

Abstract
The activation of mixed lineage kinase‐like (MLKL) by receptor‐interacting protein kinase‐3 (RIPK3) controls the execution of necroptosis, a regulated form of necrosis that occurs in apoptosis‐deficient conditions. Active oligomerized MLKL triggers the exposure of phosphatidylserine residues on the cell surface and disrupts the plasma membrane integrity by forming lytic pores. MLKL also governs endosomal trafficking and biogenesis of small extracellular vesicles as well as the production of proinflammatory cytokines during the early steps of necroptosis; however, the molecular basis continues to be elucidated. Here, we find that MLKL oligomers activate Pannexin‐1 (PANX1) channels, concomitantly to the loss of phosphatidylserine asymmetry. This plasma membrane “leakiness” requires the small GTPase RAB27A and RAB27B isoforms, which regulate intracellular vesicle trafficking, docking, and fusion with the plasma membrane. Although cells in which PANX1 is silenced or inhibited normally undergo necroptotic death, they display enhanced production of cytokines such as interleukin‐8, indicating that PANX1 may tamper with inflammation. These data identify a novel signaling nexus between MLKL, RAB27, and PANX1 and propose ways to interfere with inflammation associated with necroptosis.
Keywords: cytokines, inflammation, MLKL, necroptosis, Pannexin‐1
Subject Categories: Autophagy & Cell Death, Immunology, Membrane & Intracellular Transport
Introduction
Necroptosis is a programmed lytic cell death pathway that occurs in apoptosis‐deficient conditions 1. In contrast to apoptosis, which is thought to be immunologically silent as cellular content remains impermeable and is rapidly engulfed by surrounding phagocytic cells, necroptotic cells produce proinflammatory cytokines and release cytosolic proinflammatory material in the extracellular milieu as a consequence of the loss of plasma membrane integrity 2. Although the engagement of several immunoreceptors culminates in necroptosis, this form of death has been best studied upon ligation of the death receptor family member TNF receptor 1 (TNFR1) 2. When caspases are blocked, TNFR1 signaling assembles a filamentous death‐inducing complex of the Ser/Thr kinases RIPK1, RIPK3, and the pseudokinase MLKL coined necrosome, which culminates in the phosphorylation of MLKL by RIPK3 3, 4, 5, 6, 7, 8, 9. This causes MLKL to adopt a conformational change and to bind negatively charged phospholipids such as high‐order inositol phosphates, leading to its multimerization 10. As a consequence, MLKL oligomers insert into the plasma membrane, where they trigger the exposure of phosphatidylserine residues at the cell surface, acting as “eat‐me” signals for the surrounding immune cells 11, 12. Ultimately, oligomerized MLKL compromises cell‐membrane integrity by forming lytic pores leading to the cell's demise 10, 13, 14, 15, 16.
Cells with phosphorylated MLKL and flipped phosphatidylserine are not necessarily committed to death and can “resuscitate” provided the stimulus is removed in a timely manner 12, 17. During this commitment phase, necroptotic cells mount a potent immune response, and MLKL promotes the cell‐autonomous production of proinflammatory cytokines such as interleukin‐8 (IL‐8, CXCL8) 18. Active MLKL also governs endosomal trafficking, as well as the dynamic biogenesis and extrusion of small extracellular vesicles (EVs) 11, 19, 20. This requires the RAB27 small GTPase subfamily of RAB27A and RAB27B, which facilitates the trafficking of intracellular vesicles, as well as their docking and fusion to the plasma membrane 19, 21.
How exactly MLKL controls these diverse functions continues to be elucidated. Here, we report that MLKL oligomers promote the activation of the channel‐forming glycoprotein PANX1 via the RAB27 isoforms, in cells otherwise viable. PANX1 is a single‐membrane large‐pore channel, which is activated by a variety of stimuli, such as membrane stretches, ATP, flux of ions (K+, Ca2+), or receptor‐induced signaling 22. In its open‐state, PANX1 allows the passage of nucleotides (ATP, UTP), chloride, and small molecules, as well as the uptake of dyes such as the carbocyanine monomeric TO‐PRO‐3 from the extracellular medium 22. Recently, effector caspases were shown to cleave a COOH‐terminal latch in PANX1 to irreversibly open the channel in apoptotic and pyroptotic cells 23, 24, 25. This promotes cell clearance and inflammation, as PANX1 channels conduct the release of nucleotide‐based “find‐me” signals and of potassium, which favors the assembly of the NLRP3 inflammasome 25. However, the contribution of PANX1 to necroptosis has not been investigated. By combining genetic and chemical tools, we show that PANX1 is dispensable for the proper execution of the necroptotic program, but it restrains the production of proinflammatory cytokines such as IL‐8.
Results and Discussion
MLKL promotes “leakiness” of the plasma membrane during necroptosis
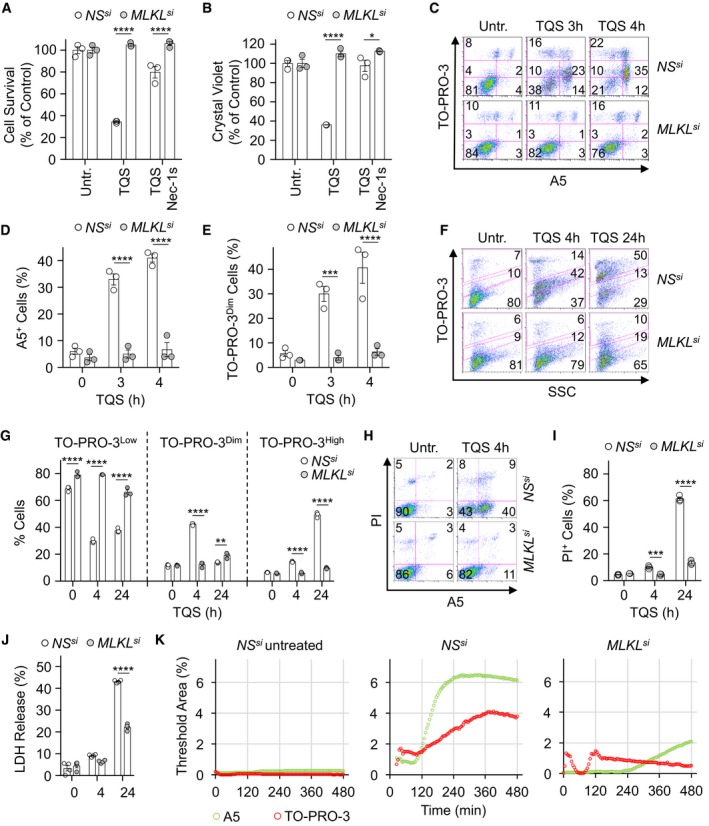
HT‐29 colon cancer cells, which constitute a classical model for necroptosis 4, 5, were treated with TNFα (T) in the presence of the pan‐caspase inhibitor QVD‐OPh (Q), together with the Smac mimetic Birinapant (S) 26. Necroptosis was then visualized by depletion of intracellular ATP, crystal violet staining, staining of phosphatidylserine residues on the outer face of the plasma membrane with Annexin V (A5), and uptake of the vital cell‐impermeant dye TO‐PRO‐3 4, 5, 11, 12, 17 (Fig 1A–D). As expected 3, 6, cell death was efficiently prevented by either the RIPK1 inhibitor Nec‐1s or by silencing MLKL (Figs 1A–D and EV1A). Supporting an initial observation by Gong et al 17, staining with TO‐PRO‐3 discriminated a population of “leaky” cells with a dim staining from dead cells with a high incorporation of the dye (Fig 1C and E). Of note, those TO‐PRO‐3Dim cells were mostly A5‐positive (Fig 1C). Silencing of MLKL significantly reduced both exposure of phosphatidylserine residues and TO‐PRO‐3 uptake (Figs 1C and EV1B). Moreover, “leakiness” preceded full permeabilization to the dye seen after 24 h, which likely reflects the loss of plasma membrane integrity (Ref. 17, Fig 1F and G). Importantly, the early uptake of TO‐PRO‐3 was selective, as it was not accompanied by a staining with propidium iodide, although of smaller molecular weight (Fig 1H and I). Further militating against a general rupture of the plasma membrane, cytosolic lactate dehydrogenase was only modestly released in the extracellular milieu during early steps of necroptosis (Fig 1J). Time‐lapse microscopy to monitor A5 staining and TO‐PRO‐3 uptake further unveiled that kinetics of phosphatidylserine exposure and TO‐PRO‐3 uptake during necroptosis slightly differed (Fig 1K, and [Link], [Link]). Although both events started 2 h post‐stimulation, phosphatidylserine exposure occurred suddenly as compared to the gradual uptake of TO‐PRO‐3. Altogether, this suggests that the plasma membrane of cells becomes “leaky” to TO‐PRO‐3 in an MLKL‐dependent manner during the early onset of necroptosis.
Figure 1. MLKL controls the selective permeabilization of the plasma membrane during the early stages of necroptosis.
-
A, BCell viability was assessed by CellTiter‐Glo (A), and Crystal Violet (B) after 16 h. Data are means ± SEM of three independent experiments. *P < 0.1, ****P < 0.0001 (ANOVA).
-
C–EFlow cytometric analysis of cells stained with TO‐PRO‐3 and Annexin V (A5). Histograms show means ± SEM of three independent experiments. ***P < 0.001, ****P < 0.0001 (ANOVA).
-
F–ICells were collected after 4 and 24 h of treatment and stained with TO‐PRO‐3 (F and G), or with propidium iodide (PI) and A5 (H and I). Data are means ± SEM of three independent experiments. **P < 0.01, ***P < 0.001, ****P < 0.0001 (ANOVA).
-
JMeasurement of lactate dehydrogenase (LDH) released from necroptotic cells, as indicated (means ± SEM, n = 4 biological replicates, ****P < 0.0001, ANOVA).
-
KTime lapsed microscopy analysis of A5 and TO‐PRO‐3 staining upon induction of necroptosis. The presented data are representative of at least three independent experiments.
Figure EV1. Impact of MLKL silencing on cell survival and TO‐PRO‐3 uptake.
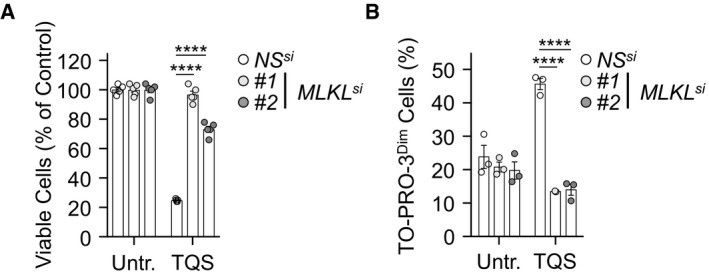
- Cell survival was evaluated by CellTiter‐Glo after 24 h of treatment. Data are means ± SEM, n = 4 biological replicates, ****P < 0.0001 (ANOVA).
- TO‐PRO‐3 uptake was analyzed by flow cytometry in cells treated with TQS for 4 h. Data are means ± SEM of three independent experiments. ****P < 0.0001 (ANOVA).
Pannexin‐1 governs plasma membrane “leakiness”
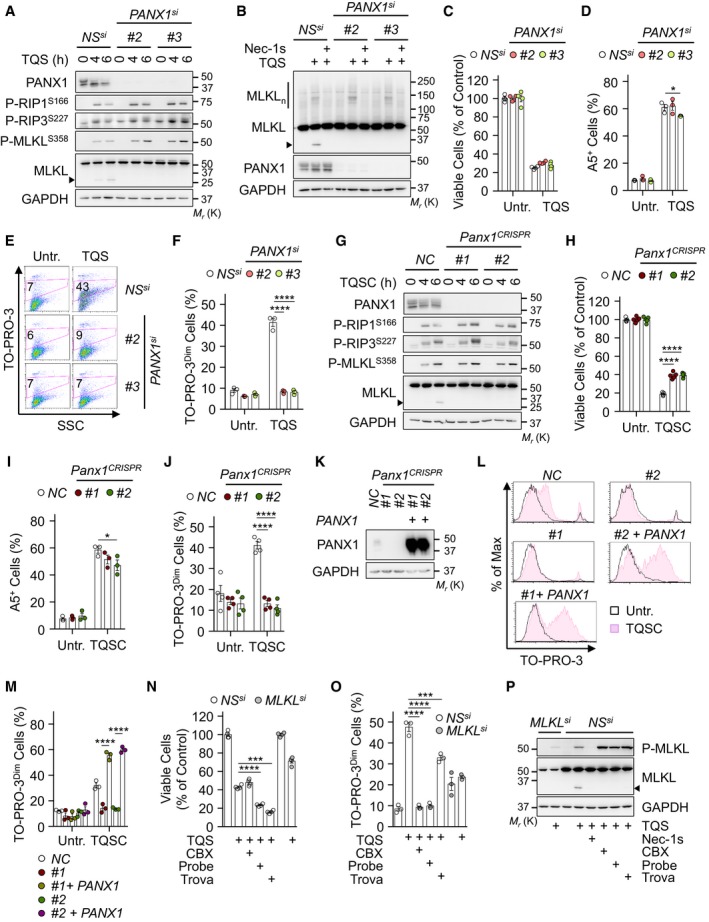
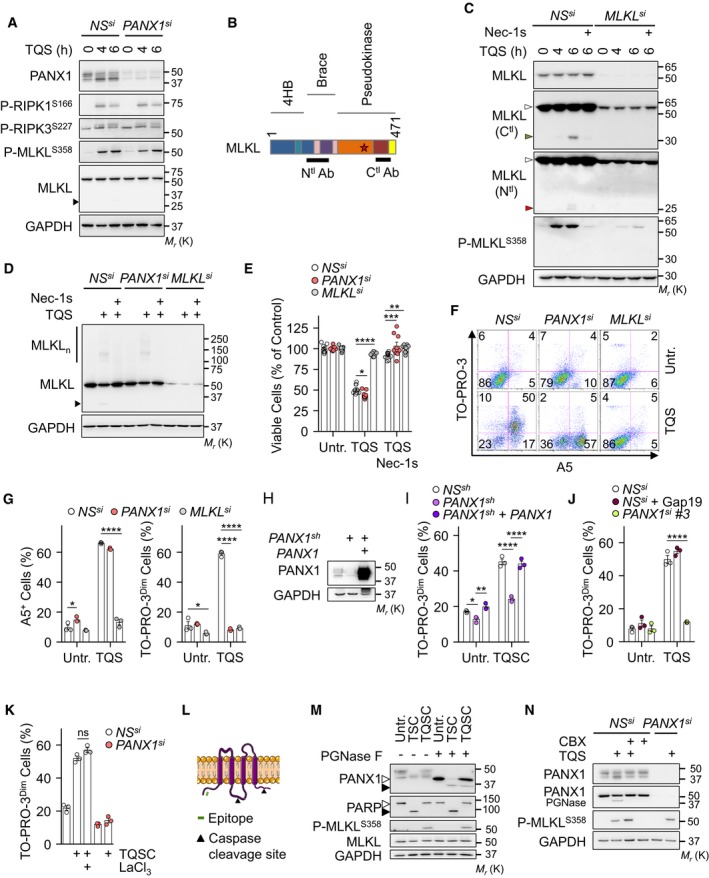
This “leakiness” of necroptotic cells is reminiscent of the caspase‐mediated opening of the plasma membrane PANX1 channels during apoptosis, as evidenced by the selective uptake of TO‐PRO‐3 23. This results in the release of purines, such as ATP in the extracellular space, which serve as a “find‐me” signal for surrounding phagocytes 23. To test the impact of PANX1 on “leakiness” during necroptosis, we first knocked down its expression with three individual siRNA oligoribonucleotides and found no overt defect on the phosphorylation of the necrosome core components (Figs 2A and EV2A). Of note, we observed the appearance of a shorter MLKL fragment 4–6 h post‐stimulation, which was PANX1‐dependent (Figs 2A and EV2A). The size of the generated fragment and the specificity of antibodies used (Fig EV2B and C) suggested a cleavage between the brace region and the pseudokinase domain of MLKL 27. Nevertheless, oligomerization of MLKL occurred normally in PANX1‐silenced cells and was efficiently blocked by pre‐treating cells with Nec‐1s (Figs 2B and EV2D). Moreover, PANX1 silencing did not alter necroptotic death, and MLKL‐mediated flip of phosphatidylserine occurred normally, suggesting that PANX1 is mostly dispensable for cell death by necroptosis (Figs 2C and D, and EV2E–G). In contrast, silencing PANX1 was as efficient as knocking down MLKL in preventing TO‐PRO‐3 uptake (Figs 2D–F, and EV2F and G). We further deployed a CRISPR/Cas9 strategy to knock out PANX1, and two single‐guide RNA (sgRNA) were used to deplete endogenous PANX1 in HT‐29 cells without single‐cell cloning. Although cell survival was slightly increased in knockout cells compared to control, no clear defect in necroptosis signaling and phosphatidylserine asymmetry was seen (Fig 2G–I). Nevertheless, plasma membrane “leakiness” was abolished in the absence of PANX1 (Fig 2J). Importantly, the re‐expression of PANX1 in those knockout cells was sufficient to restore the uptake of TO‐PRO‐3 in response to the necroptotic insult (Fig 2K–M). Similar results were obtained in cells expressing a shRNA targeting the 5′‐UTR region of PANX1 (Fig EV2H and I). Lastly, we found that cells treated with the PANX1 channel blockers carbenoxolone (CBX), Probenecid, and Trovafloxacin 28 also displayed impaired uptake of TO‐PRO‐3 (Fig 2N and O). This was however not the case when connexins, another family of large‐pore channels that shares similarities with Pannexins 22, were inhibited with Gap19 or LaCl3 (Fig EV2J and K). Of note, a greater phosphorylation of MLKL was observed without PANX1 or when its activity was inhibited (Fig 2A, G, and P). Collectively, these data demonstrate that MLKL initiates “leakiness” of the plasma membrane via PANX1 and that PANX1 activation is dispensable for the execution of cell death.
Figure 2. Pannexin‐1 controls the uptake of TO‐PRO‐3 during necroptosis.
-
AHT‐29 cells were transfected with two individual siRNA for PANX1, or scramble non‐specific (NS) siRNA for 72 h. Cells were pre‐treated with 10 μM QVD‐OPh (Q) together with 5 μM Birinapant (S), prior stimulation with 10 ng ml−1 of TNFα (T), as indicated. Western blotting for hallmarks of necroptosis, as indicated. The arrowhead indicates MLKL cleaved fragment. Molecular weight markers (Mr) are shown.
-
BCells as in (A) were exposed to TQS for 5 h. Necrostatin‐1 (Nec‐1s, 20 μM) was also used. MLKL oligomers (MLKLn) were resolved by non‐reducing SDS–PAGE after cross‐linking.
-
CThe cell viability was assessed by CellTiter‐Glo after 24 h of treatment (means ± SEM, n = 4 biological replicates).
-
D–FFlow cytometric analysis of cells as in (A) treated with TQS for 4 h and stained with Annexin V (D), or with TO‐PRO‐3 (E and F). Data are means ± SEM of three independent experiments. *P < 0.1, ****P < 0.0001 (ANOVA).
-
G–JHT‐29 cells were infected with lentiviruses containing two individual sgRNA to deplete endogenous PANX1. A control non‐specific sequence (NC) was also used. Cells were treated as in (A) with the addition of 1 μM cycloheximide (C) to enhance cell death and analyzed by Western blotting (G). Cell survival was assessed by CellTiter‐Glo after 24 h of treatment (H). Histogram shows means ± SEM, n = 4 biological replicates, ****P < 0.0001, ANOVA. Annexin V staining and TO‐PRO‐3 uptake was analyzed by flow cytometry after 4 h of stimulation (I and J). Shown are means ± SEM, n = 3 biological replicates, *P < 0.1, ****P < 0.0001, ANOVA.
-
K–MPANX1 expression was restored in PANX1 knockout cells after an infection with a lentivirus containing a PANX1 cDNA and expressing GFP. Cell lysates were analyzed by Western blotting as indicated (K). TO‐PRO‐3 uptake was analyzed by flow cytometry (L and M). Shown are means ± SEM, n = 3 biological replicates, ****P < 0.0001 ANOVA.
-
N, ONS‐ and MLKL‐silenced cells were pre‐treated with 100 μM carbenoxolone (CBX), 2 mM Probenecid (Probe), or 30 μM Trovafloxacin (Trova) and stimulated as in (A). Cell survival was evaluated by CellTiter‐Glo after 24 h (N). Data are means ± SEM, n = 4 biological replicates, ***P < 0.001, ****P < 0.0001, ANOVA. TO‐PRO‐3 uptake was analyzed by flow cytometry (O). Data are means ± SEM, n = 3 biological replicates, ***P < 0.001, ****P < 0.0001, ANOVA.
-
PCells as in (N) were analyzed by Western blotting. The presented data are representative of at least three independent experiments.
Source data are available online for this figure.
Figure EV2. Uptake of TO‐PRO‐3 via Pannexin‐1 during necroptosis.
-
AHT‐29 cells were transfected with a siRNA for PANX1 (#1), or scramble non‐specific (NS) siRNA for 72 h. Cells were pre‐treated with 10 μM QVD‐OPh (Q) together with 5 μM Birinapant (S), prior stimulation with 10 ng ml−1 of TNFα (T), as indicated. Cell lysates were analyzed by Western blotting, as indicated. Molecular weight markers (Mr) are shown.
-
BSchematical representation of MLKL. The position of the epitopes recognized by the two antibodies used is shown. The star indicates the S358 position.
-
CHT‐29 cells were treated as indicated. Necrostatin‐1 (Nec‐1s, 20 μM) was also used. Cell lysates were prepared and analyzed by Western blotting. White arrowheads show intact MLKL, and green or red arrowheads show cleaved MLKL.
-
DCells as in (A) were exposed to TQS for 5 h. Necrostatin‐1 (Nec‐1s, 20 μM) was also used. MLKL oligomers (MLKLn) were resolved by non‐reducing SDS–PAGE after cross‐linking.
-
ECell viability assay by CellTiter‐Glo in cells treated with TQS for 24 h (mean ± SEM, n = 9 biological replicates, *P < 0.1, **P < 0.01, ***P < 0.001, ****P < 0.0001, ANOVA).
-
F, GCells were treated with TQS for 4 h, stained with TO‐PRO‐3 and Annexin V (A5), and analyzed by flow cytometry. Dead cells (TO‐PRO‐3high) were not analyzed. Data are means ± SEM of three independent experiments. *P < 0.1, ****P < 0.0001 (ANOVA).
-
H, IHT‐29 cells were infected with a lentivirus containing a shRNA targeting PANX1 in the 5′‐UTR region. PANX1 was reintroduced after an infection with a lentivirus containing a PANX1 cDNA. Cell lysates were analyzed by Western blotting (H). Flow cytometric analysis of cells treated with TQS and cycloheximide for 4 h and stained with TO‐PRO‐3 (I). Data are means ± SEM of three independent experiments. *P < 0.1, **P < 0.01, ****P < 0.0001 (ANOVA).
-
J, KTO‐PRO‐3 uptake was analyzed by flow cytometry (mean ± SEM, n = 3 biological replicates, ****P < 0.0001, ANOVA). Gap19 (20 μM) and LaCl3 (1.5 mM) were used.
-
LSchematical representation of PANX1. The position of the epitopes recognized by the antibodies used and the cleavage sites by caspases are shown.
-
MWestern blotting analysis as indicated. PGNase F was used to remove glycosylation. Open and close symbols represent full‐length and cleaved proteins, respectively.
-
NWestern blotting analysis as indicated. The presented data are representative of at least three independent experiments.
During apoptosis, effector caspases remove a COOH‐terminal inhibitory domain of PANX1 to irreversibly open the channels 23. The analysis of PANX1 by immunoblotting revealed multiple species, consistent with N‐glycosylation, as they were efficiently removed with a treatment with PNGase F (Ref. 29 and Fig EV2M). Although less dramatic than during apoptosis 23, 30, we noticed a modest cleavage of PANX1 during necroptosis at a site close to the characterized caspase site, as evidenced by a slight decrease in full‐length PANX1 combined with the presence of a cleaved fragment in PNGase F‐treated samples (Fig EV2L and M). The canonical caspase substrate PARP remained intact, arguing against residual caspase activity (Fig EV2M). This modest PANX1 proteolysis occurred simultaneously with MLKL processing, and both were significantly reduced in cells treated with PANX1 inhibitors (Figs 2A, B, G, P, and EV2N). Hence, the cleavage of MLKL and PANX1 likely results from the opening of the channel in necroptotic cells.
MLKL oligomerization and vesicular trafficking control plasma membrane “leakiness”
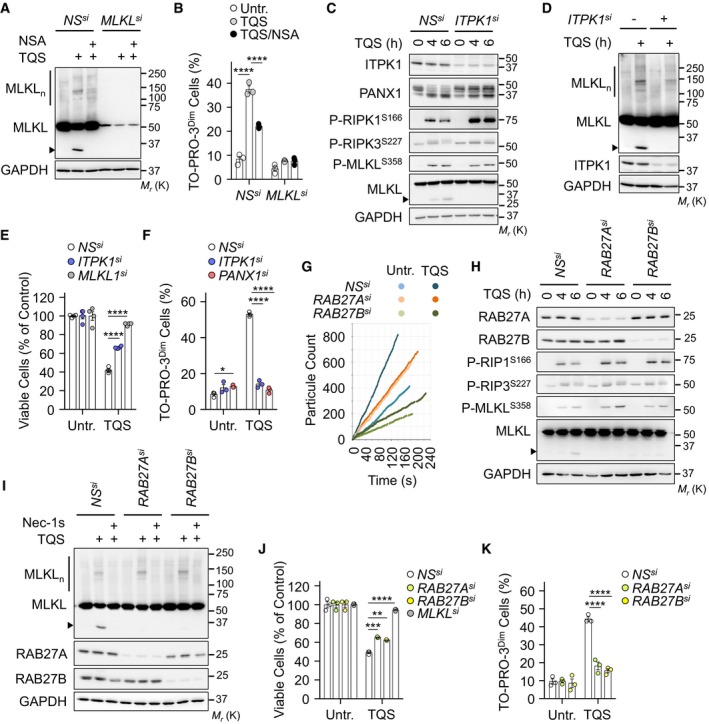
Once phosphorylated by RIPK3, MLKL binds high‐order inositol phosphates 13. This displaces MLKL's auto‐inhibitory domain and allows MLKL to oligomerize and reach the plasma membrane to exert its lethal function 13, 14. As expected 3, 14, the treatment of cells with the small molecule necrosulfonamide (NSA) prevented MLKL oligomerization (Fig 3A). NSA also significantly reduced the uptake of TO‐PRO‐3 (Fig 3B). We next silenced inositol‐tetrakisphosphate 1‐kinase (ITPK1), a crucial kinase for the generation of inositol phosphates 13. Although an increase in the phosphorylation of RIPK1 was noticed without ITPK1, the phosphorylation of RIPK3 and MLKL was normal, in agreement with Dovey et al 13 (Fig 3C). However, silencing ITPK1 precluded MLKL oligomerization (Fig 3D). This led to a reduction in the exposure of phosphatidylserine residues together with a significant inhibition of TO‐PRO‐3 uptake (Figs 3E and F, and EV3A). Thus, MLKL oligomerization is instrumental for PANX1‐mediated “leakiness” of the plasma membrane.
Figure 3. MLKL oligomerization and vesicular trafficking enable plasma membrane “leakiness”.
-
A, BNS‐ and MLKL‐silenced cells were treated with 5 μM necrosulfonamide (NSA). MLKL oligomers (MLKLn) were visualized by non‐reducing SDS–PAGE after cross‐linking proteins (A). Cells were exposed to TQS for 5 h. Molecular weight markers (Mr) are shown. TO‐PRO‐3 uptake was analyzed by flow cytometry in cells treated with TQS for 4 h (B). Data are means ± SEM of three independent experiments. ****P < 0.0001 (ANOVA).
-
CCell lysates from NS‐ and ITPK1‐silenced cells were analyzed by Western blotting as indicated.
-
DWestern blotting analysis of MLKL oligomerization as in (A).
-
ECell survival was evaluated by CellTiter‐Glo after 24 h of treatment (means ± SEM, n = 4 biological replicates, ****P < 0.0001, ANOVA).
-
FMeasurement of TO‐PRO‐3 uptake as in (B). Dead TO‐PRO‐3high cells were discarded. Data are means ± SEM of three independent experiments. *P < 0.1, ****P < 0.0001 (ANOVA).
-
GSmall EVs fractions were purified from the culture medium by ultracentrifugation. Shown is a representative diagram of particle count obtained with tunable resistive pulse sensing analysis (TRPS, qNano, IZON).
-
HCells as in (G) were subjected to Western blotting analysis as indicated.
-
IMLKL oligomerization was determined as in (A).
-
JCell survival by CellTiter‐Glo after 24 h (mean ± SEM, n = 4 biological replicates, **P < 0.01, ***P < 0.001, ****P < 0.0001, ANOVA).
-
KTO‐PRO‐3 uptake as in (B). Data are means ± SEM, n = 3 biological replicates, ****P < 0.0001 (ANOVA). The presented data are representative of at least three independent experiments.
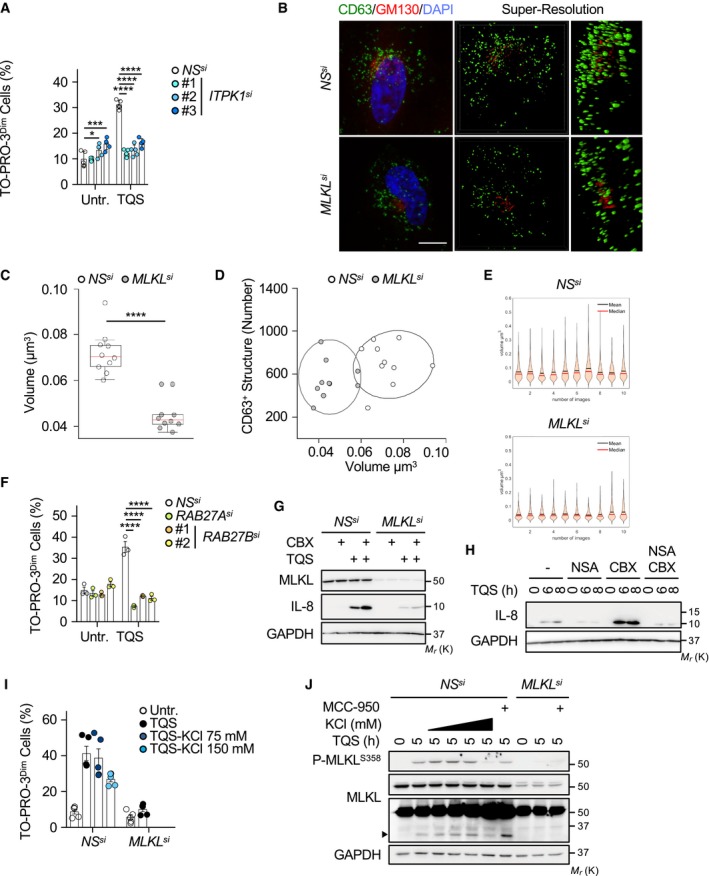
Figure EV3. MLKL controls intracellular vesicles trafficking.
-
AHT‐29 cells were transfected with the indicated siRNA for 72 h. Cells were pre‐treated with 10 μM QVD‐OPh (Q) together with 5 μM Birinapant (S), prior stimulation with 10 ng ml−1 of TNFα (T) for 4 h, and TO‐PRO‐3 uptake was analyzed by flow cytometry. Data are means ± SEM of three independent experiments. *P < 0.1, ***P < 0.001, ****P < 0.0001 (ANOVA).
-
B–DConfocal microscopic analysis of CD63 and GM130 in HeLa cells, as indicated. The staining was analyzed by Structure Illumination microscopy (SIM). Nuclei were counterstained with DAPI. Scale bar, 10 μm. The volumes of CD63‐positive structures, or scatter plots of numbers and mean volumes of CD63‐positive structures analyzed by SIM (n = 10 cells per condition; ****P < 0.0001; ANOVA), were quantified. Ellipses show 95% confidence. On each box, the central mark indicates the median, and the bottom and top edges of the box indicate the 25th and 75th percentiles, respectively. The whiskers extend to the most extreme data points not considered outliers.
-
EDensity plots showing dispersion of the volume of CD63‐positive structures for each cells.
-
FFlow cytometric analysis of cells treated as in (A) and stained with TO‐PRO‐3. Shown is mean ± SEM (n = 3 biological replicates, ****P < 0.0001, ANOVA).
-
GHT‐29 cells were transfected with siRNA for MLKL, or scramble non‐specific (NS) siRNA for 72 h. Cells were pre‐treated with 10 μM QVD‐OPh (Q) together with 5 μM Birinapant (S), and with 100 μM CBX, prior stimulation with 10 ng ml−1 of TNFα (T) for 6 h. Cell lysates were analyzed by Western blotting as indicated. Molecular weight markers (Mr) are shown.
-
HHT‐29 cells were treated as in (G), and 5 μM NSA was also used. Western blotting analysis was performed with the indicated antibodies.
-
IHT‐29 cells were transfected with siRNA for MLKL, or scramble non‐specific (NS) siRNA for 72 h. Cells were pre‐treated with 10 μM QVD‐OPh (Q) together with 5 μM Birinapant (S) and KCl, prior stimulation with 10 ng ml−1 of TNFα (T) for 4 h. Flow cytometric analysis of cells stained with TO‐PRO‐3 (mean ± SEM, n = 5 biological replicates).
-
JWestern blotting of cell lysates as in (A). MCC‐950 (1 μM) was also used. The arrowhead indicates MLKL cleaved fragment.
Source data are available online for this figure.
In addition to the plasma membrane 3, active MLKL targets intracellular vesicles and was demonstrated to regulate constitutive endosomal trafficking 19, 20. In agreement, our examination by super‐resolution microscopy of CD63, a tetraspanin enriched in late endosomes, lysosomes, and secretory vesicles confirmed a drop in the number and volume of vesicles without MLKL (Fig EV3B–E). Because MLKL‐mediated trafficking and release of small EVs require the small RAB GTPases RAB27A and RAB27B 19, we next assess their impact on TO‐PRO‐3 uptake during necroptosis. First, 100,000 × g (100k) sedimented membrane vesicle fractions, purified from culture media of RAB27A‐ or RAB27B‐silenced cells, were analyzed by tunable resistive pulse sensing technology. Supporting previous works 11, 19, the triggering of necroptosis increased small EVs release and this induction was markedly reduced when RAB27A or RAB27B was knocked down (Fig 3G). Silencing RAB27A or RAB27B did not alter classical hallmarks of necroptosis, such as RIPK1 and MLKL phosphorylation or MLKL oligomerization, whereas phosphatidylserine exposure and overall cell death were only slightly diminished (Figs 3H–J and EV3F). Yet, the uptake of TO‐PRO‐3 was drastically reduced without RAB27 isoforms (Fig 4K). Further paralleling PANX1 inhibition, the residual proteolysis of MLKL was also impaired. Combined, these data suggest that MLKL oligomerization and RAB27‐dependent vesicular trafficking control “leakiness” of the plasma membrane.
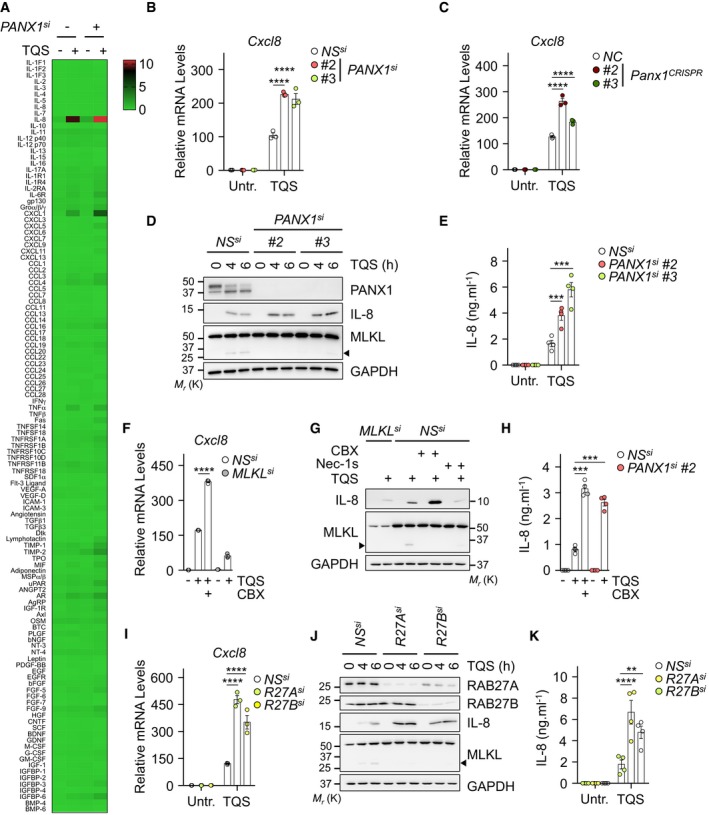
Figure 4. Pannexin‐1 restrains the production of cytokines associated with necroptosis.
-
AHT‐29 cells were transfected with a siRNA for PANX1 (#3), or scramble non‐specific (NS) siRNA for 72 h. Cells were pre‐treated with 10 μM QVD‐OPh (Q) plus 5 μM Birinapant (S) and exposed to 10 ng ml−1 of TNFα (T) for 6 h. Shown is a normalized densitometric analysis for the presence of 120 cytokines in cell supernatants with an antibody array. The color scale (0–10) represents the means of normalized densitometry values (n = 2 technical replicates).
-
B, CHT‐29 transfected as indicated (B) or PANX1 knockout HT‐29 cells (C) were treated with TQS for 6 h. The mRNA levels of Cxcl8 relative to untreated NS or NC samples were measured by qPCR (means ± SEM, n = 3 biological replicates, ****P < 0.0001, ANOVA).
-
DLysates from cells treated as indicated were prepared and subjected to Western blotting analysis as indicated. Molecular weight markers (Mr) are shown.
-
EAbundance of IL‐8, measured by ELISA, in the supernatants of cells treated as indicated after 6 h of stimulation (means ± SEM, n = 4 biological replicates, ***P < 0.001, ANOVA).
-
F–HHT‐29 cells were treated as indicated. Cxcl8 mRNA levels relative to untreated NS samples were measured by qPCR after 6 h of treatment (F). Cell lysates were analyzed by Western blotting (G). The release of IL‐8 in cell culture media was analyzed by ELISA (H). Data are means ± SEM of three (F) or four (H) independent experiments (***P < 0.001, ****P < 0.0001, ANOVA).
-
I–KHT‐29 cells were treated as indicated. Cxcl8 mRNA levels relative to untreated NS samples were measured by qPCR after 6 h of treatment (I). Cell lysates were analyzed by Western blotting (J). IL‐8 release was analyzed in cell culture media by ELISA (K). Data are means ± SEM of three (I) or four (K) independent experiments (**P < 0.01, ****P < 0.0001, ANOVA).
Pannexin‐1 restrains the production of proinflammatory cytokines during necroptosis
MLKL was recently shown to promote, in a cell‐autonomous manner, the production of proinflammatory cytokines during the early stages of necroptosis 18. We therefore investigated whether interfering with PANX1 had an effect on inflammation. To this end, we first carried out an unbiased antibody array for the presence of 120 cytokines in the culture media from control and PANX1‐silenced cells engaged in the necroptotic process. Supporting the results from Zhu et al 18, triggering necroptosis promoted the expression and release of a subset of proinflammatory cytokines such as IL‐8/CXCL8 and Groα/CXCL1 (Fig 4A). This was further enhanced when PANX1 was silenced, suggesting a role for PANX1 in tampering with inflammation. In line with this, quantitative polymerase chain reaction (qPCR) analysis revealed that more IL‐8 mRNA was produced without PANX1 (Fig 4B and C). This was accompanied by an increase in the abundance in IL‐8 protein, as measured by immunoblotting and ELISA (Fig 4D and E). Importantly, the same was true in cells treated with the PANX1 inhibitor CBX (Fig 4F–H). Of note, silencing MLKL or inhibiting its activity with NSA abolished CBX‐mediated effects (Fig EV3G and H). Lastly, we assessed the impact of RAB27 proteins, which participate in PANX1‐mediated “leakiness” of the plasma membrane, on IL‐8 synthesis. Paralleling the situation with PANX1, the silencing of RAB27A or RAB27B significantly increased the production of IL‐8 both at the mRNA and at the protein level (Fig 4I–K). Altogether, these findings support the idea that PANX1 and the RAB27 proteins play a negative role in inflammation by dampening the production of cytokines.
In conclusion, we provide evidence that PANX1 channels operate downstream of MLKL to constrict the production of proinflammatory cytokines during necroptosis. Our data also illustrate how the plasma membrane of necroptotic cells undergoes drastic rearrangements before it collapses. This includes the insertion of MLKL oligomers 10, 13, 14, 15, 16, scrambling of phosphatidylserine residues, ions fluxes 15, 31, activation of cell surface proteases 32, the release of so‐called “bubbles” of broken plasma membrane and of small EVs 12, and now the activation of the channel‐forming PANX1. Although PANX1 silencing only marginally affected phosphatidylserine asymmetry, additional work is required to elucidate whether these “eat‐me” signals constitute a prerequisite for plasma membrane “leakiness”, or if MLKL signaling bifurcates.
How exactly is MLKL linked to PANX1? Hints may come from the discovery that MLKL oligomers target intracellular compartments, in addition to the plasma membrane 14, 19, 20. Whether MLKL's location dictates its apparently independent functions remains unknown. Nevertheless, our results indicate that MLKL oligomer‐mediated activation of PANX1 channels requires RAB27 GTPase family. Future investigations will define how RAB27‐dependent intracellular trafficking of vesicles to the PM is propitious for PM “leakiness”. In addition to mechanical distortion, ATP, K+ efflux, Ca2+ influx, phosphorylation, or caspase‐mediated processing was shown to open PANX1 channels 33. Of note, oligomerized MLKL promotes a drastic efflux of potassium in bone marrow‐derived macrophages, which culminates in the activation of the NLRP3 inflammasome 34. However, buffering the concentration of K+ with KCl at concentrations that did not interfere with MLKL phosphorylation 34 had no apparent impact on PANX1‐mediated uptake of TO‐PRO‐3 and PANX1‐dependent processing of MLKL (Fig EV3I and J). Although not formally ruled out here, MLKL‐dependent Ca2+ flux was shown to occur rather lately in necroptotic HT‐29 cells, concomitantly to the loss of plasma membrane integrity 31. Interestingly, MLKL phosphorylation and phosphatidylserine asymmetry do not mark a point‐of‐no‐return and both events can be reversed if the insult is removed 11, 12, 17. Because “leakiness” occurs concomitantly to the exposure of phosphatidylserine residues, it is tempting to speculate that PANX1 opening during necroptosis is also reversible.
During apoptosis, the presence of phosphatidylserine residues on the outer leaflet of the plasma membrane, combined with the opening of PANX1 channels, serves as “eat‐me” and “find‐me” signals to favor silent phagocytosis 23, 28, 35. Caspase‐mediated activation of PANX1 also coordinates NLRP3 inflammasome assembly, apoptotic bodies release and prevents the formation of “string‐like” protrusion called apoptopodia 28, 36. During necroptosis, MLKL oligomers govern the production of cytokines, the secretion of small EVs, and the subsequent release in the extracellular milieu of “danger signals” from dying cells drive inflammation 37. Our data indicate that MLKL simultaneously initiates a negative feedback loop via PANX1 to finely tune the production of cytokines and therefore shape the necroptotic microenvironment. Future work will elucidate how PANX1 and its interplay with MLKL regulate this inflammatory response. The necroptotic machinery has been linked to several inflammatory conditions such as ischemia/reperfusion, inflammatory neurodegenerative diseases, intestinal inflammation, and cancer 1, 38, 39. Strategies aimed at targeting PANX1 might be relevant to selectively modulate early inflammation associated with necroptosis.
Materials and Methods
Cell culture and reagents
HEK293T, HT‐29, and HeLa cells were purchased from the American Type Culture Collection. Bafilomycin A1, carbenoxolone (CBX) Concanamycin A, Cycloheximide, LaCl3, and Trovafloxacin were purchased from Sigma. Gap19, QVD‐OPh, and TNFα were obtained from R&D Systems. Birinapant, Necrostatin‐1 (Nec‐1s), and Necrosulfonamide (NSA) were from Selleckchem. MCC‐950 (Invivogen), Probenecid, TO‐PRO‐3, Alexa‐488 Annexin V, and propidium iodide (Life Technologies), CellTiter‐Glo and PGNase F (Promega), and Cytotoxicity Detection kitPLUS (Roche Diagnostics) were also used.
siRNA and transfections
Cells were transfected with 10 pmol of siRNA using the Lipofectamine RNAiMAX Transfection Reagent (Invitrogen) as previously described 40. The following sequences were obtained from Life Technologies: ITPK1#1, GCAGCACGCCGUCAUUGACAUCAAU (HSS105601); ITPK1#2, GCCUACAUGGAAGACGACAGGAUCU (HSS105602); ITPK1#3, GCACCAACUCUCACGAGAUGGCUAU (HSS105603); MLKL#1, UCGAAUCUCCCAACAUCCUGCGUAU (HSS136795); MLKL#2, CAAGGAAACUUUGAGGCAGUAUUUA (HSS176401); MLKL#3, GCAACGCAUGCCUGUUUCACCCAUA (HSS136796); PANX1#1,UCAGGCUGCCUUUGUGGAUUCAUAU (HSS119236); PANX1#2, ACCUCCCACUGUGGCUGCAUAAGUU (HSS119237); PANX1#3, CAUAUUUGCUCAGACUUGAAGUUUA (HSS177598).
A mixture of siRNAs targeting RAB27A was purchased from Sigma (EHU91501). RAB27B was silenced as previously published 41.
shRNA, CRISPR, and infections
A lentiviral shRNA vector containing a sequence targeting the 5′‐UTR region of PANX1 was used (Dharmacon, Cat#V2LHS‐73947, ATGTGTAAGTCCTCATCAA). For CRISPR, single‐guide RNAs (sgRNA) targeting PANX1 were chosen in the sgRNA library (Ref. 42 and cloned into a lentiviral lentiCRISPRv2 (GeCKO, ZhangLab) backbone 43) . The sgRNA used are the following: PANX1#1CRISPR forward: CACCGACTGTTAGCGTGTATCTACC; PANX1#1CRISPR reverse: AAACGGTAGATACACGCTAACAGTC; PANX1#2CRISPR forward: CACCGGCTGCGAAACGCCAGAACAG; PANX1#2CRISPR reverse: AAACCTGTTCTGGCGTTTCGCAGCC. For infections, lentiviral particles were produced in HEK293T by co‐transfection of the various constructs together with pVSV‐G and psPAX2 plasmids. Supernatants containing lentiviral particles were collected after 48 h and applied on HT‐29 during a 1,250 × g centrifugation for 90 min in presence of 8 μg ml−1 of polybrene (Sigma). For CRISPR, cells were cultured with 1 μg ml−1 of puromycin to select infected cells. Knockout or shRNA‐expressing cells were reconstituted after transduction with a pHIV‐derived lentivirus encoding PANX1 and for GFP, kindly provided by G. Núñez 24.
Cell death induction and measurement
To induce necroptosis, HT‐29 cells were pre‐treated 30–60 min with 10 μM QVD‐OPH plus 5 μM Birinapant prior stimulation with 0–10 ng ml−1 of TNFα. In some experiments, CBX (100 μM), Probenecid (2 mM), Gap19 (20 μM), LaCl3 (1.5 mM), Nec‐1s (20 μM), Trovafloxacin (30 μM), NSA (5 μM), and cycloheximide (1 μM) were used. Cell viability was assessed using CellTiter‐Glo, staining with Crystal violet, and staining with Annexin V and TO‐PRO‐3, following the manufacturer's instructions.
Western blotting analysis and antibodies
Cells were washed with ice‐cold PBS and lysed with TNT buffer [50 mM Tris‐HCl (pH 7.4), 150 mM NaCl, 1% Triton X‐100, 1% Igepal, 2 mM EDTA] or with RIPA buffer [25 mM Tris‐HCl (pH 7.4), 150 mM NaCl, 0.1% SDS, 0.5% Na‐Deoxycholate, 1% NP40, 1 mM EDTA] supplemented with protease inhibitors (ThermoFisher Scientific) for 30 min on ice. Extracts were cleared by centrifugation at 9,000 × g, and protein concentration was determined by BCA (ThermoFisher Scientific). 5–10 μg of proteins was resolved by SDS–polyacrylamide gel electrophoresis (SDS–PAGE) and transferred onto nitrocellulose membranes (GE Healthcare). To visualize MLKL oligomers, protein cross‐linking was performed as previously described 44. Briefly, cells were fixed in PBS containing 0.4% paraformaldehyde for 12 min at room temperature and lysed with PBS containing 1% Triton‐X100 and proteases inhibitors. Proteins were incubated with 2× Laemmli buffer (Life Technologies) at room temperature and separated by SDS–PAGE.
Antibodies specific for the following proteins were purchased from Cell Signaling Technology: MLKL (D2I6N), P‐MLKL S358 (D6H3V), PANX1 (D9M1C), RAB27A (D7V6B), P‐RIPK1 S166 (D1L3S), RIPK3 (E1Z1D), and P‐RIPK3 S227 (D6W2T). Antibodies specific for GAPDH (6C5) and PARP (F‐2) were purchased from Santa Cruz Biotechnology. Antibodies specific for MLKL (ab183770) were purchased from Abcam. Antibodies against IL‐8 (R&D, MAB208), against RIPK1 (BD Biosciences, 51), against MLKL (Millipore, MABC604), against ITPK1 (Sigma, HPA055230), and against RAB27B (Proteintech, 13412‐1‐AP) were also used.
qPCR
RNA was extracted using the Nucleospin RNAplus (Macherey‐Nagel) following manufacturer's instructions. 2 μg of RNA was reverse transcribed using the Maxima Reverse Transcriptase kit (ThermoFisher), and 50 ng of the resulting cDNA was amplified by qPCR using PerfeCTa SYBR Green SuperMix Low ROX (QuantaBio). Data were analyzed using the equation and normalized by the housekeeping genes ACTB, HPRT1.
The following primers were used: ACTB forward: GGACTTCGAGCAAGAGATGG; ACTB reverse: AGCACTGTGTTGGCGTACAG; HPRT1 forward: TGACACTGGCAAAACAATGCA; HPRT1 reverse: GGTCCTTTTCACCAGCAAGCT; Cxcl8 forward: CAGTTTTGCCAAGGAGTGCT; Cxcl8 reverse: ACTTCTCCACAACCCTCTGC.
Immunofluorescence microscopy
HT‐29 grown on coverslips were fixed with PBS containing 4% paraformaldehyde for 30 min at room temperature, and staining was performed as previously described 13. Briefly, fixed samples were incubated at 4°C overnight with primary antibodies and 60 min with secondary antibodies in PBS containing 3% BSA, 1% saponin, and 1% Triton X‐100. The following antibodies were used: anti‐CD63 (556019, BD Biosciences) and anti‐GM130 (Abcam, ab52649). Coverslip was sealed with Prolong gold anti‐fade mounting media (Life Technologies) that allowed illumination of the nuclei by DAPI staining. For confocal microscopy, images were acquired with a Nikon A1R confocal microscope using the NIS‐Element Software and processed using the FIJI Software. Structure illumination microscopy (SIM) images were acquired with a Nikon N‐SIM microscope. Z‐stacks of 0.12 μm were performed using a 100× oil‐immersion lens with a 1.49 numerical aperture and reconstructed in 3D using the NIS‐Element Software. Reconstructed images with a maximal NIS score of 8 were further analyzed. For live imaging, transfected HT‐29 cells were pre‐treated with QVD‐OPh, Birinapant, and incubated with TO‐PRO‐3 and Annexin V. After 30 min, TNFα was added and images were recorded every 5 min for 8 h. Acquisition and analysis were performed on a Leica Timelapse DMI‐8 using the MetaMorph software (version 7.5; MicroPicell, SFR François Bonamy, France).
Image analysis
Reconstructed images validated with the SIMcheck plugin 1.2 were analyzed with Fiji ImageJ2 v1.52i 45. A binary mask was applied to select images with a modulation contrast‐to‐noise ratio (MCNR) > 8. Individual spots corresponding to single CD63‐positive structure were isolated with the classic watershed function of the MorpholibJ plugin 46, and pixel intensity was measured. Only structures with the MCNR > 12 were retained.
IL‐8 secretion and cytokine array
IL‐8 was measured from cell culture supernatants by ELISA (R&D, Quantikine ELISA kit, D8000C). Cytokine presence in culture media from HT‐29 cells undergoing necroptosis was assessed using a Human Cytokine Array C1000 (RayBiotech) followed by a densitometric analysis (ImageJ, NIH, USA).
Small extracellular vesicle analysis
Small EVs were purified as previously described 47. Briefly, to avoid contamination with extracellular vesicles (EVs) from the FBS, serum was centrifuged at 100,000 × g for 16 h on a Beckman Coulter ultracentrifuge using the SW‐41 Ti rotor. EVs were purified from the conditional media of 0.4 × 106 transfected HT‐29 cells via successive centrifugations (300 × g 5 min, 2,000 × g 10 min, 10,000 × g 30 min, 100,000 × g 2 h). Pellets were washed in PBS‐0.22 μm filtered, re‐centrifuged at 100,000 × g for 2 h, and resuspended in a small volume of PBS‐0.22 μm filtered. 100,000 × g steps were performed on a Beckman Coulter Centrifuge using the MLA‐130 rotor. Diameter and concentration of the particles were determined by Tunable Resistive Pulse Sensing (TRPS) using the qNano system (IZON). Particles within the range of 50–330 nm were detected using the pore “NP 100”. A stretch 46 mm was applied to the pores with an appropriate voltage (0.80 Ω–1.00 V) to reach a 120 nA baseline with noise < 10 pA. We have submitted all relevant data of our experiments to the EV‐TRACK knowledgebase (EV‐TRACK ID: EV180073) 48.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 7 software using one‐way and two‐way analysis (ANOVA).
Author contributions
Conceptualization: TD, GA‐G, KT, AT, AP, MF, PH, and NB; Methodology: TD, GA‐G, KT, AT, AP, MF, PH, and NB; Investigation: TD, GA‐G, KT, AT, AP, MF, PH, and NB; Supervision: DC and JG; Writing‐Original Draft: TD and NB; Writing‐Review & Editing: TD, GA‐G, MF, PH, JG, and NB; and Funding Acquisition: JG and NB.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Movie EV1
Movie EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
We thank Kathryn Jacobs for critically reading the article; Gabriel Núñez for kindly providing reagents; and Micropicell and Cytocell facilities (SFR Santé François Bonamy, Nantes, France). This research was funded by an International Program for Scientific Cooperation (PICS, CNRS), Fondation pour la Recherche Medicale (Equipe labellisée DEQ20180339184), Fondation ARC contre le Cancer (NB), Ligue nationale contre le cancer comités de Loire‐Atlantique, Maine et Loire, Vendée (JG, NB), Région Pays de la Loire et Nantes Métropole under Connect Talent Grant (JG), the National Research Agency under the Programme d'Investissement d'Avenir (ANR‐16‐IDEX‐0007), and the SIRIC ILIAD (INCa‐DGOS‐Inserm_12558). TD is a PhD fellow funded by Nantes Métropole; GAG holds postdoctoral fellowships from Fondation de France; and AT holds postdoctoral fellowship from Fondation ARC.
EMBO Reports (2019) 20: e47840
References
- 1. Shan B, Pan H, Najafov A, Yuan J (2018) Necroptosis in development and diseases. Genes Dev 32: 327–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weinlich R, Oberst A, Beere HM, Green DR (2017) Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol 18: 127–136 [DOI] [PubMed] [Google Scholar]
- 3. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X et al (2012) Mixed lineage kinase domain‐like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148: 213–227 [DOI] [PubMed] [Google Scholar]
- 4. He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X (2009) Receptor interacting protein kinase‐3 determines cellular necrotic response to TNF‐alpha. Cell 137: 1100–1111 [DOI] [PubMed] [Google Scholar]
- 5. Zhang D‐W, Shao J, Lin J, Zhang N, Lu B‐J, Lin S‐C, Dong M‐Q, Han J (2009) RIP3, an energy metabolism regulator that switches TNF‐induced cell death from apoptosis to necrosis. Science 325: 332–336 [DOI] [PubMed] [Google Scholar]
- 6. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1: 112–119 [DOI] [PubMed] [Google Scholar]
- 7. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK‐M (2009) Phosphorylation‐driven assembly of the RIP1‐RIP3 complex regulates programmed necrosis and virus‐induced inflammation. Cell 137: 1112–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, Liu Z‐G (2012) Mixed lineage kinase domain‐like is a key receptor interacting protein 3 downstream component of TNF‐induced necrosis. Proc Natl Acad Sci USA 109: 5322–5327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mompean M, Li W, Li J, Laage S, Siemer AB, Bozkurt G, Wu H, McDermott AE (2018) The structure of the necrosome RIPK1‐RIPK3 core, a human hetero‐amyloid signaling complex. Cell 173: 1244–1253.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang J‐G, Alvarez‐Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI et al (2013) The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39: 443–453 [DOI] [PubMed] [Google Scholar]
- 11. Zargarian S, Shlomovitz I, Erlich Z, Hourizadeh A, Ofir‐Birin Y, Croker BA, Regev‐Rudzki N, Edry‐Botzer L, Gerlic M (2017) Phosphatidylserine externalization, ‘necroptotic bodies’ release, and phagocytosis during necroptosis. PLoS Biol 15: e2002711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gong Y‐N, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, Linkermann A, Green DR (2017) ESCRT‐III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 169: 286–300.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dovey CM, Diep J, Clarke BP, Hale AT, McNamara DE, Guo H, Brown NWJ, Cao JY, Grace CR, Gough PJ et al (2018) MLKL requires the inositol phosphate code to execute necroptosis. Mol Cell 70: 936–948.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang H, Sun L, Su L, Rizo J, Liu L, Wang L‐F, Wang F‐S, Wang X (2014) Mixed lineage kinase domain‐like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 54: 133–146 [DOI] [PubMed] [Google Scholar]
- 15. Cai Z, Jitkaew S, Zhao J, Chiang H‐C, Choksi S, Liu J, Ward Y, Wu L‐G, Liu Z‐G (2014) Plasma membrane translocation of trimerized MLKL protein is required for TNF‐induced necroptosis. Nat Cell Biol 16: 55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW et al (2014) MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 7: 971–981 [DOI] [PubMed] [Google Scholar]
- 17. Gong Y‐N, Guy C, Crawford JC, Green DR (2017) Biological events and molecular signaling following MLKL activation during necroptosis. Cell Cycle 16: 1748–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhu K, Liang W, Ma Z, Xu D, Cao S, Lu X, Liu N, Shan B, Qian L, Yuan J (2018) Necroptosis promotes cell‐autonomous activation of proinflammatory cytokine gene expression. Cell Death Dis 9: 500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yoon S, Kovalenko A, Bogdanov K, Wallach D (2017) MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity 47: 51–65.e7 [DOI] [PubMed] [Google Scholar]
- 20. Fan W, Guo J, Gao B, Zhang W, Ling L, Xu T, Pan C, Li L, Chen S, Wang H et al (2019) Flotillin‐mediated endocytosis and ALIX–syntenin‐1–mediated exocytosis protect the cell membrane from damage caused by necroptosis. Sci Signal 12: eaaw3423 [DOI] [PubMed] [Google Scholar]
- 21. Colombo M, Raposo G, Thery C (2014) Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 30: 255–289 [DOI] [PubMed] [Google Scholar]
- 22. Esseltine JL, Laird DW (2016) Next‐generation connexin and pannexin cell biology. Trends Cell Biol 26: 944–955 [DOI] [PubMed] [Google Scholar]
- 23. Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS et al (2010) Pannexin 1 channels mediate ‘find‐me’ signal release and membrane permeability during apoptosis. Nature 467: 863–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang D, He Y, Muñoz‐Planillo R, Liu Q, Núñez G (2015) Caspase‐11 requires the Pannexin‐1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity 43: 923–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, Pelczar P, Broz P (2019) Extrinsic and intrinsic apoptosis activate pannexin‐1 to drive NLRP3 inflammasome assembly. EMBO J 38: e101638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Galluzzi L, Kepp O, Chan FK‐M, Kroemer G (2017) Necroptosis: mechanisms and relevance to disease. Annu Rev Pathol 12: 103–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, Pierotti C, Garnier J‐M, Dobson RCJ, Webb AI et al (2014) Activation of the pseudokinase MLKL unleashes the four‐helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci USA 111: 15072–15077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Poon IKH, Chiu Y‐H, Armstrong AJ, Kinchen JM, Juncadella IJ, Bayliss DA, Ravichandran KS (2014) Unexpected link between an antibiotic, pannexin channels and apoptosis. Nature 507: 329–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boassa D, Ambrosi C, Qiu F, Dahl G, Gaietta G, Sosinsky G (2007) Pannexin1 channels contain a glycosylation site that targets the hexamer to the plasma membrane. J Biol Chem 282: 31733–31743 [DOI] [PubMed] [Google Scholar]
- 30. Boyd‐Tressler AM, Lane GS, Dubyak GR (2017) Up‐regulated ectonucleotidases in Fas‐associated death domain protein‐ and receptor‐interacting protein kinase 1‐deficient Jurkat leukemia cells counteract extracellular ATP/AMP accumulation via Pannexin‐1 channels during chemotherapeutic drug‐induced apoptosis. Mol Pharmacol 92: 30–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ros U, Pena‐Blanco A, Hanggi K, Kunzendorf U, Krautwald S, Wong WW‐L, Garcia‐Saez AJ (2017) Necroptosis execution is mediated by plasma membrane nanopores independent of calcium. Cell Rep 19: 175–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cai Z, Zhang A, Choksi S, Li W, Li T, Zhang X‐M, Liu Z‐G (2016) Activation of cell‐surface proteases promotes necroptosis, inflammation and cell migration. Cell Res 26: 886–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chiu Y‐H, Schappe MS, Desai BN, Bayliss DA (2018) Revisiting multimodal activation and channel properties of Pannexin 1. J Gen Physiol 150: 19–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Nunez G, Masters SL, Murphy JM, Schroder K, Vaux DL et al (2017) Active MLKL triggers the NLRP3 inflammasome in a cell‐intrinsic manner. Proc Natl Acad Sci USA 114: E961–E969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Segawa K, Nagata S (2015) An apoptotic ‘eat me’ signal: phosphatidylserine exposure. Trends Cell Biol 25: 639–650 [DOI] [PubMed] [Google Scholar]
- 36. Atkin‐Smith GK, Tixeira R, Paone S, Mathivanan S, Collins C, Liem M, Goodall KJ, Ravichandran KS, Hulett MD, Poon IKH (2015) A novel mechanism of generating extracellular vesicles during apoptosis via a beads‐on‐a‐string membrane structure. Nat Commun 6: 7439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kearney CJ, Martin SJ (2017) An inflammatory perspective on necroptosis. Mol Cell 65: 965–973 [DOI] [PubMed] [Google Scholar]
- 38. Seehawer M, Heinzmann F, D'Artista L, Harbig J, Roux P‐F, Hoenicke L, Dang H, Klotz S, Robinson L, Dore G et al (2018) Necroptosis microenvironment directs lineage commitment in liver cancer. Nature 562: 69–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Strilic B, Yang L, Albarran‐Juarez J, Wachsmuth L, Han K, Muller UC, Pasparakis M, Offermanns S (2016) Tumour‐cell‐induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 536: 215–218 [DOI] [PubMed] [Google Scholar]
- 40. Douanne T, André‐Grégoire G, Thys A, Trillet K, Gavard J, Bidère N (2019) CYLD regulates centriolar satellites proteostasis by counteracting the E3 ligase MIB1. Cell Rep 27: 1657–1665.e4 [DOI] [PubMed] [Google Scholar]
- 41. Hendrix A, Maynard D, Pauwels P, Braems G, Denys H, Van den Broecke R, Lambert J, Van Belle S, Cocquyt V, Gespach C et al (2010) Effect of the secretory small GTPase Rab27B on breast cancer growth, invasion, and metastasis. J Natl Cancer Inst 102: 866–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG et al (2014) Genome‐scale CRISPR‐Cas9 knockout screening in human cells. Science 343: 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sanjana NE, Shalem O, Zhang F (2014) Improved vectors and genome‐wide libraries for CRISPR screening. Nat Methods 11: 783–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chiu Y‐H, Jin X, Medina CB, Leonhardt SA, Kiessling V, Bennett BC, Shu S, Tamm LK, Yeager M, Ravichandran KS et al (2017) A quantized mechanism for activation of pannexin channels. Nat Commun 8: 14324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ball G, Demmerle J, Kaufmann R, Davis I, Dobbie IM, Schermelleh L (2015) SIMcheck: a toolbox for successful super‐resolution structured illumination microscopy. Sci Rep 5: 15915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Legland D, Arganda‐Carreras I, Andrey P (2016) MorphoLibJ: integrated library and plugins for mathematical morphology with ImageJ. Bioinformatics 32: 3532–3534 [DOI] [PubMed] [Google Scholar]
- 47. Andre‐Gregoire G, Bidere N, Gavard J (2018) Temozolomide affects extracellular vesicles released by glioblastoma cells. Biochimie 155: 11–15 [DOI] [PubMed] [Google Scholar]
- 48. Van Deun J, Mestdagh P, Agostinis P, Akay O, Anand S, Anckaert J, Martinez ZA, Baetens T, Beghein E, Bertier L et al (2017) EV‐TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods 14: 228–232 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Movie EV1
Movie EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4