

Abstract
Histamine plays an important role in neuromodulation and the biological immune response. Although many electrochemical methods have been developed for histamine detection, the mechanism of its redox reaction has not been directly investigated. Here, we studied the mechanism of histamine oxidation at carbon electrodes and used that mechanistic information to design better fast-scan cyclic voltammetry (FSCV) methods for histamine. Using amperometry, cyclic voltammetry (CV), and X-ray photoelectron spectroscopy (XPS), we demonstrate that histamine oxidation requires a potential of at least +1.1 V vs Ag/AgCl. We propose that histamine undergoes one-electron oxidation on an imidazole nitrogen that produces a radical. The radical species dimerize and continue to undergo oxidation, leading to electropolymerization, which fouls the electrode. CV shows a peak at 1.3 V, that is pH dependent, consistent with a one proton, one electron oxidation reaction. This mechanism is confirmed using 1- and 3-methylhistamine, which do not electropolymerize, compared to Nα-methylhistamine, which does. XPS also revealed a nitrogen-containing product adsorbed on the electrode surface after histamine oxidation. For FSCV detection of histamine at carbon-fiber microelectrodes, histamine oxidation was adsorption-controlled, and the anodic peak was observed at +1.2 V on the backward scan because of the rapid scan rate. However, the oxidation fouled the electrode and convoluted the FSCV temporal response; therefore, we implemented Nafion coating to alleviate the electrode fouling and preserve the time response of FSCV. Knowing the mechanism of histamine oxidation will facilitate design of better electrochemical methods for real-time monitoring of histamine.
Graphical Abstract
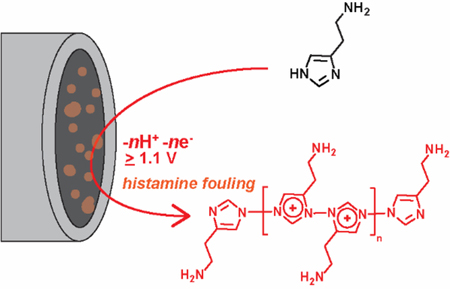
Introduction
Histamine is known for its role in immune responses as an inflammatory agent causing allergic reactions.1,2 It also functions as a neurotransmitter and neuromodulator, regulating the sleep cycle.1 Many methods have been developed for histamine analysis, including microdialysis coupled to liquid chromatography (LC) with fluorescence detection, which measures histamine over long time frames.3 For single cell content analysis, capillary LC with amperometric detection was used to detect histamine content in single mast cells.2 Histamine was quantified in the ventral nerve cord of fruit flies using capillary electrophoresis with fast-scan cyclic voltammetry (FSCV) detection.4 FSCV is also used to measure real-time release of histamine. Wightman’s group used FSCV to demonstrate that histamine and serotonin are co-released from vesicles in mast cells,5,6 and FSCV was also used in vivo to study histamine and serotonin co-release in the brain.7 Despite the fact that there are many papers using electrochemistry to detect histamine, there is no literature proposing a mechanism of histamine oxidation. Understanding the mechanism of oxidation is important to help design electrochemical methods to detect histamine and interpret electrochemical data.
While the redox mechanism of histamine oxidation has not been thoroughly investigated, there is one study on the oxidation of imidazole in organic solvents. Histamine (2-(1H-Imidazol-4-yl)ethanamine) is an imidazole derivative, so its mechanism of oxidation might be similar. Imidazole oxidation occurred at 1.5 V vs. Ag/Ag+ and the mechanism was the generation of a radical cation on a ring nitrogen, which subsequently dimerized and underwent electropolymerization.8 Similarly, most studies that have detected histamine in aqueous solution at carbon electrodes have reported high oxidation potentials. The histamine anodic peak is at 1.2 V vs. saturated calomel electrode (SCE) at glassy carbon electrodes (GCE), 1.4 V vs. SCE at boron-doped diamond electrodes, and 1.1 V vs. SCE at carbon-fiber microelectrodes (CFME).2,9 These high oxidation potentials are almost out of the potential window for carbon electrodes and make histamine difficult to study in aqueous solutions. The FSCV community has developed several waveforms to monitor histamine dynamics in vivo. The Wightman group used a +0.1 V holding potential and +1.4 V switching potential with an 800 V/s scan rate and found a strong histamine primary anodic peak around +1.3 V vs SCE and broad secondary anodic peak at +0.9 V vs SCE.2 Because of the fast scan rate, the time required for the histamine electron transfer process cannot catch up with the potential ramping,10 and the primary peak is located on the backward scan. The Lee group used a waveform from –0.4 V to +1.4 V vs Ag/AgCl at a scan rate of 400 V/s, and both primary and secondary anodic peaks were at the same positions as Wightman’s work.11 However, the Hashemi lab recently developed a FSCV waveform that only scans to 1.1 V, and they claim the histamine faradaic peak is at 0.3 V vs Ag/AgCl and that previous reports of peaks at 1.3 V vs Ag/AgCl correspond to histamine adsorption.12 Therefore, there is controversy in the FSCV field over the potential of histamine oxidation, which needs more mechanistic insight.
In this work, we use amperometry, cyclic voltammetry (CV), X-ray photoelectron spectroscopy (XPS), and FSCV to propose a mechanism of histamine oxidation and electropolymerization at carbon electrodes. Using amperometry, we show that histamine is not oxidized until 1.1 V. CV with a 50 mV/s scan rate shows a peak at 1.3 V that is pH dependent, consistent with a one proton, one electron oxidation reaction. CV of histamine derivatives with methyl groups on the nitrogens confirm that histamine electropolymerizes after oxidation, forming dimers with the nitrogens on the imidazole rings, and this causes electrode fouling. XPS reveals a large increase in surface nitrogen content after histamine oxidation, consistent with electropolymerization of histamine that fouls the surface. With FSCV, potentials over 1.1 V are necessary to observe faradaic oxidation of histamine but fouling is observed, particularly with a 1.45 V switching potential. However, Nafion coating alleviates fouling and provides better current vs time traces for histamine detection.13 This improved understanding of the mechanism of histamine oxidation, and particularly its electropolymerization, will help in future development of electrochemical methods to detect histamine in biological systems.
Experimental Section
Chemicals
Histamine dihydrochloride, 1-methylhistamine dihydrochloride, 3-methylhistamine dihydrochloride, and Nα-methylhistamine dihydrochloride were purchased from Millipore Sigma (Burlington, MA). A stock solution of each chemical was prepared in 0.1 M HClO4. Final working solutions were prepared by diluting the stock solution in a phosphate buffered saline (PBS) (131.25 mM NaCl, 3.00 mM KCl, 10 mM NaH2PO4, 1.2 mM MgCl2, 2.0 mM Na2SO4, and 1.2 mM CaCl2 with pH adjusted to 7.4) or 0.1 M phosphate buffer to the desired concentration. In some case, high-concentration solutions were prepared by directly dissolving the chemicals in the buffer and adjusting the pH and volume of the solution appropriately.
Microelectrodes Preparation
A cylindrical carbon-fiber microelectrode (CFME) was prepared as described elsewhere.14 Briefly, a 7-μm diameter T-650 carbon fiber (Cytec Engineering Materials, West Patterson, NJ) was pulled into a 1.28-mm inner diameter × 0.68-mm outer diameter glass capillary (A-M Systems, Sequim, WA) by an aspirating pump. The capillary was then pulled by a vertical puller (Narishige, Tokyo, Japan) to get two electrodes. The fiber was cut to a length of 100 μm. The electrode was epoxied by dipping in a solution of 14% m-phenylenediamine hardener (Acros Organics, Morris Plains, NH) in Epon Resin 828 (Miller-Stephenson, Danbury, CT) at 80oC for 30 s to seal the fiber with the glass capillary. The electrode was then left at room temperature overnight, cured at 100oC for 2 h, and 150oC for overnight.
Nafion-modified CFMEs (Nafion/CFME) was prepared by applying a pulsed chronoamperometry waveform between 0.0 V for 1 s and 1.0 V for 1 s for 60 cycles in a 5% Nafion in methanol solution (LQ-1105-MeOH, Ion Power, New Castle, DE) to CFME working electrode vs Ag/AgCl reference electrode and Pt counter electrode. The modified electrode was baked at 70oC for 10 min and left at room temperature overnight before use.
Electrochemical Instrumentation
Amperometry and CV measurements were taken at a potentiostat (Gamry Instruments, Warminster, PA) using a 3-mm diameter glassy carbon working electrode (GCE), Ag/AgCl reference electrode, and Pt counter electrode (CH Instruments, Austin, TX). GCE was polished with 1.0-μm and then 0.3-μm alumina polishing powder before use. Amperometry was performed by applying a constant potential in a stirred solution. CV measurements were performed in a stagnant solution with a scan rate of 50 mV/s if not specified.
FSCV experiments were performed using a two-electrode system including CFME or Nafion/CFME working electrode backfilled with 1 M KCl and a Ag/AgCl reference electrode. All electrodes were connected to a ChemClamp potentiostat and headstage (Dagan, Minneapolis, MN). The FSCV waveform, unless stated otherwise, was applied to a CFME with a holding potential of –0.4 V, a switching potential of +1.3 V or +1.45 V, a scan rate of 400 V/s, and a repetition rate of 10 Hz. The buffer and test solutions were flowed by the electrode in a flow cell at a 2 mL/min by a syringe pump (Harvard Apparatus, Holliston, MA). The flow-injection system consists of a six-port loop injector with an air actuator (VIVI Valco Instruments, Houston, TX). The data were collected with HDCV Analysis software (Department of Chemistry, University of North Carolina at Chapel Hill).
XPS
Characterization of histamine oxidation product was performed by applying a repeated CV waveform to a screen-printed carbon electrode (SPCE) (Pine Instrument, Grove City, PA) in a histamine solution. SPCEs were characterized by using an X-ray photoelectron spectrometer (Physical Electronics, Chanhassen, MN) at the UVa Nanoscale Materials Characterization Facility to obtain elemental composition and electronic states information. The Al Kα monochromatic X-ray source (1486.6 eV) was used with a pass energy of 224 eV for elemental composition and 55 eV for electronic state information. The XPS spectra were analyzed with MultiPak software which came with the instrument. All spectra were corrected for the charging effect by shifting the C 1s peak to the binding energy of 284.8 eV.
Statistics
All values in this work are the mean ± standard deviation (SD) for n number of measurements, except for the XPS experiment from which the average value from two electrodes are reported. Statistics were performed in GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA), and significance was defined at p < 0.05.
Results and Discussion
Amperometry of Histamine
To determine the required potential for histamine oxidation at carbon electrodes, amperometry was performed at a GCE. GCE has a similar graphitic structure15 to a CFME but has a larger surface area, which makes the measurements easier. Three different applied potentials are plotted: 0.9 V (Fig. 1A), 1.1 V (Fig. 1B), and 1.3 V (Fig. 1C). For each experiment, in a stirred solution, PBS was injected twice and then histamine was injected to raise the concentration 50 μM twice. PBS injections did not change the measured current at any potential. For histamine injections, the current did not increase at the applied potential of 0.9 V; thus, histamine cannot be oxidized at a potential lower than 0.9 V. At 1.1 V, histamine increased the current noticeably above the baseline. However, the second injection increased the current less than the first injection and the current slowly fell back to the baseline, despite histamine being present and constant stirring. The current increase was larger at an applied potential of 1.3 V, but the decrease was also more dramatic and the histamine faradaic current was much higher for the first injection than the second injection.
Fig. 1.

Amperograms of PBS and histamine (HA) at GCE held at different constant potentials vs Ag/AgCl. (A) 0.9 V, (B) 1.1 V, and (C) 1.3 V. To a stirred solution of PBS pH 7.4, PBS was injected twice as a control, followed by two injections of 10 mM histamine to raise the concentration 50 μM in the whole solution.
Based on these amperometry experiments, we conclude that histamine oxidation occurs around 1.1 V and is not observed before 0.9 V. We also hypothesize that oxidation of histamine fouls the electrode surface because the signal for histamine decreases, and there is a lower current from the second histamine injection compared to the first injection. Imidazole undergoes an oxidation and then polymerizes,8 and histamine could similarly undergo oxidation and polymerize to form the polymer that adheres to the electrode surface. To confirm the mechanism and fouling hypothesis, cyclic voltammetry and electrode surface characterization were performed.
CV of Histamine at GCE
To obtain more information about the histamine oxidation at carbon electrodes, CV was performed at a GCE with a voltage triangular waveform of –0.5 V to +1.5 V at 50 mV/s for 5 cycles (Fig. 2A). Histamine oxidation was studied at a high concentration (50 mM), to distinguish its anodic peak, and physiological pH (7.4). In the first CV cycle, the histamine anodic current started to rise at 0.96 V vs Ag/AgCl and peaked with a current (ip,a) of 87 μA at a peak potential (Epa) of 1.33 V on the forward scan. Cathodic currents were not detected, indicating the reaction is not reversible. The ip,a was higher than background current (21 μA at 1.5 V, Fig. S1A). No anodic peak was detected before 1.0 V, consistent with the amperometry data that found no faradaic processes before that potential. In subsequent cycles, the anodic peak dramatically diminished, and CV of the last cycle was almost identical to the background CV. Hence, the oxidation of histamine in PBS occurs after 1.0 V and is irreversible, and the oxidation product fouls the electrode.
Fig. 2.

(A) Background-subtracted cyclic voltammogram of 50 mM histamine in PBS pH 7.4 at GCE. Scan rate 50 mV/s. (B) Cyclic voltammogram of 1 mM [Fe(CN)6]3– in 1 M KCl at GCE performed (i) at a fresh electrode, (ii) after GCE was scanned in 50 mM histamine from −0.5 to +1.5 V, (iii) electrode polished after (ii), and (iv) after electrode scanned in 50 mM histamine from −0.5 to +1.0 V. Scan rate 100 mV/s. Polymerization of histamine at waveform to 1.5 V prevents histamine detection, but electrode can be regenerated by polishing. Scanning to 1.0 V does not produce polymerization or fouling. (C) Cyclic voltammogram of 50 mM histamine in phosphate buffer at different pH. Inset illustrates the pH-dependence of anodic peak potential. Scan rate 50 mV/s.
The existence and fouling of the histamine oxidation product on the GCE was also confirmed by performing CV of 1 mM [Fe(CN)6]3–, a standard inner sphere redox probe, on GCE before and after histamine oxidation (Fig. 2B). A fresh GCE gave the quasi-reversible CV with equal anodic and cathodic currents (i) higher than background (Fig. S1B). However, after histamine was oxidized, no ferricyanide CV was observed (ii, Fig. 2B) because the histamine oxidation product fouled the electrode and prevented ferricyanide redox at the GCE surface. The electrode was polished to renew the surface and the reversible ferricyanide CV returned with identical anodic and cathodic currents (iii). To prove that histamine was not oxidized at lower potentials, CV was applied between –0.5 V and +1.0 V in 50 mM histamine solution and the ferricyanide CV resembled that of the freshly polished GCE because histamine was not polymerized (iv). This result supports the previous findings that no faradaic oxidation of histamine occurs in this potential range.
The effect of pH on histamine oxidation at GCE was also investigated using CV in a 0.1 M phosphate buffer prepared at different pH (Fig. 2C). Lower pH shifted the peaks positively, and the full anodic peak of histamine cannot be observed with scanning limits of 1.5 V when the pH was lower than 6. The electrode fouling caused by histamine oxidation was also slower at lower pH (Fig. S2A-B). In contrast, the histamine peak shifted negatively when the pH increased (Fig. S2C). Since pH affected the oxidation of histamine, its oxidation mechanism involves a proton transfer step from the oxidation intermediate. The slope between Epa and pH is –70 ± 17 mV/decade (Fig. 2C inset), indicating the ratio of transferred electron to proton to be approximately 1, despite non-ideal Nernstian behavior because of its irreversible oxidation. This pH dependence means that for in vivo measurements, shifts in pH during biological experiments would also change the histamine oxidation current, but these changes are expected to be small because the brain is well buffered and pH shifts are generally only 0.05 units.16
From the data, we propose the mechanism of the electrochemical oxidation of histamine (Scheme 1), based on the proposed mechanism of imidazole oxidation.8 Histamine undergoes one-electron oxidation to become a radical cation with a positive charge on an imidazole nitrogen. The radical stabilizes itself by dimerization with another histamine radical, and electron delocalization allows the dimer to lose two imidazole nitrogen protons. The number of protons and electrons is consistent with the pH data because it is a one proton and one electron loss per histamine. Also, the loss of a proton causes the whole reaction to be more difficult at lower pH, as we observed. The proposed mechanism shows a loss of one proton per histamine molecule, so higher pH drives the equilibrium to the product formation. The dimer then goes through electropolymerization, a series of one-electron oxidations and dimerizations to form a “polyhistamine” polymer. This polyhistamine adsorbs on the carbon electrode surface and fouls the electrode.
Scheme 1.
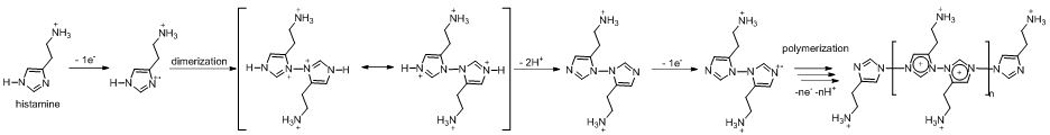
Mechanism of the oxidative electropolymerization of histamine.
CV of Histamine Derivatives at GCE
To further verify the proposed mechanism of histamine electropolymerization, several derivatives of histamine were examined. Because the proposed electropolymerization of histamine needs chain elongation at positions 1 and 3 of the imidazole ring, any functionalization on these positions should inhibit polymerization and electrode fouling. Three derivatives were tested, 1-methylhistamine (1-MeHA), 3-methylhistamine (3-MeHA), and Nα-methylhistamine (Nα-MeHA). CV was performed by scanning the potential between –0.5 V to +1.5 V for 10 cycles to examine electrode fouling. For 2 mM histamine at pH 7.4, fouling is observed and the CVs from cycle 1 to cycle 10 decrease by about 75 % in current (Fig. 3A). Here, a lower concentration was used so the fouling was slower than that for 50 mM histamine in Fig. 2A. The anodic peaks in the CVs of 2 mM 1-MeHA (Fig. 3B) and 3-MeHA (Fig. 3C) have similar features and did not decrease from cycle 1 to cycle 10. Thus, 1-MeHA and 3-MeHA undergo one-electron oxidation and likely form a dimer, but the methyl group on position 1 or 3 prevents polymerization (Scheme 2A). The final oxidation product of these two derivatives should be just their dimer, and this dimer did not cause electrode fouling. The anodic peak of both compounds also shifted positively compared to histamine because the dimer product might be unstable or the methyl group causes the oxidation to be more difficult. In contrast, the CV current from Nα-MeHA (Fig. 3D) progressively declined from cycle 1 to 10, indicating electrode fouling. The amine aliphatic chain is not involved in the reaction, therefore, methyl substitution at that position still allows the oxidative electropolymerization of Nα-MeHA to occur, as illustrated in Scheme 2B. However, the methyl group on the amine aliphatic chain could cause steric hindrance and may either slow down the electropolymerization process or shrink the polymer size, so the current decrease and electrode fouling from Nα-MeHA electropolymerization is slower than histamine.
Fig. 3.

Cyclic voltammogram of 2 mM (A) histamine, (B) 1-MeHA, (C) 3-MeHA, and (D) Nα-MeHA in PBS pH 7.4 at GCE. Scan rate 50 mV/s, repeated for 10 cycles.
Scheme 2.
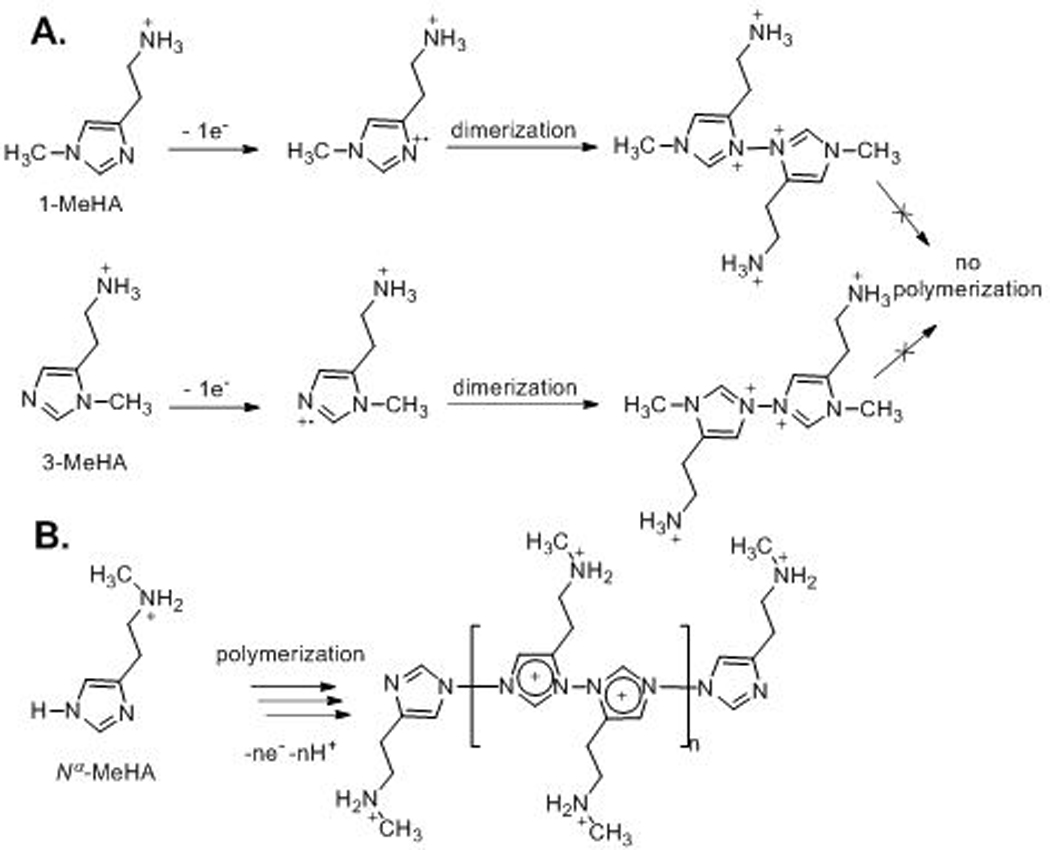
Mechanism of (A) electrochemical oxidation of 1-MeHA and 3-MeHA, and (B) oxidative electropolymerization of Nα-MeHA.
XPS Confirms Histamine Polymerization.
To test the proposed polymerization, we also performed XPS to measure surface elemental composition and electronic states of each element. Screen-printed carbon electrodes (SPCEs) were used as the working electrode instead of GCEs because they are flat and easily analyzed using XPS. Four samples of SPCE were characterized: new SPCE, SPCE scanned in PBS from −0.5 V to +1.5 V, SPCE scanned in 50 mM histamine from −0.5 V to +1.0 V, and SPCE scanned in 50 mM histamine from −0.5 V to +1.5 V. Table 1 summarizes the elemental composition and nitrogen peak position of each SPCE (Fig. S3 plots the actual XPS spectra with peaks fittings). The nitrogen composition of new SPCE, SPCE scanned in histamine to +1.0 V, and SPCE scanned in PBS were low, not above 2–3%. This nitrogen on the SPCE, which had the binding energy of 399.8 eV, may due to impurities on the electrode, and the electrode exposed to histamine may have a little physisorption of histamine on the SPCE surface, but it is not much. In contrast, the nitrogen content of SPCE scanned in histamine to +1.5 V is 15.5%. Moreover, the overall nitrogen peak shape changed and there was a new subpeak at 400.9 eV in addition to the original 399.8 eV peak (Fig. S3D). The nitrogen binding energy of 400.9 eV is consistent with a nitrogen in an aromatic ring like imidazole.17 This higher nitrogen content and new peak position indicates the existence of nitrogen-containing polymer on the SPCE surface after histamine oxidation, which is consistent with the proposed mechanism that histamine undergoes electropolymerization. This nitrogen peak only occurs when the electrode is scanned to higher potentials in histamine, and not when it is scanned to +1.0 V or when histamine is not present.
Table 1.
XPS spectral information
| Samples | Atomic elemental compositiona |
N position (eV)b | ||
|---|---|---|---|---|
| %C | %O | %N | ||
| New SPCE | 67.9 | 16.5 | 2.4 | 399.8 |
| SPCE in PBS to +1.5 V | 62.1 | 21.9 | nd | N/A |
| SPCE in histamine to +1.0 V | 66.3 | 17.2 | 3.1 | 399.9 |
| SPCE in histamine to +1.5 V | 51.4 | 22.6 | 15.5 | 399.8, 400.9 |
“nd” = not detected.
Values shown are average values from two samples. Other atoms might be present, so atomic compositions do not add to 100 %.
N positions are corrected by shifting the C 1s peak to 284.8 eV.
FSCV of Histamine at CFMEs
For real-time measurements of neurotransmitters, FSCV is predominantly used,18,19 so we investigated the detection of histamine using FSCV. FSCV parameters were investigated to understand how they influence the oxidation current for histamine. First, the switching potential was varied. For lower switching potentials: 0.5 V, 0.9 V, and 1.0 V (Fig. 4A), the current dips below baseline on the forward scan at negative potentials and then there was a weak, broad peak of only 3 nA around 0.4 V on the forward scan. The small peak is not likely a faradaic peak, since it occurs almost a volt lower than the faradaic peaks in slow scan CV or amperometry. Instead, this peak is a background current change that arises when the background capacitance changes from histamine adsorption. The log-log plot of this peak current at 0.5 V vs scan rate has a slope close to 1 (Fig. S4), indicating adsorptive capacitance change. Similarly, adsorption of dopamine to a carbon electrode can cause small, broad non-faradaic peaks in background-subtracted CVs.20 The 1.0 V scan has a small peak on the back scan near the switching potential which is likely still an adsorption peak (given the slow scan and XPS results) but may also be a small faradaic peak.
Fig. 4.

FSCV of 1 μM histamine at CFME using different waveform parameters. (A)-(B) CV from different switching potential at a holding potential of −0.4 V and scan rate of 400 V/s. Fig. 4A inset shows the enlarged CV from 0.5, 0.9, and 1.0-V switching potential. (C) CV from different scan rate using −0.4 V holding potential and 1.3 V switching potential, and (D) log-log plot between normalized anodic peak current (ipa) and scan rate (n = 4). Fig. 4C inset shows the enlarged CV from 50-V/s scan rate.
Faradaic peaks are better observed with higher switching potentials (Fig. 4B). The peak current increased with switching potential from 7 nA at 1.1 V, to 22 nA at 1.2 V, to 30 nA at 1.3 V. Lower currents were expected for the lower holding potentials of 1.1 V and 1.2 V because the CV of histamine at slower scan rates (Fig. 2A) had a peak maximum at 1.3 V. With a switching potential of 1.45 V, the anodic peak is near the switching potential on the forward scan, but the current is lower than with 1.3 V switching potential. While more oxidation is expected to occur at higher potentials because more time is spent above the E0, the fouling from electropolymerization likely happens immediately and decreases the current.
To investigate scan rate dependence, we varied the FSCV scan rate from 50 V/s to 1000 V/s. A scan rate of 50 V/s resulted in the Epa of 1.26 V on the forward scan (Fig. 4C), but a higher scan rate shifted the peak to the backward scan: 1.19 V for 400 V/s and 1.07 V for 1000 V/s, because of the time for electron transfer. The log-log plot of normalized histamine anodic peak current (ipa) vs scan rate (Fig. 4D) had a slope of 0.96 ± 0.05 (n = 4), which is close to 1 and indicates adsorption-controlled oxidation of histamine at CFMEs. Histamine has a pKa of the aliphatic amine group of 9.11,21 so it is cationic at physiological pH. The negatively charged oxygen surface functional groups of CFMEs adsorb the cationic histamine by electrostatic interactions before electron transfer. However, the response is linear only to 800 V/s and not 1000 V/s because the time of the scan is too fast to fully complete the oxidation at that fast scan rate.
Our results show that it is necessary to scan to higher switching potentials (1.3 V or above) in order to fully detect faradaic peaks for histamine. These results are consistent with early FSCV literature, which observed a histamine peak above 1.1 V on the back scan when higher switching potentials were used (vs Ag/AgCl or SCE).2,9,11 However, this work and mechanism is in contrast to the waveform developed by the Hashemi group,12 which scans only to 1.1 V (vs Ag/AgCl) and reports that the broad FSCV peak at 0.3 V is the faradaic process. All of the mechanistic data from amperometry, slow scan CV, XPS, and fast scan CV point to no faradaic reactions occurring before 1.0 V. Thus, the small peaks the Hashemi group observed at 0.3 V are broad and likely due to background changes due to adsorption. Waveform manipulation alone will not be able to shift the peak significantly unless there is an electrocatalyst on the electrode surface. One advantage of not using a high switching potential is that there will not be fouling due to electropolymerization, but this is also indicative that no faradaic reaction takes place.
Histamine Fouling with FSCV
FSCV is also a good technique to study the effect of fouling on the electrode surface because many scans are collected over time. Fig. 5A shows cyclic voltammograms and false color plots of 1 μM histamine FSCV with a switching potential of 1.3 V. Histamine was continuously flowed by the electrode surface for 5 s, and the CV changes significantly during that time. Initially (0.5 s after histamine exposure), there was an anodic peak of 24 nA around +1.21 V (“primary anodic peak”, labelled “1°” in Fig. 5) on the backward scan. At 1.0 s, the primary anodic peak reached a maximum current about 32 nA. When histamine had been present for 5.0 s, the primary peak shifted later, to +1.15 V on the backward scan, and the anodic current decreased to 24 nA. The peak current decreased because of the electrode fouling from polyhistamine, and the peak potential shifted because of the sluggish electron transfer kinetics after the fouling.
Fig. 5.

FSCV of 1 μM histamine at CFME. Current-time trace, false color plot, CVs at 0.5-s, 1.0-s and 5.0-s injection from different FSCV switching potential: (A) 1.30 V and (B) 1.45 V. Black lines under the color plots indicate histamine was flowing by the electrode. 1º and 2º indicates primary and secondary anodic peak, respectively. More detailed peak labelling can be found in Fig. S5.
During the histamine injection, there was also a small secondary peak (“secondary anodic peak”, labelled “2°” in Fig. 5) of 8 nA that appeared at +0.80 V on the forward scan in later CVs, but was not present in the first CVs. This secondary peak grew to 11 nA after 5-s of histamine. The 0.8 V secondary peak on the front scan may be due to oxidation of imidazole rings in the polymer coating, as the oxidation potential is lower for an adsorbed species, because it is on the electrode surface. The extended conjugated system in the adsorbed polyhistamine may also stabilize the radical cation generated from polyhistamine oxidation and decrease the oxidation potential.
In addition to the faradaic peaks, at later times there were other small, broader peaks near −0.17 V and 0.4 V on the forward scan and 0.5 V on the backward scan. Fig. S5 labels these peaks on a color plot. These currents are likely due to background subtraction errors due to capacitance changes when the polymer adsorbed. The extra peaks grow in over time and are present after histamine is washed out of the flow cell, indicating that they are due to a polymer build up on the surface.
FSCV of 1 μM histamine was also performed using a switching potential of 1.45 V (Fig. 5B), which is the waveform used to detect high oxidation potential analytes such as adenosine.22–24 Initially, the primary anodic peak was located on the forward scan at 1.42 V and had a current of 25 nA. However, after 5 s of histamine exposure, the peak had diminished greatly, losing about 75% of the signal (8 nA). Interestingly, the peak did not shift in voltage, which may be due to the CFME etching at high potentials, which renews the carbon surface.25 Histamine FSCV using 1.45 V switching potential also had the secondary anodic peak (0.8 V forward scan) and small broad adsorption peaks (labelled in Fig. S5). Many of these small peaks are likely due to changes in the background current from adsorption of the polymer. In vivo, pH changes also cause broad peaks due to background subtraction errors16 and data processing techniques, such as principal component regression (PCR), can be implemented to separate the contribution from pH shift.26
Although the advantage of using FSCV for real-time measurement is to obtain the molecular fingerprint from the CV, FSCV data are usually analyzed by monitoring the peak current from one potential in the CV over time. The ideal shape of a current vs time trace is square, since flow injection is used to expose the electrode to a square bolus of histamine. However, the current vs time trace in Fig. 5 shows the peak current at the oxidation potential decreases when histamine is present and even appears to drop below baseline after it is washed out. These errors are caused by histamine fouling, which changes the background charging current. These current vs time traces are not be amenable to kinetic modeling and the fouling would need to be deconvoluted, so we explored methods to alleviate the electrode fouling.
Nafion Prevents Electrode Fouling from Histamine Electropolymerization
Electropolymerization of histamine causes electrode fouling and the current vs time responses are not square, but convoluted by the fouling peaks (Fig. 5A and 5B). Nafion is a negatively-charged perfluorosulfonate polymer used to eliminate interferences and prevent electrode fouling via electrostatic repulsion and size exclusion.27–29 Nafion-coated CFMEs (Nafion/CFME) were prepared by electrodeposition30 of Nafion from a solution in methanol (the chronoamperogram from electrodeposition is shown in Fig. S6). At Nafion-coated electrodes, the CV and anodic current for histamine stayed the same throughout the injection for the 1.3 V switching potential and the peak position stayed at 1.18 V on the back scan (Fig. 6A). The color plot also shows some small, broad currents at other peaks, similar to bare CFMEs, but the current-time trace is much more square and more faithful to the time course of the histamine change. Nafion prevents electrode fouling because a polymer is already present on the surface, thus the histamine polymer cannot build up on the surface. There are also electrostatic and size exclusion effects of Nafion to prevent fouling as well.29 The histamine anodic current was smaller with Nation coating than that of the bare CFME, likely because Nafion coating restricted histamine diffusion to the electrode surface or the inhibition of polyhistamine formation decreased the current for electron transfer.
Fig. 6.

FSCV of 1 μM histamine at Nafion/CFME. Current-time trace, false color plot, CVs at 1.0-s and 5.0-s injection from different FSCV switching potential: (A) 1.30 V and (B) 1.45 V. Black lines under the color plots indicate histamine was flowing by the electrode.
After Nafion, with a 1.45 V switching potential, the color plot is much cleaner than with a 1.3 V potential and the current vs time curve is more square (Fig. 6B). The false color plot, CVs, and current-time trace illustrate less electrode fouling, similar to when 1.3 V switching potential was used. Currents obtained with the 1.45 V switching potential and Nafion are larger than with the 1.3 V potential, which would increase electrode sensitivity. Overall, Nafion coating prevents electrode fouling caused by the polymer build-up from histamine oxidation. Nafion should be considered for future studies examining histamine in vivo to prevent electrode fouling by histamine polymerization.
Conclusions
Histamine undergoes oxidative electropolymerization with a required oxidation potential of at least 1.1 V at carbon electrodes, including GCE, SPCE, and CFME. The polymer product fouls the electrode, reducing the sensitivity for histamine. FSCV of histamine had an anodic peak at 1.2 V on the backward scan and secondary peaks due to histamine polymer being oxidized on the surface. Nafion coating helps eliminate the electrode fouling observed with FSCV. Future research can utilize the fundamental understanding of histamine electrochemistry presented here to develop better electrochemical sensors and methods for real-time monitoring of histamine.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health (NIH) grant R01 EB026497.
Footnotes
Associated Content
Supporting Information
Supplemental figures include background CV of GCE in different supporting electrolytes, CV of 50 mM histamine at different pHs, XPS spectra of screen-printed electrode from different conditions, FSCV scan rate experiment using 1.0-V switching potential, and chronoamperogram from Nafion electrodeposition.
References
- (1).Haas HL; Sergeeva OA; Selbach O. Histamine in the Nervous System. Physiol. Rev 2008, 88, 1183–1241. 10.1152/physrev.00043.2007. [DOI] [PubMed] [Google Scholar]
- (2).Pihel K; Hsieh S; Jorgenson JW; Wightman RM Electrochemical Detection of Histamine and 5-Hydroxytryptamine at Isolated Mast Cells. Anal. Chem 1995, 67 (24), 4514–4521. 10.1021/ac00120a014. [DOI] [PubMed] [Google Scholar]
- (3).Flik G; Folgering JHA; Cremers TIHF; Westerink BHC; Dremencov E. Interaction Between Brain Histamine and Serotonin, Norepinephrine, and Dopamine Systems: In Vivo Microdialysis and Electrophysiology Study. J. Mol. Neurosci 2015, 56 (2), 320–328. 10.1007/s12031-015-0536-3. [DOI] [PubMed] [Google Scholar]
- (4).Denno ME; Privman E; Borman RP; Wolin DC; Venton BJ Quantification of Histamine and Carcinine in Drosophila Melanogaster Tissues. ACS Chem. Neurosci 2016, 7 (3), 407–414. 10.1021/acschemneuro.5b00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pihel K; Hsieh S; Jorgenson JW; Wightman RM Quantal Corelease of Histamine and 5-Hydroxytryptamine from Mast Cells and the Effects of Prior Incubation. Biochemistry 1998, 37 (4), 1046–1052. 10.1021/bi9714868. [DOI] [PubMed] [Google Scholar]
- (6).Travis ER; Wang Y-M; Michael DJ; Caron MG; Wightman RM Differential Quantal Release of Histamine and 5-Hydroxytryptamine from Mast Cells of Vesicular Monoamine Transporter 2 Knockout Mice. Proc. Natl. Acad. Sci 2000, 97 (1), 162–167. 10.1073/pnas.97.1.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Hashemi P; Dankoski EC; Wood KM; Ambrose RE; Wightman RM In Vivo Electrochemical Evidence for Simultaneous 5-HT and Histamine Release in the Rat Substantia Nigra Pars Reticulata Following Medial Forebrain Bundle Stimulation. J. NeuroChem 2011, 118 (5), 749–759. 10.1111/j.1471-4159.2011.07352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wang HL; O’Malley RM; Fernandez JE Electrochemical and Chemical Polymerization of Imidazole and Some of Its Derivatives. Macromolecules 1994, 27 (4), 893–901. 10.1021/ma00082a003. [DOI] [Google Scholar]
- (9).Sarada BV; Rao TN; Tryk DA; Fujishima A. Electrochemical Oxidation of Histamine and Serotonin at Highly Boron- Doped Diamond Electrodes. Anal. Chem 2000, 72 (7), 1632–1638. 10.1021/ac9908748. [DOI] [PubMed] [Google Scholar]
- (10).Stamford JA Effect of Electrocatalytic and Nucleophilic Reactions on Fast Voltammetric Measurements of Dopamine at Carbon Fiber Microelectrodes. Anal. Chem 1986, 58 (6), 1033–1036. 10.1021/ac00297a011. [DOI] [PubMed] [Google Scholar]
- (11).Chang S-Y; Jay T; Muñoz J; Kim I; Lee KH Wireless Fast-Scan Cyclic Voltammetry Measurement of Histamine Using WINCS -- a Proof-of-Principle Study. Analyst 2012, 137 (9), 2158–2165. 10.1039/c2an16038b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Samaranayake S; Abdalla A; Robke R; Wood KM; Zeqja A; Hashemi P. In Vivo Histamine Voltammetry in the Mouse Premammillary Nucleus. Analyst 2015, 140 (11), 3759–3765. 10.1039/c5an00313j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Qi L; Thomas E; White SH; Smith SK; Lee CA; Wilson LR; Sombers LA Unmasking the Effects of L-DOPA on Rapid Dopamine Signaling with an Improved Approach for Nafion Coating Carbon-Fiber Microelectrodes. Anal. Chem 2016, 88 (16), 8129–8136. 10.1021/acs.analchem.6b01871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Huffman ML; Venton BJ Electrochemical Properties of Different Carbon-Fiber Microelectrodes Using Fast-Scan Cyclic Voltammetry. Electroanalysis 2008, 20 (22), 2422–2428. 10.1002/elan.200804343. [DOI] [Google Scholar]
- (15).McCreery RL Advanced Carbon Electrode Materials for Molecular Electrochemistry. Chem. Rev 2008, 108 (7), 2646–2687. 10.1021/cr068076m. [DOI] [PubMed] [Google Scholar]
- (16).Venton BJ; Michael DJ; Wightman RM Correlation of Local Changes in Extracellular Oxygen and pH That Accompany Dopaminergic Terminal Activity in the Rat Caudate-Putamen. J. NeuroChem 2003, 84 (2), 373–381. 10.1046/j.1471-4159.2003.01527.x. [DOI] [PubMed] [Google Scholar]
- (17).Al-Hinai M; Hassanien R; Watson SMD; Wright NG; Houlton A; Horrocks BR Metal-Conductive Polymer Hybrid Nanostructures: Preparation and Electrical Properties of Palladium–polyimidazole Nanowires. Nanotechnology 2016, 27 (9), 095704. 10.1088/0957-4484/27/9/095704. [DOI] [PubMed] [Google Scholar]
- (18).Venton BJ; Wightman RM Psychoanalytical Electrochemistry: Dopamine and Behavior. Anal. Chem 2003, 75 (19), 414 A–421 A. 10.1021/ac031421c. [DOI] [Google Scholar]
- (19).Ganesana M; Lee ST; Wang Y; Venton BJ Analytical Techniques in Neuroscience: Recent Advances in Imaging, Separation, and Electrochemical Methods. Anal. Chem 2017, 89 (1), 314–341. 10.1021/acs.analchem.6b04278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bath BD; Michael DJ; Trafton BJ; Joseph JD; Runnels PL; Wightman RM Subsecond Adsorption and Desorption of Dopamine at Carbon-Fiber Microelectrodes. Anal. Chem 2000, 72 (24), 5994–6002. 10.1021/ac000849y. [DOI] [PubMed] [Google Scholar]
- (21).Altun Y; Köseoĝlu F. Stability of Copper(II), Nickel(II) and Zinc(II) Binary and Ternary Complexes of Histidine, Histamine and Glycine in Aqueous Solution. J. Solution Chem 2005, 34 (2), 213–231. 10.1007/s10953-005-2763-7. [DOI] [Google Scholar]
- (22).Swamy BEK; Venton BJ Subsecond Detection of Physiological Adenosine Concentrations Using Fast-Scan Cyclic Voltammetry. Anal. Chem 2007, 79 (2), 744–750. 10.1021/ac061820i. [DOI] [PubMed] [Google Scholar]
- (23).Nguyen MD; Lee ST; Ross AE; Ryals M; Choudhry VI; Venton BJ Characterization of Spontaneous, Transient Adenosine Release in the Caudate-Putamen and Prefrontal Cortex. PLoS One 2014, 9 (1), e87165. 10.1371/journal.pone.0087165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ganesana M; Venton BJ Early Changes in Transient Adenosine during Cerebral Ischemia and Reperfusion Injury. PLoS One 2018, 13 (5), e0196932. 10.1371/journal.pone.0196932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Takmakov P; Zachek MK; Keithley RB; Walsh PL; Donley C; McCarty GS; Wightman RM Carbon Microelectrodes with a Renewable Surface. Anal. Chem 2010, 82 (5), 2020–2028. 10.1021/ac902753x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Keithley RB; Wightman RM; Heien ML Multivariate Concentration Determination Using Principal Component Regression with Residual Analysis. TrAC - Trends Anal. Chem 2009, 28 (9), 1127–1136. 10.1016/j.trac.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Mauritz KA; Moore RB State of Understanding of Nafion. Chem. Rev 2004, 104 (10), 4535–4585. 10.1021/cr0207123. [DOI] [PubMed] [Google Scholar]
- (28).Vreeland RF; Atcherley CW; Russell WS; Xie JY; Lu D; Laude ND; Porreca F; Heien ML Biocompatible PEDOT:Nafion Composite Electrode Coatings for Selective Detection of Neurotransmitters in Vivo. Anal. Chem 2015, 87 (5), 2600–2607. 10.1021/ac502165f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Cao Q; Puthongkham P; Venton BJ Review: New Insights into Optimizing Chemical and 3D Surface Structures of Carbon Electrodes for Neurotransmitter Detection. Anal. Methods 2019, 11 (3), 247–261. 10.1039/C8AY02472C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Hashemi P; Dankoski EC; Petrovic J; Keithley RB; Wightman RM Voltammetric Detection of 5-Hydroxytryptamine Release in the Rat Brain. Anal. Chem 2009, 81 (22), 9462–9471. 10.1021/ac9018846 . [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.