

Abstract

Developing visible-light-regulated controlled/living radical polymerization techniques for the synthesis of polymers with a predictable molecular weight, spatial and temporal control, and well-defined end-group functionality is being pursued by the macromolecular community worldwide. In this study, a new metal-free photoinduced electron transfer-reversible addition-fragmentation chain transfer (PET-RAFT) polymerization system was developed for controlled macromolecular synthesis in both heterogeneous and homogeneous systems by structural engineering of graphitic carbon nitrides (g-C3N4) to improve the textural, optical, and electronic properties. A heteroatom-mediated synthesis enabled the preparation of g-C3N4 with improved structural properties and increased absorption in the visible light region. Enhanced PET-RAFT polymerization of vinyl monomers with low dispersity (Đ < 1.2), temporal control, and high chain-end fidelity was achieved under mild blue light irradiation (λmax = 465 nm, 3 mW/cm2). Moreover, we demonstrate, for the first time, that the g-C3N4-catalyzed RAFT polymerization could be realized in a homogeneous system after structural evolution of bulk g-C3N4 into soluble nanosheets with enhanced photocatalytic efficiency up to high monomer conversion. This study provides new insights into the structure–performance relationship of g-C3N4 for photoregulated PET-RAFT polymerization under visible light. Moreover, the development of a homogeneous g-C3N4-catalyzed photosynthesis system should broaden the application scope of these fascinating photocatalysts while benefiting synthetic upscaling by continuous flow and/or microfluidic reactors.
1. Introduction
The booming development of photocatalysis and artificial photosynthesis has inspired polymer chemists to exploit light to regulate macromolecular synthesis for better control over the polymerization process.1 During the past few years, photomediated controlled/living radical polymerizations (CRPs) have aroused tremendous research enthusiasm.2−6 These techniques enable the tailored synthesis of polymers with a determined molecular weight (MW), low dispersity, and chain-end fidelity. More importantly, by employing light as an external stimulus to manipulate the deactivation–activation equilibrium of the propagating and dormant chains, these techniques afford spatial and temporal control over the polymerization process.7,8
One of the major challenges to establish a photoregulated CLRP system is to develop a highly efficient and robust photocatalyst, which can initiate polymerization through either an energy transfer or a photoinduced electron transfer (PET) pathway while regulating the activation–deactivation equilibrium for the controlled growth of polymer chains.9 Taking advantage of the research advances of photoredox catalysis, the pioneering groups of photoregulated CLRPs, such as Hawker and Fors,10−13 Matyjaszewski,14−16 Boyer,17−24 Yagci,25−28 Miyake,29−31 and others, have identified a series of photocatalysts including metal-containing complexes (fac-Ir(ppy)3, Ru(bpy)3Cl2, ZnTPP), organic dyes, semiconductors, and naturally derived compounds applicable for atom transfer radical polymerization (ATRP) and/or reversible addition-fragmentation chain transfer (RAFT) polymerization.32−39 To prevent metal contamination in the product, more attention has been paid to the development of metal-free and environmentally benign photocatalytic systems.
Graphitic carbon nitride, usually denoted as g-C3N4, represents a class of attractive metal-free photocatalysts.40,41 Due to the appealing electronic band property, high physicochemical stability, “earth-abundant” nature, and facile synthesis, graphitic carbon nitride has been a research hotspot in the arena of solar energy conversion since the creative work of Antonietti in 2009, reporting the potential of the conjugated semiconductor material for visible-light-induced water splitting for H2 production.42 Yagci and co-workers then bridged graphitic carbon nitrides and macromolecular synthesis, achieving successful photoinduced free radical polymerization43 and ATRP44 under UV light. Recently, the group led by Qiao developed a robust UV-light-regulated RAFT polymerization system using urea-derived g-C3N4 that showed improved oxygen tolerance in the presence of an amine cocatalyst.45 These successes have drawn considerable interest in the field, however, for pristine g-C3N4, no absorption above 460 nm, low electronic conductivity, high recombination rate of charge carriers, and poor dispersibility remain obstacles to their applications in visible-light-mediated CLRPs.46,47
Achievable strategies to improve the photoactivity of graphitic carbon nitrides include tailored synthesis (elemental doping, postfunctionalization, supramolecular preorganization, etc.), nanoarchitecture fabrication, and electronic structure modulation.48−53 Regarding the synthetic protocols, heteroatom-mediated synthesis generated g-C3N4 with improved textural, optical, and electronic properties.54−56 Meanwhile, when subject to controllable thermal–chemical etching treatment, bulk g-C3N4 could be converted to nanosheets, nanoribbons, and even quantum dots, making them versatile for more photocatalytic systems.57
We present here a new and efficient metal-free PET-RAFT polymerization system based on structural tuning of a sulfur-containing precursor-derived graphitic carbon nitride. The presence of sulfur species regulated the thermal condensation process of the precursor, resulting in g-C3N4 products with improved porosity, electronic structure, and absorption in visible light range. Thus, enhanced PET-RAFT polymerization of vinyl monomers with temporal control, low dispersity, and chain-end fidelity was achieved. Moreover, the g-C3N4 could be further transformed into nanosheets with good optical properties and solubility in polar solvents, enabling the development of the first homogeneous g-C3N4-catalyzed PET-RAFT polymerization system under visible light irradiation.
2. Results and Discussion
2.1. Graphitic Carbon Nitride Synthesis and Characterization
Photoactivity of graphitic carbon nitrides in photoredox catalysis depends on their microstructure, optical and electronic properties, which can be improved by rationally controlling the synthesis procedure. In this work, a sulfur-mediated synthetic protocol, using trithiocyanuric acid (TCA) as the precursor, was employed to prepare the graphitic carbon nitride (denoted as “TCA-g-C3N4”) used for visible-light-regulated RAFT polymerizations. It has been reported that precursors containing heteroatoms in addition to nitrogen exhibit positive influence in the thermal polycondensation. The big-sized sulfur atom is expected to modify the conformation and connection of the resultant graphitic carbon nitrides while tuning their textural and physicochemical properties.40 Meanwhile, a conventional melamine-derived graphitic carbon nitride (denoted “MA-g-C3N4”) was also prepared for a comparative study.
The morphology of the as-prepared g-C3N4 was characterized by transmission electron microscopy (TEM). Typically, MA-g-C3N4 exhibits irregular particles and sheets without determinate size (Figure 1a), while a well-developed porous structure with thin walls was observed for TCA-g-C3N4 (Figure 1b), suggesting that the nature of precursor significantly influenced the morphological evolution during thermal polycondensation. Basically, for TCA-g-C3N4, the presence of sulfur atoms in the precursor enabled the generation of volatile species such as H2S and CS2 that were released during the reaction (Figure 1f).54,55 This offered a template tool to mediate the modification of the texture, resulting in g-C3N4 with improved porosity. The digital photos showed that the as-prepared MA-g-C3N4 and TCA-g-C3N4 were light yellow and brown powders, respectively (Figure 1c,d). Both of the g-C3N4 samples could be well-dispersed in polar solvents, such as dimethyl sulfoxide (DMSO), after moderate ultrasonication (Figure 1e).
Figure 1.

Morphologies of graphitic carbon nitrides (g-C3N4) made from melamine (MA-g-C3N4) (a) and trithiocyanuric acid (TCA-g-C3N4) (b) under TEM; photographs of MA-g-C3N4 (c) and TCA-g-C3N4 (d) in the solid form and dispersed in DMSO (left, MA-g-C3N4; right, TCA-g-C3N4) (e); and reaction pathway of thermal polycondensation of trithiocyanuric acid into g-C3N4 at high temperature in air (f).
The X-ray diffraction (XRD) patterns in Figure 2a suggest a graphitic-like layer structure for MA-g-C3N4 and TCA-g-C3N4. The latter shows intensity decrease and broadening of the signal peak, which is associated with the volatile sulfur species-mediated formation of porous structure in TCA-g-C3N4. The strongest XRD peak located at ca. 27° is determined as the signal of (002) interlayer reflection, corresponding to an interlayer distance of d = 0.342 nm. Another identifiable XRD peak at ca. 13° is ascribed to in-plane repeated units in the layer structure. The Fourier transform infrared (FT-IR) spectra show a similar signal peak for the two kinds of g-C3N4 (Figure 2b). The strong bands at 1600–1200 cm–1 correspond to the stretching vibration of the heptazine heterocyclic ring (C6H7) system, while the sharp peak at 805 cm–1 is assigned to the breathing mode of the triazine units.58 Besides, the broad absorbance at 3300–2900 cm–1 is assigned to the stretching vibration of residual N–H components.
Figure 2.

XRD patterns (a) and FT-IR spectra (b) of MA-g-C3N4 and TCA-g-C3N4.
X-ray photoelectron spectroscopy (XPS) was used to examine the surface chemistry of the graphitic carbon nitrides. According to the wide scan spectra in Figure 3a,b, only C 1s, N 1s, and O 1s signals were found for both the samples, suggesting that the sulfur atoms were not incorporated into the frameworks of the graphitic carbon nitrides. This is in accordance with the previous report that the role of the sulfur species in the precursor is only to mediate the thermal condensation instead of doping the g-C3N4.54,55 The presence of O 1s signal might be due to the adsorbed H2O or CO2 molecules on the surfaces of the g-C3N4 samples. High-resolution XPS spectra show that there are mainly two carbon species observed in the C 1s spectrum with the binding energy of 284.9 and 288.2 eV, respectively (Figure 3c,d). The former peak is generally assigned to the sp2 C–C bonds, while the latter peak is typically ascribed to sp2-bonded carbons in the heptazine ring system (N–C=N), which is considered the major carbon species in the g-C3N4.59 Besides, deconvolution of high-resolution XPS N 1s spectra of MA-g-C3N4 (Figure 3e) and TCA-g-C3N4 (Figure 3f) suggests that the nitrogen species consisted of four components. For MA-g-C3N4, the component signals show peaks at 398.7, 401.0, 401.2, and 404.5 eV. The main source of the peak at 398.7 eV is from the sp2-bonded nitrogen in the triazine rings. While the weak peak at 401.0 eV originates from the tertiary nitrogen N–(C)3 groups. Both of them are the main components of the heptazine heterocyclic ring (C6N7) units, which are the basic substructure units of the g-C3N4. Another weak peak at 401.2 eV indicates the presence of residue amino functions (C–N–H) in the melon structures. In addition, the component signal at 404.5 eV is attributed to the charging effects or positive charge localization in the heterocycles.59 The component signals of TCA-g-C3N4 after deconvolution are coincident with those of MA-g-C3N4 except for a slight difference in specific binding energy. It is worth mentioning that a careful scanning of both the graphitic carbon nitrides shows no signal attributed to S 2p species, which further confirms that there are no sulfur dopants in the structure of the graphitic carbon nitrides. Table 1 summarizes the content of corresponding elements in MA-g-C3N4 and TCA-g-C3N4 based on XPS analysis. Higher nitrogen content was found in the structure of TCA-g-C3N4.
Figure 3.
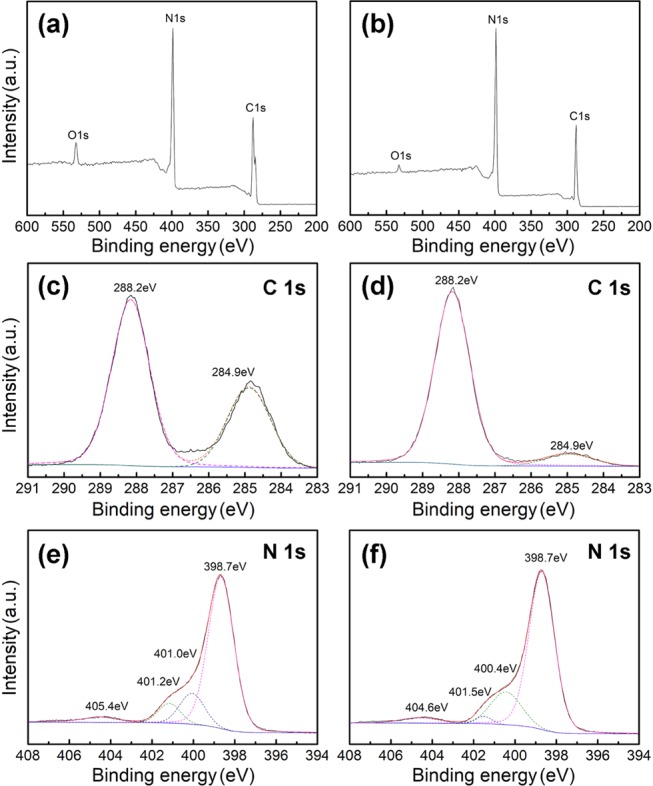
XPS spectra of MA-g-C3N4 (a) and TCA-g-C3N4 (b), high-resolution C 1s XPS spectra of MA-g-C3N4 (c) and TCA-g-C3N4 (d), and deconvolution of XPS N 1s spectra of MA-g-C3N4 (e) and TCA-g-C3N4 (f).
Table 1. XPS Elemental Analysis and Nitrogen Adsorption–Desorption Measurements of g-C3N4.
| samples | O, wt % | C, wt % | N, wt % | S, wt % | specific surface area, m2/g | pore size, nm | pore volume, cm3/g |
|---|---|---|---|---|---|---|---|
| MA-g-C3N4 | 4.7 | 46.9 | 48.4 | 12 | 1.9 | 0.03 | |
| TCA-g-C3N4 | 2.2 | 38.6 | 59.2 | 79 | 9.8 | 0.15 |
The graphitic carbon nitrides show typical UV–vis absorption spectra (Figure 4a) of semiconductors that originate from electron transition from the valence band to the conduction band. Compared to MA-g-C3N4, TCA-g-C3N4 shows intensified absorption in both UV and visible light regions. A red shift of absorption edge is observed for TCA-g-C3N4, corresponding to a reduced band gap energy. This might be explained by the extension of electron delocalization in the aromatic sheets with enhanced structural connections.54 The photoluminescence spectra of TCA-g-C3N4 dispersed in DMSO exhibit similar emission profile yet higher intensity to that of MA-g-C3N4 under the excitation wavelength of 365 nm (Figure 4b). The emission peaks are located at 434, 544, and 576 nm. Nitrogen adsorption–desorption analysis (Figure 4c) reveals that the texture of the g-C3N4 samples is evidently influenced by the precursor type. TCA-g-C3N4 derived from trithiocyanuric acid exhibits a Brunauer–Emmett–Teller (BET) specific surface area of 79 m2/g, much higher than that (12 m2/g) of the melamine-derived counterpart (Table 1). Besides, a higher pore volume (0.15 cm3/g) with a larger pore size (9.8 nm) (Figure 4d) was also found for TCA-g-C3N4. These results confirmed that the sulfur-containing precursor-mediated synthesis benefited the formation of g-C3N4 product with improved specific surface area and porosity. This synthetic protocol to prepare highly porous g-C3N4 is facile in operation that prevents the introduction of extra templates such as silica nanoparticles.43 The highly porous texture of g-C3N4 is supposed to decrease the length of photoinduced charge transportation while providing more active sites for photocatalytic reactions.40
Figure 4.

UV–vis absorption spectra of MA-g-C3N4 and TCA-g-C3N4 dispersed in DMSO (0.4 g/mL) (a), photoluminescence spectra in DMSO (0.4 g/mL) under the excitation wavelength of 365 nm (b), nitrogen adsorption–desorption measurements (c), and pore size distribution (d) of MA-g-C3N4 and TCA-g-C3N4.
2.2. Blue Light-Regulated RAFT Polymerizations
Photoregulated CRP has been recognized as an enabling tool for controlled synthesis of polymers with a predetermined MW, narrow MW distribution, and well-defined end-group functionality. Previous studies demonstrated that graphitic carbon nitrides were of great potential to catalyze CRPs under UV light irradiation via a photoinduced electron transfer (PET) pathway.44,45 Here, taking advantage of the structurally enhanced TCA-g-C3N4, a new PET-RAFT polymerization system was established under mild blue light-emitting diode (LED) light (λmax = 465 nm) irradiation (Figure S1). Besides, the structure–performance relationship of g-C3N4 was evaluated by a comparative study on the photocatalytic ability of different precursor-derived g-C3N4.
First, RAFT polymerization of methyl methacrylate (MMA) was carried out using MA-g-C3N4 and TCA-g-C3N4 as photocatalysts. Blank experiments showed that, in the absence of either g-C3N4 (Table 2, entry 1) or light irradiation (Table 2, entries 2 and 6), the RAFT polymerization was not initiated. Meanwhile, the possibility of thermally induced initiation under light irradiation was ruled out by conducting the experiment in a dark environment at 45 °C, which resulted in no polymer product (Table 2, entries 3 and 7). Under blue light irradiation, the MA-g-C3N4-catalyzed RAFT polymerization achieved a conversion of 33%. Gel permeation chromatography (GPC) analysis showed a Mn ∼ 6800 for the polymer product, which was in agreement with the theoretical value (Table 2, entry 4). In comparison, the TCA-g-C3N4-catalyzed RAFT polymerization system exhibited enhanced photocatalytic efficiency. A higher Mn ∼ 10 300 for the polymer product with a lower dispersity (Đ ∼ 1.18) was achieved (Table 2, entry 8).
Table 2. g-C3N4-Catalyzed RAFT Polymerizations under Blue LED (λmax = 465 nm) Irradiation.
| entry | [MMA]/[CPADB]a | g-C3N4b | α (%)c | Mn,theod | Mn,GPCe | Đe |
|---|---|---|---|---|---|---|
| 1 | 200:0 | |||||
| 2f | 200:1 | MA, 5 mg | ||||
| 3g | 200:1 | MA, 5 mg | ||||
| 4 | 200:1 | MA, 5 mg | 33 | 6600 | 6800 | 1.25 |
| 5 | 200:0 | MA, 5 mg | 45 | 8900 | 127 500 | 1.95 |
| 6f | 200:1 | TCA, 5 mg | ||||
| 7g | 200:1 | TCA, 5 mg | ||||
| 8 | 200:1 | TCA, 5 mg | 50 | 10 100 | 10 300 | 1.18 |
| 9 | 200:0 | TCA, 5 mg | 93 | 18 700 | 274 600 | 1.61 |
| 10 | 200:1 | TCA-NS, 5 mg | 72 | 14 400 | 14 800 | 1.08 |
The polymerizations were performed in 1 mL of DMSO using 4-cyanopentanoic acid dithiobenzoate (CPADB) as a chain transfer agent (CTA) for 16 h under irradiation of blue LED light (λmax = 465 nm, 3 mW/cm2). The molar ratio of [MMA]/[CPADB] is 200:1.
“MA” represents melamine-derived g-C3N4, “TCA” represents trithiocyanuric acid-derived g-C3N4, and “TCA-NS” represents the soluble TCA-g-C3N4 nanosheets.
Monomer conversion was determined by online Fourier transform near-infrared (FT-NIR) spectroscopy.
The theoretical molecular weight was calculated according to the equation Mn,theo = [M]0/[CPADB]0 × MWM × α + MWCPADB, where [M]0, [CPADB]0, MWM, α, and MWCPADB represent the initial monomer concentration, initial CPADB concentration, molar mass of the monomer, conversion, and molar mass of CPADB.
The molecular weight and dispersity were determined by GPC using tetrahydrofuran (THF) as an eluent.
The polymerization was performed in the dark.
The polymerization was performed at 45 °C in the dark.
A g-C3N4-catalyzed photo-RAFT polymerization started by the activation of the chain transfer agents (CTAs) via electron transfer under light irradiation. The activated CTAs initiated the polymerization of monomers. The RAFT process was maintained by chain transfer to other CTA molecules, which resulted in dormant polymer chains and other propagating chains.45 According to this mechanism, the enhanced photocatalytic efficiency of TCA-g-C3N4 can be attributed to its improved optical property and specific surface area. The former benefits the absorption in the blue light range, resulting in more photogenerated electrons. The latter provides more active sites and facilitates the charge transport, which would accelerate the activation of the CTAs. Besides, it is worth noting that, in the absence of CPADB, there were polymer products generated in the reaction systems catalyzed by either MA-g-C3N4 or TCA-g-C3N4. However, the polymerization was not controlled due to the absence of a RAFT process to maintain the reversible degenerate transfer equilibrium. The polymer product showed a Mn much higher than the theoretical value and a larger dispersity (Table 2, entries 5 and 9).
2.3. Kinetics and “On/Off” Experiments
To fully evaluate the TCA-g-C3N4 catalyzed RAFT polymerization system, a kinetic study was performed under blue LED irradiation. A linear relationship between ln([M]0/[M]t) and the irradiation time was observed, corresponding to pseudo-first-order polymerization kinetics with an apparent rate constant (kapp) of 6.2 × 10–2 h–1 (Figure 5a and Table S1). This indicated that the concentration of the propagating radicals was maintained during the polymerization process. A comparative study on the MA-g-C3N4-catalyzed RAFT polymerization of MMA under the same conditions resulted in relatively poor kinetic control (Figure S2 and Table S2), which further confirmed the enhanced photocatalytic efficiency of TCA-g-C3N4. GPC profiles with monomodal distribution were obtained for the polymer product, showing a continuous shift to the high-MW region with the increase in exposure time (Figure 5b). A linear increase in experimental Mn with monomer conversion was observed, which was in line with the theoretical estimation (Figure 5c). Meanwhile, increasing monomer conversion also resulted in a decrease in dispersity and, for the final product, quite narrow distribution was obtained, indicating a controlled radical polymerization process.
Figure 5.

Kinetic study of TCA-g-C3N4-catalyzed PET-RAFT polymerization of MMA using CPADB as the CTA under blue LED irradiation (λmax = 465 nm, 3 mW/cm2). The molar ratio of [MMA]/[CPADB] = 200:1 in 1 mL of DMSO: (a) ln([M]0/[M]t) vs irradiation time and the fitted kinetic curve. (b) GPC profiles of poly(methyl methacrylate) (PMMA) synthesized in the kinetic study. (c) Number average molecular weight (Mn) and dispersity (Đ) of PMMA obtained in the kinetic study.
One of the significant advantages of photoregulated RAFT polymerization is the temporal control with the polymerization process effectively switching between active and dormant states by controlling the light “On” and “Off”. To examine whether the TCA-g-C3N4-catalyzed polymerization of MMA in DMSO exhibited temporal control, the On/Off light switch experiment was carried out. Figure 6a shows that the Mn of PMMA increased linearly with the exposure time in the initial 5 h. The light was then turned off and the reaction system was maintained in dark for 1 h. It was found that the polymerization was completely suppressed. Switching on the light resumed the linear chain propagation. Repeated On/Off experiments suggested that the RAFT polymerization could be effectively manipulated by light, demonstrating a good temporal control over the polymerization process. The final polymer product exhibited a symmetric GPC profile with low dispersity (Đ = 1.10) (Figure 6b).
Figure 6.

On/Off light switch experiment catalyzed by TCA-g-C3N4 under blue LED irradiation (λmax = 465 nm, 3 mW/cm2) (a) and the GPC profile of the PMMA prepared in the On/Off experiment (b).
To further demonstrate the activity of functional end groups of the polymers prepared by the TCA-g-C3N4-catalyzed polymerization, chain extension of a PMMA product was attempted. A macromolecular chain transfer agent (macro-CPADB) (Mn = 9200, Đ = 1.14) was first prepared by a TCA-g-C3N4-catalyzed RAFT polymerization under blue LED light irradiation. The preservation of functional end groups was confirmed by 1H NMR (Figure 7a). The chain extension experiment resulted in a diblock copolymer with a Mn of 24 600 and a Đ of 1.25 (Figure 7b), suggesting the chain-end fidelity and living character of the PMMA product.
Figure 7.

1H NMR spectrum of PMMA macroinitiator used for the chain extension experiment (a) and GPC profiles of PMMA and the diblock copolymer after chain extension (b).
2.4. Soluble g-C3N4-Catalyzed RAFT Polymerization
Furthermore, previous research has proved that 3D bulk g-C3N4 can be transformed into soluble nanoribbons or nanosheets by facile thermal oxidation and acidic etching treatment.57 This offers an opportunity to broaden the application of g-C3N4 in homogeneous photoregulated CRP systems that are expected to achieve uniform illumination with decreased light scattering. To reach this end, the as-synthesized TCA-g-C3N4 was subjected to further structural transformation based on an established procedure57 to generate the nanosized counterpart, as shown in the TEM image (Figure 8a). The inset suggests that the bulk TCA-g-C3N4 was cut into fragmentized sheets with a preserved porous structure. A brown solid of the g-C3N4 nanosheets (Figure 8b) could be obtained by dialysis purification and redissolved in DMSO (Figure 8c). The XRD pattern of the TCA-g-C3N4 nanosheets in Figure 9a still shows the signal of (002) interlayer reflection at ca. 27°. Meanwhile, the characteristic signal of the triazine units at 805 cm–1 is preserved in the FT-IR spectrum (Figure 9b).
Figure 8.

TEM image of TCA-g-C3N4 nanosheets after thermal oxidation and acidic etching treatment (a) and photographs of TCA-g-C3N4 nanosheets in the solid form (b) and dissolved in DMSO (c).
Figure 9.

XRD pattern (a) and FT-IR spectrum (b) of TCA-g-C3N4 nanosheets.
Finally, the capability of the TCA-g-C3N4 nanosheets for RAFT polymerization of MMA in a homogeneous system was evaluated. The polymerization was carried out following the same recipe as that used in the bulk g-C3N4-catalyzed heterogeneous systems under blue LED irradiation. An increased monomer conversion of 72% was achieved after the same irradiation time. The PMMA product showed a Mn ∼ 14 800 close to the theoretical value and a symmetric GPC profile (Figure 10) with quite narrow distribution (Đ ∼ 1.08). These results demonstrated the photocatalytic ability of the TCA-g-C3N4 nanosheets in homogeneous PET-RAFT polymerization. The improved monomer conversion and a higher MW of the polymer product in the homogeneous system might be attributed to more efficient mass transfer and better spatial illumination homogeneity. Besides, the enhanced photocatalytic ability of g-C3N4 in the homogeneous system would pave a way for performing photoregulated CLRPs in microfluidic reactors or continuous flow reactors.
Figure 10.
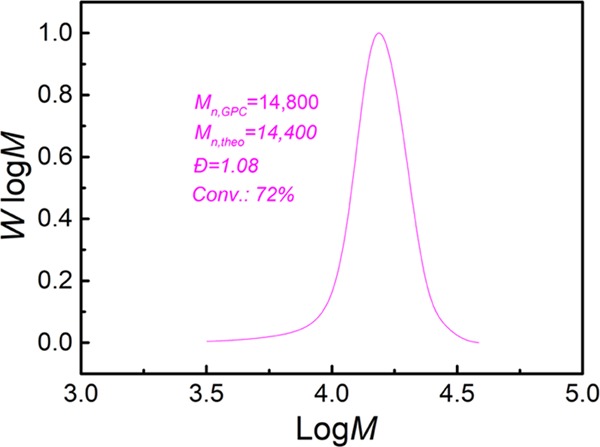
GPC profile of PMMA prepared by TCA-g-C3N4 nanosheets catalyzed PET-RAFT polymerization. The polymerization was performed in 1 mL of DMSO using CPADB as CTA under irradiation of blue LED light (λmax = 465 nm, 3 mW/cm2) for 16 h. The molar ratio of [MMA]/[CPADB] was 200:1.
3. Conclusions
The development of new and efficient metal-free photoinduced electron transfer (PET) RAFT polymerization systems regulated by visible light based on the structural engineering of graphitic carbon nitrides is presented herein. Taking advantage of a heteroatom-modulated synthetic protocol, graphitic carbon nitrides with improved textural, optical, and electronic properties were obtained, enabling an enhanced PET-RAFT polymerization of vinyl monomers under mild blue light irradiation (λmax = 465 nm, 3 mW/cm2). Well-defined polymers with good control over the molecular weight (MW) and narrow MW distribution were obtained. The On/Off light switch experiment showed good temporal control of the photoregulated polymerization system. Successful chain extension proved the high chain-end fidelity of the polymer product. Moreover, the graphitic carbon nitride photocatalyst could be transformed into soluble nanosheets when subject to thermal oxidation and acidic etching. Thus, homogeneous photocatalysis of PET-RAFT polymerizations using graphitic carbon nitrides was achieved. Overall, this work provides new insights into the structure–performance relationship of graphitic carbon nitrides for catalyzing visible-light-regulated PET-RAFT polymerization. The demonstration of PET-RAFT polymerizations in a homogeneous system catalyzed by graphitic carbon nitride can offer new opportunities for broadening their applications in continuous flow reactors.
4. Experimental Section
4.1. Materials
Melamine (99%), trithiocyanuric acid (98%), dimethyl sulfoxide (DMSO, 99.7%, Superdry), and tetrahydrofuran (THF) were purchased from J&K Scientific Ltd. and used as received. Triethanolamine (TEA, Vetec reagent-grade, 97%), 4-cyano-4-(thiobenzoylthio)pentanoic acid (CPADB, >97%), and methyl methacrylate (MMA, contains ≤30 ppm MEHQ as an inhibitor, 99%) were purchased from Sigma-Aldrich; MMA was passed over a neutral alumina column before use for the removal of the inhibitor. Graphitic carbon nitrides (g-C3N4) used as photocatalysts in this study were prepared according to a previously reported method55 by melamine (MA) and trithiocyanuric acid (TCA) as precursors, respectively.
4.2. Characterizations
Transmission electron microscopy (TEM) was performed on JEM-2010 (JEOL). Fourier transform infrared (FT-IR) spectra were recorded on a Nicolet iN10 infrared microscope. Powder X-ray diffraction (XRD) patterns were obtained on a diffractometer with Cu Kα radiation, with a scan step of 0.2° and the scan range between 10 and 40°. Nitrogen sorption isotherms were measured at 77 K on a NOVA 3200e surface area and pore size analyzer. Before measurement, the samples were degassed in vacuum at 300 °C for 3 h. The Brunauer–Emmett–Teller (BET) method was utilized to calculate the specific surface areas. Gel permeation chromatography (GPC) analysis was performed in a Shimadzu LC-20A GPC system using tetrahydrofuran (THF) as an eluent. The NIR absorption spectra between 1600 and 1700 nm (0.1 nm interval) were collected at room temperature on an Agilent Cary6000i UV–vis–NIR spectrophotometer. X-ray photoelectron spectroscopy (XPS) was performed by a 250XI spectrometer with a mono X-Ray source Al K excitation (1361 eV). Binding energy calibration was based on C 1s at 284.6 eV.
4.3. General Procedure of g-C3N4-Catalyzed RAFT Polymerization
To perform the RAFT polymerizations, typically, g-C3N4 (5 mg), MMA (1 mL, 9.35 mmol), CPADB (13 mg, 0.047 mmol), and DMSO (1 mL) were mixed in a 20 mL vial glass sealed with a plug-type rubber sleeve stopper. The molar ratio of [MMA]/[CPADB] was 200:1. After ultrasonication for 2 min, argon was bubbled to exclude the oxygen in the polymerization system. Then, the reactant mixture was irradiated under a blue LED light source (λmax = 465 nm, 3 mW/cm2) placed in a photo studio lighting tent at room temperature. After a predetermined polymerization time, the resulting solution was separated and precipitated in ethanol under stirring. The product was collected by filtration and redissolved in a minimum amount of THF. After the second precipitation in ethanol, the final polymer product was separated and dried in a 50 °C oven. The monomer conversion was determined according to the method reported by Boyer et al. using online Fourier transform near-infrared (FT-NIR) spectroscopy.20 GPC was utilized to analyze the molecular weight (Mn) and dispersity (Mw/Mn) of the polymers.
4.4. Kinetic Study of g-C3N4-Catalyzed RAFT Polymerization
A 20 mL glass vial charged with g-C3N4 (5 mg), MMA (1 mL, 9.35 mmol), CPADB (13 mg, 0.047 mmol), and DMSO (1 mL) was sealed with a plug-type rubber sleeve stopper and then protected from light by covering with aluminum foil. After degassed by gentle Ar bubbling for 20 min, the reaction solution was irradiated under a blue LED light source (λmax = 465 nm, 3 mW/cm2) at room temperature. Aliquots of samples were taken at predetermined time intervals for analysis. The monomer conversions were determined by FT-NIR spectroscopy mentioned above. The molecular weight (Mn) and dispersity (Mw/Mn) of the polymer products were analyzed by GPC.
4.5. On/Off Light Switch Experiment
Temporal control of the TCA-g-C3N4-catalyzed RAFT polymerization under blue LED irradiation was verified by the light switch experiment. The experimental conditions and pretreatment of the reaction system were the same as those described in the kinetic study. At a predetermined reaction time, the light source was turned off and the polymerization system was maintained in the dark for a period of time. The reaction was then reactivated by turning on the LED light. This On/Off cycle was repeated during the polymerization process. Samples were taken with a degassed syringe at timed intervals under both light and dark conditions for FT-NIR measurements and GPC analysis.
Acknowledgments
The study was supported by the National Natural Science Foundation of China (under Project 51673109), the National Science Foundation (CHE 1707490), and the National Science Fund for Distinguished Young Scholars (51425403). This work is also supported by MOE Key Laboratory of Resources and Environmental Systems Optimization (NCEPU), China.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b02597.
Photographs of the setup for PET-RAFT polymerization; kinetic study of MA-g-C3N4-catalyzed PET-RAFT polymerization of MMA using CPADB as the chain transfer agent under blue LED irradiation (λmax = 465 nm, 3 mW/cm2); and kinetic study on PET-RAFT polymerization of MMA catalyzed by TCA-g-C3N4 under 3 mW/cm2 blue LED irradiation (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Corrigan N.; Shanmugam S.; Xu J.; Boyer C. Photocatalysis in Organic and Polymer Synthesis. Chem. Soc. Rev. 2016, 45, 6165–6212. 10.1039/C6CS00185H. [DOI] [PubMed] [Google Scholar]
- Shanmugam S.; Xu J.; Boyer C. Photocontrolled Living Polymerization Systems with Reversible Deactivations through Electron and Energy Transfer. Macromol. Rapid Commun. 2017, 38, 1700143 10.1002/marc.201700143. [DOI] [PubMed] [Google Scholar]
- Dadashi-Silab S.; Doran S.; Yagci Y. Photoinduced Electron Transfer Reactions for Macromolecular Syntheses. Chem. Rev. 2016, 116, 10212–10275. 10.1021/acs.chemrev.5b00586. [DOI] [PubMed] [Google Scholar]
- Pan X.; Tasdelen M. A.; Laun J.; Junkers T.; Yagci Y.; Matyjaszewski K. Photomediated Controlled Radical Polymerization. Prog. Polym. Sci. 2016, 62, 73–125. 10.1016/j.progpolymsci.2016.06.005. [DOI] [Google Scholar]
- Kreutzer J.; Yagci Y. Metal Free Reversible-Deactivation Radical Polymerizations: Advances, Challenges, and Opportunities. Polymers 2018, 10, 35. 10.3390/polym10010035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot J. C.; Mccarthy B. G.; Lim C.; Miyake G. M. Organocatalyzed Atom Transfer Radical Polymerization: Perspectives on Catalyst Design and Performance. Macromol. Rapid Commun. 2017, 38, 1700040 10.1002/marc.201700040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X.; Fang C.; Fantin M.; Malhotra N.; So W. Y.; Peteanu L. A.; Isse A. A.; Gennaro A.; Liu P.; Matyjaszewski K. Mechanism of Photoinduced Metal-Free Atom Transfer Radical Polymerization: Experimental and Computational Studies. J. Am. Chem. Soc. 2016, 138, 2411–2425. 10.1021/jacs.5b13455. [DOI] [PubMed] [Google Scholar]
- Pan X.; Fantin M.; Yuan F.; Matyjaszewski K. Externally Controlled Atom Transfer Radical Polymerization. Chem. Soc. Rev. 2018, 47, 5457–5490. 10.1039/C8CS00259B. [DOI] [PubMed] [Google Scholar]
- Shanmugam S.; Boyer C. Polymer Synthesis Organic Photocatalysts for Cleaner Polymer Synthesis. Science 2016, 352, 1053–1054. 10.1126/science.aaf7465. [DOI] [PubMed] [Google Scholar]
- Treat N. J.; Sprafke H.; Kramer J. W.; Clark P. G.; Barton B. E.; Read De Alaniz J.; Fors B. P.; Hawker C. J. Metal-Free Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2014, 136, 16096–16101. 10.1021/ja510389m. [DOI] [PubMed] [Google Scholar]
- Poelma J. E.; Fors B. P.; Meyers G. F.; Kramer J. W.; Hawker C. J. Fabrication of Complex Three-Dimensional Polymer Brush Nanostructures through Light-Mediated Living Radical Polymerization. Angew. Chem., Int. Ed. 2013, 52, 6844–6848. 10.1002/anie.201301845. [DOI] [PubMed] [Google Scholar]
- Fors B. P.; Hawker C. J. Control of a Living Radical Polymerization of Methacrylates by Light. Angew. Chem., Int. Ed. 2012, 51, 8850–8853. 10.1002/anie.201203639. [DOI] [PubMed] [Google Scholar]
- Treat N. J.; Fors B. P.; Kramer J. W.; Christianson M.; Chiu C.; Alaniz J. R. D.; Hawker C. J. Controlled Radical Polymerization of Acrylates Regulated by Visible Light. ACS Macro Lett. 2014, 3, 580–584. 10.1021/mz500242a. [DOI] [PubMed] [Google Scholar]
- Pan X.; Lamson M.; Yan J.; Matyjaszewski K. Photoinduced Metal-Free Atom Transfer Radical Polymerization of Acrylonitrile. ACS Macro Lett. 2015, 4, 192–196. 10.1021/mz500834g. [DOI] [PubMed] [Google Scholar]
- Pan X.; Malhotra N.; Simakova A.; Wang Z.; Konkolewicz D.; Matyjaszewski K. Photoinduced Atom Transfer Radical Polymerization with ppm-Level Cu Catalyst by Visible Light in Aqueous Media. J. Am. Chem. Soc. 2015, 137, 15430–15433. 10.1021/jacs.5b11599. [DOI] [PubMed] [Google Scholar]
- Konkolewicz D.; Schröder K.; Buback J.; Bernhard S.; Matyjaszewski K. Visible Light and Sunlight Photoinduced ATRP with ppm of Cu Catalyst. ACS Macro Lett. 2012, 1, 1219–1223. 10.1021/mz300457e. [DOI] [PubMed] [Google Scholar]
- Xu J.; Jung K.; Atme A.; Shanmugam S.; Boyer C. A Robust and Versatile Photoinduced Living Polymerization of Conjugated and Unconjugated Monomers and its Oxygen Tolerance. J. Am. Chem. Soc. 2014, 136, 5508–5519. 10.1021/ja501745g. [DOI] [PubMed] [Google Scholar]
- Shanmugam S.; Xu J.; Boyer C. Photoinduced Electron Transfer-Reversible Addition-Fragmentation Chain Transfer (PET-RAFT) Polymerization of Vinyl Acetate and N-Vinylpyrrolidinone: Kinetic and Oxygen Tolerance Study. Macromolecules 2014, 47, 4930–4942. 10.1021/ma500842u. [DOI] [Google Scholar]
- Xu J.; Jung K.; Boyer C. Oxygen Tolerance Study of Photoinduced Electron Transfer-Reversible Addition-Fragmentation Chain Transfer (PET-RAFT) Polymerization Mediated by Ru(Bpy)3Cl2. Macromolecules 2014, 47, 4217–4229. 10.1021/ma500883y. [DOI] [Google Scholar]
- Xu J.; Shanmugam S.; Duong H. T.; Boyer C. Organo-Photocatalysts for Photoinduced Electron Transfer-Reversible Addition-Fragmentation Chain Transfer (PET-RAFT) Polymerization. Polym. Chem. 2015, 6, 5615–5624. 10.1039/C4PY01317D. [DOI] [Google Scholar]
- Shanmugam S.; Xu J.; Boyer C. Aqueous Raft Photopolymerization with Oxygen Tolerance. Macromolecules 2016, 49, 9345–9357. 10.1021/acs.macromol.6b02060. [DOI] [Google Scholar]
- Shanmugam S.; Xu J.; Boyer C. Light-Regulated Polymerization Under Near-Infrared/Far-Red Irradiation Catalyzed by Bacteriochlorophylla. Angew. Chem., Int. Ed. 2016, 55, 1036–1040. 10.1002/anie.201510037. [DOI] [PubMed] [Google Scholar]
- Shanmugam S.; Xu J.; Boyer C. Photoinduced Oxygen Reduction for Dark Polymerization. Macromolecules 2017, 50, 1832–1846. 10.1021/acs.macromol.7b00192. [DOI] [Google Scholar]
- Yeow J.; Chapman R.; Xu J.; Boyer C. Oxygen Tolerant Photopolymerization for Ultralow Volumes. Polym. Chem. 2017, 8, 5012–5022. 10.1039/C7PY00007C. [DOI] [Google Scholar]
- Kütahya C.; Schmitz C.; Strehmel V.; Yagci Y.; Strehmel B. Near-Infrared Sensitized Photoinduced Atom-Transfer Radical Polymerization (ATRP) with a Copper(II) Catalyst Concentration in the Ppm Range. Angew. Chem., Int. Ed. 2018, 57, 7898–7902. 10.1002/anie.201802964. [DOI] [PubMed] [Google Scholar]
- Aydogan C.; Ciftci M.; Kumbaraci V.; Talinli N.; Yagci Y. Hyperbranced Polymers by Photoinduced Self-Condensing Vinyl Polymerization Using Bisbenzodioxinone. Macromol. Chem. Phys. 2017, 218, 1700045 10.1002/macp.201700045. [DOI] [Google Scholar]
- Ciftci M.; Yoshikawa Y.; Yagci Y. Living Cationic Polymerization of Vinyl Ethers through a Photoinduced Radical Oxidation/Addition/Deactivation Sequence. Angew. Chem., Int. Ed. 2017, 56, 519–523. 10.1002/anie.201609357. [DOI] [PubMed] [Google Scholar]
- Kutahya C.; Aykac F. S.; Yilmaz G.; Yagci Y. LED and Visible Light-Induced Metal Free Atrp Using Reducible Dyes in the Presence of Amines. Polym. Chem. 2016, 7, 6094–6098. 10.1039/C6PY01417H. [DOI] [Google Scholar]
- Theriot J. C.; Lim C.; Yang H.; Ryan M. D.; Musgrave C. B.; Miyake G. M. Organocatalyzed Atom Transfer Radical Polymerization Driven by Visible Light. Science 2016, 352, 1082–1086. 10.1126/science.aaf3935. [DOI] [PubMed] [Google Scholar]
- Pearson R. M.; Lim C.; Mccarthy B. G.; Musgrave C. B.; Miyake G. M. Organocatalyzed Atom Transfer Radical Polymerization Using N-Aryl Phenoxazines as Photoredox Catalysts. J. Am. Chem. Soc. 2016, 138, 11399–11407. 10.1021/jacs.6b08068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan M. D.; Pearson R. M.; French T. A.; Miyake G. M. Impact of Light Intensity on Control in Photoinduced Organocatalyzed Atom Transfer Radical Polymerization. Macromolecules 2017, 50, 4616–4622. 10.1021/acs.macromol.7b00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Zhu Y.; Egap E. Semiconductor Quantum Dots as Photocatalysts for Controlled Light-Mediated Radical Polymerization. ACS Macro Lett. 2018, 7, 184–189. 10.1021/acsmacrolett.7b00968. [DOI] [PubMed] [Google Scholar]
- Fu Q.; Xie K.; Mckenzie T. G.; Qiao G. G. Trithiocarbonates as Intrinsic Photoredox Catalysts and Raft Agents for Oxygen Tolerant Controlled Radical Polymerization. Polym. Chem. 2017, 8, 1519–1526. 10.1039/C6PY01994C. [DOI] [Google Scholar]
- Chen M.; Macleod M. J.; Johnson J. A. Visible-Light-Controlled Living Radical Polymerization From a Trithiocarbonate Iniferter Mediated by an Organic Photoredox Catalyst. ACS Macro Lett. 2015, 4, 566–569. 10.1021/acsmacrolett.5b00241. [DOI] [PubMed] [Google Scholar]
- Chen M.; Johnson J. A. Improving Photo-Controlled Living Radical Polymerization from Trithiocarbonates through the Use of Continuous-Flow Techniques. Chem. Commun. 2015, 51, 6742–6745. 10.1039/C5CC01562F. [DOI] [PubMed] [Google Scholar]
- Anastasaki A.; Nikolaou V.; Nurumbetov G.; Truong N. P.; Pappas G. S.; Engelis N. G.; Quinn J. F.; Whittaker M. R.; Davis T. P.; Haddleton D. M. Synthesis of Well-Defined Poly(Acrylates) in Ionic Liquids Via Copper(II)-Mediated Photoinduced Living Radical Polymerization. Macromolecules 2015, 48, 5140–5147. 10.1021/acs.macromol.5b01192. [DOI] [Google Scholar]
- Anastasaki A.; Nikolaou V.; Brandford-Adams F.; Nurumbetov G.; Zhang Q.; Clarkson G. J.; Fox D. J.; Wilson P.; Kempe K.; Haddleton D. M. Photo-Induced Living Radical Polymerization of Acrylates Utilizing a Discrete Copper(II)-Formate Complex. Chem. Commun. 2015, 51, 5626–5629. 10.1039/C4CC09916H. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Ye G.; Wang Z.; Lu Y.; Chen J.; Matyjaszewski K. Heteroatom-Doped Carbon Dots (Cds) as a Class of Metal-Free Photocatalysts for PET-RAFT Polymerization Under Visible Light and Sunlight. Angew. Chem., Int. Ed. 2018, 57, 12037–12042. 10.1002/anie.201807385. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Ye G.; Lorandi F.; Liu Z.; Liu Y.; Hu T.; Chen J.; Lu Y.; Matyjaszewski K. Localized Surface Plasmon Resonance Meets Controlled/Living Radical Polymerization: An Adaptable Strategy for Broadband Light Regulated Macromolecular Synthesis. Angew. Chem., Int. Ed. 2019, 58, 12096–12101. 10.1002/anie.201906194. [DOI] [PubMed] [Google Scholar]
- Ong W.; Tan L.; Ng Y. H.; Yong S.; Chai S. Graphitic Carbon Nitride (g-C3N4)-Based Photocatalysts for Artificial Photosynthesis and Environmental Remediation: Are We a Step Closer to Achieving Sustainability?. Chem. Rev. 2016, 116, 7159–7329. 10.1021/acs.chemrev.6b00075. [DOI] [PubMed] [Google Scholar]
- Cao S.; Low J.; Yu J.; Jaroniec M. Polymeric Photocatalysts Based on Graphitic Carbon Nitride. Adv. Mater. 2015, 27, 2150–2176. 10.1002/adma.201500033. [DOI] [PubMed] [Google Scholar]
- Wang X.; Maeda K.; Thomas A.; Takanabe K.; Xin G.; Carlsson J. M.; Domen K.; Antonietti M. A Metal-Free Polymeric Photocatalyst for Hydrogen Production from Water Under Visible Light. Nat. Mater. 2009, 8, 76–80. 10.1038/nmat2317. [DOI] [PubMed] [Google Scholar]
- Kiskan B.; Zhang J.; Wang X.; Antonietti M.; Yagci Y. Mesoporous Graphitic Carbon Nitride as a Heterogeneous Visible Light Photoinitiator for Radical Polymerization. ACS Macro Lett. 2012, 1, 546–549. 10.1021/mz300116w. [DOI] [PubMed] [Google Scholar]
- Dadashi-Silab S.; Tasdelen M. A.; Kiskan B.; Wang X.; Antonietti M.; Yagci Y. Photochemically Mediated Atom Transfer Radical Polymerization Using Polymeric Semiconductor Mesoporous Graphitic Carbon Nitride. Macromol. Chem. Phys. 2014, 215, 675–681. 10.1002/macp.201400063. [DOI] [Google Scholar]
- Fu Q.; Ruan Q.; Mckenzie T. G.; Reyhani A.; Tang J.; Qiao G. G. Development of a Robust PET-RAFT Polymerization Using Graphitic Carbon Nitride (g-C3N4). Macromolecules 2017, 50, 7509–7516. 10.1021/acs.macromol.7b01651. [DOI] [Google Scholar]
- Ye C.; Li J.; Li Z.; Li X.; Fan X.; Zhang L.; Chen B.; Tung C.; Wu L. Enhanced Driving Force and Charge Separation Efficiency of Protonated g-C3N4 for Photocatalytic O2 Evolution. ACS Catal. 2015, 5, 6973–6979. 10.1021/acscatal.5b02185. [DOI] [Google Scholar]
- Zhang S.; Li J.; Wang X.; Huang Y.; Zeng M.; Xu J. Rationally Designed 1D Ag@AgVO3 Nanowire/Graphene/Protonated g-C3N4 Nanosheet Heterojunctions for Enhanced Photocatalysis Via Electrostatic Self-Assembly and Photochemical Reduction Methods. J. Mater. Chem. A 2015, 3, 10119–10126. 10.1039/C5TA00635J. [DOI] [Google Scholar]
- Chen L.; Song J. Tailored Graphitic Carbon Nitride Nanostructures: Synthesis, Modification, and Sensing Applications. Adv. Funct. Mater. 2017, 27, 1702695 10.1002/adfm.201702695. [DOI] [Google Scholar]
- Jiang L.; Yuan X.; Pan Y.; Liang J.; Zeng G.; Wu Z.; Wang H. Doping of Graphitic Carbon Nitride for Photocatalysis: A Reveiw. Appl. Catal., B 2017, 217, 388–406. 10.1016/j.apcatb.2017.06.003. [DOI] [Google Scholar]
- Han Q.; Wang B.; Zhao Y.; Hu C.; Qu L. A Graphitic-C3N4 “Seaweed” Architecture for Enhanced Hydrogen Evolution. Angew. Chem., Int. Ed. 2015, 54, 11433–11437. 10.1002/anie.201504985. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Lin L.; Ye X.; Guo F.; Wang X. Helical Graphitic Carbon Nitrides with Photocatalytic and Optical Activities. Angew. Chem., Int. Ed. 2014, 53, 11926–11930. 10.1002/anie.201407319. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Zhang M.; Yang C.; Wang X. Nanospherical Carbon Nitride Frameworks with Sharp Edges Accelerating Charge Collection and Separation at a Soft Photocatalytic Interface. Adv. Mater. 2014, 26, 4121–4126. 10.1002/adma.201400573. [DOI] [PubMed] [Google Scholar]
- Niu P.; Yin L.; Yang Y.; Liu G.; Cheng H. Increasing the Visible Light Absorption of Graphitic Carbon Nitride (Melon) Photocatalysts by Homogeneous Self-Modification with Nitrogen Vacancies. Adv. Mater. 2014, 26, 8046–8052. 10.1002/adma.201404057. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Zhang J.; Zhang M.; Wang X. Polycondensation of Thiourea into Carbon Nitride Semiconductors as Visible Light Photocatalysts. J. Mater. Chem. 2012, 22, 8083. 10.1039/c2jm00097k. [DOI] [Google Scholar]
- Zhang J.; Sun J.; Maeda K.; Domen K.; Liu P.; Antonietti M.; Fu X.; Wang X. Sulfur-Mediated Synthesis of Carbon Nitride: Band-Gap Engineering and Improved Functions for Photocatalysis. Energy Environ. Sci. 2011, 4, 675–678. 10.1039/C0EE00418A. [DOI] [Google Scholar]
- Martin D. J.; Qiu K.; Shevlin S. A.; Handoko A. D.; Chen X.; Guo Z.; Tang J. Highly Efficient Photocatalytic H2 Evolution From Water Using Visible Light and Structure-Controlled Graphitic Carbon Nitride. Angew. Chem., Int. Ed. 2014, 53, 9240–9245. 10.1002/anie.201403375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Yu J. C.; Shen Z.; Chan D. K. L.; Gu T. G-C3N4 Quantum Dots: Direct Synthesis, Upconversion Properties and Photocatalytic Application. Chem. Commun. 2014, 50, 10148–10150. 10.1039/C4CC02543A. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Chen X.; Takanabe K.; Maeda K.; Domen K.; Epping J. D.; Fu X.; Antonietti M.; Wang X. Synthesis of a Carbon Nitride Structure for Visible-Light Catalysis by Copolymerization. Angew. Chem., Int. Ed. 2010, 49, 441–444. 10.1002/anie.200903886. [DOI] [PubMed] [Google Scholar]
- Thomas A.; Fischer A.; Goettmann F.; Antonietti M.; Müller J.; Schlögl R.; Carlsson J. M. Graphitic Carbon Nitride Materials: Variation of Structure and Morphology and their Use as Metal-Free Catalysts. J. Mater. Chem. 2008, 18, 4893. 10.1039/b800274f. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.