

Abstract
S‐acylation (palmitoylation) is the only fully reversible lipid modification of proteins; however, little is known about how protein S‐acyltransferases (PATs) that mediate it are regulated. DHHC5 is a PAT that is mainly localised at the plasma membrane with roles in synaptic plasticity, massive endocytosis and cancer cell growth/invasion. Here, we demonstrate that DHHC5 binds to and palmitoylates a novel accessory protein Golga7b. Palmitoylation of Golga7b prevents clathrin‐mediated endocytosis of DHHC5 and stabilises it at the plasma membrane. Proteomic analysis of the composition of DHHC5/Golga7b‐associated protein complexes reveals a striking enrichment in adhesion proteins, particularly components of desmosomes. We show that desmoglein‐2 and plakophilin‐3 are substrates of DHHC5 and that DHHC5 and Golga7b are required for localisation of desmoglein‐2 to the plasma membrane and for desmosomal patterning. Loss of DHHC5/Golga7b causes functional impairments in cell adhesion, suggesting these proteins have a wider role in cell adhesion beyond desmosome assembly. This work uncovers a novel mechanism of DHHC5 regulation by Golga7b and demonstrates a role for the DHHC5/Golga7b complex in the regulation of cell adhesion.
Keywords: cell adhesion, desmosome, DHHC5, Golga7b, palmitoylation
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Post-translational Modifications, Proteolysis & Proteomics
Introduction
S‐acylation (palmitoylation) is unique among post‐translational modifications as it is the only fully reversible modification that involves the attachment of a lipid to a protein, in this case to the side chain of cysteine residues via a thioester bond. While palmitoylation was discovered several decades ago as a modification of viral proteins 1, the progress in our understanding of palmitoylation has been slower than that for other modifications such as phosphorylation. For example, the regulation of the family of enzymes which attach the fatty acid onto proteins, palmitoyl acyltransferases (PATs), is still poorly understood. There are 23 PATs encoded in the human genome, all of which are integral membrane proteins 2, with a range of cellular locations and tissue expression patterns 3. They are also referred to as DHHC proteins as their characteristic active site motif contains the sequence Asp‐His‐His‐Cys, which is required for palmitoylation of substrates. However, it has been shown that in some cases, additional proteins are required for PAT activity. This was first demonstrated in yeast, where the palmitoylation of Ras, which is essential for its signalling and function 4, requires a complex of two proteins localised to the endoplasmic reticulum, Erf2 and Erf4 5. Subsequently, it was discovered that in mammalian cells, DHHC9 requires the protein GCP16, also known as Golga7, to form a complex capable of palmitoylating Ras 6. The mechanism by which Erf4 acts on Erf2 was subsequently determined to involve stabilisation of the intermediate state where the acyl chain is transferred to the PAT before the final transfer of the acyl chain to the substrate 7.
In a large‐scale affinity purification–mass spectrometry study, Golga7b was identified as a putative interactor of DHHC5 8. Golga7b is a member of the Erf4 protein family which is closely related to GCP16 (Golga7) with 61% sequence identity. This interaction has yet to be confirmed, and it is unknown whether Golga7b could regulate DHHC5 in a manner analogous to how DHHC9 is regulated by GCP16. Interestingly, Golga7b was also identified as a putative palmitoylated protein in both humans and mice 9, 10, although confirmation of this palmitoylation and the identity of the cognate PAT is lacking.
While the majority of PATs are localised to the Golgi, DHHC5 is localised to the plasma membrane 3, and in neurons, it cycles from the synaptic membrane to dendritic shafts in response to neuronal activity 11. It is expressed in all tissues in mammals, but it is highly enriched in the heart and the brain. It plays a role in the growth and invasion of certain types of non‐small‐cell lung cancer, and DHHC5 knockdown reduces the growth and invasion of these types of tumours 12. DHHC5 has few confirmed substrates, but it is known to palmitoylate the plasma membrane protein flotillin‐2 (Flot2), which is enriched in cholesterol‐rich microdomains 13. In the heart, DHHC5 is a key player in the phenomenon of massive palmitoylation‐dependent endocytosis (MEND) 14. This process occurs when oxygen is reintroduced to cardiac muscle cells starved of oxygen and involves the endocytosis of a large proportion of the plasma membrane, giving DHHC5 a role in the recovery of heart muscle from periods of anoxia 15. Additionally, an important role for DHHC5 in the regulation of the cardiac sodium pump through palmitoylation of phospholemman has been established 16.
In neurons, DHHC5 is present at the post‐synaptic density, and knockout mice have defects in learning and memory 17. DHHC5 is retained at the plasma membrane of the post‐synaptic density through interactions with the scaffolding protein PSD‐95 and the tyrosine kinase Fyn 11. When neurons are activated by long‐term potentiation, phosphorylation of DHHC5 by Fyn is reduced, and DHHC5 is lost from the complex and is endocytosed to dendritic spines where it palmitoylates δ‐catenin before both proteins are trafficked back to the plasma membrane at the post‐synaptic density 11. This type of controlled endocytic movement of DHHC5 has not been described for any other PAT, but does leave a number of open questions including the mechanism of endocytosis from the plasma membrane and whether a similar system of trafficking of DHHC5 to and from the plasma membrane is present in other cell types or it is unique to neurons.
We and others have identified a cluster of three palmitoylation sites in the C‐terminus of DHHC5 (located just after its fourth transmembrane domain), some of which appear to be conserved in several other PATs 10, 18. In brain extracts, DHHC5 exists in a predominantly palmitoylated form with several palmitoylated states apparent 10. The function of palmitoylation at these sites is unknown, but we hypothesise that they may regulate DHHC5 localisation and/or stability. Indeed, recent work has established an important role for palmitoylation of the C‐terminus of DHHC6 by DHHC16 in the regulation of its activity and turnover 19. This region of DHHC5 is also involved in substrate recruitment; DHHC5 binds to and palmitoylates the sodium pump regulator phospholemman, and this is dependent on a region of the C‐terminus of DHHC5 just after its fourth transmembrane domain 16.
A number of studies have reported that several proteins involved in both cell:cell and cell:matrix adhesion are palmitoylated and that this palmitoylation regulates localisation and function of these proteins 20, 21, 22. Among adhesion protein complexes, desmosomes are notable as several components of the complex are palmitoylated 23. Palmitoylation of plakophilin‐3 (Pkp3) is both essential for its inclusion into desmosomes and is required for the efficient assembly of desmosomes. One of the two desmosomal cadherins, desmoglein‐2 (Dsg2), is palmitoylated on a pair of cysteines 24. Mutation of the palmitoylation sites of Dsg2 does not prevent the incorporation of Dsg2 into desmosomes but does cause a trafficking defect with a portion of Dsg2 localised to lysosomes. This shows that it is likely that palmitoylation plays a central role in desmosomal assembly, turnover and trafficking; however, it is unknown which PAT or PATs regulate this process.
Here, we demonstrate that Golga7b interacts with DHHC5 and Golga7b palmitoylation regulates plasma membrane localisation of DHHC5. We have determined that the interaction between DHHC5 and Golga7b is controlled by the palmitoylation of the C‐terminal tail of DHHC5. We also show that palmitoylation of Golga7b prevents internalisation of DHHC5 by AP2‐regulated clathrin‐mediated endocytosis. Using affinity purification–mass spectrometry analysis, we demonstrate that Golga7b defines the interactome of DHHC5 and contains known and novel substrates as well as regulators of DHHC5. Investigation of selected novel substrates revealed a central role for DHHC5 and Golga7b in cell adhesion; DHHC5 palmitoylates both Dsg2 and Pkp3 and loss of DHHC5 or Golga7b causes defects in cell adhesion. These results uncover a novel palmitoylation‐dependent mechanism to control the localisation and function of DHHC5 and reveal an important new role for DHHC5 in cell adhesion.
Results
DHHC5 interacts with and palmitoylates a novel accessary protein Golga7b
Initially, we set out to confirm whether Golga7b is indeed palmitoylated and if Golga7b interacts with DHHC5, as both of these results had come from large‐scale studies and had not been validated by orthogonal methods 8, 10. Initial attempts to express a palmitoylation‐deficient mutant Golga7b in which cysteines in the sequence were mutated to alanines resulted in undetectable levels of expression of the construct by immunoblotting and immunofluorescence microscopy. As palmitoylation regulates the stability of some proteins, this can lead to degradation of palmitoylation‐deficient mutants 25. Expression of mutant Golga7b with the addition of MG132 prevented degradation of mutant Golga7b (Fig 1A), and therefore, we included MG132 in all subsequent experiments with this and the WT Golga7b construct. To measure palmitoylation, we used an acyl‐biotin exchange (ABE) strategy 26 in which palmitoyl groups are selectively released from cysteine residues by hydroxylamine treatment at neutral pH, and these cysteine residues are biotinylated and enriched using streptavidin resin. Using this approach, we found that a C‐terminally FLAG‐tagged full‐length wild‐type (WT) Golga7b is palmitoylated when expressed in HeLa cells, but when the cysteines in the sequence were mutated to alanines, this palmitoylation was lost (Fig 1A). Next, we sought to test whether DHHC5 could palmitoylate Golga7b using ABE plus/minus siRNA‐mediated knockdown of DHHC5 (Appendix Fig S1A). When compared to cells that are treated with a non‐targeting siRNA, there was a significant reduction (to below the limit of detection) in the palmitoylation state of Golga7b when DHHC5 is depleted (Fig 1B). Ectopic expression of a WT DHHC5 construct in cells depleted of endogenous DHHC5 can rescue palmitoylation of Golga7b back to endogenous levels while expression of a catalytically dead DHHC5 mutant is unable to rescue the palmitoylation of Golga7b (Fig 1B–D). Although we cannot rule out that Golga7b may also be a target for other PATs, our data clearly indicate that that DHHC5 is the major PAT for Golga7b.
Figure 1. A palmitoylation‐dependent interaction between DHHC5 and Golga7b.
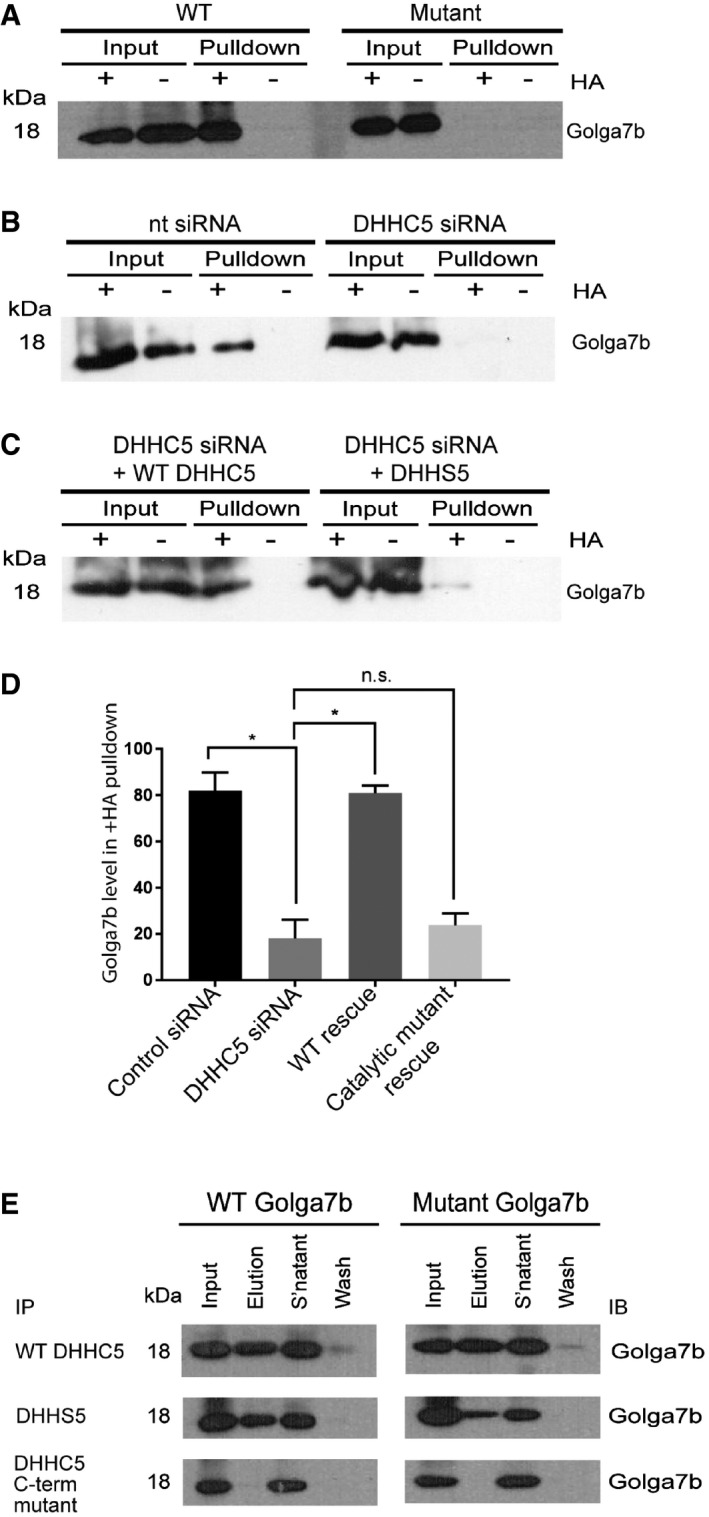
- Immunoblots of acyl‐biotin exchange purifications from Hela cells transiently transfected with WT Golga7b or a palmitoylation‐deficient mutant form of the protein with cysteines replaced with alanine residues. The hydroxylamine sensitivity of the Golga7b signal in the +HA condition of the WT pull‐down and not in either the −HA condition or the +HA condition of the mutant shows that Golga7b is a palmitoylated protein.
- Immunoblots of acyl‐biotin exchange purifications from Hela cells treated with either DHHC5 siRNA or control non‐targeting siRNA.
- Immunoblots showing rescue of Golga7b palmitoylation in DHHC5‐depleted cells after transfection of WT DHHC5 but not by the DHHC5 catalytic mutant.
- A significant reduction in the Golga7b signal is observed in the +HA condition of the pull‐down when DHHC5 is depleted with siRNA. The graph displays mean ± SEM. N = 4, *P < 0.05, paired t‐test.
- Immunoblots of WT and non‐palmitoylatable mutant Golga7b from immunoprecipitations of WT DHHC5 and a pair of DHHC5 mutants (DHHS5 = catalytically inactive mutant where catalytic cysteine has been mutated to serine; C‐terminal mutant = mutant where the 3 C‐terminal palmitoylation sites of DHHC5 have been mutated to alanine residues). WT and DHHS5 immunoprecipitates both WT and mutant Golga7b, but the C‐terminal mutant is unable to immunoprecipitate either form of Golga7b, showing that the palmitoylation of the C‐terminus of DHHC5 is essential for the stability of this interaction.
We sought to confirm the interaction between DHHC5 and Golga7b by co‐immunoprecipitation using N‐terminally HA‐tagged DHHC5 and FLAG‐tagged WT and palmitoylation‐deficient mutant Golga7b constructs co‐expressed in HeLa cells. DHHC5 immunoprecipitated both WT and mutant Golga7b (Fig 1E), indicating that the palmitoylation state of Golga7b does not affect its interaction with DHHC5. The same is also true for a catalytically inactive form of DHHC5 where the catalytic cysteine is mutated to a serine residue (DHHS5; Fig 1E). However, when the three palmitoylation sites present in the C‐terminal tail of DHHC5 10 are mutated, this mutant DHHC5 was unable to immunoprecipitate either the WT or mutant Golga7b (Fig 1E), suggesting that palmitoylation of DHHC5 is directly required for this interaction.
To confirm that the interaction of DHHC5 and Golga7b occurs in vivo, we performed immunoprecipitations of endogenous DHHC5 and Golga7b from a mouse forebrain lysate. This demonstrated that DHHC5 is immunoprecipitated by endogenous Golga7b (Fig EV1A) and that endogenous DHHC5 can reciprocally immunoprecipitate Golga7b (Fig EV1B). We next confirmed that endogenous Golga7b is palmitoylated in this context using an ABE assay (Fig EV1C). These results demonstrate that DHHC5 and Golga7b interact in vivo and that DHHC5 is the major PAT for Golga7b.
Figure EV1. Reciprocal DHHC5 and Golga7b co‐IP in brain tissue lysates.
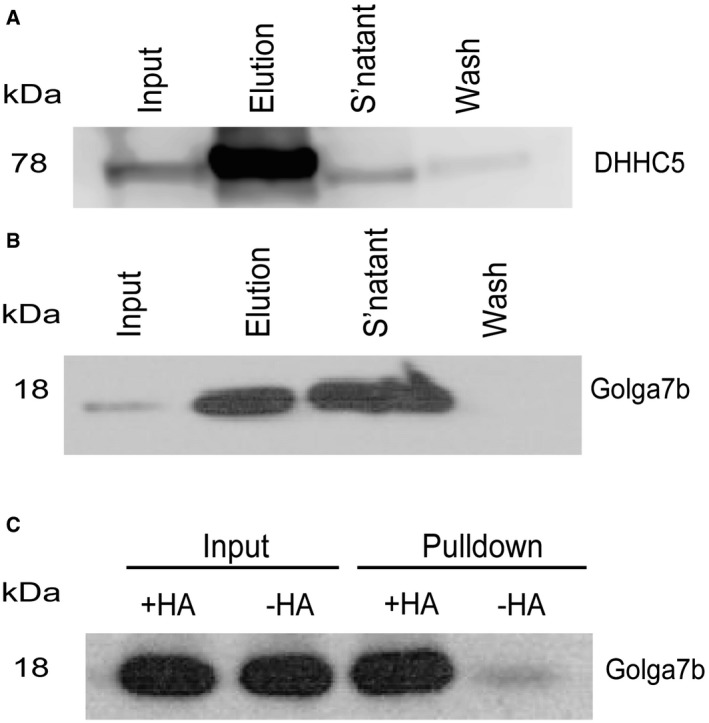
- DHHC5 immunoblot of a Golga7b immunoprecipitation from mouse forebrain extract.
- Golga7b immunoblot of a DHHC5 immunoprecipitation from mouse forebrain extract.
- Immunoblot of an ABE assay of Golga7b from mouse forebrain extract showing a hydroxylamine sensitive signal for Golga7b confirming that it is palmitoylated in vivo.
Co‐expression of Golga7b regulates plasma membrane localisation of DHHC5
We next confirmed that DHHC5 is localised to the plasma membrane 3 by staining for the endogenous protein in HeLa cells (Fig EV2A). However, when we over‐express DHHC5, we observe staining throughout the cell (Fig EV2B), indicating that mislocalisation occurs upon over‐expression. It has been reported that the over‐expression of PATs can lead to their mislocalisation 27, which could suggest that factors that regulate their localisation need to be present in sufficient quantities to ensure proper localisation. We hypothesised that Golga7b might act as a regulator of DHHC5 and may be involved in the targeting or stabilisation of DHHC5 at the plasma membrane. To investigate this, we co‐expressed DHHC5 with either WT or a palmitoylation‐deficient mutant Golga7b and performed immunofluorescence microscopy. When endogenous DHHC5 is depleted and a tagged version is co‐expressed with WT Golga7b, the majority of DHHC5 is present at the plasma membrane (Fig 2A). This indicates that when DHHC5 is over‐expressed alone, there is insufficient endogenous Golga7b present to either target DHHC5 to the plasma membrane or stabilise DHHC5 protein at the plasma membrane. Strikingly, when DHHC5 is co‐expressed with mutant Golga7b which cannot be palmitoylated, the majority of DHHC5 is lost from the plasma membrane (Fig 2B). This indicates that palmitoylation of Golga7b is required for the maintenance of DHHC5 at the plasma membrane, and when this is prevented, DHHC5 is mislocalised.
Figure EV2. Over‐expression of DHHC5 causes a localisation defect.

- Confocal images of endogenous DHHC5 in HeLa cells.
- Confocal images of HeLa cells expressing WT DHHC5 C‐terminally tagged with a FLAG epitope.
- Confocal images of HeLa cells expressing a C‐terminally FLAG‐tagged catalytic mutant of DHHC5 where the active site cysteine of DHHC5 has been replaced with a serine (DHHS5).
- Confocal microscopy images of HeLa cells expressing DHHC5 with the three palmitoylated cysteines in the C‐terminal tail mutated to serine residues. Antibody detecting endogenous FLAG epitope.
- Quantification of membrane signal of DHHC5 seen in (A‐D). All experiments were repeated three individual times; all n numbers refer to the number of cells quantified. Endogenous DHHC5, n = 45; DHHC5, n = 48; DHHS5 only, n = 38; C‐terminal mutant, n = 35, ****P < 0.001, unpaired t‐test. Error bars represent SEM.
Figure 2. Regulation of DHHC5 localisation by palmitoylation of Golga7b and the C‐terminus of DHHC5.
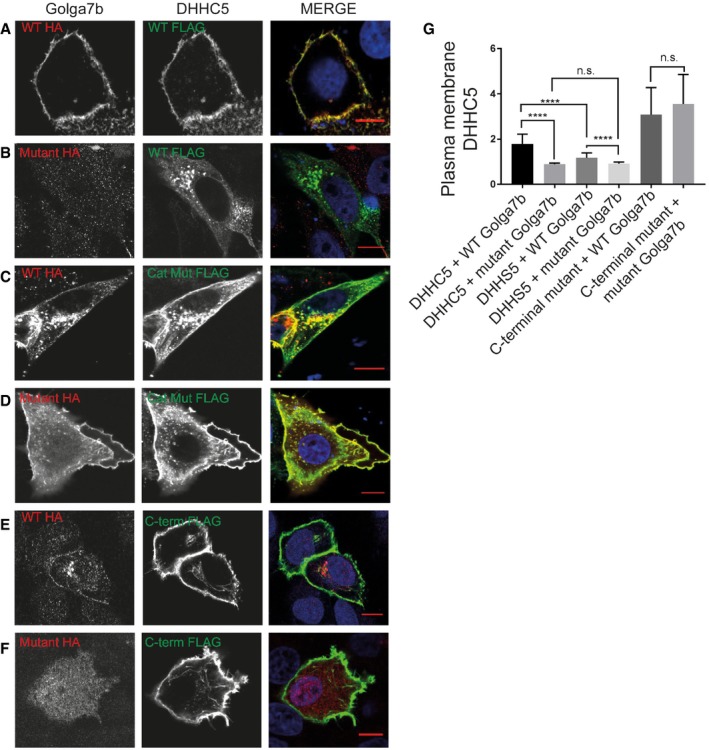
- Confocal images of HeLa cells co‐expressing WT Golga7b (1st column) and DHHC5 (2nd column), with a merged image (3rd column).
- Confocal images of HeLa cells co‐expressing palmitoylation‐deficient mutant Golga7b (1st column) and DHHC5 (2nd column), with a merged image (3rd column).
- Confocal images of HeLa cells co‐expressing WT Golga7b (1st column) and the catalytic mutant DHHS5 (2nd column), with a merged image (3rd column).
- Confocal images of HeLa cells co‐expressing palmitoylation‐deficient mutant Golga7b (1st column) and DHHS5 (2nd column), with a merged image (3rd column).
- Confocal images of HeLa cells co‐expressing WT Golga7b (1st column) and DHHC5 C‐terminal palmitoylation‐deficient mutant (2nd column), with a merged image (3rd column).
- Confocal images of HeLa cells co‐expressing palmitoylation‐deficient mutant Golga7b (1st column) and DHHC5 C‐terminal palmitoylation‐deficient mutant (2nd column), with a merged image (3rd column).
- Quantification of the plasma membrane signal of DHHC5 in each of the images (A–F). The graph displays mean ± SEM. ****P < 0.001, n.s. = not significant, unpaired t‐test. N = 35, DHHC5 + WT Golga7b. N = 41, DHHC5 + mutant Golga7b. N = 35, DHHS5 + WT Golga7b. N = 31, DHHS5 + Golga7b mutant. N = 29, C‐terminal mutant + WT Golga7b. N = 30, C‐terminal mutant + mutant Golga7b. Co‐expression of palmitoylation‐deficient mutant Golga7b significantly reduces DHHC5 as well as DHHS5 signal at the plasma membrane. The majority of the DHHC5 C‐terminal palmitoylation‐deficient mutant is localised to the plasma membrane in a Golga7b‐independent manner. All N numbers refer to the number of cells imaged and quantified, all experiments repeated four times.
Co‐expression of wild‐type Golga7b with the catalytic mutant of DHHC5 (DHHS5), in cells depleted of endogenous DHHC5, causes a significant reduction in DHHS5 at the plasma membrane compared to WT DHHC5 (Fig 2C). This mislocalisation is likely caused by a reduction of Golga7b palmitoylation as its palmitoylation level is significantly reduced in cells expressing the catalytic mutant DHHS5 compared to WT DHHC5 (Fig 1B–D). Expression of DHHS5 with mutant Golga7b mimicked what we observed with WT DHHC5 (Fig 2D) in that there is significantly more DHHS5 at the plasma membrane when co‐expressed with WT Golga7b compared to mutant Golga7b. However, the effect size (difference in plasma membrane localised DHHC5) is smaller than that observed for WT DHHC5 (Fig 2G), and there is significantly less DHHS5 localised to the plasma membrane compared to WT DHHC5 when both are expressed with WT Golga7b (Appendix Fig S5). The fact that any DHHS5 is localised to the plasma membrane could be due to low levels of palmitoylated Golga7b remaining after DHHC5 depletion (Fig 1B) because of residual endogenous DHHC5 activity or compensation by other PATs that palmitoylate Golga7b. However, when endogenous DHHC5 is not depleted, the DHHS5 catalytic mutant when co‐expressed with WT Golga7b is localised at the plasma membrane (Fig EV3C). This demonstrates that over‐expressed Golga7b palmitoylated by endogenous DHHC5 can localise the DHHS5 catalytic mutant to the plasma membrane. The localisation of WT DHHC5 co‐expressed with WT Golga7b is unaffected by the presence or depletion of endogenous DHHC5 as Golga7b is palmitoylated in both of these conditions (Fig EV3A).
Figure EV3. Regulation of DHHC5 localisation by palmitoylation of Golga7b and the C‐terminus of DHHC5.
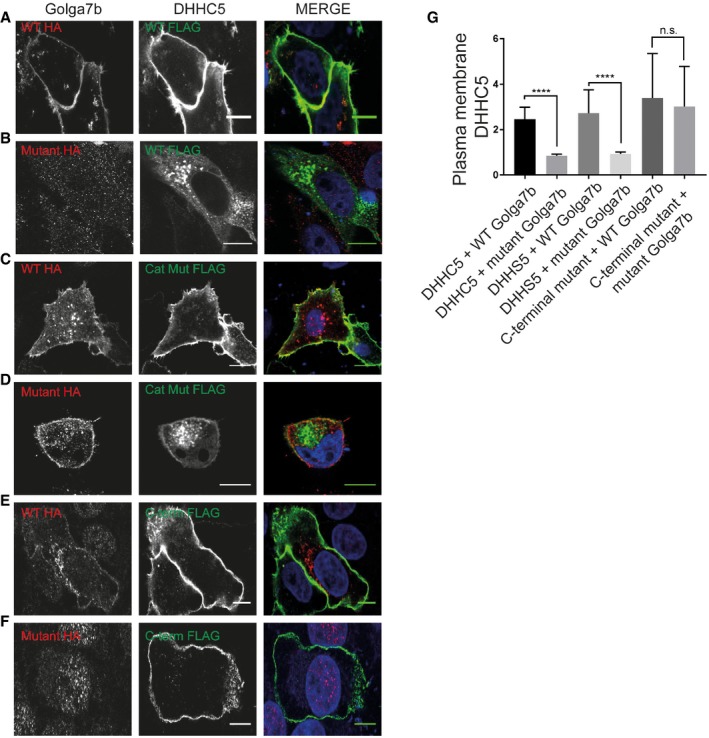
- Confocal images of HeLa cells co‐expressing WT Golga7b (1st column) and DHHC5 (2nd column), with a merged image (3rd column).
- Confocal images of HeLa cells co‐expressing palmitoylation‐deficient mutant Golga7b (1st column) and DHHC5 (2nd column), with a merged image (3rd column).
- Confocal images of HeLa cells co‐expressing WT Golga7b (1st column) and DHHS5 (2nd column), with a merged image (3rd column).
- Confocal images of HeLa cells co‐expressing palmitoylation‐deficient mutant Golga7b (1st column) and DHHS5 (2nd column), with a merged image (3rd column).
- Confocal images of HeLa cells co‐expressing WT Golga7b (1st column) and DHHC5 C‐terminal palmitoylation‐deficient mutant (2nd column), with a merged image (3rd column).
- Confocal images of HeLa cells co‐expressing palmitoylation‐deficient mutant Golga7b (1st column) and DHHC5 C‐terminal palmitoylation‐deficient mutant (2nd column), with a merged image (3rd column).
- Quantification of the membrane signal of DHHC5 in each of the images (A–F). ****P < 0.001, n.s. = not significant, unpaired t‐test. N = 49, DHHC5 + WT Golga7b. N = 53, DHHC5 + mutant Golga7b. N = 40, DHHS5 + WT Golga7b. N = 28, DHHS5 + Golga7b mutant. N = 42, C‐terminal mutant + WT Golga7b. N = 31, C‐terminal mutant + mutant Golga7b. All experiments were repeated 3 individual times; N number refers to the number of cells, and error bars represent SEM.
Next, we investigated the role of palmitoylation at the C‐terminus of DHHC5 and its effect on DHHC5 localisation given that the palmitoylation‐deficient mutant DHHC5 is unable to interact with Golga7b. Unexpectedly, when we co‐expressed the DHHC5 C‐terminal palmitoylation‐deficient mutant in DHHC5‐depleted cells, with either wild‐type or mutant Golga7b (Fig 2E and F), it was localised to the plasma membrane. Furthermore, it was localised to the plasma membrane when over‐expressed alone without Golga7b (Fig EV2D). These data suggest that palmitoylation at the C‐terminus of DHHC5 and the potential changes in local protein structure, as a result, may regulate the internalisation of DHHC5 from the plasma membrane.
Palmitoylation of Golga7b regulates plasma membrane localisation of endogenous DHHC5
In order to demonstrate that Golga7b regulates the localisation of endogenous DHHC5, we expressed WT or mutant Golga7b in HeLa cells and used immunofluorescence microscopy to probe the localisation of endogenous DHHC5 (Fig 3). Expression of WT Golga7b significantly increases levels of endogenous DHHC5 at the plasma membrane compared to endogenous DHHC5 without Golga7b over‐expression (Fig 3A, C and E), indicating that the expression level of Golga7b regulates the amount of plasma membrane localised DHHC5. Interestingly, over‐expression of mutant Golga7b reduces endogenous DHHC5 localisation at the plasma membrane (Fig 3B and F), similar to what we observe when both DHHC5 and mutant Golga7b are over‐expressed. This indicates that the expression of a palmitoylation‐deficient form of Golga7b prevents stabilisation of DHHC5 at the plasma membrane in a dominant negative manner.
Figure 3. Palmitoylated Golga7b stabilises endogenous DHHC5 at the plasma membrane.
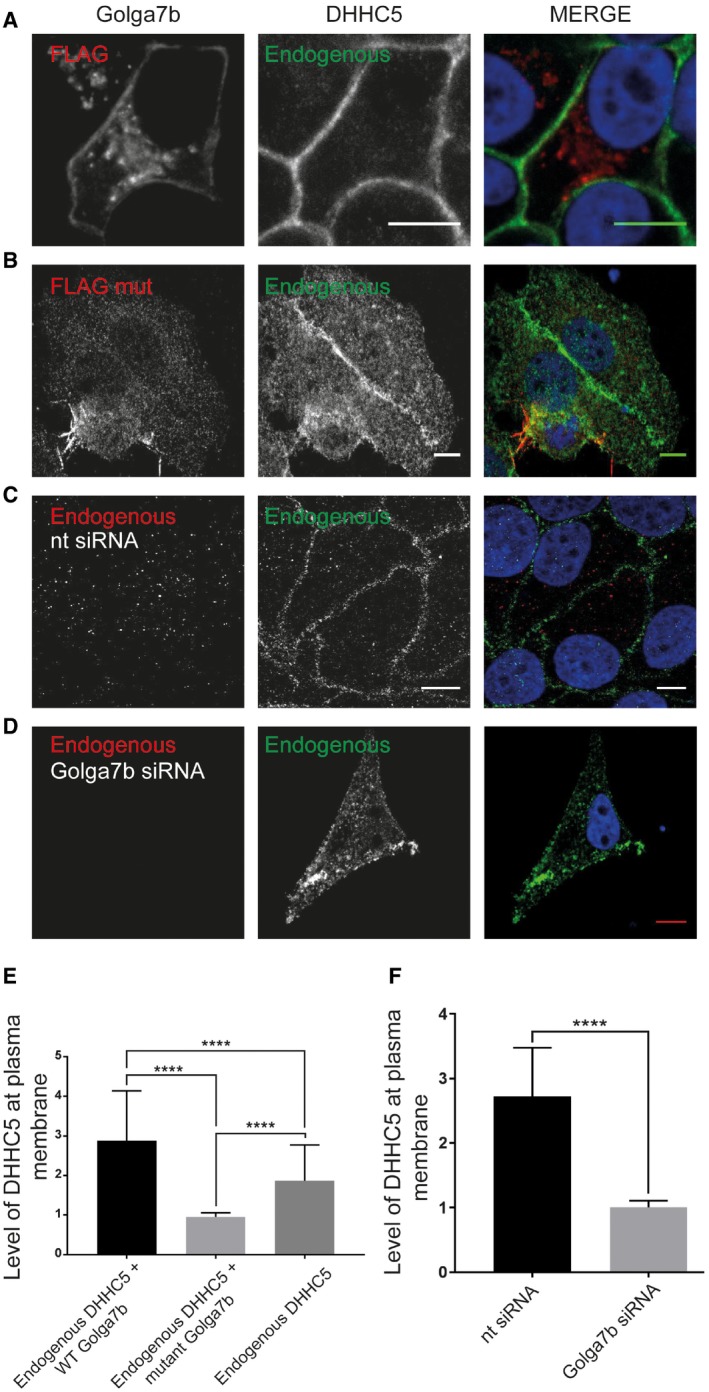
- Confocal images of endogenous DHHC5 in HeLa cells expressing FLAG‐tagged WT Golga7b.
- Confocal images of endogenous DHHC5 in HeLa cells expressing FLAG‐tagged mutant Golga7b.
- Confocal images of endogenous Golga7b and DHHC5 in HeLa cells treated with negative control non‐targeting siRNA.
- Confocal images of endogenous Golga7b and DHHC5 in HeLa cells treated with Golga7b siRNA.
- Quantification of plasma membrane signal of endogenous DHHC5 when co‐expressed with WT or mutant Golga7b or without any transfected Golga7b. ****P < 0.001, t‐test, endogenous + WT Golga7b n = 37, endogenous + mutant Golga7b n = 23, endogenous n = 42. Expression of the palmitoylation‐deficient mutant Golga7b causes endogenous DHHC5 to be internalised in the cell, while WT Golga7b stabilises DHHC5 at the plasma membrane.
- Quantification of the plasma membrane signal of DHHC5 in (C and D), ****P < 0.001 t‐test, control siRNA n = 23, Golga7b siRNA n = 23.
In order to more definitively show that over‐expressed DHHC5 is mislocalised because of insufficient Golga7b levels (Fig EV2B), we depleted Golga7b with siRNA in HeLa cells (Fig 3D). We observe that the localisation of endogenous DHHC5 is significantly reduced at the plasma membrane upon Golga7b depletion, compared to cells treated with a non‐targeting negative control siRNA, where DHHC5 was present almost exclusively at the plasma membrane (Fig 3C and D). This confirms that sufficient levels of Golga7b are required to localise DHHC5 to the plasma membrane. To ensure this was an effect specific to Golga7b, we attempted to rescue the localisation defect of DHHC5 due to Golga7b depletion by expression of the closely related Golga7 (GCP16) (Appendix Fig S2). Over‐expression of Golga7 in Golga7b‐depleted cells failed to restore DHHC5 to the plasma membrane, suggesting that this effect is specific to Golga7b and cannot be significantly rescued by related proteins (Appendix Fig S2).
Golga7b defines DHHC5 interactomes
As Golga7b has such a profound effect on the localisation of DHHC5, we exploited this to investigate whether the interactions of DHHC5 are different when it is present at the plasma membrane compared to when it is localised to endomembranes. To achieve this, a C‐terminal triple‐FLAG‐tagged DHHC5 construct was co‐expressed with either WT or mutant Golga7b in HeLa cells before performing immunoprecipitations. Initially, we sought to prove that DHHC5 would immunoprecipitate proteins differentially when present at the plasma membrane or within the cell. We chose Flot2 to test this as it is a known DHHC5 substrate 13 and is present at the plasma membrane. DHHC5 is able to immunoprecipitate significantly more Flot2 when it is present at the plasma membrane (co‐expressed with WT Golga7b) compared to when it is present at endomembranes (co‐expressed with palmitoylation‐deficient mutant Golga7b) (Fig 4A).
Figure 4. Proteomic analysis of context‐dependent DHHC5‐associated protein complexes.
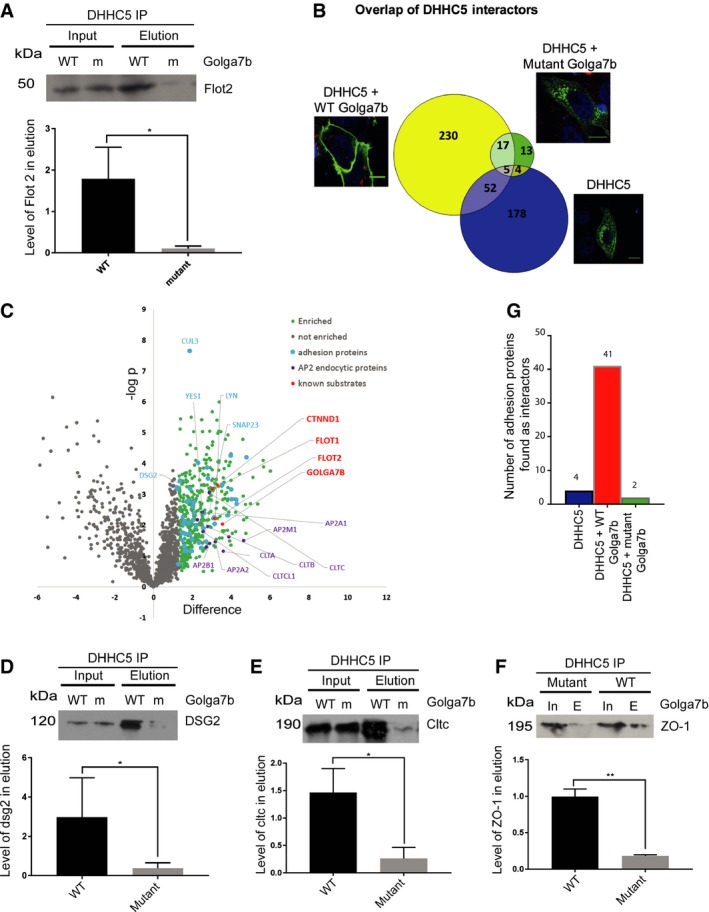
- FLAG‐DHHC5 immunoprecipitations were performed using lysates from Hela cells co‐expressing either wild‐type (WT) or palmitoylation‐deficient mutant (m) Golga7b. The interaction of DHHC5 with its substrate Flot2 was measured by immunoblotting and is significantly reduced in the presence of palmitoylation‐deficient mutant Golga7b, quantification normalised to input, *P < 0.05, ratio paired t‐test, n = 3.
- Volcano plot of DHHC5 interactors from AP‐MS analysis from HeLa cells co‐expressing FLAG‐DHHC5 and WT Golga7b highlighting interacting proteins involved in clathrin‐mediated endocytosis, cell:cell adhesion and known substrates of DHHC5.
- Validation of novel DHHC5 interactors by co‐IP: immunoblots of Dsg2 from Hela cells co‐expressing DHHC5 and either wild‐type (WT) or mutant (m) Golga7b and quantification normalised to input, *P < 0.05, ratio paired t‐test, n = 3.
- Co‐IP immunoblots of clathrin heavy chain from Hela cells co‐expressing DHHC5 and either wild‐type (WT) or mutant (m) Golga7b and quantification normalised to input, *P < 0.05, ratio paired t‐test, n = 3.
- Co‐IP immunoblots of ZO‐1 protein from Hela cells co‐expressing DHHC5 and either wild‐type (WT) or mutant (m) Golga7b and quantification normalised to input, **P < 0.01 ratio paired t‐test, n = 3.
- Bar graph comparing the number of the cell:cell adhesion proteins identified in each protein interaction dataset.
In order to explore this more widely, we used an affinity purification–mass spectrometry (AP‐MS) approach in which DHHC5 complexes were purified and competitively eluted by triple‐FLAG peptide, separated by SDS–PAGE, subjected to in‐gel digestion and analysed by quantitative tandem mass spectrometry. We defined sets of DHHC5 interactors based on their quantitative enrichment in IPs versus control (non‐transfected) purifications when (i) DHHC5 was localised to the plasma membrane (co‐expressed with WT Golga7b), (ii) DHHC5 was localised at endomembranes (co‐expressed with palmitoylation‐deficient mutant Golga7b) or (iii) DHHC5 was localised at endomembranes and the plasma membrane (expressed alone). In order to rule out any effects of addition of MG132 when we co‐expressed DHHC5 with palmitoylation‐deficient mutant Golga7b, we also generated and analysed IPs from cells which we co‐expressed DHHC5 with wild‐type Golga7b ± MG132. Comparison of enriched proteins in these IPs ± MG132 identified only one protein differentially enriched by the addition of MG132, demonstrating that it did not significantly affect the set of DHHC5 interactors.
Data from these experiments revealed that DHHC5 interacts with many more proteins when localised at the plasma membrane by wild‐type Golga7b (304 proteins; Fig 4B, Dataset EV1) compared to when it is expressed with mutant Golga7b when it is localised to endomembranes (39 proteins; Dataset EV2), with a small number of proteins common to both conditions (22 proteins) (t‐test with a permutation‐based FDR 0.05). DHHC5 does interact with a large number of proteins at the plasma membrane; however, this is not uncommon for enzymes which may have a large number of substrates. We identified several known substrates of DHHC5 including delta‐catenin and Flot2, which suggests that our AP‐MS approach allows the discovery of new substrates of PATs. However, this may not be viable in all cases as it is known that some PATs only interact weakly with some of their substrates 28.
When DHHC5 is expressed alone (Dataset EV3), it interacts with some proteins also identified when it was co‐expressed with WT Golga7b (52 proteins). Among these are members of the AP2 complex, which is involved in certain types of clathrin‐mediated endocytosis and has been shown to be involved in endocytosis of DHHC5 11. When DHHC5 is expressed alone, there are many more cytoplasmic proteins identified as interactors compared to when it is co‐expressed with WT Golga7b; this is likely caused by the mislocalisation of DHHC5 in the absence of exogenous Golga7b.
When we directly compare the interaction datasets from DHHC5 co‐expressed with WT or mutant Golga7b, a number of proteins are differentially enriched; nine were enriched in the DHHC5/mutant Golga7b dataset and 163 in the DHHC5/WT Golga7b dataset (t‐test with a permutation‐based FDR 0.05). This clearly demonstrates the profound effect that Golga7b has on the interactome of DHHC5. To validate this, we chose three proteins that were differently enriched between the WT and mutant Golga7b conditions that were not known as DHHC5 interactors or substrates. Dsg2, a desmosomal cadherin; clathrin heavy chain; and tight junction protein ZO‐1 interacted with DHHC5 significantly more when DHHC5 was at the plasma membrane (co‐expressed with WT Golga7b) compared to when it was at endomembranes (co‐expressed with mutant Golga7b) (Fig 4D–F), demonstrating that these proteins are bona fide DHHC5 interactors and that our AP‐MS results are reliable.
Among the set of proteins that were significantly enriched when DHHC5 was expressed with WT Golga7b compared to mutant Golga7b were a number of proteins associated with clathrin‐mediated endocytosis, including three major members of the AP2 complex (Fig 4C), plus the clathrin light and heavy chains. This indicates that the endocytosis of DHHC5 may be mediated by this complex, as DHHC5 is known to be actively trafficked away from the plasma membrane in neurons 11 and has a number of endocytic sequences in its C‐terminal tail. It is likely that DHHC5 interacts with the AP2 complex more when it is at the plasma membrane compared to when it is within the cell, as the clathrin coat and associated proteins are shed from the endocytic vesicle rapidly after the vesicle buds from the membrane 29. When looking at the proteins that interact with DHHC5 when it is co‐expressed with WT Golga7b, nine of the top ten over‐represented gene ontology terms are involved with cell:cell or cell:matrix adhesion (Fig 4G). While it is known that a number of proteins involved in adhesion are palmitoylated 21, 22, 23, the PATs responsible are mostly unknown. Given the large number of proteins immunoprecipitated by DHHC5, particularly in the desmosomal cell adhesion complex, we hypothesised that DHHC5 is the PAT responsible for the palmitoylation of these proteins and could be a key player in regulating cellular adhesion. In particular, it was noted that the desmosomal cadherin Dsg2, which is a known palmitoylated protein 23, and the tight junction protein ZO‐1 are both interactors of DHHC5, which opens up the possibility that palmitoylation by DHHC5 is an important regulatory process in these two different types of cell adhesion junctions.
Palmitoylation of Golga7b prevents endocytosis of DHHC5
As several of the clathrin‐mediated endocytosis complex members were found as interactors of DHHC5 in our AP‐MS study (Fig 4C), we decided to further investigate the mechanism of DHHC5 internalisation and endocytosis in the context of Golga7b. We first investigated whether it was possible to rescue mutant Golga7b‐induced mislocalisation of DHHC5 by inhibiting endocytosis with the dynamin inhibitor dynasore. When HeLa cells expressing WT DHHC5 and mutant Golga7b are treated with 25 μM dynasore, the majority of DHHC5 is stabilised at the plasma membrane after a relatively short time (within 30 min) (Fig 5A, B and E). This shows that DHHC5 is rapidly trafficked to and from the plasma membrane in other cell types besides neurons 11. It also suggests a role for Golga7b in stabilising DHHC5 at the plasma membrane by preventing endocytosis.
Figure 5. Inhibition of endocytosis restores DHHC5 to the plasma membrane in the presence of mutant Golga7b.
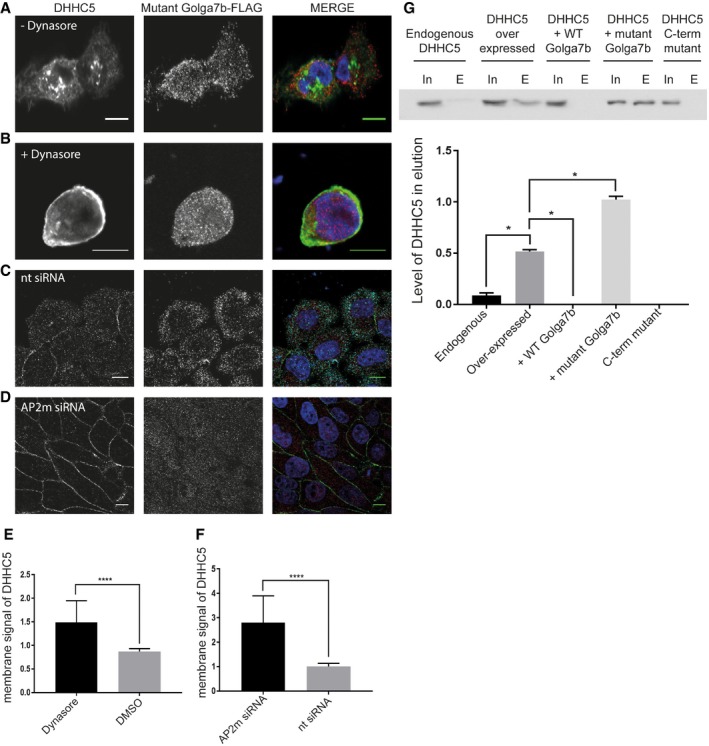
- Thirty‐minute DMSO treatment of HeLa cells co‐expressing WT DHHC5 (FLAG‐tagged) and mutant Golga7b (HA‐tagged).
- Thirty‐minute dynasore treatment (25 μM) of HeLa cells co‐expressing WT DHHC5 (FLAG‐tagged) and mutant Golga7b (HA‐tagged).
- Non‐targeting control siRNA treatment (10 nM) of HeLa cells expressing mutant Golga7b (HA‐tagged). Images are detecting endogenous DHHC5 and the HA‐tagged Golga7b.
- AP2μ siRNA treatment (10 nM) of HeLa cells that were subsequently transfected with mutant Golga7b (HA‐tagged). Images are detecting endogenous DHHC5 and the HA‐tagged Golga7b.
- Quantification of plasma membrane signal of DHHC5 in (A and B). ****P < 0.001, unpaired t‐test. N = 37, +dynasore. N = 28, +DMSO.
- Quantification of plasma membrane signal of DHHC5 in (C and D). ****P < 0.001, unpaired t‐test.
- Biotin endocytosis assays of HeLa cells with endogenous DHHC5, over‐expression of DHHC5 alone, co‐expression of DHHC5 and WT Golga7b, co‐expression of DHHC5 and mutant Golga7b or expression of the DHHC5 C‐terminal mutant. Biotinylated cell surface proteins endocytosed within 15 min were purified using streptavidin resin and blots probed with an antibody to DHHC5. The lack of signal in the pull‐down of the DHHC5 C‐terminal mutant or when DHHC5 and WT Golga7b are co‐expressed indicates that DHHC5 is stabilised at the plasma membrane in these conditions. The robust signal in the pull‐down of endocytosed proteins from cells co‐expressing DHHC5 and mutant Golga7b shows a high rate of endocytosis of DHHC5 in this condition. N = 3, *P < 0.05, paired t‐test.
As dynasore may have some off‐target effects 30 and does target not only clathrin‐mediated endocytosis but all types of endocytosis that require dynamin, we confirmed that DHHC5 is trafficked by clathrin‐mediated endocytosis using siRNA depletion of the AP2μ subunit, which we found as an interactor of DHHC5 in the AP‐MS experiments. When we depleted AP2μ in HeLa cells co‐expressing WT DHHC5 and mutant Golga7b, DHHC5 was restored to the plasma membrane, while in cells treated with a control non‐targeting siRNA, DHHC5 remained mislocalised (Fig 5C, D and F). This confirms that DHHC5 requires AP2μ for endocytosis from the plasma membrane 11 and that palmitoylation of Golga7b prevents endocytosis of DHHC5. Importantly, this demonstrates conclusively that palmitoylation‐deficient Golga7b does not prevent DHHC5 trafficking to, or association with, the plasma membrane, but instead that palmitoylated Golga7b acts by stabilising DHHC5 at the plasma membrane.
As the localisation of DHHC5 can be altered by preventing the palmitoylation of Golga7b or by DHHC5 itself (Fig 2), we wanted to directly investigate whether the changes observed were due to changes in the endocytosis of DHHC5. A biotin‐based endocytosis assay showed that mutant Golga7b significantly increases the rate of endocytosis whereas WT Golga7b reduces endocytosis to essentially zero over the 30‐min time course used in the assay (Fig 5G). Furthermore, preventing palmitoylation of DHHC5 abolished endocytosis of DHHC5, which bolsters the idea that palmitoylation of DHHC5 is essential for the retrieval of DHHC5 from the plasma membrane. This shows that palmitoylation of Golga7b and depalmitoylation of DHHC5 both prevent endocytosis of DHHC5.
DHHC5 and Golga7b regulate desmosome trafficking and assembly
As a number of proteins involved in cell adhesion were found as DHHC5 interactors in our AP‐MS study (Fig 4C), we wanted to investigate whether any of these proteins were substrates of DHHC5. Among these is the important desmosomal cadherin Dsg2 (Fig 4D). Palmitoylation of Dsg2 regulates its localisation and trafficking to the plasma membrane 24, but the identity of the PAT that mediates this is unknown. Because of this, we decided to investigate whether DHHC5 is the PAT responsible for Dsg2 palmitoylation by performing ABE assays on DHHC5‐depleted and control siRNA‐treated HeLa cells (Fig 6A). We found that upon DHHC5 depletion, Dsg2 palmitoylation is almost completely abolished, showing that DHHC5 is the PAT responsible for the majority, if not all, of Dsg2 palmitoylation. This contradicts previous work which hypothesised that Dsg2 is palmitoylated at the Golgi or somewhere within the trafficking pathway 24. To confirm that DHHC5 palmitoylates Dsg2 at the plasma membrane, we performed an ABE experiment where A431 cells were cultured in normal conditions or in low calcium, where desmosomal components are internalised and no longer at the plasma membrane. At low calcium, there was a significant decrease in palmitoylation of Dsg2, which is consistent with Dsg2 being palmitoylated at the plasma membrane, where DHHC5 is resident (Fig 6F).
Figure 6. Desmoglein‐2 palmitoylation by DHHC5/Golga7b regulates its plasma membrane localisation and incorporation into desmosomes.
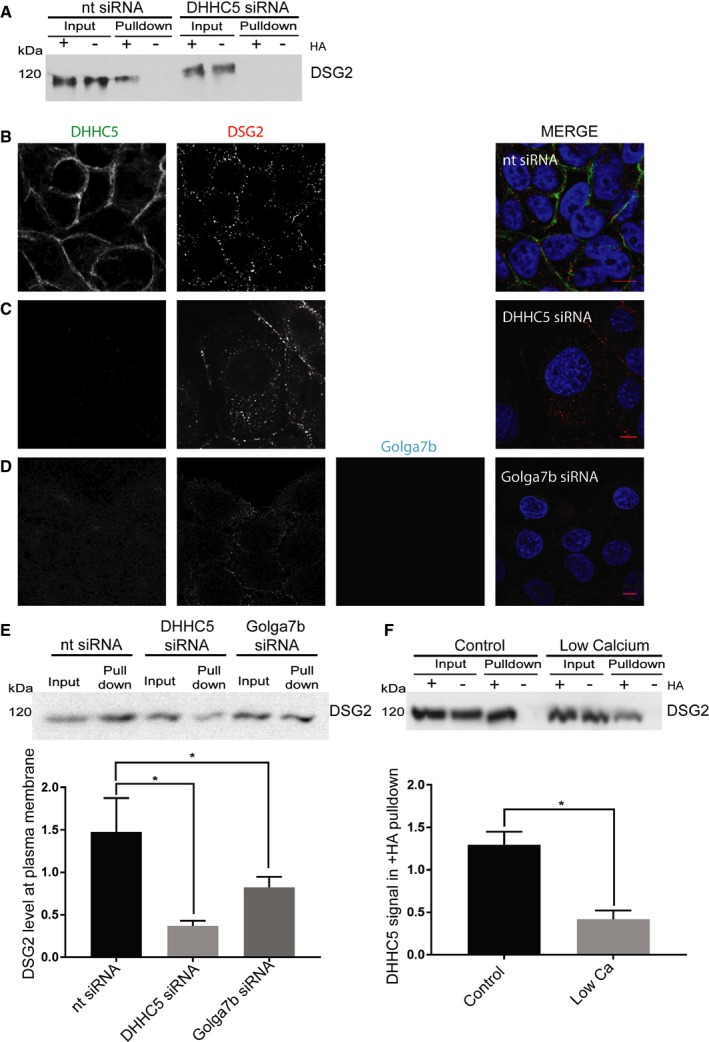
- ABE purifications from HeLa cells treated with 10 nM non‐targeting siRNA (nt siRNA) or DHHC5 siRNA were immunoblotted for Dsg2. Loss of the band in the +HA pull‐down in the DHHC5 siRNA condition shows that Dsg2 palmitoylation is dependent on DHHC5.
- Confocal images of DHHC5 and Dsg2 in nt siRNA‐treated A431 cells 3 h after calcium was reintroduced to the medium in calcium switch experiments.
- Confocal images of DHHC5 and Dsg2 in DHHC5‐depleted A431 cells 3 h after calcium was reintroduced to the medium in calcium switch experiments.
- Confocal images of DHHC5, Dsg2 and Golga7b in Golga7b‐depleted A431 cells 3 h after calcium was reintroduced to the medium in calcium switch experiments.
- Immunoblots and quantification of Dsg2 pulled down in cell surface biotinylation assays in nt siRNA, DHHC5 siRNA or Golga7b siRNA treated conditions. Quantification of the intensity of the Dsg2 signal in the pull‐down is normalised to input. A significant reduction of cell surface Dsg2 occurs when DHHC5 or Golga7b is depleted by siRNA. *P < 0.05, paired t‐test. N = 4; error bars represent SEM.
- Immunoblot of an ABE purification for Dsg2 in cells with either normal medium or low‐calcium (5 μM calcium) medium, with quantification of the intensity of the DSG2 signal in the pull‐down normalised to input. There is a significant reduction in the Dsg2 signal in the +HA pull‐down from low‐calcium cells, showing that Dsg2 is significantly less palmitoylated in this condition. *P < 0.05, paired t‐test, N = 4; error bars represent SEM.
Given that palmitoylation of Dsg2 regulates its localisation to the plasma membrane and its trafficking to desmosomes, we investigated the effect of DHHC5 knockdown on the localisation and trafficking of Dsg2 using immunofluorescence microscopy of A431 cells with DHHC5 depletion and calcium switch assays. In this assay, the calcium dependence of desmosome assembly is exploited; cells are cultured in a medium that is free from calcium so that desmosomes are rapidly internalised and then trafficked back to the plasma membrane when calcium is reintroduced. In A431 cells treated with a non‐targeting negative control siRNA, Dsg2 returned to the plasma membrane gradually over the 3‐h time course when calcium was added to the medium (Figs 6B and EV4). However, when DHHC5 was depleted, Dsg2 signal remained within the cell, with no significant change in the level of Dsg2 at the plasma membrane during the 3‐h time course (Figs 6C and EV5, quantification in Appendix Fig S6). This shows that depletion of DHHC5 significantly inhibits the trafficking to or stabilisation of Dsg2 at the plasma membrane. While in our experiments DSG2 was never completely depleted from the plasma membrane at low calcium, a significant portion of the protein was internalised from the plasma membrane which allowed us to assay the return of DSG2 to the plasma membrane over time. This residual plasma membrane signal has been observed in other calcium switch studies of DSG2 31.
Figure EV4. Calcium switch assay of A431 cells treated with non‐targeting siRNA.
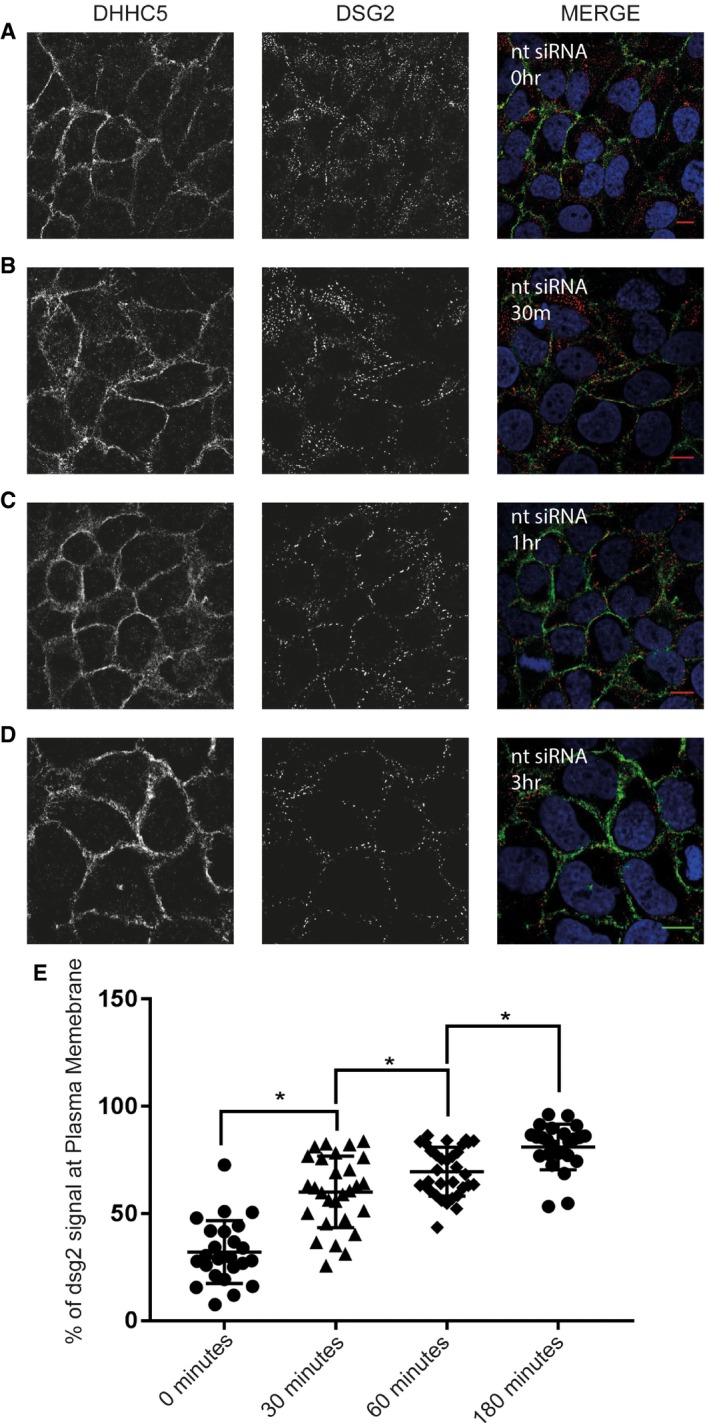
- Time zero after calcium chloride addition.
- Thirty minutes after calcium chloride addition.
- One hour after calcium chloride addition.
- Three hours after calcium chloride addition.
- Percentage of Dsg2 signal seen at the plasma membrane at each time point, *P < 0.05, unpaired t‐test. N = 3 individual experiments, n = 25 cells at 0‐h time point, n = 30 cells at 0‐h time point, n = 34 cells at 1‐h time point, n = 26 cells at 3‐h time point. Error bars represent SEM.
Figure EV5. Calcium switch assay of A431 cells treated with DHHC5 siRNA.

- Time zero after calcium chloride addition.
- One hour after calcium chloride addition.
- Three hours after calcium chloride addition.
- Percentage of Dsg2 signal seen at the plasma membrane at each time point. N = 3 individual experiments, n = 23 cells at 0‐h time point, n = 16 cells at 1‐h time point, n = 24 cells at 3‐h time point. Error bars represent SEM.
In order to establish if Golga7b plays a functional role in desmosome assembly through its regulation of DHHC5 localisation, we determined the effect of Golga7b depletion on the localisation of Dsg2. As with DHHC5, A431 cells were treated with Golga7b siRNA and a calcium switch assay performed. Again, after 3 h of calcium being returned to the medium, Dsg2 had not returned to the plasma membrane at the same level as cells treated with a non‐targeting siRNA (Fig 6D). This links DHHC5 localisation to the correct trafficking of Dsg2 and implicates Golga7b in the regulation of DHHC5 substrates. In order to validate this finding, we performed cell surface biotinylation experiments 3 h after calcium reintroduction (Fig 6E). This showed very similar results to the quantification of the confocal images showing that depletion of DHHC5 leads to a decrease in Dsg2 in the plasma membrane. Removal of Golga7b by siRNA also causes a significant drop in Dsg2 at the plasma membrane, although to a lesser extent observed with DHHC5 depletion.
The Dsg2 localisation phenotype is similar to that seen when palmitoylation of the desmosomal protein Pkp3 is prevented 23. Pkp3 was not found as an interactor of DHHC5, but the localisation of Dsg2 after DHHC5 depletion is similar to palmitoylation‐deficient Pkp3 mutants 23. We treated HeLa cells with either DHHC5 siRNA or non‐targeting control siRNA, performed ABE assays to measure palmitoylation levels of Pkp3 and found that there was a significant and almost total loss of palmitoylation of Pkp3 compared to a non‐targeting siRNA (Appendix Fig S3). This shows that Pkp3 is a DHHC5 substrate and makes it likely that the phenotype seen with Dsg2 localisation and trafficking is caused by a combination of the loss of palmitoylation of both Dsg2 and Pkp3.
DHHC5 and Golga7b regulate cell adhesion
To investigate whether DHHC5 plays a more general role in cell adhesion, or if the localisation and palmitoylation defects observed in desmosomes are sufficient to cause a loss of cell adhesion, we used a modified form of a cell scatter assay. When cells are exposed to a growth factor, they internalise their cell: cell and cell:matrix adhesion proteins which cause cells to scatter away from the colonies they naturally form and mimic the epithelial‐to‐mesenchymal transition. All cell adhesion proteins are targeted in this assay, but a deficiency in one of more aspects of the adhesion machinery will lead to a change in scattering. The speed and distance of this scattering are dependent on cell adhesion, with reduced adhesion leading to enhanced cell scattering 32. When A431 cells were subject to DHHC5 depletion before treatment with low levels of EGF (5 ng/ml), cells scattered noticeably (Fig 7B and C), whereas control non‐targeting siRNA‐treated cells remained in colonies when treated with the same concentration of EGF (Fig 7A and C). This shows that depletion of DHHC5 leads to a reduction of adhesion of cells to one another and implicates DHHC5 in a wider role in cell:cell adhesion. However, it must also be noted that this increase in scattering could also be caused by the inhibition of cell:matrix adhesion as well as cell:cell adhesion.
Figure 7. Cell adhesion is regulated by DHHC5/Golga7b.
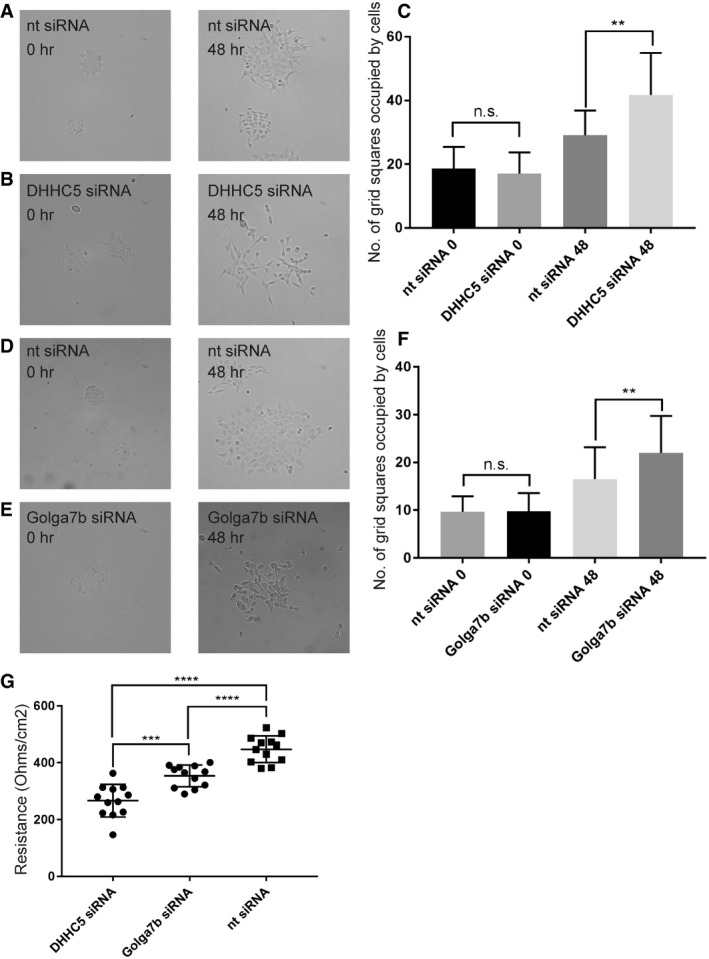
- 10× brightfield images of A431 cells treated with non‐targeting siRNA at time 0 (left) and after 48 h (right) of 5 ng/ml EGF treatment.
- 10× brightfield images of A431 cells treated with DHHC5 siRNA at time 0 (left) and after 48 h (right) of 5 ng/ml EGF treatment.
- Quantification of cell scattering of A431 cells, showing a significant increase in cell scatter after 48 h of DHHC5 knockdown and therefore a decrease in cell adhesion. N = 3 individual repeats, n = 31 fields of view. **P < 0.005, unpaired t‐test.
- 10× brightfield images of A431 cells treated with non‐targeting siRNA at time 0 (left) and after 48 h (right) of 5 ng/ml EGF treatment.
- 10× brightfield images of A431 cells treated with Golga7b siRNA at time 0 (left) and after 48 h (right) of 5 ng/ml EGF treatment.
- Quantification of cell scattering of A431 cells, showing a significant increase in cell scatter after 48 h of Golga7b knockdown and therefore a decrease in cell adhesion. N = 3 individual repeats, n = 31 fields of view. **P < 0.005, unpaired t‐test.
- Plot showing the resistance of A431 cells from a trans‐epithelial electrical resistance assay treated with either DHHC5 siRNA, Golga7b siRNA or non‐targeting siRNA. A significant reduction in TEER demonstrates a decrease in cell adhesion after both DHHC5 and Golga7b depletion. N = 12, ****P < 0.001, unpaired t‐test, ***P < 0.01, unpaired t‐test.
To test if depletion of Golga7b mirrors the effects seen with DHHC5 depletion in the calcium switch assay, we performed cell scatter assays on Golga7b‐depleted A431 cells (Fig 7D, E and F). We observed enhanced scattering of cells upon Golga7b depletion compared to control cells exposed to a non‐targeting siRNA. This shows Golga7b has a functional role in cell adhesion which is likely to be caused by its regulation of DHHC5 localisation. Given that we have shown that DHHC5 interacts not only with Dsg2 but also with tight junction protein ZO‐1 (Dataset EV2, Fig 4F), it is possible that the wider effects that DHHC5 depletion has on cell adhesion could be caused by disruption of both desmosomes and tight junctions.
To further demonstrate a functional role of DHHC5 and Golga7b in cell adhesion, we performed trans‐epithelial electrical resistance (TEER) assays. This involves the growth of cells on a semi‐permeable membrane support and measurement of the electrical resistance across this membrane; the stronger the cell adhesion, the higher the resistance measurement will be 33. When we performed this assay on confluent A431 cells that were treated with either DHHC5 or negative control non‐targeting siRNA, we observed a significant reduction in electrical resistance in cell cultures treated with DHHC5 or Golga7b siRNA (Fig 7G). This shows that the adhesion between the cells is reduced in these conditions, highlighting that the DHHC5/Golga7b complex is an important regulator of the cell adhesion machinery. Given that cell:cell adhesion is measured in TEER assays, this finding supports the idea that that the changes observed in the cell scatter assays were due to changes in the cell:cell rather than cell:matrix adhesion machinery.
Discussion
The regulation of subcellular localisation of many PATs is poorly understood, and DHHC5 is unique among them as it is the only known PAT to exhibit dynamic changes in localisation in response to external stimuli 11. While it has been shown in neurons that the localisation of DHHC5 is regulated by phosphorylation, we have shown that the localisation of DHHC5 is also regulated by the interaction and palmitoylation of an accessory protein, Golga7b. It is notable that the over‐expression of palmitoylation‐deficient mutant Golga7b causes mislocalisation of endogenous DHHC5, which suggests that Golga7b has a dominant negative effect on DHHC5. Over‐expression of WT Golga7b increases localisation of endogenous DHHC5 at the plasma membrane, which further strengthens the argument that the level of Golga7b within the cell is an important determinant of DHHC5 localisation. The catalytically dead DHHC5 (DHHS5) mutant localises to the plasma membrane when co‐expressed with Golga7b in the presence of endogenous DHHC5, but this localisation is significantly reduced in cells depleted of endogenous DHHC5. This shows that DHHC5‐mediated palmitoylation of Golga7b is required for stabilisation of DHHC5 at the plasma membrane.
Palmitoylation of sites in the C‐terminus of DHHC5 is required for its interaction with Golga7b and stabilisation at the plasma membrane, and mutation of these sites prevents its internalisation (Fig 8). Sorting signals in the C‐terminal tails of PATs are important factors in controlling their localisation and are able to switch the localisation of a PAT from the Golgi to the ER 34. Our results suggest that palmitoylation of DHHC5 is not required for correct trafficking of DHHC5 to the plasma membrane nor for the interaction between DHHC5 and the endocytic machinery, as the known endocytic interaction motifs are unaffected by these mutations 11. The data presented here suggest that when palmitoylated DHHC5 is not bound to Golga7b, it is endocytosed. Preventing palmitoylation of the GluR1 subunit of the AMPA receptor inhibits its endocytosis 35, which is similar to what we propose for DHHC5. Interestingly, the three palmitoylation sites on the C‐terminal tail of DHHC5 map to an amphipathic alpha‐helix in the corresponding crystal structure of DHHC20 36, and this palmitoylation could potentially pin this helix to the plasma membrane, thereby altering the conformation of the C‐terminal tail. As the majority of DHHC5 is palmitoylated on two or more sites in vivo 10, it suggests that palmitoylation could play a major role in the regulation of cell surface expression of DHHC5.
Figure 8.
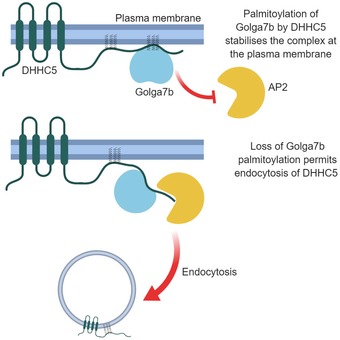
Proposed model of how palmitoylation of Golga7b regulates plasma membrane localisation of DHHC5.
The observation that DHHC5 requires interaction with Golga7b for stabilisation at the plasma membrane appears to be in conflict with the finding that C‐terminal DHHC5 mutant, which does not interact with Golga7b, is stabilised at the plasma membrane. The DHHC5 C‐terminal mutant, while being unable to interact with Golga7b, is unable to be internalised because it is not palmitoylated on its C‐terminus. This raises the possibility that the interaction with Golga7b functions to protect C‐terminally palmitoylated DHHC5 from endocytosis and suggests that palmitoylation of the C‐terminal tail of DHHC5 is an endocytic trigger for the protein (Fig 8).
Activity‐dependent trafficking of DHHC5 has been described to occur in neurons after long‐term potentiation 11. Here, we have confirmed that DHHC5 is endocytosed from the plasma membrane by the AP2‐regulated clathrin‐mediated endocytosis pathway in HeLa cells. Interestingly, the results presented here suggest that DHHC5 is trafficked as part of its normal life cycle and does not require stimulation of cells in any way, as DHHC5 is restored to the plasma membrane within 30 min of dynasore treatment (Fig 5). This could be a general feature of DHHC5 in all cell types, or it could have different cycling dynamics in various specialised cell types. Importantly, inhibition of endocytosis corrects the localisation defect when DHHC5 is co‐expressed with mutant Golga7b. This demonstrates that the palmitoylation‐deficient Golga7b does not prevent trafficking of DHHC5 to the plasma membrane. Instead, Golga7b acts to stabilise DHHC5 at the plasma membrane by preventing its internalisation.
Our unbiased AP‐MS experiments uncovered a new role for DHHC5 in cell adhesion. Palmitoylation regulates several proteins involved in cell:cell and cell:matrix adhesion 20, 21, 22, 23, and for many of them, palmitoylation regulates their localisation. Among cell adhesion complexes, desmosomes are notable for the number of proteins in the complex that are palmitoylated, with at least 6 members of the complex palmitoylated in A431 cells 23. In this work, we have shown that DHHC5 is the PAT responsible for the palmitoylation of the key complex members Dsg2 and Pkp3. We have also demonstrated that knockdown of DHHC5 affects the trafficking and localisation of Dsg2, which had been demonstrated previously to be dependent on Dsg2 palmitoylation, albeit with a noticeably milder phenotype than we observed 24. It is also notable that the knockdown of Golga7b also caused defects in Dsg2 localisation and trafficking, demonstrating a functional role for Golga7b in cell adhesion. Our data imply that plasma membrane localisation of DHHC5 is required for efficient Dsg2 palmitoylation and either trafficking or incorporation into desmosomes, or possibly both of these processes. However, these results contradict previous work which suggested that palmitoylation of Dsg2 occurs at the Golgi, which facilitates trafficking to the plasma membrane 24. The reasons for this apparent contradiction are unclear, but the same study showed that palmitoylation‐deficient Dsg2 is still able to reach the plasma membrane. This indicates that palmitoylation of Dsg2 is not strictly required for trafficking to the plasma membrane and that once Dsg2 reaches the plasma membrane, it is palmitoylated by DHHC5, which facilitates its incorporation and therefore stabilisation in desmosomal complexes. However, DHHC5 appears to cycle between the plasma membrane and endomembranes, making it possible that Dsg2 is palmitoylated by DHHC5 at non‐plasma membrane sites, similar to what has been demonstrated for δ‐catenin at dendritic spines 11. This is a less likely scenario, however, when we consider our data from low‐calcium conditions, which cause Dsg2 to be localised away from the plasma membrane and therefore away from DHHC5, leading to a significant reduction in palmitoylation of Dsg2.
Furthermore, we have determined that Pkp3 is also a DHHC5 substrate, and as it has been shown previously that palmitoylation of Pkp3 regulates the localisation of Dsg2 as well as a number of other desmosomal components 23, this would indicate that DHHC5 regulates the localisation and assembly of desmosomes more generally. The finding that Pkp3 is a DHHC5 substrate also helps to explain why the localisation and trafficking phenotype of Dsg2 upon DHHC5 knockdown is more severe than when Dsg2 palmitoylation alone is prevented. Loss of Pkp3 palmitoylation has a profound effect on the localisation of Dsg2 24. These findings reveal a completely novel role for DHHC5 in the regulation of desmosome function.
We established a broader role for DHHC5 in cell adhesion by observing increased scattering of DHHC5‐depleted cells due to a reduction in cell adhesion. To our knowledge, this is the first report of a specific PAT that directly regulates the palmitoylation of essential adhesion proteins and that its depletion leads to defects in cell adhesion. The link between DHHC5 and adhesion is not without precedent, however, as a number of known DHHC5 substrates have roles in cell adhesion. Flot2 regulates the stability of Dsg3 at the plasma membrane, and depletion of Flot2 leads to reduced desmosomal adhesion 37. As DHHC5 can palmitoylate Flot2 palmitoylation 13, it is possible that DHHC5 could have a role in desmosomal adhesion through this pathway in addition to palmitoylation of Pkp3 and Dsg2 as demonstrated in this work. However, palmitoylation of Flot2 was not completely lost upon depletion of DHHC5 in our experiments, and our data from PEG switch assays suggest that Flot2 is still palmitoylated at one to two sites (Appendix Fig S4). This is in contrast to the complete loss (below limit of detection) of palmitoylation of Dsg2 and Pkp3.
Another DHHC5 substrate, δ‐catenin, competes with p120ctn to interact with the adhesion protein E‐cadherin 38. δ‐Catenin binding to E‐cadherin reduces p120ctn at cell junctions and inhibits adhesion, and E‐cadherin binds to desmogleins 2 and 3 to promote desmosome formation 39. This suggests that DHHC5 could influence the assembly, stability and location of desmosomes through a number of mechanisms, and this work shows that it has a direct regulatory role for at least two members of the desmosomal protein complex.
The regulation of cell adhesion by DHHC5 has some potentially interesting implications for DHHC5 in disease. Disruption of desmosomes in the heart is known to lead to the serious heart condition arrhythmogenic right ventricular cardiomyopathy (ARVC) 40. To the best of our knowledge, palmitoylation of desmosomal proteins has not been implicated in ARVC to date, but given that DHHC5 has been shown to be enriched in the heart 3, it is possible that palmitoylation of desmosomal proteins by DHHC5 in the heart could play a role in this condition. It also opens up the possibility of DHHC5 being used as a marker for potential cancer metastasis, as the results presented here suggest that a lower level of DHHC5 in a cell would lead to an increased chance of metastasis due to reduced cell adhesion.
In conclusion, the results presented in this work establish a new mechanism of DHHC5 localisation through the action of Golga7b and implicate DHHC5 and Golga7b as important members of cellular adhesion machinery. Going forward, the elucidation of exactly how the interaction between Golga7b and DHHC5 is mediated will be an important question to address to fully understand this novel regulatory mechanism. It will also be important to investigate the triggers that control the endocytosis of DHHC5 and how palmitoylation and phosphorylation of DHHC5 work in concert to control its presence at the plasma membrane.
Materials and Methods
Cell culture
All cells were grown in normal DMEM (Gibco) supplemented with 1% penicillin/streptomycin (Gibco), except for during calcium switch experiments, when A431 cells were grown in calcium‐free DMEM (Gibco). All transfection was carried out using polyethylenimine (PEI, 25 kDa linear; Alfa Aesar) using a ratio of 1:3 of DNA:PEI with cells allowed to grow for 24 h after transfection before harvesting or fixing unless otherwise stated. For siRNA treatments, siRNA was mixed with the MISSION siRNA Transfection Reagent (Sigma‐Aldrich) and incubated for 30 min at room temperature before the mixture was added to cells. The siRNAs used were DHHC5, Golga7b, AP2μ, and universal non‐targeting control (all from Dharmacon, SMARTpool).
Constructs
The N‐terminally HA‐tagged DHHC5 construct was a kind gift from Dr Andrew Peden. All other constructs were ordered from GenScript with either human DHHC5 (Gene ID 25921) or human Golga7b (Gene ID 401647) as template. All ORF clones were under the CMV promoter in the pcDNA3.1+ vector, which had either a FLAG or HA tag following the protein coding region before the stop codon. siRNA was obtained from Dharmacon. Cysteine residues mutated in the Golga7b constructs were at positions 33, 78, 81, 90 and 141. DHHC5 residues mutated were at position 134 for catalytic mutant and positions 236, 237 and 245 for C‐terminal mutant.
Antibodies
Anti‐DHHC5 (Atlas Antibodies), for ICC 1:250, for immunoblotting 1:1,000. Anti‐Golga7b (Bioss Antibodies), for ICC 1:100, for immunoblotting 1:1,000. Anti‐FLAG (Sigma‐Aldrich), for ICC 1:500, for immunoblotting 1:1,000. Anti‐HA (Sigma‐Aldrich), for ICC 1:200. Anti‐AP2μ (GeneTex), for ICC 1:150, for immunoblotting 1:1,000. Anti‐Dsg2 (Santa Cruz Biotech), for ICC 1:200, for immunoblotting 1:250. Anti‐Pkp3 (Santa Cruz Biotech), for immunoblotting 1:200. Anti‐Flot2 (Cell Signalling Technology), for immunoblotting 1:1,000. Anti‐actin (Cell Signalling Technology), for immunoblotting 1:2,500. Anti‐rabbit HRP (Thermo Fisher Scientific), for immunoblotting 1:2,500. Anti‐mouse HRP (Cell Signalling Technology), for immunoblotting 1:2,500. Anti‐mouse Alexa Fluor 488 (Invitrogen), for ICC 1:2,500. Anti‐rabbit Alexa Fluor 594 (Invitrogen), for ICC 1:2,500. Anti‐chicken Alexa Fluor 647 (Invitrogen), for ICC 1:2,500.
Acyl‐biotin exchange
Cell pellets were collected and resuspended in lysis buffer (2.5% SDS, 50 mM Tris pH 7.2, 50 mM NaCl, 1 mM EDTA) and then homogenised using an electric homogeniser for 15 s. Lysates were heated at 70°C for 15 min before being centrifuged at 16,000× g for 10 min and the supernatants retained. After cooling, maleimide (Sigma‐Aldrich) was added to 100 mM final concentration and incubated for 3 h at 40°C with shaking. Samples were then acetone‐precipitated by the addition of 4× volume of −20°C acetone and resuspended twice in lysis buffer with five washes with ice‐cold acetone after each precipitation to remove any excess maleimide. Lysates were split into + and − hydroxylamine (HA) treatment conditions. The +HA samples had 2M hydroxylamine/50 mM Tris pH 7.4 added to a final hydroxylamine concentration of 1M, while the −HA samples had an equal volume of 50 mM Tris pH 7.4 added. HA was made fresh immediately before use and prepared by dissolving hydroxylamine in distilled water before the pH was adjusted up to 7.4 with NaOH. EZ‐Link Biotin‐HPDP (Thermo Fisher) was dissolved in DMSO, mixed with N,N‐dimethylformamide and then added to samples at a final concentration of 0.5 mM. Samples were then incubated for 1 h at 37°C with shaking. Proteins were precipitated with acetone and then washed 4 times with ice‐cold acetone to remove any excess biotin. Protein was resuspended in IP buffer (1% Triton X‐100, 50 mM Tris pH 7.2, 150 mM NaCl, 1 mM EDTA, protease inhibitor cocktail 1:200 (Sigma)) and incubated with 30 μl of streptavidin‐Sepharose resin (Thermo Fisher), which was prewashed twice with IP buffer, for 1 h at room temperature with end‐over‐end rotation. The resin was washed 4 times in the IP buffer before elution of bound proteins by the addition of 20 mM TCEP for 20 min at 37°C with shaking. Eluted proteins were collected by centrifugation at 800× g for 5 min, and the supernatants were retained. Samples were stored at −20°C until further use.
Immunoprecipitation
Cell pellets were collected and immediately resuspended in lysis buffer (1% Triton X‐100, 50 mM Tris pH 7.2, 150 mM NaCl, 1 mM EDTA, protease inhibitor cocktail 1:200 (Sigma)). Samples were then rotated end‐over‐end at 4°C for 30 min before spinning at 16,000× g for 5 min. The supernatants were retained. 100 μl of anti‐FLAG M2 resin (Sigma–Aldrich, A2220) was washed in lysis buffer without protease inhibitors three times, added to the samples and then left overnight with end‐over‐end rotation at 4°C. Samples were then spun at 800× g for 2 min to collect the resin before being washed five times for 5 min each at room temperature with end‐over‐end rotation with lysis buffer without protease inhibitors. Immunoprecipitated proteins were eluted by three times the resin bed volume of 2 mg/ml triple‐FLAG peptide (Sigma‐Aldrich) dissolved in lysis buffer without protease inhibitors for 30 min at room temperature with end‐over‐end rotation. The samples were then centrifuged at 800× g for 5 min and the supernatants retained. Eluted proteins were stored at −20°C until further use. For immunoprecipitations of endogenous proteins from mouse forebrain, a whole mouse forebrain was added to a lysis buffer (1% Triton X‐100, 1% sodium deoxycholate, 50 mM Tris pH 7.2, 150 mM NaCl, 1 mM EDTA, protease inhibitor cocktail 1:200 (Sigma)) and homogenised 10 times with a Dounce homogeniser on ice. Samples were spun at 16,000× g and the supernatant retained as above. Samples were pre‐cleared with protein G‐Sepharose before centrifugation and retention of the supernatant. Samples were then incubated with either anti‐DHHC5 or anti‐Golga7b antibody and protein G‐Sepharose overnight at 4°C. The resin was then washed five times with lysis buffer before elution of bound proteins by heating the resin for 20 min at 70°C.
Immunocytochemistry
Cells were seeded onto glass coverslips in a 12‐well plate and were grown for 24 h. The medium was changed and cells were transfected with a 3:1 ratio of PEI:cDNA for 24 h as described previously. Medium was removed, coverslips were washed briefly with 1–2 ml of PBS, and cells were fixed with 4% PFA for 10 min at room temperature. Coverslips were washed again with PBS before permeabilisation with 0.3% Triton X‐100–PBS (vol/vol) for 15 min at room temperature with gentle agitation. After washing with PBS, coverslips were blocked with 0.2% fish skin gelatin (Sigma‐Aldrich)/0.01% Triton X‐100 (vol/vol) in PBS (blocking solution) for an hour at room temperature with agitation. Primary antibody was incubated at the concentrations mentioned previously diluted in blocking solution for 1 h at room temperature. Coverslips were washed three times for 5 min with 1 ml of PBS with gentle agitation before incubation with secondary antibodies diluted in blocking solution for 2 h at room temperature, protected from light by wrapping plates in tinfoil with gentle agitation. Coverslips were then washed with 1 ml of PBS three times for 5 min with gentle agitation before mounting on glass slides with DAPI Fluoromount‐G mounting medium (Southern Biotech) before being kept in the dark at 4°C before confocal imaging.
Calcium switch assays
A431 cells were seeded on glass coverslips in a 12‐well plate with either normal DMEM or low‐calcium (5 μM calcium) DMEM and placed back into the incubator for 18 h. Control or targeted siRNA was then added as described previously, and cells were placed back into the incubator for 72 h. CaCl₂ was then added to the low‐calcium medium to a final concentration of 2 mM for 30 min or 1 or 3 h. Control cells were left in low‐calcium medium. Cells were then fixed with 4% PFA and prepared as described previously in the immunocytochemistry section.
Confocal microscopy
All confocal microscopy was performed on a Nikon A1 confocal microscope using the following laser wavelengths: 405 nm (blue), 488 nm (green), 561 nm (red) and 640 nm (far‐red). Images were acquired using a 60× objective lens and the NIS‐Elements program (Nikon Instruments) in an ND2 format. For all samples, no primary and no secondary controls were used to threshold the exposure at each individual laser wavelength. All images were analysed using the FIJI 41 and ImageJ 42 software packages. To assess plasma membrane or internal protein signal, the colour channels were split to isolate the channel of interest. The free draw tool was used to draw around a single cell, and the mean intensity, raw integrated density and integrated density were measured. The shape was then moved just within the plasma membrane of the cell and the same measurements recorded. The ratio of the two means was then used to calculate the level of protein signal at the plasma membrane (higher ratio indicates more signal at the plasma membrane). In calcium switch assays, the remaining DSG2 signal at the plasma membrane was used to identify the plasma membrane for image quantification purposes.
Immunoblotting
Samples were run on 10% Tris–glycine acrylamide gels unless otherwise stated, and proteins were transferred to 0.2‐μm polyvinylidene difluoride membranes (Thermo Fisher) for 90 min at 30 V. Membranes were blocked for 1 h at room temperature on a roller in PBS 0.1%–Tween‐20 (vol/vol, PBS‐T)–5% skim milk powder (weight/vol), before overnight incubation with primary antibody diluted in PBS‐T–1% skim milk powder (weight/vol) at 4°C on a roller. Membranes were then washed five times with 10 ml PBS‐T for 5 min each on a roller at room temperature, before incubation with secondary antibody diluted in PBS‐T–1% milk for 1 h at room temperature. Membranes were washed again five times with 10 ml PBS‐T for 5 min before a final wash with 10 ml PBS. Clarity ECL solution (Bio‐Rad) was added in a 1:1 ratio of peroxide solution:luminol/enhancer solution with a final volume of 10 ml for 5 min at room temperature on a roller before another wash with PBS on a roller at room temperature. Blots were imaged by exposure on CL‐XPosure X‐ray film (Thermo Fisher) prior to development to visualise protein bands.
Mass spectrometry
In‐gel digestion was performed as reported previously 43. Extracted peptides were resuspended in 0.5% formic acid and analysed by nanoflow LC‐MS/MS using an Orbitrap Elite (Thermo Fisher) hybrid mass spectrometer equipped with a nanospray source, coupled to an Ultimate RSLCnano LC System (Dionex). The system was controlled by Xcalibur 2.1 (Thermo Fisher) and DCMSLink 2.08 (Dionex). Peptides were desalted on‐line using a Micro‐Precolumn cartridge (C18 PepMap 100, LC Packings) and then separated using a 130‐min gradient from 3 to 40% buffer B (0.5% formic acid in 80% acetonitrile) on an EASY‐Spray column, 50 cm × 50 μm ID, PepMap C18, 2 μm particles, 100 Å pore size (Thermo). The LTQ‐Orbitrap Elite was operated with a cycle of one MS (in the Orbitrap) acquired at a resolution of 60,000 at m/z 400, with the top 20 most abundant multiply charged (2+ and higher) ions in a given chromatographic window subjected to MS/MS fragmentation in the linear ion trap. An FTMS target value of 1e6 and an ion trap MSn target value of 1e4 were used with the lock mass (445.120025) enabled. Maximum FTMS scan accumulation time of 500 ms and maximum ion trap MSn scan accumulation time of 100 ms were used. Dynamic exclusion was enabled with a repeat duration of 45 s with an exclusion list of 500 and an exclusion duration of 30 s.
Mass spectrometry data analysis
All raw mass spectrometry data were analysed with MaxQuant version 1.5.6 44. Data were searched against a human UniProt sequence database (downloaded June 2015) using the following search parameters: digestion set to Trypsin/P, methionine oxidation and N‐terminal protein acetylation as variable modifications, cysteine carbamidomethylation as a fixed modification, match between runs enabled with a match time window of 0.7 min and a 20‐min alignment time window, label‐free quantification enabled with a minimum ratio count of 2, minimum number of neighbours of 3 and an average number of neighbours of 6. PSM and protein match thresholds were set at 0.1 ppm. A protein FDR of 0.01 and a peptide FDR of 0.01 were used for identification level cut‐offs. For immunoprecipitation data, protein group files generated by MaxQuant were loaded into the Perseus platform 45 with the LFQ intensity from each individual run as main columns. The matrix was filtered to remove all proteins that were potential contaminants, only identified by site and reverse sequences. The LFQ intensities were then transformed by log2(x), and individual LFQ columns were grouped by experiment (e.g. all control experiments grouped, all DHHC5 pull‐downs grouped). Rows without valid values in 3 out of 4 runs in at least one of the groups were removed, and all the remaining missing values were replaced from the normal distribution with each column imputed individually. Following this, a two‐tailed Student's t‐test with S0 set to 3 and a permutation‐based FDR of 0.05 were performed and all non‐significant proteins were removed, and the matrix was exported in a.txt format for further analysis. For further filtering of results, files were analysed using the online Contaminant Repository for Affinity Purification Mass Spectrometry Data program 46 with additional controls (CC130, CC135, CC136, CC137). Proteins with an SP score above 0.95 were considered bona fide interactors. Gene ontology analysis was performed using the WebGestalt platform 47 with the following parameters: species set to Homo sapiens; reference gene set being “genome protein coding”; and FDR set to 0.01 with the Benjamini–Hochberg multiple test adjustment.
Cell scatter assay
A total of 10,000 A431 cells were seeded in single wells of 6‐well plates 24 h after treatment with either DHHC5 or non‐targeting siRNA. Cells were grown for 24 h before medium was removed and replaced with serum‐free DMEM, and cells were incubated for 8 h. Cells were imaged at 10× magnification on a Nikon Widefield microscope system, positions of fields of view were recorded, EGF was added to a final concentration of 5 ng/ml, and cells were incubated for 48 h. Cells were then imaged again at the same positions as previously. To quantify scattering, the grid tool of ImageJ was used, with the number of grid squares occupied by cells compared at 0 and 48 h.
Trans‐epithelial electrical resistance
A total of 125,000 A431 cells were seeded in transwell inserts in 24‐well plates which had a 0.4 μm pore size. Cells were allowed to grow for 12 h until confluency before treatment with siRNA, which was performed for 24 h. The resistance in each well was measured three times with an epithelial voltohmmeter. The measurements for each well were averaged, and a blank value of a transwell insert without cells was subtracted from each measurement before statistical analysis.
Cell surface biotinylation
The procedure was performed essentially as described previously 11. In brief, cells were washed with PBS supplemented with 0.1 mM magnesium chloride and 1 mM calcium chloride (PBS‐CM), before treatment for 30 min at 4°C with PBS‐CM with 1 mg/ml NHS‐SS‐Biotin (Thermo). Cells were then washed with PBS‐CM, then twice with PBS‐CM with 50 mM glycine at 4°C. Cells were then lysed in a buffer containing 50 mM Tris pH 7.2, 1 mM EDTA, 1% IGEPAL CA‐630 and protease inhibitors (1:200; Sigma). Lysates were then incubated with streptavidin‐agarose resin (Sigma) overnight at 4°C with agitation. The resin was then washed three times in lysis buffer supplemented with 150 mM NaCl before lysis with PAGE buffer with 50 mM TCEP at 70°C.
Endocytosis assay
The method was performed as previously described 48 with some minor changes. In short, cells were washed with PBS‐CM twice for 15 min at 4°C before the addition of PBS‐CM supplemented with 1 mg/ml NHS‐SS‐Biotin for 30 min at 4°C. Cells were then moved back to 37°C while being washed three times with PBS‐CM for 5 min each. Cells were then treated four times with glutathione buffer (75 mM NaCl, 1 mM MgCl2, 0.1 mM CaCl2, 50 mM GSH, 80 mM NaOH and 10% FBS) at 4°C. Cells were then lysed and a pull‐down performed with streptavidin‐agarose exactly as described for the cell surface biotinylation assay.
PEG switch
PEG switch assay was performed essentially as described previously 49. In short, cells were lysed in a lysis buffer containing 2.5% SDS, 1 mM EDTA and 50 mM Tris pH 7.2. Lysates were heated at 70°C for 20 min and DNA was sheared by passing the lysates through a fine‐gauge needle. After cooling, HEPES buffer and maleimide were both added to 100 mM final concentration and samples were incubated at 40°C for 3 h with shaking to block all free cysteines. Protein was precipitated with a 4× volume of ice‐cold acetone, and the pellet was washed five times with 80% acetone to remove any excess maleimide. Pellets were resuspended in 1% SDS, 1 mM EDTA and 50 mM Tris pH 7.2 and split into ± hydroxylamine samples. To the +HA sample, 2M HA pH 7.2/50 mM Tris pH 7.2 was added to a final concentration of 1M HA, and to the −HA sample, the same volume of 50 mM Tris pH 7.2 was added. Samples were left at room temperature for 1 h with end‐over‐end rotation. Acetone precipitation of protein was performed as described above. Pellets were resuspended in 1% SDS/50 mM Tris pH 7.2, and mPEG 5 kDa was added to 5 mM final concentration. Samples were incubated at 25°C for 1 h with shaking. Protein was acetone‐precipitated as described above. Pellets were resuspended in 2.5% SDS/50 mM Tris, before Western blotting.
Author contributions
KTW and MOC conceived the experiments and wrote the manuscript, and KTW performed experiments and data analysis.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Review Process File
Source Data for Figure 5
Acknowledgements
M.O.C. is supported by grants from the BBSRC (BB/P021689/1 and BB/R003491/1). K. T. W. was funded by a University of Sheffield scholarship. We would like to thank Darren Robinson from the Wolfson Light Microscopy Facility and Adelina Acosta‐Martin from the biOMICS mass spectrometry facility for providing technical assistance. We would also like to thank Andrew Wood and Dr Kai Erdmann for advice about TEER experiments, Dr Elena Rainero for advice about cell scatter assays, and Dr Andrew Peden and Maria Davies for useful discussions and critical reading of the manuscript.
EMBO Reports (2019) 20: e47472
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (https://www.ebi.ac.uk/pride/archive/) 50 partner repository with the dataset identifier PXD011553.
References
- 1. Schmidt MF, Schlesinger MJ (1979) Fatty acid binding to vesicular stomatitis virus glycoprotein: a new type of post‐translational modification of the viral glycoprotein. Cell 17: 813–819 [DOI] [PubMed] [Google Scholar]
- 2. Korycka J, ?ach A, Heger E, Bogus?awska DM, Wolny M, Toporkiewicz M, Augoff K, Korzeniewski J, Sikorski AF (2012) Human DHHC proteins: a spotlight on the hidden player of palmitoylation. Eur J Cell Biol 91: 107–117 [DOI] [PubMed] [Google Scholar]
- 3. Ohno Y, Kihara A, Sano T, Igarashi Y (2006) Intracellular localization and tissue‐specific distribution of human and yeast DHHC cysteine‐rich domain‐containing proteins. Biochim Biophys Acta 1761: 474–483 [DOI] [PubMed] [Google Scholar]
- 4. Hancock JF, Paterson H, Marshall CJ (1990) A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 63: 133–139 [DOI] [PubMed] [Google Scholar]
- 5. Lobo S, Greentree WK, Linder ME, Deschenes RJ (2002) Identification of a Ras palmitoyltransferase in Saccharomyces cerevisiae . J Biol Chem 277: 41268–41273 [DOI] [PubMed] [Google Scholar]
- 6. Swarthout JT, Lobo S, Farh L, Croke MR, Greentree WK, Deschenes RJ, Linder ME (2005) DHHC9 and GCP16 constitute a human protein fatty acyltransferase with specificity for H‐ and N‐Ras. J Biol Chem 280: 31141–31148 [DOI] [PubMed] [Google Scholar]
- 7. Mitchell DA, Hamel LD, Ishizuka K, Mitchell G, Schaefer LM, Deschenes RJ (2012) The Erf4 subunit of the yeast Ras palmitoyl acyltransferase is required for stability of the Acyl‐Erf2 intermediate and palmitoyl transfer to a Ras2 substrate. J Biol Chem 287: 34337–34348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K et al (2015) The bioplex network: a systematic exploration of the human interactome. Cell 162: 425–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Morrison E, Kuropka B, Kliche S, Brugger B, Krause E, Freund C (2015) Quantitative analysis of the human T cell palmitome. Sci Rep 5: 11598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Collins MO, Woodley KT, Choudhary JS (2017) Global, site‐specific analysis of neuronal protein S‐acylation. Sci Rep 7: 4683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brigidi GS, Santyr B, Shimell J, Jovellar B, Bamji SX (2015) Activity‐regulated trafficking of the palmitoyl‐acyl transferase DHHC5. Nat Commun 6: 8200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Breusegem SY, Seaman MNJ (2014) Genome‐wide RNAi screen reveals a role for multipass membrane proteins in endosome‐to‐golgi retrieval. Cell Rep 9: 1931–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li Y, Martin BR, Cravatt BF, Hofmann SL (2012) DHHC5 protein palmitoylates flotillin‐2 and is rapidly degraded on induction of neuronal differentiation in cultured cells. J Biol Chem 287: 523–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hilgemann DW, Fine M, Linder ME, Jennings BC, Lin MJ (2013) Massive endocytosis triggered by surface membrane palmitoylation under mitochondrial control in BHK fibroblasts. Elife 2: e01293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin MJ, Fine M, Lu JY, Hofmann SL, Frazier G, Hilgemann DW (2013) Massive palmitoylation‐dependent endocytosis during reoxygenation of anoxic cardiac muscle. Elife. 2: e01295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Howie J, Reilly L, Fraser NJ, Vlachaki Walker JM, Wypijewski KJ, Ashford ML, Calaghan SC, McClafferty H, Tian L, Shipston MJ et al (2014) Substrate recognition by the cell surface palmitoyl transferase DHHC5. Proc Natl Acad Sci USA 111: 17534–17539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Y, Hu J, Hofer K, Wong AM, Cooper JD, Birnbaum SG, Hammer RE, Hofmann SL (2010) DHHC5 interacts with PDZ domain 3 of post‐synaptic density‐95 (PSD‐95) protein and plays a role in learning and memory. J Biol Chem 285: 13022–13031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang W, Di Vizio D, Kirchner M, Steen H, Freeman MR (2010) Proteome scale characterization of human S‐acylated proteins in lipid raft‐enriched and non‐raft membranes. Mol Cell Proteomics 9: 54–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Abrami L, Dallavilla T, Sandoz PA, Demir M, Kunz B, Savoglidis G, Hatzimanikatis V, van der Goot FG (2017) Identification and dynamics of the human ZDHHC16‐ZDHHC6 palmitoylation cascade. Elife 6: e27826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Little EB, Edelman GM, Cunningham BA (1998) Palmitoylation of the cytoplasmic domain of the neural cell adhesion molecule N‐CAM serves as an anchor to cellular membranes. Cell Adhes Commun 6: 415–430 [DOI] [PubMed] [Google Scholar]
- 21. Lievens PM, Kuznetsova T, Kochlamazashvili G, Cesca F, Gorinski N, Galil DA, Cherkas V, Ronkina N, Lafera J, Gaestel M et al (2016) ZDHHC3 tyrosine phosphorylation regulates neural cell adhesion molecule palmitoylation. Mol Cell Biol 36: 2208–2225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aramsangtienchai P, Spiegelman NA, Cao J, Lin H (2017) S‐Palmitoylation of junctional adhesion molecule c regulates its tight junction localization and cell migration. J Biol Chem 292: 5325–5334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roberts BJ, Johnson KE, McGuinn KP, Saowapa J, Svoboda RA, Mahoney MG, Johnson KR, Wahl JK III (2014) Palmitoylation of plakophilin is required for desmosome assembly. J Cell Sci 127: 3782–3793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roberts BJ, Svoboda RA, Overmiller AM, Lewis JD, Kowalczyk AP, Mahoney MG, Johnson KR, Wahl JK III (2016) Palmitoylation of desmoglein 2 is a regulator of assembly dynamics and protein turnover. J Biol Chem 291: 24857–24865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ampah KK, Greaves J, Shun‐Shion AS, Asnawi AW, Lidster JA, Chamberlain LH, Collins MO, Peden AA (2018) S‐acylation regulates the trafficking and stability of the unconventional Q‐SNARE STX19. J Cell Sci 131: jcs212498 [DOI] [PubMed] [Google Scholar]
- 26. Drisdel RC, Green WN (2004) Labeling and quantifying sites of protein palmitoylation. Biotechniques 36: 276–285 [DOI] [PubMed] [Google Scholar]
- 27. Hou H, John Peter AT, Meiringer C, Subramanian K, Ungermann C (2009) Analysis of DHHC acyltransferases implies overlapping substrate specificity and a two‐step reaction mechanism. Traffic 10: 1061–1073 [DOI] [PubMed] [Google Scholar]
- 28. Lemonidis K, Gorleku OA, Sanchez‐Perez MC, Grefen C, Chamberlain LH (2014) The Golgi S‐acylation machinery comprises zDHHC enzymes with major differences in substrate affinity and S‐acylation activity. Mol Biol Cell 25: 3870–3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Massol RH, Boll W, Griffin AM, Kirchhausen T (2006) A burst of auxilin recruitment determines the onset of clathrin‐coated vesicle uncoating. Proc Natl Acad Sci USA 103: 10265–10270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Park RJ, Shen H, Liu L, Liu X, Ferguson SM, De Camilli P (2013) Dynamin triple knockout cells reveal off target effects of commonly used dynamin inhibitors. J Cell Sci 126: 5305–5312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ungewiss H, Vielmuth F, Suzuki ST, Maiser A, Harz H, Leonhardt H, Kugelmann D, Schlegel N, Waschke J (2017) Desmoglein 2 regulates the intestinal epithelial barrier via p38 mitogen‐activated protein kinase. Sci Rep 7: 6329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen HC (2005) Cell‐scatter assay. Methods Mol Biol 294: 69–77 [DOI] [PubMed] [Google Scholar]
- 33. Chen S, Einspanier R, Schoen J (2015) Transepithelial electrical resistance (TEER): a functional parameter to monitor the quality of oviduct epithelial cells cultured on filter supports. Histochem Cell Biol 144: 509–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gorleku OA, Barns AM, Prescott GR, Greaves J, Chamberlain LH (2011) Endoplasmic reticulum localization of DHHC palmitoyltransferases mediated by lysine‐based sorting signals. J Biol Chem 286: 39573–39584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hayashi T, Rumbaugh G, Huganir RL (2005) Differential regulation of AMPA receptor subunit trafficking by palmitoylation of two distinct sites. Neuron 47: 709–723 [DOI] [PubMed] [Google Scholar]
- 36. Rana MS, Kumar P, Lee CJ, Verardi R, Rajashankar KR, Banerjee A (2018) Fatty acyl recognition and transfer by an integral membrane S‐acyltransferase. Science 359: eaao6326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vollner F, Ali J, Kurrle N, Exner Y, Eming R, Hertl M, Banning A, Tikkanen R (2016) Loss of flotillin expression results in weakened desmosomal adhesion and Pemphigus vulgaris‐like localisation of desmoglein‐3 in human keratinocytes. Sci Rep 6: 28820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brigidi GS, Sun Y, Beccano‐Kelly D, Pitman K, Mobasser M, Borgland SL, Milnerwood AJ, Bamji SX (2014) Palmitoylation of delta‐catenin by DHHC5 mediates activity‐induced synapse plasticity. Nat Neurosci 17: 522–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shafraz O, Rubsam M, Stahley SN, Caldara AL, Kowalczyk AP, Niessen CM, Sivasankar S (2018) E‐cadherin binds to desmoglein to facilitate desmosome assembly. Elife 7: e37629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kant S, Krusche CA, Gaertner A, Milting H, Leube RE (2016) Loss of plakoglobin immunoreactivity in intercalated discs in arrhythmogenic right ventricular cardiomyopathy: protein mislocalization versus epitope masking. Cardiovasc Res 109: 260–271 [DOI] [PubMed] [Google Scholar]
- 41. Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bayes A, Collins MO, Reig‐Viader R, Gou G, Goulding D, Izquierdo A, Choudhary JS, Emes RD, Grant SG (2017) Evolution of complexity in the zebrafish synapse proteome. Nat Commun 8: 14613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- 45. Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- 46. Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St‐Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY et al (2013) The CRAPome: a contaminant repository for affinity purification‐mass spectrometry data. Nat Methods 10: 730–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang B, Kirov S, Snoddy J (2005) WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res 33: W741–W748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cihil KM, Swiatecka‐Urban A (2013) The cell‐based L‐glutathione protection assays to study endocytosis and recycling of plasma membrane proteins. J Vis Exp e50867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yokoi N, Fukata Y, Sekiya A, Murakami T, Kobayashi K, Fukata M (2016) Identification of PSD‐95 depalmitoylating enzymes. J Neurosci 36: 6431–6444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Martens L, Hermjakob H, Jones P, Adamski M, Taylor C, States D, Gevaert K, Vandekerckhove J, Apweiler R (2005) PRIDE: the proteomics identifications database. Proteomics 5: 3537–3545 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Review Process File
Source Data for Figure 5
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (https://www.ebi.ac.uk/pride/archive/) 50 partner repository with the dataset identifier PXD011553.