

Abstract
Iron overload, a common clinical occurrence, is implicated in the metabolic syndrome although the contributing pathophysiological mechanisms are not fully defined. We show that prolonged iron overload results in an autophagy defect associated with accumulation of dysfunctional autolysosomes and loss of free lysosomes in skeletal muscle. These autophagy defects contribute to impaired insulin‐stimulated glucose uptake and insulin signaling. Mechanistically, we show that iron overload leads to a decrease in Akt‐mediated repression of tuberous sclerosis complex (TSC2) and Rheb‐mediated mTORC1 activation on autolysosomes, thereby inhibiting autophagic‐lysosome regeneration. Constitutive activation of mTORC1 or iron withdrawal replenishes lysosomal pools via increased mTORC1‐UVRAG signaling, which restores insulin sensitivity. Induction of iron overload via intravenous iron‐dextran delivery in mice also results in insulin resistance accompanied by abnormal autophagosome accumulation, lysosomal loss, and decreased mTORC1‐UVRAG signaling in muscle. Collectively, our results show that chronic iron overload leads to a profound autophagy defect through mTORC1‐UVRAG inhibition and provides new mechanistic insight into metabolic syndrome‐associated insulin resistance.
Keywords: ALR, autophagy, insulin resistance, iron overload, mTORC1
Subject Categories: Autophagy & Cell Death, Membrane & Intracellular Transport, Metabolism
Introduction
Iron is an essential element involved in multiple cellular processes such as erythropoiesis, mitochondrial respiration, and growth/differentiation 1, 2, 3. The total amount of labile iron must be precisely regulated and in circulation iron typically exists bound to transferrin (TF) 4, 5. However, when iron exceeds TF capacity, iron homeostasis becomes imbalanced 6 and non‐TF bound iron contributes to pathophysiological processes, including insulin resistance and diabetes 7, 8, 9, 10. Interventions to reduce iron have been reported to improve insulin sensitivity and delay the onset of type 2 diabetes (T2D). These include use of chelators 11, 12, 13, blood‐letting 14, 15, 16, and iron restriction diet 17. However, the molecular mechanisms linking iron overload (IO) to T2D are poorly understood. Cellular labile iron, which contains chelatable redox‐active Fe2+/Fe3+, has been implicated in iron‐mediated cellular toxicity by increasing oxidative stresses. Excess accumulation of intracellular iron leads to the generation of reactive oxygen species (ROS) and tissue damage 2, 18.
We have previously shown that autophagy plays an important role in regulating insulin sensitivity and metabolism in skeletal muscle 19. Autophagy is a stress‐sensitive cellular degradative process capable of clearing and recycling potential substrates such as damaged mitochondria and protein aggregates 20. Indeed, autophagy is generally considered to play a protective role against T2D, recycling nutrients to maintain energy homeostasis and remove damaged organelles 21, 22. IO has been described to regulate the activity of both AMP‐activated protein kinase (AMPK) and mTOR complex 1 (mTORC1) 23, 24, which are both established upstream regulators of the autophagy pathway 25, 26. However, the potential mechanisms linking IO‐mediated autophagy to insulin resistance remain to be elucidated.
Here, we established in vitro and in vivo models to examine direct effects of IO on autophagy flux in skeletal muscle and its significance in insulin resistance. We describe a mechanistic link between chronic IO and autophagy dysfunction, which alters insulin sensitivity in skeletal muscle. We identified the regulation of mTORC1 by IO as a double‐edged sword that initially leads to transient autophagy activation, but ultimately causes an autophagy defect through loss of autophagic‐lysosome regeneration (ALR), a newly discovered membrane recycling mechanism that to date has not been identified as a contributor to pathophysiology 27. The data presented here provide new mechanistic knowledge to enhance our understanding of the pathogenic mechanisms of IO, which may have widespread consequences in insulin resistance, metabolic dysfunction, and beyond.
Results
Iron overload (IO) induced insulin resistance in L6 cells
Skeletal muscle is a primary consumer of glucose, yet the effects of IO on glucose uptake and insulin sensitivity are not known. Therefore, we first sought to determine whether IO induces insulin resistance in rat L6 skeletal myoblasts. An experimental model of IO was established by treating cells with ferrous labile iron for up to 24 h. The extent of IO was then determined via dose and temporal analysis using biochemical intracellular iron measurement (Fig 1A). The concentration selected for subsequent use, 250 μM, is intended to mimic IO and is consistent with iron concentrations used in the literature for other cell types 28, 29. Conventional iron overload response 5 by skeletal muscle cells was monitored by using an iron response element (IRE)‐driven reporter construct tagged with cyan fluorescence protein (CFP) transfected in L6 cells as well as the use of fluorescence‐based indicators of iron levels (IP‐1). We observed that iron treatment for 24 h increased the expression of CFP and fluorescence of iron probe 1 (IP‐1) without adversely affecting viability (Figs 1B and EV1A). Analyzing increased intracellular iron via quenching of the iron‐sensitive fluorescent probe phen green SK (PGSK), we confirmed elevated iron in our model and that it could be effectively blocked by the iron chelator, 2,2′ di‐pyridyl (DPD; Fig 1C). We further tested iron‐responsive transcription by performing qPCR against ferritin (FTH and FTL, H‐heavy and L‐light chain), ferroportin (SLC40A1), and Tfr1 (transferrin receptor 1, TFRC). Iron treatment for 24 h significantly increased the expression of ferritins and ferroportin and decreased the expression of Tfr1 (Fig 1D). Transcriptional changes aligned with protein levels indicating that iron treatment significantly increased ferritin heavy chain and decreased TfR1 expression (Fig 1E). Together, these results indicate that our in vitro IO model in skeletal muscle line recapitulates the key hallmarks of IO 30.
Figure 1. Validation of iron overload (IO) model and insulin resistance induction after IO in L6 muscle cells.
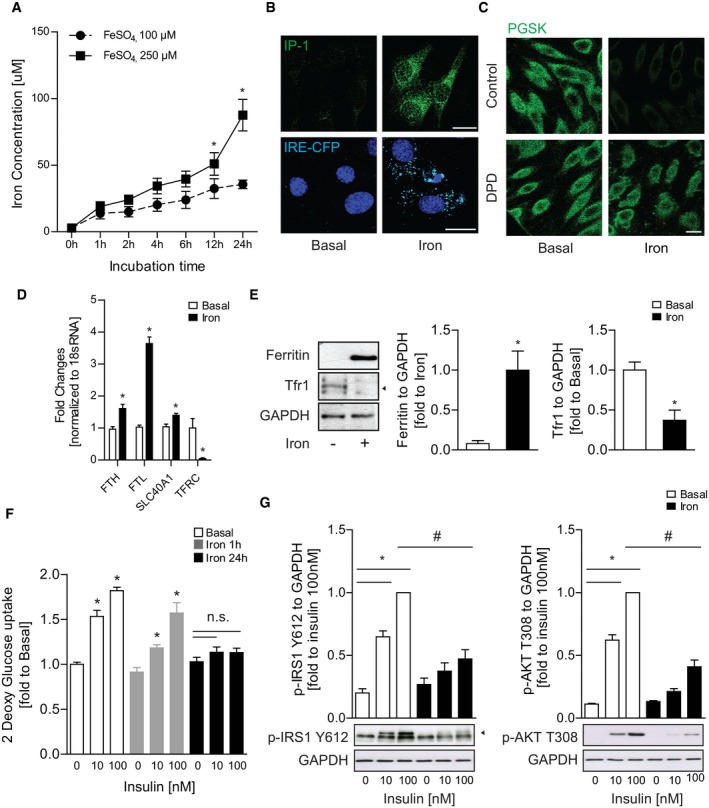
- Ferrozine‐based intracellular iron concentration measurement in L6 cells after temporal analysis of iron treatment at 100 or 250 μM for multiple time points. *P < 0.05 (unpaired Student's t‐test versus each time points with FeSO4, 100 μM).
- Representative confocal microscope images of L6 cells stained with IP‐1 (iron probe 1) or transfected with IRE‐CFP (iron regulatory element) reporter after iron treatment (FeSO4, 250 μM) for 24 h.
- Representative confocal images of L6 cells using iron‐sensitive fluorescent PGSK dye after iron treatment (250 μM, 24 h) with iron chelator DPD (500 μM).
- Relative gene expressions—ferritin heavy chain (FTH), ferritin light chain (FTL), ferroportin (SLC40A1, transferrin receptor 1 (tfr1, TFRC)—normalized to 18S rRNA expression after iron treatment (FeSO4, 250 μM) for 24 h. *P < 0.05 (unpaired Student's t‐test versus basal).
- Representative Western blot images and quantification of ferritin (heavy chain) and Tfr1 (indicated with arrowhead) over GAPDH after iron treatment (250 μM, 24 h). *P < 0.05 (unpaired Student's t‐test versus basal).
- Glucose uptake of L6 cells with insulin stimulation (10 or 100 nM, 20 min) after iron treatment (250 μM, 1 or 24 h). *P < 0.05 (one‐way ANOVA with multiple comparisons).
- Representative Western blot images and quantification of phospho‐IRS1 (Y612) and phospho‐AKT (T308) over GAPDH with insulin stimulation (10 or 100 nM, 5 min) after iron treatment (250 μM, 24 h). *P < 0.05 (one‐way ANOVA with multiple comparisons). # P < 0.05 (unpaired Student's t‐test versus iron with insulin 100 nM).
Figure EV1. Prevention of IO‐induced insulin resistance in L6 cells with iron chelator.

- MTT viability assay in L6 cells after iron treatment (FeSO4, 250 μM) for 24 h.
- Time course measurement of intracellular iron concentration in L6 cells after iron treatment (250 μM) with iron chelator DPD (500 nM) for multiple time points. # P < 0.05 (unpaired Student's t‐test versus iron at each time points).
- Glucose uptake of L6 cells with insulin stimulation (100 nM, 20 min) after iron treatment (250 μM, 24 h) with DPD (500 nM). *P < 0.05, # P < 0.05 (one‐way ANOVA test with multiple comparisons).
- Representative Western blot images of phosphor‐IRS1 Y612, phosphor‐AKT T308, and GAPDH expression.
- Quantification of phosphor‐IRS1 Y612 over GAPDH and phosphor‐AKT T308 over GAPDH in L6 cells with insulin stimulation (100 nM, 5 min) after iron treatment (250 μM, 24 h) with DPD (500 nM). *P < 0.05 (unpaired Student's t‐test versus basal without insulin), # P < 0.05 (unpaired Student's t‐test versus iron with insulin).
We next analyzed insulin sensitivity after IO by measuring glucose uptake and phosphorylation of insulin signaling molecules (IRS‐1 Y612 and AKT T308) after insulin stimulation. Iron treatment for 24 h significantly reduced glucose uptake following insulin stimulation (Fig 1F). Additionally, insulin‐stimulated phosphorylation of IRS‐1 and AKT was significantly reduced after 24‐h iron treatment (Fig 1G). Furthermore, we determined that insulin resistance was due to intracellular iron accumulation since DPD significantly reduced the intracellular iron accumulation at 24‐h iron treatment and restored insulin‐stimulated glucose uptake and insulin signaling phosphorylation (Fig EV1B–E). Taken together, our results indicate that IO directly caused insulin resistance in skeletal muscle.
Prolonged iron treatment causes autophagy flux defects in skeletal muscle cells
We have previously established that autophagy is an important regulator of insulin sensitivity in skeletal muscle 19. Thus, we analyzed the temporal effect of iron treatment on autophagosome production and fusion with lysosomes by imaging L6 cells stably expressing LC3B‐eGFP‐mCherry. In this assay, eGFP fluorescence was quenched by low pH after autophagosome fusion with the lysosome, whereas mCherry is not 31. We observed a rapid increase in the number of autophagosome puncta after iron treatment, which was corroborated by Western blot analysis of LC3B that showed the lipidated form of LC3B (LC3‐II) was significantly increased after iron treatment (Figs 2A and B, and EV2A). Consistent with our analysis of LC3B, we found that p62 puncta formation and protein clearance were rapidly increased by iron treatment (Figs 2C, and EV2B and C). Surprisingly, while we observed p62 protein levels decreased significantly at early time points after iron treatment, we found they were significantly stabilized at 24 h posttreatment (Fig 2C). p62 stabilization under prolonged iron treatment potentially indicates a blockage of autophagic flux at later time points. To further analyze autophagy flux, we quantified the accumulation of lipidated LC3B in the presence of a late‐stage autophagy inhibitor (CQ, chloroquine). At 4 h after iron treatment, LC3‐II significantly increased with CQ, indicating an induction in autophagy (Fig 2D). However, 24 h after iron treatment LC3‐II levels were elevated but unchanging, indicating a lack of autophagy flux despite the presence of a large pool of autophagosomes (Fig 2E).
Figure 2. Iron treatment transiently induced autophagy yet led to flux inhibition at 24‐h iron treatment.
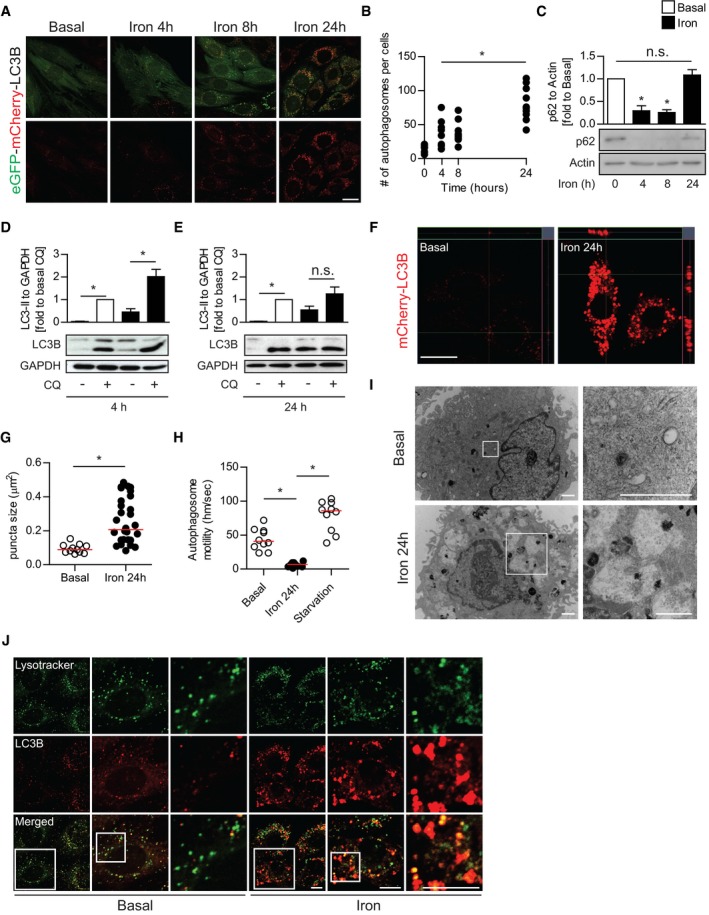
- Representative confocal microscope image of L6 cells stably expressing tandem fluorescent‐eGFP‐mCherry LC3B treated with iron (FeSO4, 250 μM) for the indicated time points (4, 8, 24 h).
- Quantification of mean LC3B puncta per cell from (A). Experiments were repeated three times, and one representative experiment is presented here. *P < 0.05 (one‐way ANOVA with Dunnett's post hoc test versus basal).
- Representative Western blot images and quantification of p62 to actin in L6 cells after iron treatment (FeSO4, 250 μM) at multiple time points. *P < 0.05 (multiple unpaired Student's t‐test versus basal).
- Representative Western blot images and quantification of LC3B‐II to GAPDH in L6 cells after 4‐h iron treatment stimulated with chloroquine (CQ, 30 μM). *P < 0.05 (unpaired Student's t‐test versus control, basal, or iron without CQ).
- Representative Western blot images and quantification of LC3B‐II to GAPDH in L6 cells after 24‐h iron treatment stimulated with CQ 30 μM. *P < 0.05 (unpaired student's t‐test versus control, basal, or iron without CQ).
- Representative confocal microscope z‐stack image of mCherry‐LC3B L6 cells after iron treatment (250 μM, 24 h).
- Quantification of LC3B puncta size from (F). Experiments were performed three times, and one representative experiment is presented here. *P < 0.05 (unpaired Student's t‐test compared to basal).
- Quantification of autophagosome motility from live cell microscopy (Movie EV1) from mCherry‐LC3B L6 cells under basal, iron (250 μM, 24 h), and starvation (amino acid free) condition. Experiments were performed three times, and one representative experiment is presented here. *P < 0.05 (unpaired Student's t‐test compared to iron 24 h).
- Representative TEM images of autophagosome and autolysosomes in L6 cells after iron treatment (250 μM, 24 h).
- Representative confocal microscope images of eGFP‐mCherry‐LC3B L6 cells with LysoTracker Deep Red after iron treatment (250 μM, 24 h).
Figure EV2. Chronic IO resulted in accumulation of abnormal autophagosomes despite normal proteolytic activity.

- Representative Western blot images and quantification of LC3B‐II to GAPDH in L6 cells after iron treatment (FeSO4, 250 μM) for multiple time points. Experiments were performed three times. *P < 0.05 (multiple unpaired Student's t‐test compared to basal).
- Representative epi‐fluorescent images of p62 puncta in L6 cells stably expressing mCherry‐eGFP p62, after iron treatment (250 μM) at time points indicated.
- Representative Western blot images and quantification of L6 transfected with HA‐p62 after iron treatment (250 μM) at time points indicated. Experiments were performed three times. *P < 0.05 (multiple unpaired Student's t‐test compared to 0 h of iron).
- Representative epi‐fluorescent images of MEF cells (wild type, and ULK1 and ULK2 dKO) immuno‐stained against LC3B.
- Quantification of LC3B puncta size from (D). Experiments were performed three times, and one representative experiment is presented here. *P < 0.05 (unpaired Student's t‐test compared to wt).
- Representative epi‐fluorescent images of autophagosomes (Cyto‐ID) and lysosome (LysoTracker) in L6 cells after iron (FeSO4, 250 μM) for 24 h.
- Representative epi‐fluorescent images of beta‐glucosidase (GlucGreen) and lysosome (LysoTracker) in L6 cells after iron (FeSO4, 250 μM) for 24 h.
- Representative confocal microscope images of L6 cells pulsed with Magic Red L after iron treatment (250 μM, 24 h) with 1 μM concanamycin A.
- Quantification mean fluorescence intensity from magic red signals in 3D puncta in (H). Experiments were performed three times, and all biological and technical replicates are plotted here. *P < 0.05 (one‐way ANOVA with multiple comparisons).
We also observed autophagosomes under prolonged IO were morphologically abnormal, with a mean diameter nearly three times those in the untreated samples (Fig 2F and G). Enlarged LC3‐positive vesicles are often observed in autophagy‐deficient backgrounds including cells with knock outs in ULK1/2 (Fig EV2D and E), Beclin‐1 32, FIP200 33, and ATG14L1 34. We next performed live cell imaging to analyze autophagosome mobility that, in addition to autophagosome number, is positively correlated with autophagic flux 35. As expected, amino acid starvation increased autophagosome motility to 78 nm/s compared to 44 nm/s in untreated samples indicating an increase in autophagy rates. In contrast, autophagosomes observed in cells with IO were nearly static, moving an average of 6.4 nm/s (Fig 2H, Movie EV1). Ultrastructural analysis by transmission electron microscopy (TEM) showed that chronic IO resulted in a striking accumulation of enlarged vesicles, characteristic of autolysosomes (Fig 2I). To further characterize the blockage of autophagy that accompanies insulin resistance under chronic IO, we sought to determine whether lysosomal fusion with autophagosomes was inhibited at this time point 31, 36. We immuno‐stained for endogenous LC3B and LAMP1 and observed large dual positive structures, which indicated that lysosomal fusion was not inhibited in the accumulated autolysosomes under prolonged IO (Fig 2J). This was confirmed by additional markers for both autophagosomes and lysosomes (Fig EV2F). Moreover, we found that chronic IO did not inhibit the proteolytic activity of lysosomal enzymes (β‐glucosidase and cathepsin B) that were still active in autolysosomes under chronic IO, indicating that autophagy defects were not caused by an inhibition of lysosomal fusion or lysosomal enzyme activity (Fig EV2G–I). However, we also observed that under IO there was a precipitous loss of “free” lysosomes (defined as LAMP1‐positive, LC3B‐negative), with nearly all LAMP1 staining detected on autophagosomes (Figs 2J, and EV3A and B). Taken together, our data indicate that IO overload results in a temporary increase in autophagy rates, followed by an accumulation of non‐functional autolysosomes and autophagy inhibition.
Figure EV3. Precipitous loss in autophagosome‐free lysosome after IO .
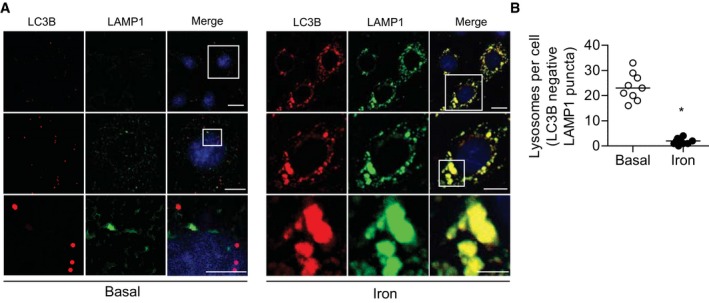
- Representative epi‐fluorescent microscope images of L6 cells immuno‐stained against LC3B (autophagosomes) and LAMP1 (lysosome) in L6 cells in response to iron treatment (250 μM, 24 h). Scale bar = 20, 10, and 5 μm.
- Quantification of number of autophagosome‐free lysosomes per cell. Experiments were performed three times, and one representative experiment was shown here. *P < 0.05 (unpaired Student's t‐tests versus control).
IO inhibits reactivation of mTOR following autophagosome degradation
The nature of the lysosomal and autophagosomal defects described above could be consistent with attenuation of the recently described membrane recycling event termed ALR 37, 38. ALR is essential to sustain prolonged periods of autophagic induction 37. ALR is an mTOR‐dependent process where membrane from the spent autophagosome is extruded, followed by scission to form protolysosomes that then mature into new lysosomes. mTORC1 promotes this scission after being activated by nutrients generated from the degradation of autophagic cargo. Importantly, this function of mTORC1 acts to promote autophagy under prolonged stress through the production of lysosomes and is independent of the autophagy‐suppressive effects mTORC1 exerts under basal conditions 37. Therefore, we next sought to monitor mTORC1 activity in iron‐treated samples. Temporal analysis of mTORC1 target phosphorylation (p‐S6K T389 and p‐ULK1 S757) upon FBS withdrawal showed a partial rescue at 8 h that was absent in the iron‐treated samples (Fig 3A and B). This result indicates that IO may prevent reactivation of mTORC1 from nutrients generated by the autolysosomes.
Figure 3. Iron treatment impaired mTOR restoration following autophagosome degradation and enforced mTOR reactivation reversed autophagy defects and insulin resistance.
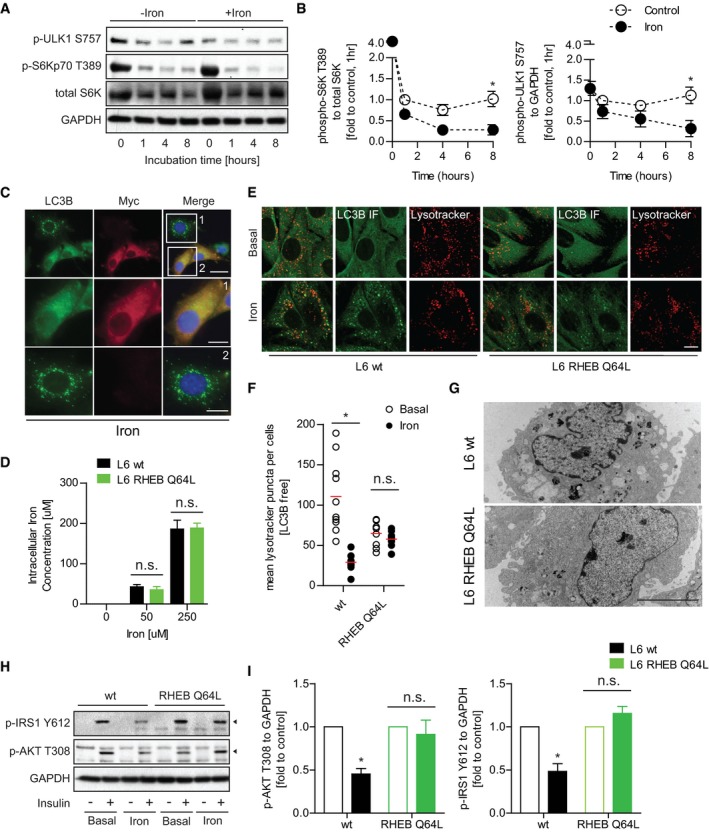
-
ARepresentative Western blot images of phospho‐ULK1 (S757), phospho‐S6Kp70 (T389), total S6K, and GAPDH after iron treatment (FeSO4, 250 μM) for multiple time points.
-
BQuantification of mTORC1 activity analyzed through phosphorylation of S6K T389 to total S6k and ULK1 S757 to GAPDH after iron treatment (FeSO4, 250 μM) for multiple time points. *P < 0.05 (multiple unpaired Student's t‐test versus control at each time point).
-
CRepresentative epi‐immunofluorescent images of L6 cells transfected with myc‐RHEB Q46L and immuno‐stained against LC3B and myc after iron treatment (FeSO4, 250 μM) for 24 h.
-
DFerrozine‐based colorimetric measurement of intracellular iron in wild‐type (wt) L6 and RHEB‐Q64L L6 cells after iron treatment (50 or 250 μM) for 24 h.
-
ERepresentative confocal images of wt L6 and RHEB‐Q64L L6 cells pulsed with LysoTracker Deep Red and immuno‐stained against LC3B after iron treatment (250 μM, 24 h).
-
FQuantification of LC3B‐free lysosome numbers in (E). Experiments were repeated three times, and one representative experiment is presented here. *P < 0.05 (multiple unpaired Student's t‐test versus basal in wt and RHEB Q64L cells).
-
GRepresentative TEM images of wt L6 and RHEB‐Q64L cells after iron treatment (250 μM, 24 h).
-
H, IRepresentative Western blot images and quantifications of phospho‐IRS1 (Y612, indicated by arrowhead) and AKT (T308, indicated by arrowhead) to GAPDH in wt and RHEB‐Q64L L6 cells stimulated with insulin (100 nM, 5 min) after iron treatment (250 μM, 24 h). *P < 0.05 (multiple unpaired Student's t‐test versus basal in wt and RHEB Q64L cells).
We next tested whether maintenance of mTORC1 activation under chronic IO was sufficient to rescue the autophagic defects observed. To do so, L6 cells were transiently transfected with a Myc‐tagged RHEB (Ras homolog enriched in brain) GTPase, which contained the Q64L mutation to remain GTP bound and can directly maintain mTORC1 activity 39, 40. Transfected cells were treated with iron for 24 h and then stained with anti‐LC3B antibodies to identify autophagosomal aggregates and anti‐Myc antibodies to mark cells transfected with Myc‐RHEB mutant. We found that cells with forced activation of mTORC1 exhibited a dramatic absence of large autolysosome accumulation under chronic iron treatment (Fig 3C, Box1) when compared to cells that were not transfected under the same conditions (Fig 3C, Box2). We next generated stable cell lines over expressing RHEB mutant (RHEB Q64L L6) and compared to wt (wild‐type) cells after treating with iron. As a control, we tested intracellular iron levels in RHEB Q64L and wt cells after 24‐h iron treatment and found them comparable (Fig 3D). After 24‐h iron treatment, wt cells developed autophagosomal aggregates and compromised LC3B‐free lysosomal pools as previously observed. Conversely, RHEB Q64L cells did not accumulate large autophagosomes and total lysosomal content was comparable with/without iron treatment (Fig 3E and F). Ultrastructure analysis by TEM further confirmed that RHEB Q64L stable cells did not develop abnormal autophagosomal structures after iron treatment, when compared to control (Fig 3G). To examine the functional significance of these observations, we tested if maintenance of mTORC1 activity affected IO‐induced insulin resistance. In RHEB Q64L cells, we observed improved insulin signaling (p‐IRS‐1 Y612 and p‐AKT T308) under IO (Fig 3H and I). Taken together, these data suggest that loss of mTORC1 activity during chronic IO is responsible for autophagy defects and insulin resistance.
IO‐induced insulin resistance and autophagy defects are reversed upon iron withdrawal
In the clinic, IO‐induced insulin resistance can be improved by iron‐restricted diets or iron chelation therapy 17, 41. Therefore, we examined whether IO‐induced insulin resistance could be rescued by withdrawing iron from treatment medium following iron treatment for 24 h. 24‐h withdrawal resulted in alleviation of IO, by significantly reducing intracellular iron levels as indicated by PGSK and ferrozine‐based colorimetric assay (Fig 4A and B). The recovery from IO occurred concurrently with insulin sensitivity restoration. Phosphorylation of insulin signaling molecules after insulin stimulation significantly recovered after 24‐h withdrawal (Fig 4C and D). We next analyzed the effects of iron withdrawal after iron overload on autophagosome and lysosomal populations. We observed that iron withdrawal after chronic IO resulted in clearance of accumulated autolysosomes and a restoration of LC3B‐negative lysosomes (Fig 4E and F). Iron overload has been described to inhibit the targeting of ferritin to autophagosomes. Therefore, we looked at ferritin protein levels under IO and after iron wash off. As expected, ferritin was stabilized by chronic IO consistent with a block in autophagic clearance and was cleared significantly by 24 h after media replacement without excess iron (Fig 4G). Taken together, our data show that iron removal results in clearance of abnormal autophagosomes, restoration of lysosomes, and increased insulin sensitivity.
Figure 4. Restoration of lysosomal pools following iron withdrawal reversed insulin resistance.
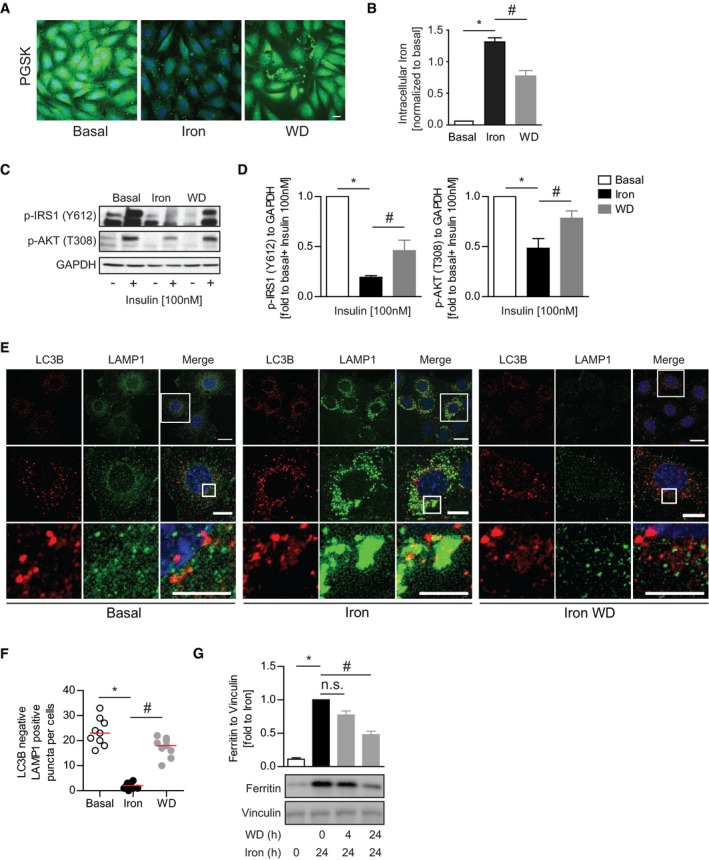
-
ARepresentative confocal microscope images of L6 cells pulsed with PGSK dye after iron treatment (250 μM, 24 h) followed by 24‐h iron withdrawal.
-
BFerrozine‐based colorimetric measurement of intracellular iron in L6 cells after iron treatment (250 μM, 24 h) followed by 24‐h withdrawal. *P < 0.05, # P < 0.05 (multiple unpaired Student's t‐test versus basal or iron).
-
C, DRepresentative Western blot images and quantification of phospho‐IRS1 (Y612) and phospho‐AKT (T308) to GAPDH in L6 cell iron treatment (250 μM, 24 h) followed by 24‐h withdrawal. *P < 0.05, # P < 0.05 (multiple unpaired Student's t‐test versus basal or iron).
-
ERepresentative epi‐immunofluorescent microscope images of L6 cells immuno‐stained against LC3B and LAMP1 after iron treatment (250 μM, 24 h) followed by 3‐h withdrawal.
-
FQuantification of autophagosome‐free lysosomes in (E). Experiments were performed three times, and one representative experiment is presented here. Red line indicates median. *P < 0.05, # P < 0.05 (multiple unpaired Student's t‐test versus basal or iron).
-
GRepresentative Western blot images and quantification of ferritin to vinculin in L6 cells after iron treatment (250 μM, 24 h) followed by withdrawal for 4 or 24 h. *P < 0.05, # P < 0.05 (multiple unpaired Student's t‐test versus basal or iron).
Chronic IO blocks mTORC1 reactivation on autolysosomes and signaling to UVRAG
We next sought to determine the mechanism underlying IO‐induced autophagic defects. We previously determined that forced mTORC1 activation was sufficient to block autolysosome accumulation (Fig 3C and G). Therefore, we first sought to characterize the effects of prolonged IO on regulatory phosphorylation of mTORC1. L6 cells were treated with iron for 24 h or 24 h plus iron withdrawal. mTORC1 was monitored by phosphorylation at S2448, which correlates with mTORC1 activity 42, 43. We observed mTORC1 phosphorylation was greatly diminished at 24‐h IO and recovered after 4 h of iron withdrawal (Fig 5A). mTORC1 phosphorylation at S2448 is dependent on localization to the lysosome or autolysosome for activation by the RHEB‐GTPase activity, which is in turn regulated by tuberous sclerosis complex (TSC)‐AKT signaling 44, 45. We stained cells for endogenous mTOR and LC3B and found that mTOR was localized to autolysosomes under prolonged IO, indicating localization defects are likely not the cause of mTORC1 activity loss (Fig EV4A). Therefore, we next looked at the effect of IO on AKT‐mediated inhibition of TSC2. We observed a decrease in inhibitory phosphorylation of TSC2 under IO, which was dramatically reversed upon wash off (Fig 5B). However, when iron withdrawal media was supplemented with AKT inhibitor, mTORC1 was not re‐activated upon iron removal (Fig 5B). Together, these data demonstrate that IO results in a decrease in AKT‐mediated repression of TSC2, resulting in a potent repression of RHEB and mTORC1.
Figure 5. Molecular mechanisms underlying IO‐induced mTOR activity suppression.
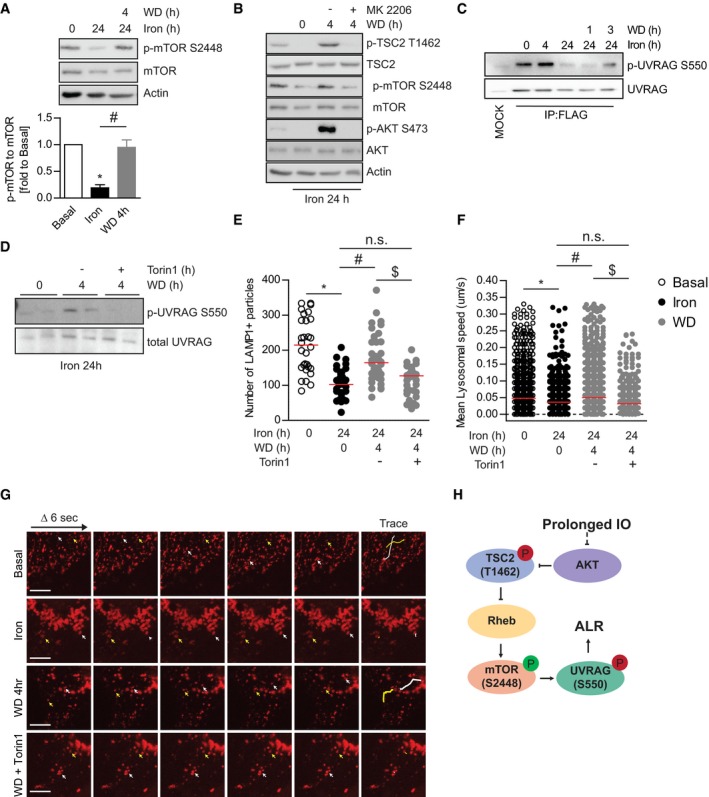
-
ARepresentative Western blot images and quantification of phospho‐mTOR S2448, total mTOR, and actin in L6 cells after iron treatment (250 μM, 24 h) followed by withdrawal for 4 h. Experiments were performed three times. *P < 0.05, # P < 0.05 (multiple unpaired Student's t‐test versus basal or iron).
-
BRepresentative Western blot images of phospho‐mTOR S2448, total mTOR phospho‐TSC2 T1462, total TSC2, phospho‐AKT S473, total AKT, and actin in L6 cells after iron treatment (250 μM, 24 h) followed by withdrawal for 4 h with or without MK2206 (AKT inhibitor).
-
CRepresentative Western blot images of phospho‐UVRAG S550 and total UVRAG in L6 cells transfected with FLAG‐UVRAG and FLAG pulldown after iron treatment (250 μM, 4 and 24 h) followed by withdrawal for 1 and 3 h.
-
DRepresentative Western blot images of endogenous phospho‐UVRAG S550 and total UVRAG expression in L6 cells after iron treatment (250 μM, 24 h) followed by withdrawal for 4 h with or without torin1 (200 nM).
-
E, FLive cell imaging analysis of L6 cells transfected with LAMP1‐RFP from Movie EV2: lysosomal number (E) and speed (F) in L6 cells after iron treatment (250 μM, 24 h) followed by withdrawal for 4 h with or without Torin1 (200 nM). Experiments were performed three times, and all biological and technical replicates are plotted here. Red line indicates median. *P < 0.05, # P < 0.05, $ P < 0.05 (one‐way ANOVA with multiple comparisons).
-
GRepresentative confocal images of time‐lapse captures of lysosomes at 6‐s intervals for 30 s. White and yellow arrows mark positions of two representative LAMP1 puncta over 30 s, and the traces of puncta were shown as lines in final panel.
-
HSchematic diagram of mTOR signaling regulation by IO.
Figure EV4. Molecular mechanisms underlying iron‐mediated mTOR suppression and restoration of lysosomal pools after iron withdrawal.
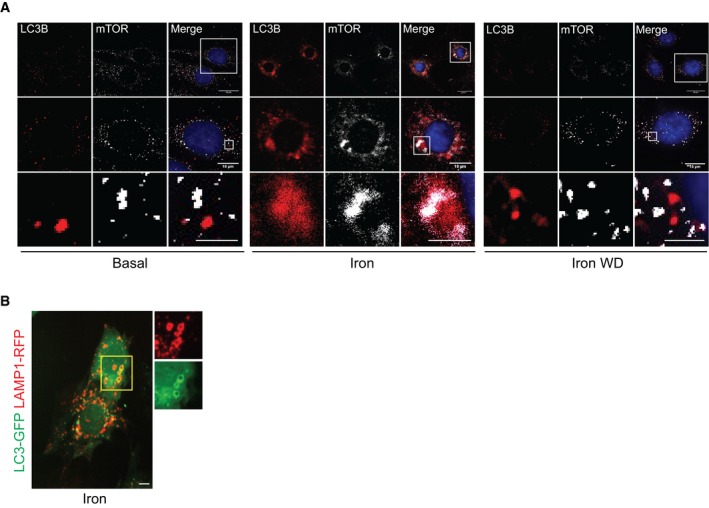
- Representative epi‐fluorescent microscope images of L6 cells immuno‐stained against LC3B and mTOR after iron treatment (250 μM, 24 h) followed by withdrawal for 4 h.
- Representative confocal images of L6 cells transfected with LC3‐GFP and LAMP1‐RFP after 24‐h iron treatment (250 μM).
The lysosomal loss, mTORC1 inhibition, and autophagy defect we observe under IO are all consistent with a defect in ALR. mTORC1 promotes ALR through scission of the autolysosome activity via direct phosphorylation of UVRAG, a component of the VPS34 lipid kinase complex 38. Phospholipid production by UVRAG‐containing VPS34 complexes is essential for the scission of the autolysosomal membrane. mTORC1‐mediated phosphorylation of UVRAG on serine S550 was monitored in L6 cells transfected with FLAG‐UVRAG that were treated with iron as indicated followed by immunoprecipitation of FLAG‐UVRAG‐containing VPS34 complexes (Fig 5C). Interestingly, at 4‐h iron treatment we saw a slight increase in mTORC1‐mediated UVRAG phosphorylation, which indicates that mTORC1 can be activated at the autolysosome even when global mTORC1 activity (as measured by S6K phosphorylation) is low (Fig 3A). This is consistent with the relatively normal functioning of autophagosomes and lysosomes that we observe at this time point (Fig 2C and D). However, at 24‐h iron treatment we observed a dramatic loss in mTORC1‐mediated phosphorylation of UVRAG (Fig 5C), indicating mTORC1 is incapable of efficiently promoting ALR under chronic iron treatment despite an overabundance substrate (mature autolysosomes). Additionally, the removal of iron resulted in a partial recovery of mTORC1‐mediated UVRAG phosphorylation, indicating that IO‐induced stress was responsible for the loss of ALR signaling (Fig 5C). We then used Torin1, a well‐established mTOR inhibitor 46, 47, and found that in its presence recovery of UVRAG signaling was completely abolished, confirming that mTORC1 ALR signaling is regulated by IO (Fig 5D). Based on the recovery of mTORC1 signaling to UVRAG upon iron withdrawal, we hypothesized that treatment of cells with Torin1 would be sufficient to block lysosomal recovery following iron withdrawal. To validate our hypothesis, we monitored lysosomal number and trafficking using cells transfected with RFP‐LAMP1 to determine whether inhibition of mTORC1 was sufficient to ablate the rescue of lysosomes upon iron withdrawal after chronic IO. RFP‐LAMP1 was present on enlarged autolysosomes similar to endogenous LAMP1 (Fig EV4B). Under basal conditions, the number of lysosomes ranged from 100 to 300 per cell, and continuously trafficked and underwent fusion events (Fig 5E). On the other hand, iron treatment significantly reduced the number of lysosomes (Fig 5E) and the motility of LAMP1 particles was reduced significantly (Fig 5F and G, Movie EV2). The lysosomal pools were significantly recovered near basal levels after 4‐h withdrawal. However, as predicted the addition of Torin1 abolished recovery of lysosomal pools and lysosomal motility following iron withdrawal (Fig 5E and F). Collectively, these data demonstrate that prolonged IO results in lysosomal loss due to ablation of mTORC1 reactivation on autolysosomes caused by alterations in AKT‐TSC‐RHEB signaling (Fig 5H).
Induction of IO in mice and development of insulin resistance following IO
As in previous studies which have adopted injections to induce IO 48, 49, we delivered iron intravenously at 15 mg/kg, a dose with minimal toxicity 50, via three injections at 2‐h intervals (Fig 6A). Twenty‐four hours after first administration, animals were sacrificed, and tissue iron accumulation examined with Perls Prussian Blue Staining. As expected, a robust increase in hepatic iron staining and more modest increase in skeletal muscle iron content was apparent (Fig 6B). Western blotting indicated that skeletal muscle ferritin levels increased while TfR1 levels decreased significantly in IO mice (Fig 6C and D). IO mice exhibited signs of peripheral insulin resistance, as shown upon examination of glucose handling via insulin and glucose tolerance tests (Fig 6E–H). To directly investigate changes in skeletal muscle insulin sensitivity, we examined insulin‐stimulated phosphorylation of IRS‐1 (Y612) and AKT (T308) and observed significantly attenuated insulin‐induced phosphorylation in IO mice (Fig 6I and J). Collectively, these data indicate that our intravenous iron injection regimen recapitulated key aspects of iron‐induced insulin resistance in skeletal muscle from our L6 cell culture model.
Figure 6. Development of acute iron overload (IO) in vivo model and validation of IO and insulin resistance in skeletal muscle.

-
ASchematic diagram of iron‐dextran injections’ experimental plan.
-
BPrussian Perl blue staining in liver and skeletal muscle after iron injections.
-
C, DRepresentative Western blot images and quantification of ferritin and Tfr1 (indicated by arrowhead) to tubulin in skeletal muscles 24 h after iron injections. *P < 0.05 (unpaired Student's t‐test versus control).
-
EInsulin tolerance test 24 h after iron injections.
-
FGlucose tolerance test (GTT) 24 h after iron injections.
-
GQuantification of area under curve in ITT (E). *P < 0.05 (unpaired Student's t‐test versus control).
-
HQuantification of area under curve for panel (F). *P < 0.05 (unpaired Student's t‐test versus control).
-
I, JRepresentative Western blot images and quantification of phospho‐IRS1 (Y612) and phospho‐AKT (T308) to GAPDH in skeletal muscles 24 h after iron injection followed by i.p. insulin injection. *P < 0.05 (unpaired Student's t‐test versus control).
IO caused reduced lysosomal pools with reduced UVRAG signaling and inhibited autophagy flux
To measure ALR events in vivo, we performed LC3B and LAMP1 immunofluorescence on tissue sections and observed a pronounced difference between the control and IO groups (Fig 7A). In control mice, there was a minimal complement of autophagosomes (LC3B puncta) yet numerous lysosomes (LAMP1 puncta), quantitatively 200–300 puncta per field of view (Fig 7B). In IO mice, the number of autophagosomes increased while the number of lysosomes decreased significantly to less than 100 puncta per field of view. TEM analysis of muscle tissue from the control group confirmed the predominant appearance of lysosomes as single membrane clear structures, with occasional observation of autophagosomes (Fig 7C). On the other hand, TEM analysis of muscle from iron group showed very sparse lysosomal content, with much smaller size than lysosomal structures observed in the control group. Also present in IO samples were tubular projections that are characteristic of reduced activity of UVRAG‐containing VPS34 complexes 38 (Fig 7C). We further analyzed autophagy flux and ALR by Western blotting of UVRAG phosphorylation, LC3B, and p62 expression. In iron‐treated mice, phosphorylation of UVRAG at S550 significantly decreased (Fig 7D), matching in vitro data (Fig 5C and D). Moreover, both LC3B‐II expression and p62 expression in the IO group increased, indicating impaired autophagy flux, compared to the control groups (Fig 7E and F). These changes were also observed in liver, indicating that IO‐induced ALR defects are not limited to skeletal muscle and may have widespread highly significant pathophysiological implications (Fig EV5). Taken together, we have identified that IO induces an mTORC1 reactivation defect in skeletal muscle which leads to an ALR defect (Fig 8). This represents a new mechanistic link connecting disturbed iron homeostasis to insulin resistance and metabolic dysfunction.
Figure 7. Evidence of ALR defects in skeletal muscles after acute IO .

-
ARepresentative confocal microscope images of skeletal muscle tissue sections immuno‐stained against LC3B (Alexa 647) and LAMP1 (Alexa 555)
-
BQuantification of lysosomes (LAMP1 puncta) in skeletal muscles 24 h after iron injections. *P < 0.05 (unpaired Student's t‐test versus control).
-
CRepresentative TEM images of skeletal muscle 24 h after iron injections.
-
DRepresentative Western blot images of phospho‐UVRAG S550 and total UVRAG in skeletal muscle 24 h after iron injections.
-
E, FRepresentative Western blot images and quantification of LC3‐B and p62 to tubulin in skeletal muscles 24 h after iron injections. *P < 0.05 (unpaired Student's t‐test versus control).
Figure EV5. ALR defects in liver after iron overload.

-
A, BRepresentative Western blot and quantification of ferritin and Tfr1 to GAPDH in liver 24 h after iron injections. P < 0.05 (unpaired Student's t‐tests versus control).
-
CConfocal microscope images of liver tissue sections immuno‐stained with against LC3B (Alex 647) and LAMP1 (Alexa 555).
-
D, ERepresentative Western blot images and quantification of phospho‐UVRAG S550 to total UVRAG in liver 24 h after iron injections.
Figure 8. Schematic diagram of IO‐mediated autophagy regulation.

Working model of autophagy regulation by IO. Acute IO leads to inhibition of mTORC1 leading to autophagy induction. Prolonged IO prevents ALR‐mediated production of new lysosomes through AKT‐TSC‐Rheb‐mTORC1‐UVRAG signaling defects. The lack of free lysosomes contributes to autophagy inhibition and insulin resistance in skeletal muscle.
Discussion
IO is a devastating and complex condition that most notably arises in individuals with beta thalassemia that require frequent blood transfusions or those with hereditary hemochromatosis, while IO in metabolic syndrome is also a common finding 10. The pathophysiological mechanism underlying IO‐induced diabetes is complicated as both insulin deficiency and insulin resistance contribute 8. The causative relationship between IO and insulin secretion defects is well established 51, yet the precise mechanisms whereby iron can elicit insulin resistance are complicated and we believe that IO in skeletal muscle is underappreciated 13, 52. Here, we used an in vitro model using L6 cells and translated this to analysis of mouse skeletal muscle using an in vivo model of iron overload. In both, we observed that skeletal muscle insulin sensitivity was significantly compromised after IO. We found that preventing excess free iron levels in L6 cells using an iron chelator could prevent IO‐induced insulin resistance and metabolic dysfunction, which is in keeping with the fact that clinical interventions to reduce free iron improved insulin sensitivity and can delay onset of T2D 11, 12, 13.
We and others have recently focused on the role of autophagy in regulation of metabolism at various levels 19, 53, 54. For example, we have previously shown that stimulation of autophagy by adiponectin was of functional significance in improving insulin sensitivity and metabolism in skeletal muscle 19. Interestingly, IO‐induced insulin resistance has been described to involve reduced adiponectin expression in adipocytes. However, systematic IO did not always translate to insulin resistance because adipocytes could enhance iron excretion to avoid intracellular iron overload 55.
The relationship between IO and autophagy has been examined, where acute IO was observed to stimulate autophagy 56, 57. Indeed, this is in agreement with our own observations in that IO exposure for up to 8 h stimulates autophagy. This increase in autophagy is likely the result of a compensatory cellular response upon detection of cellular stress. Acute IO activation of autophagy is also consistent with the observed rapid inhibition of mTORC1, the single most potent repressor of autophagy in mammals that responds to a large number of stressors including nutrient starvation and ER stress 58, 59, 60, 61. Yet, there are several problems with these conditions. First, acute IO does not reflect clinical reality of individuals that suffer from IO–chronic IO treatments and their pathophysiological effects are a more realistic case study. Second, initiation of autophagy is only the first step in a much more elaborate series of events that evolved to resolve stress and promote cell survival. Following autophagy, autophagosomes mature by fusing with lysosomes to degrade cargo and this is then followed by a third stage, whereby autolysosomal membranes are resorbed back into the endomembrane system, including reformation of lysosomes using a process now referred to as ALR. ALR is thus critical for cells to regain their degradative capacity and autophagic proficiency under prolonged stress. We now show for the first time that chronic IO causes autolysosomes to accumulate while depleting cells of free lysosomes, suggesting a defect in ALR.
Importantly, we have determined that IO prevents the reactivation of mTORC1 on autolysosomes, thereby causing a precipitous loss of a distinct lysosomal compartment. Normally, degradation of autophagic cargo locally stimulates mTORC1 on matured autolysosomes, even when the majority of mTORC1 remains inactive and is not bound to lysosomes. This localized reactivation of mTORC1 then promotes lysosomal reformation from the spent autolysosomes 38. However, since IO prevents mTORC1 activation, lysosome reformation is impaired and lysosome numbers are depleted. Conversely, iron withdrawal restored mTORC1 activity, downstream UVRAG signaling, lysosomal numbers, and insulin signaling. Furthermore, forced activation of mTORC1 by expressing the constitutively active RHEB‐GTPase not only prevented autophagosome accumulation with minimal lysosomal loss, but also restored insulin sensitivity in cells exposed to IO. Together, these data strongly indicate that the block of mTORC1 activation on autolysosomes by IO is a contributor of insulin resistance.
Overall, our study adds important new knowledge on a novel molecular mechanisms contributing to insulin resistance in response to IO and presents the first in vivo model to show an ALR defect 27. Mechanistically, we observed that chronic IO led to a decrease in Akt‐mediated repression of TSC2, resulting in a potent repression of RHEB and mTORC1, with consequent loss of ALR. Furthermore, our data indicate that mTOR‐UVRAG‐dependent lysosomal pool regeneration is an important contributor in maintaining autophagic flux and insulin sensitivity in skeletal muscle. Together, our data uncover a previously undocumented mechanism via which chronic IO limits autophagic capacity and leads to metabolic dysfunction. This observation may have implications in a wide range of disease states where cellular IO plays a pathogenic role.
Materials and Methods
Materials
The cell culture medium [α‐minimal essential medium (α‐MEM)], fetal bovine serum (FBS), and antibiotics/antimycotic solution were purchased from Wisent (St Bruno, QC, Canada). Cytochalasin B (Sigma) and human insulin (Humulin) were purchased from Eli Lilly (Toronto, ON, Canada), and deoxy‐D‐[2‐3H] glucose was purchased from PerkinElmer (Woodbridge, ON, Canada). Ferrous Sulfate heptahydrates (FeSO4·7H2O, Cat#310077), Torin1 (Cat# 475991) Monoclonal Anti‐Vinculin (Cat#V9131), and beta‐actin antibody (Cat#A5441 clone AC‐15) were purchased from Sigma‐Aldrich (Ottawa, ON, Canada). AKT inhibitor MK‐2206 was purchased from Selleck Chemicals. S6K (Cat#ab32529), LAMP1 (Cat#ab25630), and Alexa 647‐conjugated antibody (goat anti‐rabbit, Cat#ab15007) were purchased from Abcam (Cambridge, MA, USA). LC3B (Cat#2775), GAPDH (Cat#2118), tubulin (Cat#2148) antibodies, and phospho‐specific antibodies (phosphorylation sites) for AKT (T308, Cat#4056), phospho‐S6K (T389, Cat#9234), phospho‐ULK1 (S757, Cat#14202), mTOR (Cat#2983) phospho‐mTOR (S2448, Cat#2971), TSC2 (Cat#4308T) phospho‐TSC2 (T1462, Cat#3617T) phospho‐AKT (Ser473 Cat#4060), total AKT (Cat#4691), horseradish peroxidase‐conjugated secondary antibodies (anti‐rabbit‐IgG, Cat# #7074 and anti‐mouse‐IgG, Cat#7076) were purchased from Cell Signaling Technology (Beverly, MA, USA). UVRAG phospho‐specific (S550, Cat# S307D) and UVRAG antibody (Cat# S323D) were provided from MRC PPU at The University of Dundee. Anti‐transferrin receptor (Tfr1) monoclonal antibody (Cat#13‐6800 clone H68.4), phospho‐specific antibody for anti‐IRS‐1 (Y612, Cat#44‐816G), Alexa 555‐conjugated antibody (donkey anti‐rat, Cat#A‐21434), Lipofectamine 2000 (Cat# 11668019), and ProLong Gold (Cat#P36930) were purchased from Thermo Fisher Scientific (Burlington, ON, Canada). Anti‐LC3B (Cat#PM036) and Anti‐p62 (Cat# M162‐3) were purchased from MBL. Anti‐ferritin heavy chain (Cat#NBP1‐31944) was purchased from Novus Biologicals. Anti‐Myc (Cat# 9E10) was purchased from Alzforum. LAMP1 (Cat#sc‐19992, Santa Cruz) antibody and iron‐dextran dextran (a ferric hydroxide, Fe(OH)3, complex with low molecular weight dextran, Cat#9004‐66‐4) were purchased from Santa Cruz Biotechnology Inc. VECTASHIELD Antifade Mounting Medium with DAPI (Cat# H‐1200) was purchased from Vector Laboratories. Polyvinylidene difluoride membrane was from Bio‐Rad Laboratories, Inc (Burlington, ON, Canada), and chemiluminescence reagent plus was from PerkinElmer (Boston, MA). Concanamycin A was purchased from BioShop Canada Inc. (Cat# FOL202, Burlington, ON, Canada). All other reagents and chemicals used were of the highest purity available.
Cell culture and generation of L6 cell line stably overexpressing gene of interest
L6 skeletal muscle cell line (ATCC® CRL‐1458™ tested mycoplasma free) was grown to confluency in α‐MEM supplemented with 10% [volume/volume (v/v)] FBS and 1% (v/v) antibiotic/antimycotic solution under a humidified atmosphere of 95% air and 5% CO2 at 37°C. During treatment or iron withdrawal, the cells were switched to medium containing 0.5% (v/v) FBS and 1% (v/v) antibiotic/antimycotic. During iron treatment, ferrous iron stock, prepared by dissolving ferrous sulfate heptahydrate (FeSO4·7H2O) in sterile distilled water at 10 mM, was dissolved in treatment medium at appropriate concentration. L6‐GLUT4 cells stably transfected to overexpress myc‐tagged GLUT4 were a gift from Dr Amira Klip, The Hospital for Sick Children, Toronto. IRE‐CFP L6 cells stably transfected to overexpress IRE‐CFP were a gift from Dr.s’ James R Connor and Stephanie Patton, Penn State Hershey Medical Center, USA 62. eGFP‐mCherry‐LC3B, eGFP‐mCherry‐p62 stables, and myc‐tagged RHEB Q64L mutant L6 cells were infected with lentivirus, and a stable polyclonal population was obtained through puromycin (1 μg/ml) selection for stable integration.
Determination of intracellular iron
Intracellular iron concentration was estimated by ferrozine‐based assay as described previously 63. Phen green SK (PGSK) diacetate dyes 64 were used to determine the degree of di‐valent ions. Iron‐specific probe‐1 (IP‐1) 29, 65 was a kind gift from Dr Christopher J. Chang, University of California, Berkeley, and was used to determine labile iron levels in cells.
Gene expression analysis
Quantitative polymerase chain reaction (qPCR) was performed as previously described 19. Total RNA was extracted with RNEasy Mini Kit (Qiagen, Toronto, Canada) and then converted to cDNA with GoScript Reverse Transcriptase (Promega). PCR cycle was performed with iTaq™ Universal SYBR® green mixture (Bio‐Rad) at following condition: 2 min at 95°C, followed by 40 cycles of 15 s each at 95°C, 60°C, 72°C. Relative gene expression levels were normalized to 18S rRNA. Primers used in this study are summarized in Table 1.
Table 1.
Primers used in this study for PCR
| Name | Kind | Sequence (5′–3′) |
|---|---|---|
| FTH | Forward | CTTTGCAACTTCGTCGCTCC |
| Reverse | AGTCATCACGGTCAGGTTTCTTT | |
| FTL | Forward | AGACCCTCACCTCTGTGACT |
| Reverse | GGCGGTTACAAAGCTGCCTA | |
| SLC40A1 | Forward | CGTGCTATCTCCGGTTCCTC |
| Reverse | TGTCAAGAGGAGGCCGTTTC | |
| TFRC | Forward | AGCCAGATCAGCATTCTCTAACT |
| Reverse | GCCTTCATGTTATTGTCGGCAT | |
| 18S rRNA | Forward | CCATAAACGATGCCGACTG |
| Reverse | CGCTCCACCAACTAAGAAC |
Western blotting
Lysates were prepared as we described before 66. SDS–PAGE was performed, and proteins were transferred to PVDF membrane then incubated with blocking buffer (3% BSA or 5% skim milk in TBS‐T), washed with TBS‐T five times, and incubated with primary antibodies (1:1,000 in TBS‐T 2% BSA) overnight at 4°C. Next, membranes were incubated in appropriate horseradish peroxidase‐conjugated secondary antibody (1:10,000 in TBS‐T 2% BSA or skim milk) for 1 h at room temperature. Quantitation of each specific protein band was determined by densitometric scanning with correction for the respective loading control.
Insulin sensitivity test
Glucose uptake was determined by measuring uptake of 2‐deoxy‐d‐[3H] glucose exactly as described previously 67. L6‐GLUT4 cells were incubated with insulin (10 and 100 nM) for 20 min after treatment with or without iron as described. Phosphorylation of IRS1 (Y612) and AKT (T308) was determined in cells incubated with insulin (10 or 100 nM) for 5 min after appropriate treatment.
Transmission electron microscope (TEM)
TEM was performed as described previously 66. Briefly, samples were fixed in fixative (2% formaldehyde, 2% glutaaldehyde in 0.1 M sodium cacodylate buffer) for 2 h at room temperature. After washing three times with sodium cacodylate buffer, samples were fixed in 1% osmium tetroxide for 1 h at room temperature. After dehydration with ascending concentration of ethanol in series (50–100%), cells were embedded in Spurr's epoxy resin. Thin sections (60–80 nM) were cut with ultramicrotome and mounted on copper mesh grids. The sections were then contrasted with 1% uranyl acetate and lead citrate and examined with a FEI CM100 TEM and Kodak Megaplus Camera.
In vitro autophagy analysis
Activity of the autophagy pathway was monitored by Western blot and immunofluorescence‐based puncta quantification of LC3B. Flux assay was performed by measuring levels of LC3B‐I and LC3B‐II from cell lysates after appropriate treatment co‐treated with lysosomal inhibitor (chloroquine, 30 μM). LC3B immunofluorescence was performed as described previously 32. Briefly, cells were fixed, permeabilized, and blocked with PBS solution containing 1% BSA and 2% goat serum. After blocking, cells were incubated with blocking solution containing LC3B (Cat#PM036, MBL, conjugated with Cy3 Alexa Fluor 555 1:1,000) and mouse anti‐LAMP1 (Cat#A11029, 1:500). Cells were incubated with anti‐mouse Alexa Fluor 488 secondary antibody (Cat#ab25630, Thermo Fisher Scientific, 1:200) at room temperature for 1 h. After incubation, cells were mounted with DPAI after washes. Deconvoluted images were captured with an Apotome enabled Zeiss AxioObserver.Z1. For live cell imaging, L6 cells stably expressing eGFP‐mCherry‐LC3B or eGFP‐mCherry‐p62 were seeded into ibidi chambers and treated with iron (FeSO4, 250 μM) or starvation medium (without amino acid). Treatments were carried out in FluorBright™ phenol red‐free DMEM (Invitrogen) supplemented with GlutaMAX. Images were acquired and deconvolved using an environmental chamber control (DeltaVision Elite‐Olympus IX‐71 with FemtoJet Microinjector) microscopy.
Lysosome dynamics analysis
L6 cells were co‐transfected with LAMP1‐RFP and LC3‐GFP using Lipofectamine 2000 as per manufacturer's specifications. Following 8 h of transfection, cells were incubated in treatment medium then imaged live. During live cell imaging, cells were maintained in an environment set to 5% CO2 and 37°C. We used a Quorum Diskovery spinning disk confocal microscope system equipped with a Leica DMi8 microscope and connected to an Andor Zyla Megapixel sCMOS camera. Microscope and acquisition settings were controlled using Quorum Wave FX powered by MetaMorph software (Quorum Technologies, Guelph, ON). For time‐lapse imaging and determination of lysosomal dynamics, images were acquired every 3 s for 3 min. For determination of lysosomal numbers, images were acquired along the z‐plane at a defined interval of 0.3 μm.
For image analysis, lysosomal numbers were unbiasedly determined using particle detection tools in Volocity 6.3.0 software (PerkinElmer). Lysosomal numbers were determined in at least 15 cells per condition per experiment, where we repeated each experiment at least three independent times. For lysosomal track analysis, movies were analyzed using particle detection tools in Imaris (Bitplane) image analysis software. Lysosome particles were defined as having a minimum of 0.5 μm in diameter and tracked using the software's autoregressive motion track analysis function. To minimize mis‐tracking of particles, tracks were restricted to particles that moved a maximum distance of 1 μm between frames and with no more than a maximum gap distance of three frames. This analysis was completed for at least 6 cells per trial per condition with more than 100 tracks per cell. Track mean speed and displacement, which is defined as the distance between start and endpoints, were calculated.
Cathepsin activity assay
We used Magic Red Cathepsin L Kit (Bio‐Rad Technologies, Cat# ICT941) and performed experiments as follows. After 24‐h IO treatment, IO and control cells were incubated with 1× magic red cathepsin L reagent, concurrently or in the absence of 1 μM ConA, for 1 h, prior to live cell imaging. Using spinning disk confocal microscopy, confocal slices were acquired with a 0.3‐micron interval between slices. For image analysis, the total magic red fluorescence for MR‐positive puncta was determined and compared to the control counterpart.
Lysosomal β‐glucosidase activity assay
L6 cells were plated on ibidi 8‐well m‐Slide (Ibidi, Cat# 80826) overnight and treated with iron (FeSO4, 250 μM) for 24 h. Cells were then incubated in AMEM containing the β‐glucosidase substrate (5 μM) for 1 h (Marker Gene Technologies, Cat# M2775). Then, cells were washed 3 times with PBS prior to the addition of Opti‐Klear™ Live Cell Imaging Buffer. Images were acquired and deconvolved using an environmental chamber control (DeltaVision Elite‐Olympus IX‐71 with FemtoJet Microinjector) microscopy.
Cyto‐ID autophagy detection assay
L6 cells were plated on ibidi 8‐well m‐Slide (Ibidi, Cat# 80826) overnight and treated with iron (FeSO4, 250 μM). Cells were then incubated in AMEM without phenol red containing Cyto‐ID autophagy detection stain (Enzo, ENZ‐KIT175‐0050) for 30 min with/without iron and then washed with PBS. Images were acquired and deconvolved using an environmental chamber control (DeltaVision Elite‐Olympus IX‐71 with FemtoJet Microinjector) microscopy.
Iron overload animals, glucose tolerance tests (GTT), and insulin tolerance tests (ITT)
Animal facilities met the guidelines of Canadian Council on Animal Cars, and the York University Animal Care Committee approved the experimental protocols. Animals were fed ad libitum on regular chow diet and kept in temperature and humidity control rooms (21 ± 2°C, 35–40%) with a daily 12:12‐h light–dark cycle. Groups (n = 6) of 2‐month‐old C57/BL6 male mice were randomized into two groups and injected with iron‐dextran intravenously (15 mg/kg, diluted in PBS to make 150 μl injection volume) three times at 2 h of interval or with only PBS as control. After 24 h postfirst injection, GTT and ITT were performed, without blinding, as described previously 68. For phosphorylation of insulin signaling molecule analysis, mice were injected with 4 units of insulin per kg before sacrifice.
Tissue immunofluorescence
Paraffin‐embedded sections were deparaffinized and rehydrated with descending concentrations of ethanol and then brought into double distilled water. The antigens were retrieved in citrate buffer pH 6.0 in autoclave for 15 min. After three washes with PBS, sections then were permeabilized with Triton X‐100 (0.3% Triton X‐100 in PBS) and blocked with 2% BSA and 5% goat serum in PBS for 90 min. Sections were incubated with LAMP1 (Santa Cruz) and LC3B (MBL) at 1:100 in 2% BSA in PBS, followed by Alexa secondary antibody‐conjugated fluorophores (Alexa 555 donkey anti‐rat and Alexa 647 goat anti‐rabbit). Sections were mounted on coverslips with ProLong Gold and VECTASHIELD antifade mounting medium with DAPI, and images were captured with Zeiss LSM 700. The number of LAMP1 puncta was determined by setting pre‐set threshold “momentum” in ImageJ and counted the puncta (> 0.6 μm diameter, > 0.35 circularity) per field of views.
Statistical analysis
Data are expressed as mean ± standard error mean (SEM) from at least three separate experiments. The differences between groups were analyzed using Prism 5.0 (GraphPad Software Inc., San Diego, CA, USA) with one‐way analysis of variance (ANOVA) followed by Student's t‐test, with P < 0.05 considered as statistically significant. Quantification of endogenous lysosome and autophagosome number was performed using Perkin Elmer Volocity software for unbiased identification and quantification of objects. Vesicle numbers and individual vesicle sizes were identified from a minimum of nine representative cells, with a minimum of 40 vesicles per condition. Samples were compared using Student's t‐test. Autophagosome mobility was determined by blinded vesicle tracking using a minimum of 11 time points to track an average of 10 autophagosomes per sample. Mean velocity was calculated for each autophagosome, and sample sets across multiple conditions were compared using Student's t‐test.
Author contributions
JWSJ and RMA performed majority of experiments and contributed to manuscript writing. RP, ES, VEBH, and HKS made substantial experimental contributions. RJB designed lysosome dynamics experiments and edited the final manuscript. RCR and GS initiated and developed the project, discussed planning of experiments and interpretation of data, and wrote final manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Movie EV1
Movie EV2
Review Process File
Acknowledgements
GS acknowledges support from Canadian Institutes of Health Research (CIHR) and a Career Investigator Award from Heart & Stroke Foundation of Ontario. RCR also acknowledges support from CIHR (#PJT153034). JWSJ is a recipient of CIHR Frederick Banting and Charles Best Canada Graduate Scholarship. We greatly appreciate Dr.s’ James R Connor and Stephanie Patton, Penn State Hershey Medical Center for providing IRE‐CFP reporter and Dr Christopher J. Chang, University of California Berkeley for providing IP‐1 probe.
EMBO Reports (2019) 20: e47911
Contributor Information
Ryan Charles Russell, Email: ryan.russell@uottawa.ca.
Gary Sweeney, Email: gsweeney@yorku.ca.
References
- 1. Evstatiev R, Gasche C (2012) Iron sensing and signalling. Gut 61: 933–952 [DOI] [PubMed] [Google Scholar]
- 2. Wang J, Pantopoulos K (2011) Regulation of cellular iron metabolism. Biochem J 434: 365–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zimmermann MB, Hurrell RF (2007) Nutritional iron deficiency. Lancet 370: 511–520 [DOI] [PubMed] [Google Scholar]
- 4. Hentze MW, Muckenthaler MU, Galy B, Camaschella C (2010) Two to tango: regulation of Mammalian iron metabolism. Cell 142: 24–38 [DOI] [PubMed] [Google Scholar]
- 5. Pantopoulos K, Porwal SK, Tartakoff A, Devireddy L (2012) Mechanisms of mammalian iron homeostasis. Biochemistry 51: 5705–5724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sebastiani G, Pantopoulos K (2011) Disorders associated with systemic or local iron overload: from pathophysiology to clinical practice. Metallomics 3: 971–986 [DOI] [PubMed] [Google Scholar]
- 7. Brissot P, Ropert M, Le Lan C, Loreal O (2012) Non‐transferrin bound iron: a key role in iron overload and iron toxicity. Biochem Biophys Acta 1820: 403–410 [DOI] [PubMed] [Google Scholar]
- 8. Simcox JA, McClain DA (2013) Iron and diabetes risk. Cell Metab 17: 329–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhuang T, Han H, Yang Z (2014) Iron, oxidative stress and gestational diabetes. Nutrients 6: 3968–3980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McClain DA, Abraham D, Rogers J, Brady R, Gault P, Ajioka R, Kushner JP (2006) High prevalence of abnormal glucose homeostasis secondary to decreased insulin secretion in individuals with hereditary haemochromatosis. Diabetologia 49: 1661–1669 [DOI] [PubMed] [Google Scholar]
- 11. Wongjaikam S, Kumfu S, Chattipakorn SC, Fucharoen S, Chattipakorn N (2015) Current and future treatment strategies for iron overload cardiomyopathy. Eur J Pharmacol 765: 86–93 [DOI] [PubMed] [Google Scholar]
- 12. Rajpathak SN, Crandall JP, Wylie‐Rosett J, Kabat GC, Rohan TE, Hu FB (2009) The role of iron in type 2 diabetes in humans. Biochem Biophys Acta 1790: 671–681 [DOI] [PubMed] [Google Scholar]
- 13. Datz C, Felder TK, Niederseer D, Aigner E (2013) Iron homeostasis in the metabolic syndrome. Eur J Clin Invest 43: 215–224 [DOI] [PubMed] [Google Scholar]
- 14. Fernandez‐Real JM, Lopez‐Bermejo A, Ricart W (2002) Cross‐talk between iron metabolism and diabetes. Diabetes 51: 2348–2354 [DOI] [PubMed] [Google Scholar]
- 15. Bofill C, Joven J, Bages J, Vilella E, Sans T, Cavalle P, Miralles R, Llobet J, Camps J (1994) Response to repeated phlebotomies in patients with non‐insulin‐dependent diabetes mellitus. Metabolism 43: 614–620 [DOI] [PubMed] [Google Scholar]
- 16. Abraham D, Rogers J, Gault P, Kushner JP, McClain DA (2006) Increased insulin secretory capacity but decreased insulin sensitivity after correction of iron overload by phlebotomy in hereditary haemochromatosis. Diabetologia 49: 2546–2551 [DOI] [PubMed] [Google Scholar]
- 17. Cooksey RC, Jones D, Gabrielsen S, Huang J, Simcox JA, Luo B, Soesanto Y, Rienhoff H, Abel ED, McClain DA (2010) Dietary iron restriction or iron chelation protects from diabetes and loss of beta‐cell function in the obese (ob/ob lep‐/‐) mouse. Am J Physiol Endocrinol Metab 298: E1236–E1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Galaris D, Pantopoulos K (2008) Oxidative stress and iron homeostasis: mechanistic and health aspects. Crit Rev Clin Lab Sci 45: 1–23 [DOI] [PubMed] [Google Scholar]
- 19. Liu Y, Palanivel R, Rai E, Park M, Gabor TV, Scheid MP, Xu A, Sweeney G (2015) Adiponectin stimulates autophagy and reduces oxidative stress to enhance insulin sensitivity during high‐fat diet feeding in mice. Diabetes 64: 36–48 [DOI] [PubMed] [Google Scholar]
- 20. Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147: 728–741 [DOI] [PubMed] [Google Scholar]
- 21. Yang JS, Lu CC, Kuo SC, Hsu YM, Tsai SC, Chen SY, Chen YT, Lin YJ, Huang YC, Chen CJ et al (2017) Autophagy and its link to type II diabetes mellitus. BioMedicine 7: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sarparanta J, Garcia‐Macia M, Singh R (2017) Autophagy and mitochondria in obesity and type 2 diabetes. Curr Diabetes Rev 13: 352–369 [DOI] [PubMed] [Google Scholar]
- 23. Zheng Q, Zhao Y, Guo J, Zhao S, Fei C, Xiao C, Wu D, Wu L, Li X, Chang C (2018) Iron overload promotes mitochondrial fragmentation in mesenchymal stromal cells from myelodysplastic syndrome patients through activation of the AMPK/MFF/Drp1 pathway. Cell Death Dis 9: 515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Watson A, Lipina C, McArdle HJ, Taylor PM, Hundal HS (2016) Iron depletion suppresses mTORC1‐directed signalling in intestinal Caco‐2 cells via induction of REDD1. Cell Signal 28: 412–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Egan D, Kim J, Shaw RJ, Guan KL (2011) The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 7: 643–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen Y, Yu L (2018) Development of research into autophagic lysosome reformation. Mol Cells 41: 45–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu G, Ahn J, Chang S, Eguchi M, Ogier A, Han S, Park Y, Shim C, Jang Y, Yang B et al (2012) Lipocalin‐2 induces cardiomyocyte apoptosis by increasing intracellular iron accumulation. J Biol Chem 287: 4808–4817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Au‐Yeung HY, Chan J, Chantarojsiri T, Chang CJ (2013) Molecular imaging of labile iron(II) pools in living cells with a turn‐on fluorescent probe. J Am Chem Soc 135: 15165–15173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Varghese J, James J, Vaulont S, McKie A, Jacob M (2018) Increased intracellular iron in mouse primary hepatocytes in vitro causes activation of the Akt pathway but decreases its response to insulin. Biochim Biophys Acta Gen Subj 1862: 1870–1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kimura S, Noda T, Yoshimori T (2007) Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent‐tagged LC3. Autophagy 3: 452–460 [DOI] [PubMed] [Google Scholar]
- 32. Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL (2013) ULK1 induces autophagy by phosphorylating Beclin‐1 and activating VPS34 lipid kinase. Nat Cell Biol 15: 741–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N (2008) FIP200, a ULK‐interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 181: 497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama‐Noda K, Ichimura T, Isobe T et al (2009) Two Beclin 1‐binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 11: 385–396 [DOI] [PubMed] [Google Scholar]
- 35. Zhang Y, Xu M, Xia M, Li X, Boini KM, Wang M, Gulbins E, Ratz PH, Li PL (2014) Defective autophagosome trafficking contributes to impaired autophagic flux in coronary arterial myocytes lacking CD38 gene. Cardiovasc Res 102: 68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kao JK, Wang SC, Ho LW, Huang SW, Chang SH, Yang RC, Ke YY, Wu CY, Wang JY, Shieh JJ (2016) Chronic iron overload results in impaired bacterial killing of THP‐1 derived macrophage through the inhibition of lysosomal acidification. PLoS One 11: e0156713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F et al (2010) Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465: 942–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Munson MJ, Allen GF, Toth R, Campbell DG, Lucocq JM, Ganley IG (2015) mTOR activates the VPS34‐UVRAG complex to regulate autolysosomal tubulation and cell survival. EMBO J 34: 2272–2290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Parmar N, Tamanoi F (2010) Rheb G‐proteins and the activation of mTORC1. Enzymes 27: 39–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li Y, Inoki K, Guan KL (2004) Biochemical and functional characterizations of small GTPase Rheb and TSC2 GAP activity. Mol Cell Biol 24: 7965–7975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fernandez‐Real JM, Penarroja G, Castro A, Garcia‐Bragado F, Hernandez‐Aguado I, Ricart W (2002) Blood letting in high‐ferritin type 2 diabetes: effects on insulin sensitivity and beta‐cell function. Diabetes 51: 1000–1004 [DOI] [PubMed] [Google Scholar]
- 42. Chiang GG, Abraham RT (2005) Phosphorylation of mammalian target of rapamycin (mTOR) at Ser‐2448 is mediated by p70S6 kinase. J Biol Chem 280: 25485–25490 [DOI] [PubMed] [Google Scholar]
- 43. Holz MK, Blenis J (2005) Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)‐phosphorylating kinase. J Biol Chem 280: 26089–26093 [DOI] [PubMed] [Google Scholar]
- 44. Inoki K, Li Y, Xu T, Guan KL (2003) Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 17: 1829–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Inoki K, Li Y, Zhu T, Wu J, Guan KL (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4: 648–657 [DOI] [PubMed] [Google Scholar]
- 46. Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS (2009) An ATP‐competitive mammalian target of rapamycin inhibitor reveals rapamycin‐resistant functions of mTORC1. J Biol Chem 284: 8023–8032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu Q, Chang JW, Wang J, Kang SA, Thoreen CC, Markhard A, Hur W, Zhang J, Sim T, Sabatini DM et al (2010) Discovery of 1‐(4‐(4‐propionylpiperazin‐1‐yl)‐3‐(trifluoromethyl)phenyl)‐9‐(quinolin‐3‐yl)benz o[h][1,6]naphthyridin‐2(1H)‐one as a highly potent, selective mammalian target of rapamycin (mTOR) inhibitor for the treatment of cancer. J Med Chem 53: 7146–7155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Martin LE, Bates CM, Beresford CR, Donaldson JD, McDonald FF, Dunlop D, Sheard P, London E, Twigg GD (1955) The pharmacology of an iron‐dextran intramuscular haematinic. Br J Pharmacol Chemother 10: 375–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Italia K, Colah R, Ghosh K (2015) Experimental animal model to study iron overload and iron chelation and review of other such models. Blood Cells Mol Dis 55: 194–199 [DOI] [PubMed] [Google Scholar]
- 50. Heming N, Letteron P, Driss F, Millot S, El Benna J, Tourret J, Denamur E, Montravers P, Beaumont C, Lasocki S (2012) Efficacy and toxicity of intravenous iron in a mouse model of critical care anemia*. Crit Care Med 40: 2141–2148 [DOI] [PubMed] [Google Scholar]
- 51. Cooksey RC, Jouihan HA, Ajioka RS, Hazel MW, Jones DL, Kushner JP, McClain DA (2004) Oxidative stress, beta‐cell apoptosis, and decreased insulin secretory capacity in mouse models of hemochromatosis. Endocrinology 145: 5305–5312 [DOI] [PubMed] [Google Scholar]
- 52. Chen W, Huang FW, de Renshaw TB, Andrews NC (2011) Skeletal muscle hemojuvelin is dispensable for systemic iron homeostasis. Blood 117: 6319–6325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xu A, Sweeney G (2015) Emerging role of autophagy in mediating widespread actions of ADIPOQ/adiponectin. Autophagy 11: 723–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ahlstrom P, Rai E, Chakma S, Cho HH, Rengasamy P, Sweeney G (2017) Adiponectin improves insulin sensitivity via activation of autophagic flux. J Mol Endocrinol 59: 339–350 [DOI] [PubMed] [Google Scholar]
- 55. Gabrielsen JS, Gao Y, Simcox JA, Huang J, Thorup D, Jones D, Cooksey RC, Gabrielsen D, Adams TD, Hunt SC et al (2012) Adipocyte iron regulates adiponectin and insulin sensitivity. J Clin Investig 122: 3529–3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cen WJ, Feng Y, Li SS, Huang LW, Zhang T, Zhang W, Kong WD, Jiang JW (2017) Iron overload induces G1 phase arrest and autophagy in murine preosteoblast cells. J Cell Physiol 233: 6779–6789 [DOI] [PubMed] [Google Scholar]
- 57. Chen G, Jing CH, Liu PP, Ruan D, Wang L (2013) Induction of autophagic cell death in the rat brain caused by iron. Am J Med Sci 345: 369–374 [DOI] [PubMed] [Google Scholar]
- 58. Russell RC, Yuan HX, Guan KL (2014) Autophagy regulation by nutrient signaling. Cell Res 24: 42–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ganley IG, du Lam H, Wang J, Ding X, Chen S, Jiang X (2009) ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 284: 12297–12305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH (2009) ULK‐Atg13‐FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 20: 1992–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N et al (2009) Nutrient‐dependent mTORC1 association with the ULK1‐Atg13‐FIP200 complex required for autophagy. Mol Biol Cell 20: 1981–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Henderson RJ, Patton SM, Connor JR (2005) Development of a fluorescent reporter to assess iron regulatory protein activity in living cells. Biochem Biophys Acta 1743: 162–168 [DOI] [PubMed] [Google Scholar]
- 63. Riemer J, Hoepken HH, Czerwinska H, Robinson SR, Dringen R (2004) Colorimetric ferrozine‐based assay for the quantitation of iron in cultured cells. Anal Biochem 331: 370–375 [DOI] [PubMed] [Google Scholar]
- 64. Esposito BP, Epsztejn S, Breuer W, Cabantchik ZI (2002) A review of fluorescence methods for assessing labile iron in cells and biological fluids. Anal Biochem 304: 1–18 [DOI] [PubMed] [Google Scholar]
- 65. Aron AT, Reeves AG, Chang CJ (2018) Activity‐based sensing fluorescent probes for iron in biological systems. Curr Opin Chem Biol 43: 113–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jahng JW, Turdi S, Kovacevic V, Dadson K, Li RK, Sweeney G (2015) Pressure overload‐induced cardiac dysfunction in aged male adiponectin knockout mice is associated with autophagy deficiency. Endocrinology 156: 2667–2677 [DOI] [PubMed] [Google Scholar]
- 67. Palanivel R, Maida A, Liu Y, Sweeney G (2006) Regulation of insulin signalling, glucose uptake and metabolism in rat skeletal muscle cells upon prolonged exposure to resistin. Diabetologia 49: 183–190 [DOI] [PubMed] [Google Scholar]
- 68. Liu Q, Zhang J, Xu Y, Huang Y, Wu C (2013) Effect of carvedilol on cardiomyocyte apoptosis in a rat model of myocardial infarction: a role for toll‐like receptor 4. Indian J Pharmacol 45: 458–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Movie EV1
Movie EV2
Review Process File