low tension). With increasing MT attachment and tension, stochastic chromatin disruption occurs with DNA stretching (dashed line) or complete rupture of the kinetochore and surface CENP‐A nucleosomes. This loss of kinetochore/centromere stability results in either chromosome misalignment during metaphase or chromosome lagging during anaphase.
This model is supported by the findings: (i) Outer and inner kinetochore proteins are equally affected by the rupture event, (ii) centromeric chromatin disruption frequently coincides with kinetochore disruption, (iii) segregation fidelity is largely restored when Plk1 is tethered to H2B or INCENP, and (iv) the double‐stranded DNA binding helicase, PICH, is recruited to kinetochores in metaphase, and its depletion largely reverses kinetochore disruption in pre‐anaphase cells. Thus, Plk1 signaling at the centromere reinforces centromeric chromatin, anchoring CENP‐A against tension. When this signaling is lost, tension can physically extract CENP‐A in a stochastic manner. Consistent with a tension model, we find that reducing spindle microtubule tension abrogates the defect. Moreover, the defect occurs only after chromosome alignment, when microtubule attachments are most numerous 58. As expected, only one kinetochore of a sister pair is affected, since the inter‐kinetochore tension is relieved after disruption. Taken together, these data support a novel tension‐dependent mechanism of chromosome instability that is not restrained by the mitotic checkpoint.
Our results are inconsistent with other explanations for chromosomes lacking critical kinetochore proteins. The observed defects are not the result of errors in protein loading (Fig 2A and B), nor the result of aberrant protein turnover (Fig EV4A). Although Plk1 is known to regulate condensin II 49, these findings are also inconsistent with condensin II dysfunction as chromosomes condense normally (Fig 1B and C, Movies EV2 and EV3) and inter‐kinetochore distance is not lengthened (Appendix Fig S6A). Furthermore, the shortened inter‐kinetochore distances and relative preservation of ACA/CENP‐B signal suggest our observations are not artificial signal loss from diluted protein localization on hyperstretched chromatin. Finally, the specific loss of the kinetochore proteins from the pole‐distal kinetochore of misaligned chromosomes when cells are held in mitosis for equal periods of time argues against differences in cell division timing indirectly influencing kinetochore protein levels.
Plk1 activity at the centromere occurs, in part, via PICH. PICH depletion suppresses centromere disruption in pre‐anaphase cells (Fig 5J and K), and similar findings have been reported recently 46. We also observe PICH kinetochore recruitment is increased during metaphase in RPE1 cells inhibited by BI‐2536 (Appendix Fig S7B and C); however, this effect is not always recapitulated in anaphase segregated chromosomes after modest Plk1 inhibition (Fig 6C). Localizing Plk1 activity to the centromere significantly reverses segregation defects and increases the frequency of PICH(+) segregating chromosomes (Fig 6A–C). In light of these findings, we speculate that PICH involvement is indirect. Plk1 does not regulate PICH ATPase activity 59, and other proteins, such as BLM and RPA, are co‐recruited to DNA strands with PICH 46. Notably, the recently reported centromere disintegration associated with Plk1 inhibition implicated BLM as the main effector of Plk1 activity 46. We further speculate that Plk1 maintenance of kinetochore and centromere integrity includes other substrates in addition to PICH and its binding partners. This is based on our observation that PICH rarely localizes at kinetochores of lagging chromosomes after Plk1 inhibition (Fig 5E and F). Additionally, phosphoproteomic screens have identified numerous centromere‐localized proteins as putative Plk1 substrates 28, 60, 61, 62. Thus, Plk1 protection of kinetochore and centromere integrity likely exists as a spectrum, involving discrete locales and substrates, of which PICH is one intermediary.
Mitotic loss of CENP‐A is surprising for several reasons. First, little to no CENP‐A turnover is observed after incorporation into the nucleosome 6, 44, 63, 64, although recent evidence suggests that CENP‐A can undergo histone exchange in non‐dividing cells [preprint: 65]. Second, kinetochore loading of CENP‐A is restricted to G1 6, 44, 63. Importantly, we find loss of both ancestral and nascent CENP‐A pools is not biased to centromeres with diminished initial CENP‐A levels. Although Plk1 phosphorylation of the Mis18 complex during G1 is required for nascent CENP‐A loading at the centromere 12, effects of Plk1 inhibition on centromeric CENP‐A retention during G2/M have not been previously observed. Here, mitotic CENP‐A loss results in centromeres lacking their epigenetic mark.
One puzzling aspect of the phenotype reported here is that only a fraction of chromosomes are affected in each cell. The stochastic nature of kinetochore disruption is explained by residual Plk1 activity and by timing of the mitotic checkpoint. The partial loss of its activity used in experiments here allows for some centromeric phosphorylation, near the on/off threshold, which could stochastically unwind chromatin of different kinetochores to a variable extent. In a representative time‐lapse video (Fig 1B), loss of chromosome alignment occurs sequentially for chromosome pairs until anaphase onset. Moreover, partial disruption of the kinetochore and centromere may occur, as evidenced by split or elongated CENP‐C and FISH signals (Fig 6A and C). Taken as a whole, the incompletely penetrant phenotype is consistent with partial loss of Plk1 function and may depend on stochastics, on different tension on centromeres, or even on structure of the centromeres.
A tantalizing possibility is that intrinsic features of each chromosome predispose it to greater sensitivity or resistance to mitotic perturbations. For example, chromosomes with longer alpha‐satellite arrays could recruit more centromere proteins, thereby creating a greater number of microtubule binding sites that could make these chromosomes more resistant to perturbation. Evidence of expanded satellite repeats recruiting more centromere and kinetochore proteins has been demonstrated previously in mouse oocytes 66, 67. Our initial experiments identify a trend toward chromosome size, but not initial CENP‐A levels, influencing kinetochore disruption. However, more comprehensive experiments are necessary to determine how intrinsic chromosome qualities influence likelihood of chromosome disruption.
In conclusion, we have discovered that kinetochore anchoring into centromeric chromatin is stabilized against tension through signals generated by Plk1. This stabilization prevents unwinding of double‐stranded DNA and complete disruption of the kinetochore, CCAN, and CENP‐A nucleosomes. These effects are mediated, in part, by PICH, as its knockdown rescues the effect of BI‐2536. The findings reported here are surprising because centromere‐specific mechanisms, beyond condensation, have not previously been known to operate to stabilize against the spindle force, despite the well‐focused pulling force on this chromatin fragment. Consistent with our finding, tethering experiments suggest that this effect requires Plk1 activity at chromatin. We conclude that G2 or mitotic stabilization of centromeric chromatin against tension is crucial to govern genomic integrity. It will be important to identify the particular substrates and interactors of Plk1 that are required to stabilize the centromere against the pulling force of the mitotic spindle.
Materials and Methods
Cell line derivation and culture procedures
Cell culture
All cell lines were maintained at 37°C and 5% CO2 in a humidified incubator and propagated in the following media supplemented with 10% fetal bovine serum and 100 units/ml penicillin–streptomycin: Phoenix retroviral packaging line, Dulbecco's modified Eagle's medium (DMEM) supplemented with 4.0 mM l‐glutamine and 4,500 mg/l glucose; and hTERT‐RPE1‐derived cell lines, 1:1 mixture of DMEM and Ham's F‐12 media supplemented with 2.5 mM l‐glutamine. RPE1 cells were purchased from ATCC. EGFP‐Plk1as and EGFP‐Plk1as/Flag‐Plk1ΔC‐H2B RPE1 cell lines were derived as previously reported 28, 34. All cell lines were tested for mycoplasma contamination with the MycoAlert Mycoplasma Detection Kit (Lonza).
Plasmid construction
Human CENP‐B (clone 6470289) was purchased from Invitrogen. Human INCENP (EHS1001‐97551817) was purchased from Open Biosystems. Human YFP‐CENPA was a kind gift from Daniel Foltz. The pSNAPf vector was purchased from New England Biolabs. Retroviral plasmid backbones (pQCXIX, pQCXIN, pQCXIP) were purchased from Clontech. Unless otherwise stated, all constructs were created using standard restriction digest and ligation procedures. After insertion, PCR‐amplified DNA was fully sequenced to verify integrity. See Appendix Table S1 for primer list.
For YFP‐CENPA/mCh‐CENPB(1–158) RPE1 cells, YFP‐CENPA was PCR‐amplified and inserted into the pQCXIX backbone. CENPB amino acids 1–158 were PCR‐amplified and inserted C‐terminal to mCherry in the pQCXIN backbone. For CENPA‐SNAP‐3xFlag RPE1 cells, the pQCXIN backbone was linearized with NotI and BamHI digest, the SNAP‐tag and CENP‐A were PCR‐amplified, a 3x‐Flag G‐block was purchased from IDT, and all four components were assembled by Gibson Assembly. For Plk1 chimera RPE1 cells, full‐length CENP‐A and CENP‐B were PCR‐amplified and inserted C‐terminal to Flag‐tagged wild‐type Plk1 kinase domain (Flag‐Plk1ΔC) in the pQCXIN backbone as previously reported 28. INCENP amino acids 1–69 68 were PCR‐amplified and inserted C‐terminal to Flag‐tagged wild‐type Plk1 kinase domain (Flag‐Plk1ΔC) in the pQCXIN backbone via standard restriction digest and ligation procedures.
Retroviral transgenesis
For stable retroviral transduction, constructs were co‐transfected with a VSV‐G envelope plasmid into Phoenix cells. Fresh medium was applied 24 h post‐transfection. A further 24 h later, cells were clarified by centrifugation and filtration through a 0.45‐μm membrane to remove cell debris and diluted 1:1 with complete medium containing 10 μg/ml polybrene (Millipore). Target cells (RPE1 or EGFP‐Plk1as RPE1) were infected at 40–60% confluence for 24 h and then selected with 0.4 mg/ml G418 for 10–14 days. Polyclonal transductants were further purified by limiting dilution to obtain individual clones.
siRNA transfection
siRNA against PICH (AM16708; Ambion) or a control siRNA (D‐001810‐10‐05 ON‐TARGETplus; Dharmacon) was introduced using Lipofectamine RNAiMAX (Invitrogen) following the manufacturer's instructions. Cells grown at low confluency on a glass coverslip were transfected with 25 or 40 nM of siRNA twice in a 24‐h interval. Cells were fixed 72 h after the first transfection.
Chemicals
For chemicals, manufacturers, catalog numbers, and working concentrations, please refer to Appendix Table S3. Of note, two separate lots of BI‐2536 were used in this study with similar phenotypes observed at 40 nM in lot #1 and 25 nM in lot #2. Lot #1 was used for the majority of the experiments, with lot #2 for some experiments in Fig 5. Concentrations are included in the figure legends.
Immunoblotting
For siRNA experiments, cells were pelleted and lysed in buffer (4% SDS, 20% glycerol, 100 mM Tris–HCl pH 7.6). Proteins were quantified using Pierce™ BCA Protein Assay Kit (Thermo Scientific™) and were separated by SDS–PAGE, transferred to nitrocellulose membrane and blocked for 30 min in 5% milk and 0.1% Tween‐20/Tris‐buffered saline pH 7.4 (TBST + milk). Membranes were incubated with gentle agitation overnight at 4°C with primary antibodies (Appendix Table S2) diluted in TBST + milk, washed 3× with TBST, and incubated for 45 min at room temperature in secondary antibodies conjugated to horse radish peroxidase diluted 1:5,000 in TBST + milk.
Membranes were washed and developed with luminol/peroxide (Millipore) and visualized with a ChemiDoc MP imaging system, controlled by Image Lab 4.1 (Bio‐Rad). All results were obtained from single gels. To simultaneously probe for the protein of interest and the loading marker, the membrane was divided in two after transfer and incubated in separate antibody solutions.
Immunofluorescence microscopy
General procedures
Cells were seeded on #1.5 glass coverslips (Fisherbrand) at low density in 24‐well plates and, with the exception of SNAP‐labeling experiments, allowed to grow until 80–90% confluence prior to chemical challenge.
For anaphase segregation experiments, cells were challenged for 6 h with 40 nM BI‐2536 prior to fixation. For the nocodazole washout control, cells were challenged for 5 h with 0.2 μg/ml nocodazole, followed by removal, a single rinse with HBSS, and replenishment with fresh media for 45 min prior to fixation. For metaphase misalignment experiments, cells were challenged for 2 h with 10 μM MG‐132 to prevent anaphase onset and 40 nM BI‐2536, 40 ng/ml nocodazole, or 20 nM paclitaxel to generate misaligned chromosomes. For anaphase segregation restoration experiments with Plk1 chimeras, cells were challenged for 6 h with 200 nM 3‐MB‐PP1, which generates lagging chromosomes in EGFP‐Plk1as RPE1 cells 28. For experiments examining Hec1 recruitment to kinetochores, cells were challenged overnight with 100 μM monastrol or 0.2 μg/ml nocodazole + 0 nM, 40 nM, or 200 nM BI‐2536. For experiments examining forced mitotic exit, asynchronously growing cells were challenged for 30 min with 200 nM BI‐2536 alone or in combination with 2 μM AZ‐3146 or 500 nM reversine.
With the exception of chromosome misalignment experiments, coverslips were initially incubated for 15 s at room temperature (RT) in PHEM buffer (60 mM PIPES, 25 mM HEPES, 10 mM EGTA, 2 mM magnesium chloride) with 10% NP‐40 to pre‐extract cytoplasmic proteins. Otherwise, coverslips were fixed in 4% paraformaldehyde in PHEM buffer for 10 min at RT, washed three times in PBS, and then blocked for 30 min at RT in 3% bovine serum albumin (BSA) and 0.1% Triton X‐100 in PBS (PBSTx + BSA). Primary antibodies (Appendix Table S3) were pooled and diluted in PBSTx + BSA. Coverslips were incubated in primary antibodies for 1 h at RT and washed three times in PBSTx. Alexa Fluor (Molecular Probes) secondary antibodies were pooled and diluted at 1:350 in PBSTx + BSA. Coverslips were incubated in secondary antibodies for 30 min at RT and then washed twice with PBSTx. Coverslips were counterstained with DAPI (Sigma‐Aldrich) and mounted on glass slides with Prolong Diamond anti‐fade medium (Molecular Probes) and allowed to cure for 48 h.
SNAP labeling of CENPA‐SNAP‐3xFlag RPE1 cells was performed as illustrated (Fig EV4B) and previously described 69. Briefly, CENPA‐SNAP was labeled with SNAP‐Cell 505 STAR or SNAP‐Cell TMR‐STAR (NEB), diluted in cell culture media for 30 min, rinsed 2× with fresh media, and then replenished with fresh media. Please refer to Appendix Table S2 for working concentrations.
Image acquisition was performed on a Nikon Eclipse Ti inverted microscope equipped with motorized stage, LED epifluorescence light source (Spectra X), 60×/1.4NA (Plan Apo) DIC oil immersion objective, and ORCA Flash4.0 V2+ digital sCMOS camera (Hamamatsu). Optical sections were taken at 200‐nm intervals and deconvolved using the LIM 3D Deconvolution module in Nikon Elements. Panels were cropped using Photoshop CS5 (Adobe) and assembled with overlays using Illustrator CS5 (Adobe).
Analysis
All intensity analyses were performed using functions within Nikon Elements. For anaphase segregation experiments, the optical section image stacks were initially converted to maximum‐intensity projection images to identify chromosome position via DNA (DAPI) and kinetochore (ACA antibody) signals. “Lagging” chromosomes were defined by single DAPI or ACA signals located between two larger sets of DAPI/ACA signals. To measure kinetochore protein intensities and to minimize bias, all image processing was performed using the ACA channel. First, LUTs were adjusted to decrease background signal. Next, a freehand ROI was drawn loosely (to account for non‐overlapping kinetochore protein signal) around an individual target ACA signal. A duplicate ROI placed in an adjacent location not occupied by another kinetochore was used to measure background signal. Returning to the original optical section image stack, the target and background ROI intensities were measured for each channel at all optical sections where the target ACA was observed. The sum of the background section intensities was then subtracted from the sum of the target section intensities to get individual volume intensities for each kinetochore protein. This was repeated for all lagging chromosomes and eight segregated chromosomes per cell. To get relative signal intensities, individual intensity values were divided by the average intensity value of the nocodazole washout chromosomes in each experiment. A chromosome was excluded from analysis if its ACA signal overlapped with a second ACA signal.
For chromosome misalignment experiments, the optical section image stacks were converted to maximum‐intensity projection images. A cell was included for analysis if the majority of its chromosomes were aligned at the equator. Misaligned chromosomes were included for analysis if their ACA signal was (i) sufficiently separated from the equatorial pool and (ii) characterized by two distinct foci or an elongated signal, indicating that the inter‐kinetochore axis between the sisters was parallel to the imaging plane. To determine the signal distribution of Hec1, a 1 pixel intensity line with a 5 pixel width was drawn along the inter‐kinetochore axis of each misaligned pair. If the peak Hec1 intensity at one kinetochore was < 50% of the peak Hec1 intensity at its sister, the distribution was considered unequal or decreased. To determine if the diminished Hec1 signal was “pole‐proximal” or “pole‐distal”, a perpendicular line bisecting the equator was drawn to represent the spindle pole axis. The kinetochore signal closer to the spindle pole axis was considered “pole‐proximal”, the further “pole‐distal”. This was repeated for intensity and localization of BubR1, Mad1, and CENP‐A.
For Hec1 kinetochore recruitment experiments, background intensities were subtracted individually from each cell and then a common threshold intensity for each channel was applied to all cells in a given experiment. Individual freehand ROIs were drawn around each cell using DNA (DAPI) as a guide. Total volume and mean intensity measurements were collected within each ROI. To get total volume intensity, the mean intensity measurement was multiplied by the total volume within the ROI. Finally, average volume intensity for the control cells (chemical treatment without Plk1 inhibition) was determined to get relative volume intensities for all cells.
To measure ancestral and nascent CENP‐A intensities, 0.43‐μm2 ROIs were placed over the kinetochores of all lagging chromosomes and 20 segregated chromosomes in each cell. A single 0.43‐μm2 ROI was also used to measure the background intensity. Fluorescence intensities were measured for each ROI throughout the entire image stack, and the background intensity was subtracted from the target intensities to get signal intensity for each chromosome kinetochore, which was then divided by the average intensity of the segregated kinetochores to get a relative intensity of each kinetochore per cell.
For the PICH analysis, the image stacks were thresholded to remove background signal and then converted to 3D volume images. All PICH signals that overlapped with an ACA signal were counted as “PICH(+)”. Cells that exhibited numerous elongated PICH signals not associated with any lagging ACA signal (see Fig 5E,ii) were infrequently observed and excluded from analysis. For the metaphase analysis, “rounded” and “elongated” PICH(+) signals were subjectively determined. “Elongated” PICH signals were counted for aligned chromosomes, regardless of their overlap with an ACA signal.
Where appropriate, observer blinding was performed by slide label concealment. Sample size was selected for cell biology experiments based on prior experience and biologically significant effect size. Data analysis was performed using Prism 8 (GraphPad). Statistical significance was determined either by t‐test or one‐way ANOVA and is described in the figure legends.
Live‐cell microscopy
RPE1 cells stably expressing EGFP‐H2B and mCherry‐tubulin or YFP‐CENPA and mCherry‐CENPB were seeded on 35‐mm imaging dishes with 1.5# polymer coverslip bottom (Ibidi). Upon reaching 60–70% confluence, the cells were challenged with 40–50 nM BI‐2536 for 3 h.
Live‐cell imaging was performed on a Leica DMi8 inverted fluorescence microscope equipped with 488‐nm and 561‐nm excitation lasers, Yokogawa CSU‐W1 spinning‐disk confocal scanning unit, and 63×/1.4NA or 100×/1.4NA oil immersion objectives (Plan Apo), controlled by MetaMorph 6.1 software (Molecular Devices). Environmental control was maintained by stage‐top humidified chamber (Tokai Hit) set to 37°C and 5% CO2. Images were collected every 30 s at 250‐nm optical sections with an ORCA Flash4.0 V2+ digital sCMOS camera (Hamamatsu).
Analysis
To track CENP‐A/CENP‐B intensities of individual kinetochore pairs over time, the imaging file was cropped to include only the optical sections containing CENP‐A or CENP‐B signals for either kinetochore of the pair. Maximum‐intensity projection images were created for the selected optical sections of each channel for every time point in MetaMorph. To measure CENP‐A and CENP‐B intensities at individual kinetochores, image stacks were imported into Elements (Nikon). Identical circular ROIs were drawn around each kinetochore and the intensity recorded at each image plane where CENP‐A or CENP‐B was observed. Values for each image plane were combined to determine the volume intensity of each protein at each kinetochore. Intensity values of each protein were then plotted relative to initial intensity, time point 0.
Fluorescence recovery after photobleaching
RPE1 cells stably expressing YFP‐CENPA and mCherry‐CENPB growing in 35‐mm imaging dishes with 1.5# polymer coverslip bottom (Ibidi) were imaged on a A1RS point scanning confocal microscope (Nikon) equipped with 408‐nm, 488‐nm, and 561‐nm excitation lasers, 60×/1.4NA oil immersion objective (Plan Apo), controlled by Elements (Nikon). Environmental control was maintained by stage‐top humidified chamber (Tokai Hit) set to 37°C and 5% CO2. Metaphase cells were identified, and circular ROIs with a diameter of 1–2 μm were drawn around single or multiple kinetochore pairs. Single‐plane, pre‐bleach intensities were collected for 5 s. A 3‐s pulse from the 408‐nm laser at 100% intensity was used to bleach the YFP or mCherry fluorescence signals within a ROI. Single‐plane, post‐bleach intensities were collected every 5 s for 2 min.
Analysis
CENP‐A or CENP‐B intensities were measured for each ROI at each time point beginning from pre‐photobleaching. To determine the relative fluorescence intensity (RFI) for each ROI over time, pre‐bleach intensity was set to 100% and immediate post‐bleach intensity was set to 0%. To control for photobleaching during acquisition, fluorescence intensities were recorded from identical ROIs around non‐bleached kinetochore pairs. RFI vs. time graphs were plotted using Origin 2016 software (OriginLab).
Chromosome spreads and fluorescence in situ hybridization (FISH)
For chromosome spreads, cells grown to 75–80% confluency on a 12‐mm glass coverslip (VWR) or on a 4‐well glass slide (Millipore) were treated with BI‐2536 for 3 h and MG‐132 at 20 μM for two additional hours. Growth medium was replaced by a hypotonic medium (60% growth medium, 40% ddH2O) for 5 min and removed. After centrifugation (3 min, 800 × g) in a humid chamber, cells were pre‐extracted for 1 min in blocking buffer (0.2 M glycine, 2.5% FBS, 0.1% Triton X‐100 in 1× PBS) and fixed in 4% formaldehyde at room temperature for 10 min. Incubations with primary antibodies were conducted in blocking buffer for 1 h at room temperature. Immunofluorescence on chromosome spreads was done as described previously 70. Immunofluorescence images were collected using a DeltaVision Core system (Applied Precision).
Fluorescence in situ hybridization
Chromosome painting and centromere enumeration probes were purchased from MetaSystems Probes, and FISH was performed following the manufacturer's instructions. A cocktail of four probes (1:1:1:1) was used for each hybridization. See Appendix Fig S4 for chromosomes probed in this study.
Sequential FISH
After coverslip removal, slide was washed in ethanol 70% for 1 min, pre‐warmed denaturation solution (70% formamide, 2× SSC, pH 7.0) was applied, and slide was placed on a hot plate at 75°C for 2 min. Slide was then washed in 70% ethanol for 1 min and subsequently dehydrated in 90 and 100% ethanol for 1 min. Sample was air‐dried, and new probe hybridization was performed.
Analysis
Deconvolved 2D maximum‐intensity projections were saved as un‐scaled 16‐bit TIFF images. Centromeres were considered “intact” (2 round CENP‐C signals; Fig 5A, top), “elongated” (2 CENP‐C signals with one stretched; Fig 5A, white asterisk), or “fragmented” (> 2 CENP‐C signals; Fig 5A, yellow asterisk). Both elongated and fragmented centromeres were considered “disrupted”. For IF‐FISH, point coordinates were recorded for sequential FISHs.
Author contributions
RFL, RXN, MD, JM‐K, DF, and MEB designed the research. RFL, RXN, MD, AD, and JM‐K performed experiments. All authors analyzed the data. DF and MEB supervised the research. RFL and MEB drafted the article with contributions and revisions provided by all authors.
Conflict of interest
M.E.B. declares the following: medical advisory board of Strata Oncology; and research funding from Abbvie, Genentech, Puma, and Loxo Oncology. The other authors declare no competing financial interests.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Review Process File
Acknowledgements
We thank Dan Foltz for plasmids; Iain Cheeseman, Stephen Taylor, and Beth Weaver for antibodies; Lance Rodenkirch and the University of Wisconsin Optical Imaging Core for assistance with FRAP and live‐cell imaging experiments; Alka Choudhary for cloning assistance; and Lars Jansen, Kok‐Lung Chan, Aussie Suzuki, and Beth Weaver for advice and critical review of the article. This work was supported by NIH R01 GM097245 (to MEB) and University of Wisconsin Carbone Cancer Center Support Grant P30 CA014520. We also thank the Cell and Tissue Imaging facility at Institut Curie (PICT‐IBiSA, member of the French National Research Infrastructure France‐BioImaging ANR10‐INBS‐04). DF receives salary support from the CNRS. DF is also supported by the City of Paris, Emergence(s) 2018, an annual call for proposals that aims to help young researchers that cover MD salary.
EMBO Reports (2019) 20: e48711
References
- 1. Palmer DK, O'Day K, Wener MH, Andrews BS, Margolis RL (1987) A 17‐kD centromere protein (CENP‐A) copurifies with nucleosome core particles and with histones. J Cell Biol 104: 805–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Howman EV, Fowler KJ, Newson AJ, Redward S, MacDonald AC, Kalitsis P, Choo KHA (2000) Early disruption of centromeric chromatin organization in centromere protein A (Cenpa) null mice. Proc Natl Acad Sci USA 97: 1148–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Foltz DR, Jansen LET, Black BE, Bailey AO, Yates JR, Cleveland DW (2006) The human CENP‐A centromeric nucleosome‐associated complex. Nat Cell Biol 8: 458–469 [DOI] [PubMed] [Google Scholar]
- 4. Carroll CW, Milks KJ, Straight AF (2010) Dual recognition of CENP‐A nucleosomes is required for centromere assembly. J Cell Biol 189: 1143–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fachinetti D, Folco HD, Nechemia‐Arbely Y, Valente LP, Nguyen K, Wong AJ, Zhu Q, Holland AJ, Desai A, Jansen LET et al (2013) A two‐step mechanism for epigenetic specification of centromere identity and function. Nat Cell Biol 15: 1056–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jansen LET, Black BE, Foltz DR, Cleveland DW (2007) Propagation of centromeric chromatin requires exit from mitosis. J Cell Biol 176: 795–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Silva MCC, Bodor DL, Stellfox ME, Martins NMC, Hochegger H, Foltz DR, Jansen LET (2012) Cdk activity couples epigenetic centromere inheritance to cell cycle progression. Dev Cell 22: 52–63 [DOI] [PubMed] [Google Scholar]
- 8. Stankovic A, Guo LY, Mata JF, Bodor DL, Cao X‐J, Bailey AO, Shabanowitz J, Hunt DF, Garcia BA, Black BE et al (2017) A dual inhibitory mechanism sufficient to maintain cell‐cycle‐restricted CENP‐A assembly. Mol Cell 65: 231–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fujita Y, Hayashi T, Kiyomitsu T, Toyoda Y, Kokubu A, Obuse C, Yanagida M (2007) Priming of centromere for CENP‐A recruitment by human hMis18α, hMis18β, and M18BP1. Dev Cell 12: 17–30 [DOI] [PubMed] [Google Scholar]
- 10. Dunleavy EM, Roche D, Tagami H, Lacoste N, Ray‐Gallet D, Nakamura Y, Daigo Y, Nakatani Y, Almouzni‐Pettinotti G (2009) HJURP is a cell‐cycle‐dependent maintenance and deposition factor of CENP‐A at centromeres. Cell 137: 485–497 [DOI] [PubMed] [Google Scholar]
- 11. Foltz DR, Jansen LET, Bailey AO, Yates JR, Bassett EA, Wood S, Black BE, Cleveland DW (2009) Centromere‐specific assembly of CENP‐A nucleosomes is mediated by HJURP. Cell 137: 472–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McKinley KL, Cheeseman IM (2014) Polo‐like kinase 1 licenses CENP‐a deposition at centromeres. Cell 158: 397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Abrieu A, Magnaghi‐Jaulin L, Kahana JA, Peter M, Castro A, Vigneron S, Lorca T, Cleveland DW, Labbé JC (2001) Mps1 is a kinetochore‐associated kinase essential for the vertebrate mitotic checkpoint. Cell 106: 83–93 [DOI] [PubMed] [Google Scholar]
- 14. Stucke VM, Silljé HHW, Arnaud L, Nigg EA (2002) Human Mps1 kinase is required for the spindle assembly checkpoint but not for centrosome duplication. EMBO J 21: 1723–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sudakin V, Chan GKT, Yen TJ (2001) Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol 154: 925–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lampson MA, Kapoor TM (2005) The human mitotic checkpoint protein BubR1 regulates chromosome‐spindle attachments. Nat Cell Biol 7: 93–98 [DOI] [PubMed] [Google Scholar]
- 17. Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, Heckel A, Van Meel J, Rieder CL, Peters JM (2003) The small molecule hesperadin reveals a role for Aurora B in correcting kinetochore‐microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol 161: 281–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lampson MA, Renduchitala K, Khodjakov A, Kapoor TM (2004) Correcting improper chromosomes‐spindle attachments during cell division. Nat Cell Biol 6: 232–237 [DOI] [PubMed] [Google Scholar]
- 19. Cimini D, Wan X, Hirel CB, Salmon ED (2006) Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors. Curr Biol 16: 1711–1718 [DOI] [PubMed] [Google Scholar]
- 20. Knowlton AL, Lan W, Stukenberg PT (2006) Aurora B is enriched at merotelic attachment sites, where it regulates MCAK. Curr Biol 16: 1705–1710 [DOI] [PubMed] [Google Scholar]
- 21. Dai J, Sultan S, Taylor SS, Higgins JMG (2005) The kinase haspin is required for mitotic histone H3 Thr 3 phosphorylation and normal metaphase chromosome alignment. Genes Dev 19: 472–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sumara I, Giménez‐Abián JF, Gerlich D, Hirota T, Kraft C, De La Torre C, Ellenberg J, Peters JM (2004) Roles of Polo‐like kinase 1 in the assembly of functional mitotic spindles. Curr Biol 14: 1712–1722 [DOI] [PubMed] [Google Scholar]
- 23. Lénárt P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters JM (2007) The small‐molecule inhibitor BI 2536 reveals novel insights into mitotic roles of Polo‐like kinase 1. Curr Biol 17: 304–315 [DOI] [PubMed] [Google Scholar]
- 24. Liu D, Davydenko O, Lampson MA (2012) Polo‐like kinase‐1 regulates kinetochore‐microtubule dynamics and spindle checkpoint silencing. J Cell Biol 198: 491–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Qi W, Tang Z, Yu H (2006) Phosphorylation‐ and polo‐box‐dependent binding of Plk1 to Bub1 is required for the kinetochore localization of Plk1. Mol Biol Cell 17: 3705–3716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Elowe S, Hümmer S, Uldschmid A, Li X, Nigg EA (2007) Tension‐sensitive Plk1 phosphorylation on BubR1 regulates the stability of kinetochore‐microtubule interactions. Genes Dev 21: 2205–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kang YH, Park JE, Yu LR, Soung NK, Yun SM, Bang JK, Seong YS, Yu H, Garfield S, Veenstra TD et al (2006) Self‐regulated Plk1 recruitment to kinetochores by the Plk1‐PBIP1 interaction is critical for proper chromosome segregation. Mol Cell 24: 409–422 [DOI] [PubMed] [Google Scholar]
- 28. Lera RF, Potts GK, Suzuki A, Johnson JM, Salmon ED, Coon JJ, Burkard ME (2016) Decoding Polo‐like kinase 1 signaling along the kinetochore‐centromere axis. Nat Chem Biol 12: 411–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lera RF, Burkard ME (2012) High mitotic activity of Polo‐like kinase 1 is required for chromosome segregation and genomic integrity in human epithelial cells. J Biol Chem 287: 42812–42825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nicklas RB (1983) Measurements of the force produced by the mitotic spindle in anaphase. J Cell Biol 97: 542–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Waters JC, Skibbens RV, Salmon ED (1996) Oscillating mitotic newt lung cell kinetochores are, on average, under tension and rarely push. J Cell Sci 109(Pt 1): 2823–2831 [DOI] [PubMed] [Google Scholar]
- 32. Maresca TJ, Salmon ED (2009) Intrakinetochore stretch is associated with changes in kinetochore phosphorylation and spindle assembly checkpoint activity. J Cell Biol 184: 373–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cimini D, Howell BJ, Maddox P, Khodjakov A, Degrassi F, Salmon ED (2001) Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J Cell Biol 153: 517–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Burkard ME, Randall CL, Larochelle S, Zhang C, Shokat KM, Fisher RP, Jallepalli PV (2007) Chemical genetics reveals the requirement for Polo‐like kinase 1 activity in positioning RhoA and triggering cytokinesis in human cells. Proc Natl Acad Sci USA 104: 4383–4388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gascoigne KE, Cheeseman IM (2013) CDK‐dependent phosphorylation and nuclear exclusion coordinately control kinetochore assembly state. J Cell Biol 201: 23–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kwiatkowski N, Jelluma N, Filippakopoulos P, Soundararajan M, Manak MS, Kwon M, Choi HG, Sim T, Deveraux QL, Rottmann S et al (2010) Small‐molecule kinase inhibitors provide insight into Mps1 cell cycle function. Nat Chem Biol 6: 359–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maciejowski J, George KA, Terret M‐EE, Zhang C, Shokat KM, Jallepalli PV (2010) Mps1 directs the assembly of Cdc20 inhibitory complexes during interphase and mitosis to control M phase timing and spindle checkpoint signaling. J Cell Biol 190: 89–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sliedrecht T, Zhang C, Shokat KM, Kops GJPL (2010) Chemical genetic inhibition of Mps1 in stable human cell lines reveals novel aspects of Mps1 function in mitosis. PLoS One 5: e10251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Screpanti E, De Antoni A, Alushin GM, Petrovic A, Melis T, Nogales E, Musacchio A (2011) Direct binding of Cenp‐C to the Mis12 complex joins the inner and outer kinetochore. Curr Biol 21: 391–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Malvezzi F, Litos G, Schleiffer A, Heuck A, Mechtler K, Clausen T, Westermann S (2013) A structural basis for kinetochore recruitment of the Ndc80 complex via two distinct centromere receptors. EMBO J 32: 409–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nishino T, Rago F, Hori T, Tomii K, Cheeseman IM, Fukagawa T (2013) CENP‐T provides a structural platform for outer kinetochore assembly. EMBO J 32: 424–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Suzuki A, Badger BL, Salmon ED (2015) A quantitative description of Ndc80 complex linkage to human kinetochores. Nat Commun 6: 8161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Masumoto H, Masukata H, Muro Y, Nozaki N, Okazaki T (1989) A human centromere antigen (CENP‐B) interacts with a short specific sequence in alphoid DNA, a human centromeric satellite. J Cell Biol 109: 1963–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hemmerich P, Weidtkamp‐Peters S, Hoischen C, Schmiedeberg L, Erliandri I, Diekmann S (2008) Dynamics of inner kinetochore assembly and maintenance in living cells. J Cell Biol 180: 1101–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Irvine DV, Amor DJ, Perry J, Sirvent N, Pedeutour F, Choo KHA, Saffery R (2005) Chromosome size and origin as determinants of the level of CENP‐A incorporation into human centromeres. Chromosome Res 12: 805–815 [DOI] [PubMed] [Google Scholar]
- 46. Addis Jones O, Tiwari A, Olukoga T, Herbert A, Chan K‐L (2019) PLK1 facilitates chromosome biorientation by suppressing centromere disintegration driven by BLM‐mediated unwinding and spindle pulling. Nat Commun 10: 2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Samoshkin A, Arnaoutov A, Jansen LET, Ouspenski I, Dye L, Karpova T, McNally J, Dasso M, Cleveland DW, Strunnikov A (2009) Human condensin function is essential for centromeric chromatin assembly and proper sister kinetochore orientation. PLoS One 4: e6831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Samoshkin A, Dulev S, Loukinov D, Rosenfeld JA, Strunnikov AV (2012) Condensin dysfunction in human cells induces nonrandom chromosomal breaks in anaphase, with distinct patterns for both unique and repeated genomic regions. Chromosoma 121: 191–199 [DOI] [PubMed] [Google Scholar]
- 49. Kagami Y, Ono M, Yoshida K (2017) Plk1 phosphorylation of CAP‐H2 triggers chromosome condensation by condensin II at the early phase of mitosis. Sci Rep 7: 5583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Samejima K, Booth DG, Ogawa H, Paulson JR, Xie L, Watson CA, Platani M, Kanemaki MT, Earnshaw WC (2018) Functional analysis after rapid degradation of condensins and 3D‐EM reveals chromatin volume is uncoupled from chromosome architecture in mitosis. J Cell Sci 131: jcs210187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ribeiro SA, Gatlin JC, Dong Y, Joglekar A, Cameron L, Hudson DF, Farr CJ, Mcewen BF, Salmon ED, Earnshaw WC et al (2009) Condensin regulates the stiffness of vertebrate centromeres. Mol Biol Cell 20: 2371–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim S, Yu H (2015) Multiple assembly mechanisms anchor the KMN spindle checkpoint platform at human mitotic kinetochores. J Cell Biol 208: 181–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rago F, Gascoigne KE, Cheeseman IM (2015) Distinct organization and regulation of the outer kinetochore KMN network downstream of CENP‐C and CENP‐T. Curr Biol 25: 671–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Baumann C, Körner R, Hofmann K, Nigg EA (2007) PICH, a centromere‐associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell 128: 101–114 [DOI] [PubMed] [Google Scholar]
- 55. Chan KL, North PS, Hickson ID (2007) BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J 26: 3397–3409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Biebricher A, Hirano S, Enzlin JH, Wiechens N, Streicher WW, Huttner D, Wang LH‐C, Nigg EA, Owen‐Hughes T, Liu Y et al (2013) PICH: a DNA translocase specially adapted for processing anaphase bridge DNA. Mol Cell 51: 691–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Soto M, García‐Santisteban I, Krenning L, Medema RH, Raaijmakers JA (2018) Chromosomes trapped in micronuclei are liable to segregation errors. J Cell Sci 131: jcs214742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McEwen BF, Heagle AB, Cassels GO, Buttle KF, Rieder CL (1997) Kinetochore fiber maturation in PtK1 cells and its implications for the mechanisms of chromosome congression and anaphase onset. J Cell Biol 137: 1567–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kaulich M, Cubizolles F, Nigg EA (2012) On the regulation, function, and localization of the DNA‐dependent ATPase PICH. Chromosoma 121: 395–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Oppermann FS, Grundner‐Culemann K, Kumar C, Gruss OJ, Jallepalli PV, Daub H (2012) Combination of chemical genetics and phosphoproteomics for kinase signaling analysis enables confident identification of cellular downstream targets. Mol Cell Proteomics 11: O111.012351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kettenbach AN, Schweppe DK, Faherty BK, Pechenick D, Pletnev AA, Gerber SA (2011) Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo‐like kinase activities in mitotic cells. Sci Signal 4: rs5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Santamaria A, Wang B, Elowe S, Malik R, Zhang F, Bauer M, Schmidt A, Silljé HHW, Körner R, Nigg EA (2011) The Plk1‐dependent phosphoproteome of the early mitotic spindle. Mol Cell Proteomics 10: M110.004457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bodor DL, Valente LP, Mata JF, Black BE, Jansen LET (2013) Assembly in G1 phase and long‐term stability are unique intrinsic features of CENP‐A nucleosomes. Mol Biol Cell 24: 923–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Falk SJ, Guo LY, Sekulic N, Smoak EM, Mani T, Logsdon GA, Gupta K, Jansen LET, Van Duyne GD, Vinogradov SA et al (2015) CENP‐C reshapes and stabilizes CENP‐A nucleosomes at the centromere. Science 348: 699–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Swartz SZ, McKay LS, Su K‐C, Bury L, Padeganeh A, Maddox PS, Knouse KA, Cheeseman IM (2019) Quiescent cells actively replenish CENP‐A nucleosomes to maintain centromere identity and proliferative potential. Dev Cell 10.1016/j.devcel.2019.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chmátal L, Gabriel SI, Mitsainas GP, Martínez‐Vargas J, Ventura J, Searle JB, Schultz RM, Lampson MA (2014) Centromere strength provides the cell biological basis for meiotic drive and karyotype evolution in mice. Curr Biol 24: 2295–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Iwata‐Otsubo A, Dawicki‐McKenna JM, Akera T, Falk SJ, Chmátal L, Yang K, Sullivan BA, Schultz RM, Lampson MA, Black BE (2017) Expanded satellite repeats amplify a discrete CENP‐A nucleosome assembly site on chromosomes that drive in female meiosis. Curr Biol 27: 2365–2373.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ainsztein AM, Kandels‐Lewis SE, Mackay AM, Earnshaw WC (1998) INCENP centromere and spindle targeting: identification of essential conserved motifs and involvement of heterochromatin protein HP1. J Cell Biol 143: 1763–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bodor DL, Rodríguez MG, Moreno N, Jansen LET (2012) Analysis of protein turnover by quantitative SNAP‐based pulse‐chase imaging. Curr Protoc Cell Biol 55: 8.8.1–8.8.34 [DOI] [PubMed] [Google Scholar]
- 70. Fachinetti D, Han JS, McMahon MA, Ly P, Abdullah A, Wong AJ, Cleveland DW (2015) DNA sequence‐specific binding of CENP‐B enhances the fidelity of human centromere function. Dev Cell 33: 314–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Review Process File