

Abstract
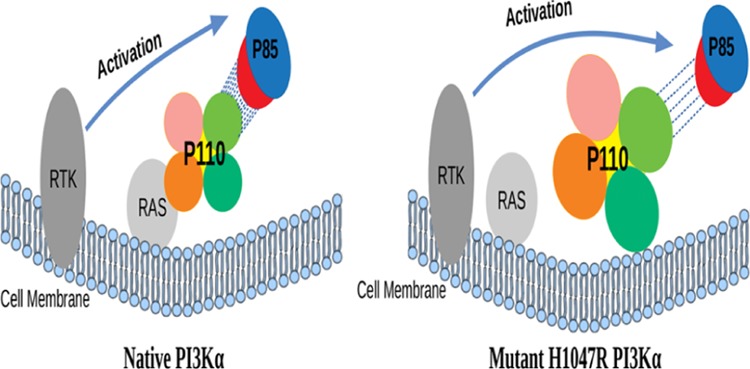
PI3Kα is a heterodimer protein consisting of two subunits (p110α and p85α) which promotes various signaling pathways. Oncogenic mutation in the catalytic subunit p110α of PI3Kα at the 1047 position in the kinase domain substitutes the histidine with arginine. This mutation brings about conformational transitions in the protein complex. These transitions expose the membrane binding region of PI3Kα, and then it independently binds to the cell membrane through its kinase domain without the involvement of the membrane-bound protein RAS. We observed notable changes between the protein complexes (p110α–p85α) of native and mutant structures at the atomic level using molecular dynamics simulations. Simulation results revealed formation of a less number of hydrogen bonds between the two subunits in the mutant protein complex which led the two subunits to move away from each other. This increase in distance between the subunits led to an expanded structure, thereby increasing the flexibility of the protein complex. Furthermore, a study of secondary structure elements and the electrostatic potential of the protein also gave a molecular insight into the change in interaction patterns of the protein with the plasma membrane. Our finding clearly indicates the role of mutation in oncogenesis and provides an insight into considering the structural aspects to handle this mutation.
Introduction
Phosphatidylinositol 3 kinase (PI3K) phosphorylates the substrate (PIP2) to generate the key signaling molecule (PIP3), which acts as a secondary messenger and promotes downstream signaling of diverse processes at the cellular level like differentiation, cell survival, metabolism, and intracellular cell signalling.1−17
There are a total of eight PI3K isoforms grouped into three definite classes (I, II, and III) on the basis of their structure, function, tissue differentiation, activation mechanism, and substrate specificities.2,7,12,16,18 A catalytic and a regulatory subunit are present in heterodimeric class I enzymes consisting of IA and IB subclasses. Class IA has p110 (α/β/δ, which are gene products of PIK3CA/PIK3CB/PIK3CD, respectively) as the catalytic subunit that interacts with p85 which acts as the regulatory subunit.4−7,12
The PI3Kα isoform of the class I PI3K is the most usually mutated in various tumors of different body parts.6,19 It has p110α as the catalytic subunit and p85α as the regulatory subunit.13,20 P110α consists of five domains: (i) N terminal adapter-binding domain (ABD) from residue 1–108, which interacts with p85α, (ii) RAS-binding domain (RBD) from residue 190–291, which regulates the interaction between p110α and RAS proteins on the plasma membrane,9,16 (iii) the C2 domain from residue 330–480 responsible for binding of lipid particles, (iv) helical domain from residue 525–696 which provides cleft for other domains, and (v) kinase domain from residue 697–1068 comprising of different binding (ATP and substrate) regions.2,13,15,21 Moreover, p85α consists of five domains including the nSH2 and the cSH2, linked by an intermediate region called the iSH2 domain from residue 431–600.2,5,6,13 P110α only requires the iSH2 domain to associate with p85α but the inhibition of the catalytic subunit can only occur when nSH2 is linked to it.22,23
PI3Kα is activated by the binding of adapter proteins like IRS-I and phosphorylated tyrosines to nSH2 and cSH2 domains of p85α.24 This binding temporarily dislodges the inhibition activity of p85 and activates p110.23,25 The activated protein phosphorylates the substrate (PIP2) to PIP3 at the cell membrane to promote the signaling cascades.2,21,26,27 To keep a check on the signaling cascade, the phosphatases, PTEN, control the level of PIP3 by dephosphorylating it.1,15,28−32
P85α plays a dual role in the p110α–p85α complex by not only stabilizing the p110α but also inhibiting its basal kinase activity.5,11,22,23,33,34 Prior to activation, the regulatory subunit generally interacts with its catalytic counterpart to inhibit membrane binding and thereby inhibiting the activity of the kinase domain.35,36 The oncogenic mutation H1047R weakens the interaction between the p110α and p85α subunits, which destabilizes the binding interface between the two subunits.2
A RAS protein is abundantly present on the cell membrane and it assists in association of the protein complex (PI3Kα) with the cell membrane by binding to the RBD of the p110α.11,21,37,38 The interaction of RAS with PI3Kα has been seen to activate PI3Kα, which is synergistic to activation by RTKs. This also plays a role in localizing PI3Kα to specific sites on the membrane where RAS is present so that p110α can access the substrate.9,11,37,38
Over 7000 mutations have been noted in the PIK3CA gene, most of which have been seen to cause tumorigenesis.2,21 The most common mutations being H1047R of the kinase domain and E542K and E545K of the helical domain.4,21 Kinase domain mutation does not require the involvement of the RBD with the membrane protein RAS but depends on p85α for activation.11,39 H1047R mutation causes structural transformations in the kinase domain which increases the membrane association of PI3Kα, thereby increasing the lipid kinase activity.2 The mutant binds with lipid molecules in a tighter association, in comparison to the native, because of the conformational changes.40 In the mutant, the arginine residue at the 1047 position points 90° away from the orientation of the histidine residue at the 1047 position in the native. The arginine residue assembles 13 residues from 1050 to 1062 to form a loop for the interaction with the cell membrane which was otherwise disordered in the native. A different loop (residues 864–874), which is already present in the kinase domain for interaction with the cell membrane in the native, also has a different conformation in the mutant.21,41 These two loops come in contact with the cell membrane, thereby giving rise to a tighter membrane association, which increases the lipid kinase activity by allowing easy access to the membrane-bound substrate.2,21 The H1047R mutant independently localizes p110α to the cell membrane because of the structural transitions.21 Attachment of p110α on the cell membrane initiates the tumerogenic cell signaling cascade by continuous phosphorylation of substrates in the absence of inhibitory effect of regulatory subunit p85α.21
The primary motives of our study were, first, to check the conformational profile of the native and mutant protein and to observe the damaging structural transition in the mutant as compared to the native. Second, to rationalize possible reason for these transitional changes in the mutant and see how they bring about changes in the interaction of the two subunits and in the association of the catalytic subunit with the cell membrane.
This study reveals that upon mutation, p110α adopts an expanded structure and has increased conformational flexibility, which dominantly affects the hydrogen-bonding pattern between the two subunits (p110α–p85α). Further, a significant secondary structural transition found in RBD of mutant protein during dynamic simulation which alters its interaction with the membrane-bound RAS protein. Also, important conformational positioning identified at two essential loops L1 and L2 of the kinase domain because of mutation which assists in oncogenic membrane localization of the mutant protein where the RAS–RBD interaction is compromised. The complete workflow of the study is represented in Figure 1.
Figure 1.

Pictorial representation of the workflow.
Results and Discussion
The protein models generated through X-ray crystallography and nuclear magnetic resonance techniques present very informative but static images of protein structures.42 However, the flexibility of a protein is important for the formation of different binding interfaces and also affects the stability of a protein.43 Proteins acquire flexibility by altering structural conformations and these perturbations in structures might be little, including just the rearrangement of a couple of amino acid side chains or it might be huge and even include folding of the whole protein. Possibly, a conformational change that alters the flexibility of a protein may meddle with its biological function.44 To get a molecular insight into all the conformational changes taking place in the protein complex, we made the native and the mutant protein structures of p110α to go through molecular dynamics (MD) simulation.45 RMSD was calculated for the C-α atoms of both the complexes, and the results are shown in Figure 2. RMSD is a quantitative method to calculate the similarity between two or more protein structures. RMSD results revealed that both the complexes at 0 ns had different starting points which was 0.25 nm for the native and 0.1 nm for the mutant. Then, there was a rise in the values of both the protein complexes from 0 to 25 ns. The mutant showed the rise of a greater extent as compared to the native structure with values reaching up to ∼0.45 nm in mutant and ∼0.38 nm in native. From 25 to 100 ns, the RMSD of both the complexes fluctuated at different rates, where the mutant showed major fluctuations between 30 and 40 ns, and the native showed major fluctuations at 100 ns. Gradually, the RMSD of both the complexes converged to similar values. The differences in the RMSD values for native and mutant protein structures of p110α indicate divergence in both the protein structures, which could possibly affect the joining of p110α and p85α subunits. These structural alterations in the catalytic subunit may also affect the biological functioning of the PI3Kα protein. The rate of fluctuations and the difference in the average values of the native and mutant protein structures provided basis for further structural analysis.
Figure 2.
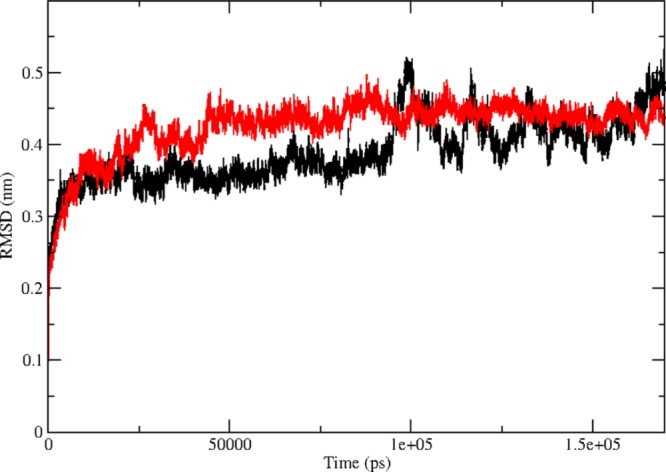
Time evolution of backbone RMSDs are shown as a function of time of the native and mutant p110α protein structures at 300 K. The symbol coding scheme is as follows: native (black) and mutant (red).
Solvent accessibility surface area (SASA) studies were carried out to evaluate the surface area of a biomolecule that is available to solvent molecules. It is the amount of surface area of the protein which is allowed for binding. A higher value of SASA denotes the relative expansion of the structure or an expanded conformation whereas a lower value denotes a compact conformation. The alteration in SASA of both the protein structures is shown in the Figure 3i. We can see that there is a sudden increase in the SASA of the mutant at 5 ns and the value reaching up to 540 nm2 as compared to the native, which shows a dip in the value of SASA during that time period with values reaching up to 495 nm2. Thereafter, a loss in SASA of the mutant structure at 10 ns but the native does not show that much fluctuation. After 10 ns, an increase in SASA of mutants could be seen and continued to fluctuate at a higher level than the native structure till 145 ns. An average value of SASA for the native is around 500 nm2 whereas for the mutant is around 530 nm2. The highest SASA value at this time interval for native was 515 nm2 whereas for the mutant structure it was 525 nm2. There is no region in the graph where the native has a greater value of SASA as compared to the mutant. These large fluctuations in the mutant protein denote significant structural transitions. Increase in the SASA value of the mutant also shows relative expansion of its structure, denoting that it has a more expanded conformation than the native. SASA results were further seconded by the trajectories of the hydrogen bond (H bond) analysis, and the alterations in the conformational geometry were also validated by radius of gyration (Rg) analysis.
Figure 3.
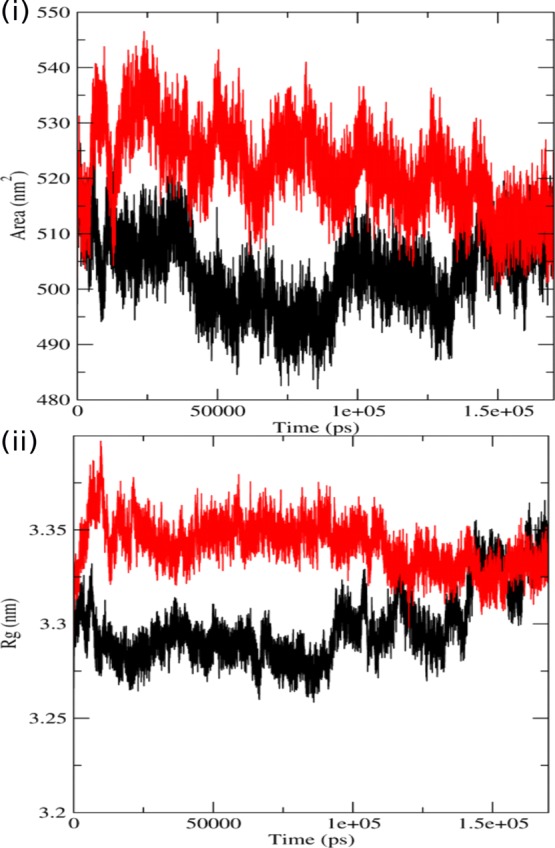
Analysis of simulation trajectory of native and mutant proteins. (i) SASA of native and mutant p110α vs time at 300 K. (ii) Radius of gyration of Cα atoms of native and mutant p110α protein vs time at 300 K. The symbol coding scheme is as follows: native (black) and mutant (red).
The Rg is described as the root-mean-square distance of the mass weight of a group of atoms from their common center of mass. It provides data concerning the dimensions of the protein. Rg analysis of mutant and native structures was carried out to complement our RMSD and SASA results. In Figure 3ii, we can see that the value of Rg for the mutant structure instantly rises at 5 ns followed by a decrease at 10 ns, and then again followed by an increase. We also observed similar fluctuations in SASA values during the same time period (until 20 ns). After 20 ns, there was no such major fluctuation in the graph, but a number of minor fluctuations were visible throughout the graph. For the native protein, a few fluctuations were visible at 5–20 ns and after 100 ns, but the overall value of Rg was less than the mutant. The average value of Rg for the native is about ∼3.31 nm whereas for the mutant is about ∼3.35 nm. The mutant has considerably higher values of Rg and fluctuates at a greater frequency throughout the simulation time that indicates that the structure of the mutant is flexible throughout the simulation, and its structure acquires an enlarged conformation. Larger fluctuations within the Rg plot additionally indicate that the mutant may be undergoing some major structural transitions. According to the Rg graph, the conformation of the native was seen to be sturdier in comparison to its counterpart which tends to adopt an expanded conformation, thereby increasing flexibility. SASA and Rg analysis performed to get an insight into the geometrical behavior of the mutant and native p110α. The expanded conformation of the p110α subunit may alter its joining interface with the p85α subunit. Modifications in protein–protein interfaces may lead to disorders or loss of functionality, and thus protein interfaces have turned out to be a standout amongst the most well-known new targets for rational drug design.46,47 Further, we carried out H bond analysis between the two subunits of PI3Kα to get an insight into the binding interface of the two subunits and the effect of mutation at the p110α subunit on the protein–protein interface.
Biological functioning of the proteins relies upon the correct orientation and binding to other bio molecules in the system, such as nucleic acids, ligands, other proteins, and lipids.48 H-bond formation is mainly responsible for maintaining a stable structure of the protein, which also contributes to protein-binding interfaces. H bonds formed by the native and mutant structures of p110α with the regulatory subunit p85α were calculated to find out the correlation between H bond formation and flexibility. In Figure 4i, we can see that the H bonds were formed between the catalytic and the regulatory subunits of both the protein complexes. The graph indicates that the native complex has greater number of H bonds because of which the two subunits are in close proximity with each other. The average number of H bonds in the native complex is ∼44 which is greater than that formed in the mutant complex which is ∼30. This signifies that the structure of the complex with the native p85α subunit is more compact than the complex with the mutant subunit. The mutant complex, differently, has a rather expanded conformation, making it more flexible than the native complex. The loss in the number of H bonds decreases the interaction between the two subunits and also suggests that the residues, which were previously involved in H-bonding, are now free to interact with other molecules. Therefore, when these mutant and native protein complexes were put in a solvent, new H bonds were formed between the protein complexes and the solvent. In Figure 4ii, we observed that the mutant formed more bonds with the solvent. This is because the residues, which were free for the interaction in the mutant, now formed H bonds with the solvent. This also suggests increased flexibility of mutant in comparison to the native structure as it has more tendency to form new bonds.
Figure 4.
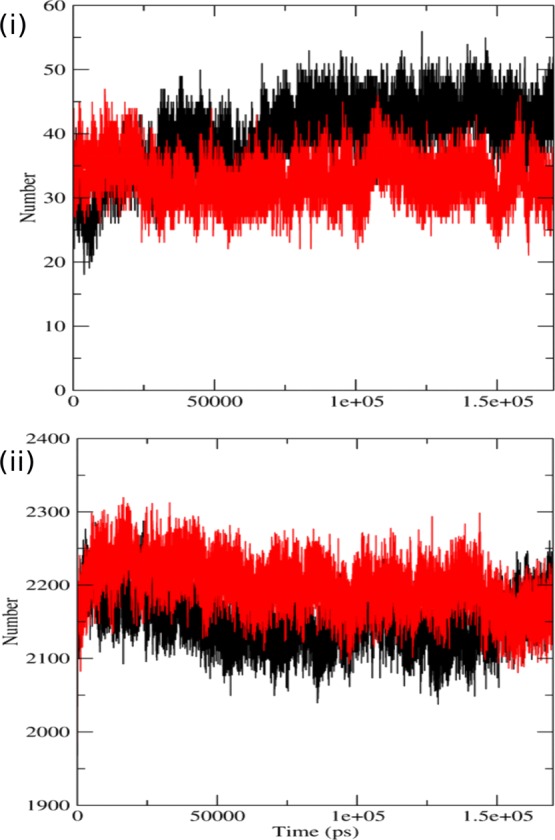
Analysis of simulation trajectory of native and mutant proteins. (i) Time evolution of number of intramolecular hydrogen bonds of the p110α and the p85α protein during the simulations. (ii) Average number of protein–solvent intermolecular hydrogen bonds in native and mutant p110α proteins vs time at 300 K. The symbol coding scheme is as follows: native (black) and mutant (red).
In order to check the effect of decreased number of H bonds in the binding interface, we calculated the number of contacts between two subunits (p110α–p85α) through MD simulations, and the results were plotted on a graph. In Figure 5i, we can see that the number of contacts in the native has increased from ∼10 000 contacts to ∼19 000 contacts. On the other hand, the number of contacts between the subunits of the mutant protein complex has decreased from ∼15 000 contacts to ∼12 000 contacts. These results are also in accordance with the other results of MD simulation, and a possible reason for these results can be narrowed down to loss of H bonds, which resulted in an expanded conformation, allowing the two subunits to move apart from each other, thereby decreasing the number of contacts between the subunits.
Figure 5.
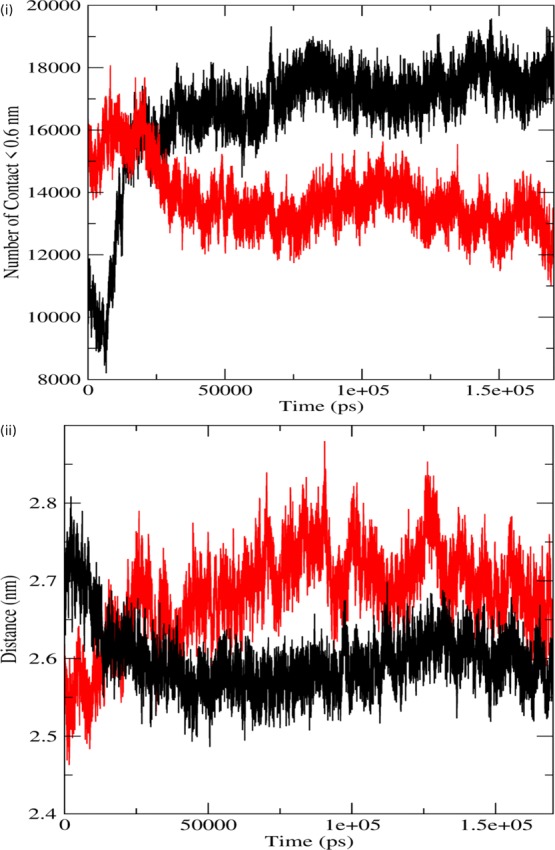
Analysis of simulation trajectory of native and mutant proteins. (i) Number of contacts between the two subunits. (ii) Distance between the two subunits. The symbol coding scheme is as follows: native (black) and mutant (red).
Further, MD simulations were performed to calculate the distance between the two subunits (p110α–p85α) of the protein, and the results are shown in Figure 5ii. The distances of the mutant are represented in red while that of the native is represented in black. Here, we observed that the distance between the subunits gradually decreased in the native protein from ∼2.7 nm to about ∼2.55 nm, whereas this distance gradually increased in the mutant protein from ∼2.5 nm to about ∼2.74 nm, denoting that the two subunits moved toward each other in the native but moved away from each other in the mutant. These MD simulation results complement previously done wet lab experiments, which reports disturbances in the binding interface between the two subunits of PI3Kα because of the H1047R mutation.2 This study provides a possible explanation based on the H bond and number of contact analysis for moving apart of the catalytic and regulatory subunits of PI3Kα. Moving away of the regulatory subunit from the catalytic subunit could also be correlated with the uncontrolled phosphorylation of the substrate, resulting in the initiation of the tumerogenic cell-signaling cascade.21
To acquire more insight into the conformational alterations of the two complexes, we used the DSSP tool which applies the H bond estimation algorithm to assign secondary structures during the simulation time period.49 The results obtained from the DSSP program are shown in Figure S1. DSSP results depicted that the α-helix, β sheet, coil, turns, and bends are present in both the protein complexes. In Figure S1i,ii, we can see that, from residue 325–450, more number of β-sheets are present in the native structure, whereas this region of the mutant shows lesser number of β-sheets and a larger number of bends and coils, throughout the simulation time. In the native, the region between residue 525–550 is dominated by α-helix whereas this region of the mutant primarily has larger number of bends with traces of coils present in it. The presence of α-helix is also seen in the catalytic region between residue 925–950, which is completely lost in the mutant. The loss in the number of α-helix and β-sheets also confirms the expanded conformation of the mutant. Coil and turn conformation tends to increase the flexibility of any protein.50 The mutant conformation has been observed to be flanked by coils and turns in regions which were otherwise inhabited by the helix and sheet conformation in the native. This is also a reason for increased flexibility of the mutant. These results indicate changes in the catalytic and other domain, which are essential for proper functioning and stability of the protein.
The protein stability mainly depends on factors such as the hydrogen bonding, packing of hydrophobic core, and secondary structures of the protein.51,52 Apart from these forces, several studies have demonstrated the importance of charge–charge interactions for the stability of proteins.53,54 In addition to providing stability to the protein structure, the local and global electrostatic properties play an essential role in protein functioning and binding.55,56 Hence, we studied the effect of mutation H1047R on the charge distribution on the p110α subunit. Figure 6 shows the adaptive Poisson–Boltzmann solver (APBS) and changes in secondary structural elements of the p110α subunit. APBS provides with long-range intermolecular interactions and effects of solvation on biomolecular interactions. We mainly emphasized on two regions, the kinase domain and the RBD domain. From Figure 6i,iii, we can see that there is a considerable loss in the number of secondary structure elements (loops) in the RBD domain of the mutant as compared to the native protein. The RBD domain of the mutant has a larger number of helices as compared to the native protein’s RBD domain. These conformational changes are also clear from Figure 6a,b. Here, we see that from residue 1–25 of the RBD domain in the native, a number of secondary structures like β-sheets, bends, turns, and coils are present. However in the same region of the mutant, the amount of β-sheets has increased significantly, and the number of bends and turns has almost declined to negligible. Another major difference is visible in the region from residue 25–65 of the RBD domain. Here again, the native has a number of secondary structures like α-helices, 3-helices, turns, bends, and coils present in the native, but the same region in the mutant is dominated by the presence of α-helices with a very insignificant amount of turns and bends. The coil and turn secondary structure elements tend to make a protein more flexible and prone to interactions with its partner. However, the secondary structure elements like α-helices and β-sheets in comparison to the loops make the protein structure more rigid and hinders it from being involved in interactions with partner proteins.50 Because the RBD domain of the native protein had abundance of coils, turns, and bends in its conformation, so it was able to interact with the RAS protein of the plasma membrane and assisted in localization of the protein complex to the membrane and the change of these secondary structure elements primarily to α-helices and β-sheets in the mutant caused the loss of interaction of the RBD domain with the RAS protein. Moreover, this increase in number of α-helices and β-sheets in the mutant causes a change in the charge distribution of its RBD domain, and it can be seen in Figure 6iii,iv. The RBD domain of the native protein has almost equal regions of negative and positive charges, and the RBD domain of the mutant protein has the majority of the region with a positive charge with very few regions of negative or neutral charge. The change in the secondary structure elements and in the charge distribution over the RBD domain could be the reason for relaxation of PI3Kα of its interaction with its target protein RBD on the plasma membrane.
Figure 6.

Pictorial representation of the p110α protein using Pymol. (i) Structure of the native p110 α protein. (ii) Electrostatic potential distribution in the native p110α protein. (iii) Structure of the mutant p110 α protein. (iv) Electrostatic potential distribution in the mutant p110α protein. (a) Time evolution of the secondary structural elements of the RBD domain of the native p110α protein at 300 K (DSSP classification). (b) Time evolution of the secondary structural elements of the RBD domain of the mutant p110α protein at 300 K (DSSP classification). The color coding scheme for DSSP classification: coil (white), β-sheets (red), β-bridge (black), bend (green), turn (yellow), α-helix (blue), 5-helix (purple), and 3-helix (gray). The color coding scheme for (i,iii) is as follows: cyan (RBD domain), pink (kinase domain), and green (ABD, C2, helical). The color coding scheme for (ii,iv) is as follows: red (region with the negative charge), blue (region with the positive charge), and white (region with the neutral charge).
In order to understand the reason for direct attachment of kinase domain of PI3Kα to the cell membrane, we further investigated the conformational changes due to mutation in the p110α subunit and observed noticeable structural change in the conformational alignment of the loops L1 and L2. Both these loops act as a hook to bind to the cell membrane in the mutated protein structure.19,39 The loop L1 of the kinase domain is tilted away from the RBD domain in the native protein while it is tilted toward the RBD domain in the mutant. Similarly, the loop L2 also has a disordered conformation in the native protein and has a more ordered structural conformation in the mutant protein. Also, the region adjacent to the L1 loop in the native is dominated by the presence of helices, whereas this region in the mutant is dominated by the presence of loops. These results are also supported by the DSSP results shown in Figures S1 and S2. The DSSP result of the mutant protein shown in Figure S2, shows that the RBD domain in the mutant has an abundance of α-helices while from Figure S1, it is clear that the RBD domain of the native has regions of coils and bends, and the α-helix region was quite diminished. Even the region around the two loops L1 and L2 show a prominent α-helix region in the native, while the mutant has an α-helix region flanked with bends and turns. This change in conformation within the kinase domain also brings about a change in the overall charge distribution of the kinase domain. From Figure 6ii,iv, it is visible that the region where loop L2 is present is dominantly negatively charged in the native while it is completely positively charged in the mutant protein. Even the area around loop L1, which was dominated by helices in the native protein, had a negative charge. Howevr in the mutant, this area was converted to the positively charged region. To concede, there are two major effects of mutation on the kinase domain of PI3Kα. Loops L1 and L2 acquires ordered conformations and orients themselves in a hook-like structure. Second, a positive charge gets developed around both the loops in the mutated structure. As we know that the cell membrane is negatively charged, hence, the acquisition of positive charge around the loops L1 and L2 in the p110α subunit would favor the direct binding of kinase domain to the cell membrane because of interactions between oppositely charged surfaces.
Conclusions
The mutation (H1047R) in PI3Kα occurs in cancers of various organs like breast, colon, uterus, stomach, and ovary.4,6,21 Studies show that this mutation promotes tumorigenesis by increasing the activity of the kinase domain which is achieved because it gets abundant supply of substrates.21 This mutation also allows the protein complex to attach directly to the cell membrane, independent of the membrane-bound RAS protein.2 We executed MD simulations of the protein complex (native and mutant) in order to understand the conformational changes at the atomic level, which leads to the abovementioned abnormalities.57,58 Our results showed a notable decrease in H bond formation between the two subunits of the protein complex in the mutant. Conformational transitions due to mutation also increased the values of Rg and SASA, which also support the results of H bond analysis, showing increased flexibility of the p110α subunit. The loss of H bonds makes the two subunits to move apart from each other, thereby increasing the distance and decreasing the number of contacts between the two chains. These results also signify that the mutant protein has an expanded conformation than the native. Increase in the flexibility of the mutant is also supported by the results of H bond analysis with the solvent. The increased distance between the catalytic subunit and the regulatory subunit due to mutation could be the possible reason for the uncontrolled phosphorylation of the substrate, resulting in the initiation of tumerogenic cell-signaling cascade. Furthermore, DSSP and charge distribution studies of the p110α subunit (native and mutant) reveals changes in the region essential for binding to the RAS protein. Moreover, mutation also encourages the loops L1 and L2 to acquire an organized, hook-like structure, surrounded by a positive charged surface which promotes the two loops L1 and L2 to be actively involved in membrane localization of proteins without the involvement of RBD and RAS proteins. These results provide an insight into the changes at the molecular level due to mutation H1047R, resulting in tumorigenesis, and could be further exploited to consider the abovementioned structural aspects to handle this mutation.
Materials and Methods
Datasets
Two protein–protein complexes were used from the RCSB Protein Data Bank, one is p110α–p85α (PDB id 4L1B)15 and second is p110α (H1047)–p85α (PDB id 3HHM).21 We considered dimer of each protein complex, where chain A holds 1068 amino acids, which were spread from residues 8–1068, and Chain B holds 277 amino acids and it was spread from residues 322–598 amino acids. Native and mutant p110α–p85α complex structures were solved at 2.5 and 2.8 Å resolution. Crystallized heteroatoms were removed from its PDB files, and the same was done to the water molecules. Missing loop regions were prepared by using “prepare protein” protocol in discovery studio package 2017R2.
MD Simulation
MPI-enabled GROMACS 5.0.6 was employed to carry out the MD simulations,59−61 which is installed in our in-house hig1h-end computer cluster machine, and also used the CHARMM27 force field.62 Starting points for the simulations were both the protein complexes (native and mutant), and they were also put into a solvated cubic box, which was filled with SPC water molecules. Because at physiological pH (approx 7.4), the proteins had a net positive charge, so we put in sodium ions (CL–) to this solvated cubic box using the “gmx genion” script to electrically neutralize the simulation system. Energy minimization protocols were further used to decrease the energy of the simulation system in GROMACS. This minimized system was then put through MD simulations which was positions restrained and had two steps. At first, within a canonical ensemble, we performed for 10 ns succeeded by another 10 ns within a constant-temperature and -pressure ensemble. In this type of MD simulations, the whole system is kept under control except for the solvent molecules. This step is carried out to ensure equilibrium of the water molecules surrounding the residues of our protein. The system then attains pressure equilibrium and then attains density equilibrium. Once the systems reached equilibrium over the time, they are then put through MD simulations without any restriction for 170 ns, and further analysis was carried out on each equilibrated part of the trajectories. A Berendsen thermostat was put to use for attaining a steady temperature of 310 K in all simulations.63 The calculations of electrostatic and coulombic interactions have been carried out by the Ewald method.64 All histidine residues were assumed neutral, and a pH of 7 was set for their ionization states. A SHAKE algorithm was used to restrict the bond lengths of H bonds, trimming both the long-range interaction (Coulomb and van der Waals) at 1.0 nm and permitting a time step of 2 fs. After every 0.5 ps, the structures were saved and at ten steps, the list of nonbonded pairs was amended. Several analyses were undertaken by making use of files in GROMACS distribution containing various scripts.59 The resultant files were examined by “gmx rms”, “gmx gyrate”, “gmx rmsf”, “gmx mindist”, and “gmx dssp” of GROMACS to get RMSD, the radius of gyration (Rg), root-mean-square fluctuation (RMSF) value, number of contact (<0.6 nm), and distance between subunits and define secondary structure of proteins plot (DSSP). We used “gmx hbond” to get a count of the number of discrete H in the protein and calculated by the donor–acceptor distance less than 0.35 nm and of donor–H–acceptor angle greater than 150°. Moreover, the simulation snapshot structures and trajectory analysis were managed by the Coot65 package. This investigation is primarily focused to study the dynamic behavior of native as compared to its oncogenic mutant proteins. We compared SASA and Rg of protein to check the three-dimensional space of the p110α protein. In order to study the conformational transitions of both the proteins, we plotted SASA, Rg, and hydrogen bonds of carbon-α of the system and distance of subunits and number of contacts of subunits for all the simulations using the Grace toolkit 5.1.22 version.
RMSD Clustering
Structural transformation of a protein can be better understood by performing ensemble clustering on the simulation data.50 RMSD conformational clustering was then performed on our protein using the GROMOS algorithm by using the “gmx cluster” script.66 These results aided us in the evaluation of three-dimensional heterogeneity in different conformers of the protein. To find a more reasonable RMSD cut-off, we first modified the C-α RMSD cut-off between 0.10 and 0.15 nm in steps of 0.02 and performed clustering analysis for each RMSD cut-off value. Here, 0.13 nm was kept to be the C-α RMSD cut-off. ∼78% of all the protein structures carried the clusters predominantly. The number of neighbors are counted by the GROMOS clustering method. It is then used as a cut-off for identifying the structure which has the largest number of neighbors. This structure is then used as the center of the cluster. The pool of clusters is then refined by eliminating all the neighbors of this structure. The same is repeated for every structure in the pool until each structure has been alloted to a cluster.66
APBS Calculation
We took APBS calculations for our native and mutant proteins to tackle the long-ranged intermolecular interactions and the effects of solvation on biomolecular processes of the protein clusters and used the APBS plugin installed in PyMOL 1.8. While using the plugin, we set all parameters to default and used the split-state script to categorize different conformers in every cluster.
Acknowledgments
The authors are thankful to the Director, CSIR-IHBT for providing necessary facilities and support. CSIR support in the form of projects MLP:0201 for this studies is highly acknowledged. This manuscript represents CSIR-IHBT communication no. 4407.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b01439.
Time evolution of the secondary structural elements of the native p110α protein at 300 K (DSSP classification) and time evolution of the secondary structural elements of the mutant p110α protein at 300 K (DSSP classification) (PDF)
Author Contributions
∥ J.S. and V.B. contributed equally to the work.
The authors declare no competing financial interest.
Supplementary Material
References
- Fyffe C.; Buus R.; Falasca M. Genetic and Epigenetic Regulation of Phosphoinositide 3-Kinase Isoforms. Curr. Pharm. Des. 2013, 19, 680–686. 10.2174/138161213804581873. [DOI] [PubMed] [Google Scholar]
- Echeverria I.; Liu Y.; Gabelli S. B.; Amzel L. M. Oncogenic mutations weaken the interactions that stabilize the p110α-p85α heterodimer in phosphatidylinositol 3-kinase α. FEBS J. 2015, 282, 3528–3542. 10.1111/febs.13365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R.; Berndt A.; Miller S.; Hon W.-C.; Zhang X. Form and flexibility in phosphoinositide 3-kinases. Biochem. Soc. Trans. 2009, 37, 615–626. 10.1042/bst0370615. [DOI] [PubMed] [Google Scholar]
- Mazloumi Gavgani F.; Smith Arnesen V.; Jacobsen R.; Krakstad C.; Hoivik E.; Lewis A. Class I Phosphoinositide 3-Kinase PIK3CA/p110α and PIK3CB/p110β Isoforms in Endometrial Cancer. Int. J. Mol. Sci. 2018, 19, 3931. 10.3390/ijms19123931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J.; McIlroy J.; Backer J. M.; Zhang Y.; Rordorf-Nikolic T.; Orr G. A. Regulation of the P85/P110 Phosphatidylinositol 3′-Kinase: Stabilization and Inhibition of the P110α Catalytic Subunit by the P85 Regulatory Subunit. Mol. Cell. Biol. 1998, 18, 1379–1387. 10.1128/mcb.18.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. S.; Maheshwari S.; McRobb F. M.; Kinzler K. W.; Amzel L. M.; Vogelstein B.; Gabelli S. B. Identification of allosteric binding sites for PI3Kα oncogenic mutant specific inhibitor design. Bioorg. Med. Chem. 2017, 25, 1481–1486. 10.1016/j.bmc.2017.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geering B.; Cutillas P. R.; Nock G.; Gharbi S. I.; Vanhaesebroeck B. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 7809–7814. 10.1073/pnas.0700373104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirola L.; Waterfield M. D.; Zvelebil M. J.; Wymann M. P.; Bulgarelli-Leva G.; Van Obberghen E. Activation Loop Sequences Confer Substrate Specificity to Phosphoinositide 3-Kinase α (PI3Kα). J. Biol. Chem. 2001, 276, 21544–21554. 10.1074/jbc.m011330200. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P.; Warne P. H.; Vanhaesebroeck B.; Waterfield M. D.; Downward J. Activation of Phosphoinositide 3-Kinase by Interaction with Ras and by Point Mutation. EMBO J. 1996, 15, 2442–2451. 10.1002/j.1460-2075.1996.tb00602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman D. A.; Meyers R. E.; Cantley L. C. PHOSPHOINOSITIDE KINASES. Annu. Rev. Biochem. 1998, 67, 481–507. 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- Shepherd P. R.; Withers D. J.; Siddle K. Phosphoinositide 3-Kinase: The Key Switch in Insulin Signalling. Biochem. J. 1998, 335, 711. 10.1042/bj3350711u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroeck B.; Guillermet-Guibert J.; Graupera M.; Bilanges B. Supplementary Information. The Emerging Mechanisms of Isoform-Specific PI3K Signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- Wang J.; Gong G. Q.; Zhou Y.; Lee W.-J.; Buchanan C. M.; Denny W. A.; Rewcastle G. W.; Kendall J. D.; Dickson J. M. J.; Flanagan J. U.; et al. High-throughput screening campaigns against a PI3Kα isoform bearing the H1047R mutation identified potential inhibitors with novel scaffolds. Acta Pharmacol. Sin. 2018, 39, 1816–1822. 10.1038/s41401-018-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan T. L.; Cantley L. C. PI3K pathway alterations in cancer: variations on a theme. Oncogene 2008, 27, 5497–5510. 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Zhang X.; Chen Y.; Lu S.; Peng Y.; Wang X.; Guo C.; Zhou A.; Zhang J.; Luo Y.; et al. Crystal Structures of PI3Kα Complexed with PI103 and Its Derivatives: New Directions for Inhibitors Design. ACS Med. Chem. Lett. 2014, 5, 138–142. 10.1021/ml400378e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L.; Vogt P. K. Helical Domain and Kinase Domain Mutations in P110 of Phosphatidylinositol 3-Kinase Induce Gain of Function by Different Mechanisms. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 2652–2657. 10.1073/pnas.0712169105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanwar G.; Mazumder A. G.; Bhardwaj V.; Kumari S.; Bharti R.; Yamini; Singh D.; Das P.; Purohit R. Target Identification, Screening and in Vivo Evaluation of Pyrrolone-Fused Benzosuberene Compounds against Human Epilepsy Using Zebrafish Model of Pentylenetetrazol-Induced Seizures. Sci. Rep. 2019, 9, 7904. 10.1038/s41598-019-44264-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman J. A.; Luo J.; Cantley L. C. The Evolution of Phosphatidylinositol 3-Kinases as Regulators of Growth and Metabolism. Nat. Rev. Genet. 2006, 7, 606–619. 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- Curran E.; Smith S. M. Phosphoinositide 3-Kinase Inhibitors in Lymphoma. Curr. Opin. Oncol. 2014, 26, 469–475. 10.1097/cco.0000000000000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsi N.; Gopalakrishnan C.; Rajendran V.; Purohit R. Biophysical Aspect of Phosphatidylinositol 3-Kinase and Role of Oncogenic Mutants (E542K & E545K). J. Biomol. Struct. Dyn. 2016, 34, 2711–2721. 10.1080/07391102.2015.1127774. [DOI] [PubMed] [Google Scholar]
- Mandelker D.; Amzel L. M.; Gabelli S. B.; Vogelstein B.; Zhu J.; Cheong I.; Huang C.-H.; Schmidt-Kittler O.; Kinzler K. W. A Frequent Kinase Domain Mutation That Changes the Interaction between PI3K and the Membrane. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 16996–17001. 10.1073/pnas.0908444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J.; Wjasow C.; Backer J. M. Regulation of the p85/p110α Phosphatidylinositol 3′-Kinase. J. Biol. Chem. 1998, 273, 30199–30203. 10.1074/jbc.273.46.30199. [DOI] [PubMed] [Google Scholar]
- Gabelli S. B.; Huang C.-H.; Mandelker D.; Schmidt-Kittler O.; Vogelstein B.; Amzel L. M.. Structural Effects of Oncogenic PI3Kα Mutations. Current Topics in Microbiology and Immunology; Springer, 2010; pp 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foukas L. C.; Beeton C. A.; Jensen J.; Phillips W. A.; Shepherd P. R. Regulation of phosphoinositide 3-kinase by its intrinsic serine kinase activity in vivo. Mol. Cell. Biol. 2004, 24, 966–975. 10.1128/mcb.24.3.966-975.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dornan G. L.; Siempelkamp B. D.; Jenkins M. L.; Vadas O.; Lucas C. L.; Burke J. E. Conformational disruption of PI3Kδ regulation by immunodeficiency mutations inPIK3CD and PIK3R1. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 1982–1987. 10.1073/pnas.1617244114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vara J. Á. F.; Cejas P.; Belda-Iniesta C.; Casado E.; de Castro J.; González-Barón M. PI3K/Akt Signalling Pathway and Cancer. Cancer Treat. Rev. 2004, 30, 193–204. 10.1016/j.ctrv.2003.07.007. [DOI] [PubMed] [Google Scholar]
- Liu S.; Knapp S.; Ahmed A. A. The Structural Basis of PI3K Cancer Mutations: From Mechanism to Therapy. Cancer Res. 2014, 74, 641–646. 10.1158/0008-5472.can-13-2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley L. C. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296, 1655–1657. 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Cully M.; You H.; Levine A. J.; Mak T. W. Beyond PTEN Mutations: The PI3K Pathway as an Integrator of Multiple Inputs during Tumorigenesis. Nat. Rev. Cancer 2006, 6, 184–192. 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- Sabbah D. A.; Simms N. A.; Wang W.; Dong Y.; Ezell E. L.; Brattain M. G.; Vennerstrom J. L.; Zhong H. A. N-Phenyl-4-Hydroxy-2-Quinolone-3-Carboxamides as Selective Inhibitors of Mutant H1047R Phosphoinositide-3-Kinase (PI3Kα). Bioorg. Med. Chem. 2012, 20, 7175–7183. 10.1016/j.bmc.2012.09.059. [DOI] [PubMed] [Google Scholar]
- Taniguchi C. M.; Kahn C. R.; Ueki K.; Tran T. T.; Cantley L. C.; Kondo T.; Luo J. Phosphoinositide 3-Kinase Regulatory Subunit P85 Suppresses Insulin Action via Positive Regulation of PTEN. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 12093–12097. 10.1073/pnas.0604628103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki A. T.; Chun C.; Takeda K.; Firtel R. A. Localized Ras Signaling at the Leading Edge Regulates PI3K, Cell Polarity, and Directional Cell Movement. J. Cell Biol. 2004, 167, 505–518. 10.1083/jcb.200406177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J. J.; Cheng H.; Wang L.; Mikami A.; Jia S.; Gjoerup O. V.; Roberts T. M. The P110 Isoform of PI3K Is Essential for Proper Growth Factor Signaling and Oncogenic Transformation. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 16296–16300. 10.1073/pnas.0607899103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann S. M.; Engelman J. A.; Kahn R. C.; Cantley L. C.; Ueki K. Phosphoinositide 3-Kinase Catalytic Subunit Deletion and Regulatory Subunit Deletion Have Opposite Effects on Insulin Sensitivity in Mice. Mol. Cell. Biol. 2005, 25, 1596–1607. 10.1128/mcb.25.5.1596-1607.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon W.-C.; Berndt A.; Williams R. L. Regulation of lipid binding underlies the activation mechanism of class IA PI3-kinases. Oncogene 2012, 31, 3655–3666. 10.1038/onc.2011.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorpe L. M.; Yuzugullu H.; Zhao J. J. PI3K in Cancer: Divergent Roles of Isoforms, Modes of Activation and Therapeutic Targeting. Nat. Rev. Cancer 2015, 15, 7–24. 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano E.; Downward J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274. 10.1177/1947601911408079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simanshu D. K.; Nissley D. V.; McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L.; Vogt P. K. Class I PI3K in Oncogenic Cellular Transformation. Oncogene 2008, 27, 5486–5496. 10.1038/onc.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke J. E.; Perisic O.; Vadas O.; Masson G. R.; Williams R. L. Oncogenic Mutations Mimic and Enhance Dynamic Events in the Natural Activation of Phosphoinositide 3-Kinase P110 (PIK3CA). Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 15259–15264. 10.1073/pnas.1205508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. S.; Bolduc D. M.; Schmidt-Kittler O.; Kinzler K. W.; Allaire M.; Jennings I. G.; Brower E. T.; Thompson P. E.; Amzel L. M.; Gabelli S. B.; et al. Structural Basis of NSH2 Regulation and Lipid Binding in PI3Kα. Oncotarget 2014, 5, 5198–5208. 10.18632/oncotarget.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran V.; Sethumadhavan R. Drug Resistance Mechanism of PncA in Mycobacterium Tuberculosis. J. Biomol. Struct. Dyn. 2014, 32, 209–221. 10.1080/07391102.2012.759885. [DOI] [PubMed] [Google Scholar]
- Yesylevskyy S. O.; Kharkyanen V. N.; Demchenko A. P. The Change of Protein Intradomain Mobility on Ligand Binding: Is It a Commonly Observed Phenomenon?. Biophys. J. 2006, 91, 3002–3013. 10.1529/biophysj.106.087866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teilum K.; Olsen J. G.; Kragelund B. B. Protein Stability, Flexibility and Function. Biochim. Biophys. Acta, Proteins Proteomics 2011, 1814, 969–976. 10.1016/j.bbapap.2010.11.005. [DOI] [PubMed] [Google Scholar]
- Rajendran V.; Gopalakrishnan C.; Sethumadhavan R. Pathological role of a point mutation (T315I) in BCR-ABL1 protein-A computational insight. J. Cell. Biochem. 2018, 119, 918–925. 10.1002/jcb.26257. [DOI] [PubMed] [Google Scholar]
- Jubb H.; Blundell T. L.; Ascher D. B. Flexibility and Small Pockets at Protein-Protein Interfaces: New Insights into Druggability. Prog. Biophys. Mol. Biol. 2015, 119, 2–9. 10.1016/j.pbiomolbio.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rask-Andersen M.; Almén M. S.; Schiöth H. B. Trends in the Exploitation of Novel Drug Targets. Nat. Rev. Drug Discovery 2011, 10, 579–590. 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- Hubbard R. E.; Kamran Haider M.. Hydrogen Bonds in Proteins: Role and Strength. Encyclopedia of Life Sciences; John Wiley & Sons, Ltd.: Chichester, UK, 2010. [Google Scholar]
- Kabsch W.; Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Purohit R. Role of ELA Region in Auto-Activation of Mutant KIT Receptor: A Molecular Dynamics Simulation Insight. J. Biomol. Struct. Dyn. 2014, 32, 1033–1046. 10.1080/07391102.2013.803264. [DOI] [PubMed] [Google Scholar]
- Baldwin E. P.; Matthews B. W. Core-Packing Constraints, Hydrophobicity and Protein Design. Curr. Opin. Biotechnol. 1994, 5, 396–402. 10.1016/0958-1669(94)90048-5. [DOI] [PubMed] [Google Scholar]
- Pace C. N.; Shirley B. A.; McNutt M.; Gajiwala K. Forces Contributing to the Conformational Stability of Proteins. FASEB J. 1996, 10, 75–83. 10.1096/fasebj.10.1.8566551. [DOI] [PubMed] [Google Scholar]
- Dow J. To Charge or Not to Charge?. J. Integr. Care 2001, 9, 38–40. 10.1108/14769018200100016. [DOI] [Google Scholar]
- Strickler S. S.; Gribenko A. V.; Gribenko A. V.; Keiffer T. R.; Tomlinson J.; Reihle T.; Loladze V. V.; Makhatadze G. I. Protein Stability and Surface Electrostatics: A Charged Relationship. Biochemistry 2006, 45, 2761–2766. 10.1021/bi0600143. [DOI] [PubMed] [Google Scholar]
- Sinha N.; Smith-Gill S. Electrostatics in Protein Binding and Function. Curr. Protein Pept. Sci. 2002, 3, 601–614. 10.2174/1389203023380431. [DOI] [PubMed] [Google Scholar]
- Bhardwaj V.; Purohit R. Computational Investigation on Effect of Mutations in PCNA Resulting in Structural Perturbations and Inhibition of Mismatch Repair Pathway. J. Biomol. Struct. Dyn. 2019, 1–12. 10.1080/07391102.2019.1621210. [DOI] [PubMed] [Google Scholar]
- Rajendran V. Structural Analysis of Oncogenic Mutation of Isocitrate Dehydrogenase 1. Mol. BioSyst. 2016, 12, 2276–2287. 10.1039/c6mb00182c. [DOI] [PubMed] [Google Scholar]
- Rajendran V.; Gopalakrishnan C.; Purohit R. Impact of Point Mutation P29S in RAC1 on Tumorigenesis. Tumor Biol. 2016, 37, 15293–15304. 10.1007/s13277-016-5329-y. [DOI] [PubMed] [Google Scholar]
- Van Der Spoel D.; Berendsen H. J. C.; Hess B.; Lindahl E.; Mark A. E.; Groenhof G. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Hess B.; Kutzner C.; Van Der Spoel D.; Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Abraham M. J.; Páll S.; Schulz R.; Hess B.; Smith J. C.; Murtola T.; Lindahl E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- MacKerell A. D.; Banavali N.; Foloppe N. Development and Current Status of the CHARMM Force Field for Nucleic Acids. Biopolymers 2000, 56, 257–265. . [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; Van Gunsteren W. F.; Dinola A.; Haak J. R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. 10.1063/1.448118. [DOI] [Google Scholar]
- Essmann U.; Lee H.; Pedersen L. G.; Darden T.; Perera L.; Berkowitz M. L. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Emsley P.; Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/s0907444904019158. [DOI] [PubMed] [Google Scholar]
- Daura X.; Gademann K.; Jaun B.; Seebach D.; van Gunsteren W. F.; Mark A. E. Peptide Folding: When Simulation Meets Experiment. Angew. Chem., Int. Ed. 1999, 38, 236–240. . [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.