

Abstract
Glioblastoma (GBM), characterized by high morbidity and mortality, is one of the most common lethal diseases worldwide. To identify the molecular mechanisms that contribute to the development of GBM, three cohort profile datasets (GSE50161, GSE90598 and GSE104291) were integrated and thoroughly analyzed; these datasets included 57 GBM cases and 22 cases of normal brain tissue. The current study identified differentially expressed genes (DEGs), and analyzed potential candidate genes and pathways. Additionally, a DEGs-associated protein-protein interaction (PPI) network was established for further investigation. Then, the hub genes associated with prognosis were identified using a Kaplan-Meier analysis based on The Cancer Genome Atlas database. Firstly, the current study identified 378 consistent DEGs (240 upregulated and 138 downregulated). Secondly, a cluster analysis of the DEGs was performed based on functions of the DEGs and signaling pathways were analyzed using the enrichment analysis tool on DAVID. Thirdly, 245 DEGs were identified using PPI network analysis. Among them, two co-expression modules comprising of 30 and 27 genes, respectively, and 35 hub genes were identified using Cytoscape MCODE. Finally, Kaplan-Meier analysis of the hub genes revealed that the increased expression of calcium-binding protein 1 (CABP1) was negatively associated with relapse-free survival. To summarize, all enriched Gene Ontology terms and Kyoto Encyclopedia of Genes and Genomes pathways may participate in mechanisms underlying GBM occurrence and progression, however further studies are required. CABP1 may be a key gene associated with the biological process of GBM development and may be involved in a crucial mechanism of GBM progression.
Keywords: glioblastoma, bioinformatical analysis, differentially expressed genes, calcium-binding protein 1
Introduction
Glioblastoma (GBM), also known as glioblastoma multiforme, comprising 30–40% of all brain tumors, is one of the most common primary brain tumors with high morbidity and mortality, and it is characterized by a high recurrence rate, invasiveness and a low cure rate (1,2). This is due to GBMs rapid growth rate, substantial molecular heterogeneity, high propensity to infiltrate vital brain structures and the regenerative capacity of treatment-resistant cancer stem cells. GBM is also the most lethal subtype of glioma, with a median survival of <2 years despite surgical resection, radiation, and chemotherapy (3–6). GBM has different histopathological features, mutations and clinical courses in an age-dependent manner (7).
All current high-grade gliomas (HGGs), such as GBM, are incurable as current standard treatments are insufficient, including maximal surgical tumor resection, radiotherapy and chemotherapy (4,8–10). Molecular approaches that target angiogenesis and single-compound-targeted therapies have also been unsuccessful (11).
Although the detailed mechanism of GBM formation and progression has been extensively studied, the exact etiology of GBM is poorly understood (12–14). The occurrence and malignant progression of GBM are associated with a variety of factors, such as gene aberrations (15). Due to the high morbidity and mortality of GBM, it is crucial to understand its etiology and potential molecular mechanisms; it is also necessary to find novel molecular biomarkers with potential diagnostic, individualized therapeutic and prognostic value. The screening of differentially expressed genes (DEGs) can be performed by using gene chips, which can easily detect all the genes within a sample and gather information at a specific time point (16). In the past few decades, the use of high-throughput microarrays to study the molecular mechanisms of GBM and to reconstruct the gene regulatory network in medical biology has made substantial progress (17). Microarrays are very valuable for screening genes associated with the occurrence, progression and targeted therapy of GBM (6). Bioinformatics analyses that use microarrays in screening have revealed that these genes are closely associated with cell signaling, cell metabolism, cytoskeleton, immunity, cell cycle and apoptosis (18,19). However, due to tissue heterogeneity in independent studies or the inherent shortcomings of the microarray technique, including small sample size, measurement error and information insufficiency, the results are always inconsistent (17,20). Therefore, unveiling the specific molecular mechanism underlying GBM remains a major challenge, although large numbers of DEGs have been identified between GBM and normal brain tissues by using microarray analysis (21). Integrated bioinformatics methods hypothesized to be capable of solving the aforementioned problems combined with expression profiling techniques have been carried out to identify the mechanisms underlying GBM (22,23). Kunkle et al (22) applied a comprehensive bioinformatics method using a genetic variation (small-scale variations and copy number variations) and environmental data integration that links with glioblastoma to distinguish genes that may be influenced by environmental exposures and associated with the development of GBM. Using this bioinformatics method, they identified 173 aberrantly expressed environmentally responsive genes that may be important to the pathogenesis of GBM. Additionally, through integrated bioinformatics methods, Li et al (23) discovered a series of gene pairs whose relationships were reversed in the progression from normal to GBM, i.e. from positive to negative correlation or vice versa, including cyclin-dependent kinase 2 and neuroblastoma RAS, fibroblast growth factor receptor and cyclin D1. DEGs were first identified from the GES4290 gene expression profile using Student t test. The relevant metabolic pathways including the cell cycle and mitogen-activated protein kinases were then extracted from the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database (https://www.kegg.jp/). A total of 432 cancer genes were subsequently obtained from the database of Cancer Gene Census (http://www.sanger.ac.uk/genetics/CGP/Census/), before the gene pairs were analyzed between DEGs and cancer genes.
In the present study, three microarray datasets (GSE50161, GSE90598 and GSE104291) were downloaded. In total, there are 57 GBM and 22 normal brain tissue datasets available. A data processing standard was used to filter the DEGs on the Morpheus website, followed by Gene Ontology (GO) and pathway enrichment analyses using Database for Annotation, Visualization and Integrated Discovery (DAVID) software. The DEGs protein-protein interaction (PPI) network and modular analysis were integrated using Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) software to identify hub genes in GBMs.
Materials and methods
Identification of DEGs and microarray data information
GBM and non-cancerous brain tissue gene expression profiles from GSE50161, GSE90598 and GSE10429 were obtained from the NCBI-Gene Expression Omnibus (NCBI-GEO; http://www.ncbi.nlm.nih.gov/geo) database, which is a publicly accessible database of next-generation sequencing and microarray/gene profiles. The microarray data in GSE50161 were based on GPL570 microarray platforms (Affymetrix Human Genome U133 Plus 2.0 Array; Affymetrix; Thermo Fisher Scientific, Inc.) and included 37 American GBM and 13 normal brain tissues (submission date, 23 Aug 2013) (24). The GSE90598 data were based on GPL17692 microarray platforms (Affymetrix Human Gene 2.1 ST Array; Affymetrix; Thermo Fisher Scientific, Inc.) and included 16 Turkish GBM and 7 normal brain tissues (submission date, 28 Nov 2016) (25). The GSE104291 data was based on GPL570 microarray platforms and included 4 Swiss GBM and 2 normal brain tissues (submission date, 26 Sep 2017) (26,27). These three datasets were chosen due to their representation of three different racial populations. This project was performed with the permission of the Institutional Review Board of Tongji University (Shanghai, China).
High-throughput functional genomic expression data from the three GSE datasets were first integrated into GEO2R for further analysis (28). Files in .TXT format, which represents the analysis results from GEO2R, were subsequently generated by the GEO2R tool. The Venn diagrams were produced using Bioinformatics and Evolutionary Genomics software (http://bioinformatics.psb.ugent.be/webtools/Venn) and the Morpheus Website (https://software.broadinstitute.org/morpheus) to process the .TXT format data. DEGs were determined by comparing their expression levels in GBM and normal brain tissues. DEGs were identified using unpaired t-test and P<0.01 was considered to indicate a statistically significant DEGs and [log Fold Change]>1 was set as the cut-off criteria using the GEO2R tool (28). GEO2R is a dataset analysis tool based on R programming language. This tool can perform ANOVA or t-tests, both of which can be applied to compare two sets of data samples under the same experimental conditions to determine differentially expressed microRNAs or genes (28).
GO and pathway enrichment analysis
DEGs GO analysis, candidate DEGs functions and pathway enrichment were analyzed using the DAVID tool (Version 6.8; http://david.ncifcrf.gov/) with P<0.05 set as the cut-off criterion (17,29–33).
Integration of the PPI network
The online database STRING (http://string-db.org) (34) was used to construct a PPI network of the proteins encoded by DEGs. Then, Cytoscape software (Version 3.7.1, National Institute of General Medical Sciences) (35) was utilized to perform protein interaction association network analysis and analyze the interaction correlation of the candidate proteins encoded by the DEGs in GBM. Next, the CentiScaPe plugin (Version 2.2) for Cytoscape was applied to calculate node degree (the number of connections to the hub in the PPI network) (36). Finally, the MCODE module (Version 1.5.1) for Cytoscape was used to collect the significant modules in the PPI network complex (20).
Survival analysis of hub genes
The online database OncoLnc (http://www.oncolnc.org/), which can link The Cancer Genome Atlas (TCGA) survival data to mRNA, miRNA or lncRNA expression levels, was employed to explore the prognostic value of the hub genes (37). Kaplan-Meier survival curves were plotted using OncoLnc. Patients with GBM were sub-classified into low- and high-expression groups according to the median expression of each hub gene. Relapse-free survival (RFS) was used for the survival endpoints. For the log-rank test, P<0.05 was considered to indicated to be statistically significant difference.
Results
Identification of DEGs in GBMs
The GSE50161, GSE90598 and GSE104291 microarray data were obtained from NCBI-GEO. Using the aforementioned cut-off criteria, 4,482, 1,355 and 1,034 DEGs were extracted from GSE50161, GSE90598 and GSE104291, respectively. Following integrated bioinformatics analysis, a total of 378 DEGs were documented (Fig. 1), including 240 and 138 genes up- and downregulated in GBM tissues compared with normal brain tissues (Table I).
Figure 1.
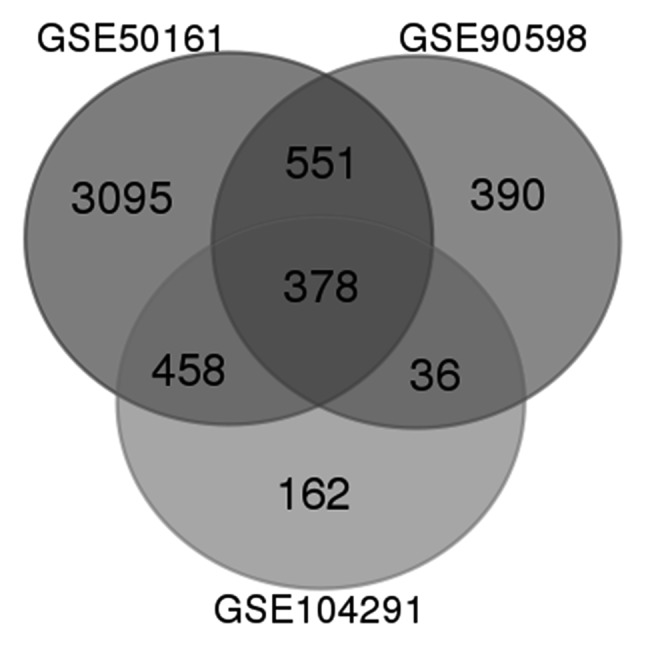
Identification of DEGs in glioblastomas. A total of 378 commonly altered DEGs were identified from three datasets (GSE50161, GSE90598 and GSE104291) using GEO2R tool. The ‘cross areas’ are common DEGs. A classical t-test was used to identify DEGs; the cut-off criteria were P<0.05 and [log Fold Change]>1. DEGs, differentially expressed genes.
Table I.
A total of 378 DEGs identified from three profile datasets, including 240 upregulated genes and 138 downregulated genes in the glioblastoma tissues compared with normal brain tissues.
| DEGs | Gene names |
|---|---|
| Upregulated | RALYL GLT1D1 STOX1 GNAL HCN1 GOT1 UNC5A RAB40B BSN PRKCB CA11 KCNA1 SRRM4 CARNS1 ZNF385B CABP1 SLC6A15 RBFOX1 RAB3A CLCN4 CRHBP NEBL HLF IGSF8 SLC39A12 IDS MFSD4A FRMPD4 ATP2B2 RIMBP2 MAP7 C1orf115 OTUD7A RAB11FIP4 NEGR1 MAP1A REPS2 CD200 GAD2 HPCAL4 SST GRM1 ERC2 TRHDE RASGEF1A RIMS1 PHF24 GRIN2A SHANK3 IL1RAPL1 KIF5C GABRA1 UBE2QL1 CACNA2D2 SYN2 GRM3 NEUROD2 KIAA0513 KIAA1644 GNG3 SLC8A2 PPFIA2 PIP4K2A MYT1L LINGO2 IPCEF1 NRG3 OSBPL1A NRGN UNC13A KCNJ3 MOBP NEURL1 NTSR2 CPLX2 FBXL16 RAPGEF5 KCNV1 DGKE KIRREL3 GABRA5 NRXN3 STXBP1 HOOK1 PPP1R1A KCNQ3 ATRNL1 TPPP ATP8A2 ATP2B3 SLIT2 OLFM3 STMN2 ADAM11 SYT4 MAP3K9 YPEL2 SLC6A17 PPIP5K1 HECW1 VAMP2 ROGDI RBFOX3 SYNGR3 GJB6 CACNA1E CNTN4 CHST1 RAP1GAP2 VAMP1 FRRS1L CREG2 CEP170B CPLX1 SYN1 MPPED1 ADCY5 MAL CACNA1B SLC7A14 OLFM1 GABRG1 EPB41L1 KIAA1549L PCLO SV2B FABP3 KCNJ9 SNAP25 ANKS1B SPOCK3 JAKMIP1 SH3GL3 RIMS3 FGFR2 GABRB2 RGS7 NPM2 SVOP ITPR1 KIF5A PKP4 CNTNAP4 PAK3 PPP2R2C RUNDC3B WBSCR17 MAP7D2 CCK DOK6 CSMD1 CDKL2 SLC17A7 S100A1 BRINP1 CELF3 RXFP1 SLC35F3 SULT4A1 KHDRBS2 RAPGEF4 PDE1A OPALIN GPR26 DNAJC6 SRCIN1 ETNPPL SYT1 EDIL3 CNNM1 DLG2 CYP4X1 PACSIN1 TBC1D30 STXBP6 CACNG8 MTURN NELL1 PLCL1 C11orf87 TBR1 ASIC2 CAMK2A FAIM2 GRM5 CACNG3 DDN ST8SIA3 NECAB1 ATP8A1 IQSEC1 LRRC7 LINC00889 CDK5R1 RYR2 FEZF2 NIPAL3 CAMK1D DYNC1I1 ARHGAP44 SLC12A5 RUSC2 PAIP2B NKAIN2 PPP1R16B KIAA0319 CHD5 MYRIP ACSL6 PITPNM3 KCNB1 UNC13C NPTX1 RIMS2 CAMKK2 KSR2 ANK3 STMN4 VIPR1 DOCK3 CHN1 SYT9 RUNDC3A JPH3 CELF4 MAL2 TUBB4A LANCL1 HHATL WASF1 DLG4 MEG3 PHYHIP AK5 SNCA CYP46A1 MIAT CBLN2 ZC3H12B KRT222 |
| Downregulated | CD44 MMP2 ABCB7 HNMT MCUB STK17A BCHE TRAM1 PHLDA1 GNG12 FCGBP ANXA2P2 FN1 PRRX1 IFI16 SYTL4 KIF4A CD93 TNFRSF19 CDH11 TIMP1 CLIC1 TWSG1 WEE1 CKS2 TRIM24 ECT2 CCDC102B FAM111A ZPR1 NAMPT SPRY1 TRIM4 GBE1 PLSCR1 RBBP8 F2R GUSB LSM8 STXBP4 CDK2 TIMELESS COL4A1 VCAN LAP3 COLGALT1 TGFB1I1 KDELR2 HLA-C TIMP4 ELK3 SERPINA3 ETV1 PARP9 LOC154761 LBR IQGAP2 ANXA1 C1R MSN KDELC2 FAM129A PTPN12 GPX7 PTPRZ1 TMEM45A APOL6 PLP2 DCBLD2 STEAP3 ABCA1 DTX3L DYNLT1 TAP1 JAG1 SMC4 CROT ZFP36L2 EMP1 ZC3HAV1 SLC43A3 LIMA1 GAS2L3 PLA2G4A VIM LGALS3BP SAMD9L PCNA CRISPLD1 TGFBR1 GBP2 NEDD1 CMTM6 RP2 ARAP3 MAP3K1 PRRC1 LAMC1 ANXA5 SNX7 RNF213 PDPN PCDHB7 IKBIP TNC LOXL2 PYGL CFI NUP160 FAM46A CALU CKAP2 PCDHB16 RIT1 TMX1 BAZ1A GBP3 TSPAN6 DDX60L DESI2 DDX58 NES SEMA5A SAMD9 SPARC PDIA4 MTMR11 ANO6 IFI44 NUSAP1 CTSC GNAI3 ARHGAP18 TCTN1 IDH1 MRC2 C21orf62 GBP1 |
Bold print indicates the 35 hub genes. DEGs, differentially expressed genes.
DEGs GO analysis in GBM
Following DEGs GO analysis using DAVID software, the DEGs were sub-classified into three functional groups including the cellular component group, the molecular function group and the biological process group (Fig. 2). The top 10 significantly enriched GO terms in each of the three groups were then determined (Fig. 2). In the biological process group, upregulated genes were involved in chemical synaptic transmission and nervous system development, and the downregulated genes were involved in cell adhesion, cell division, the positive regulation of gene expression and the innate immune response (Table II). In the molecular function group, the calcium ion binding GO term enriched both overexpressed and downregulated genes, which indicate that the molecular function of calcium binding may serve vital roles in the development of GBM. In addition, overexpressed genes were also involved in ATP binding, whilst downregulated genes were also involved in calcium and receptor binding (Table II). In the cellular component group, upregulated genes mainly included proteins integral to the plasma membrane and cell junction, while downregulated genes included those in the cytoplasm, extracellular exosomes and membrane (Table II). These results demonstrated that most of the DEGs were closely correlated with chemical synaptic transmission, protein binding, and plasma membrane. Significantly enriched GO terms containing the largest number of DEGs in GBM are listed in Table III. As shown in Fig. 3, in the biological process group, the most significant enriched GO term is chemical synaptic transmission. In the cellular component group, the most significant enriched GO term is cell junction. In the molecular function group, the most significant enriched GO term is syntaxin-1 binding.
Figure 2.

Top 10 GO terms of DEGs associated with glioblastomas. Top 10 GO terms of DEGs involved in (A) biological process (B) molecular function and (C) cellular component were obtained by GO analysis. GO, Gene Ontology; DEGs, differentially expressed genes.
Table II.
The top 3 significantly enriched GO terms in Glioblastoma stratified by different functional groups of GO analysis.
| Term | Description | Count | P-value |
|---|---|---|---|
| Upregulated | |||
| GO:0007268 (biological process) | Chemical synaptic transmission | 28 | 4.30×10−18 |
| GO:0007269 (biological process) | Nervous system development | 14 | 5.00×10−14 |
| GO:0007269 (biological process) | Synaptic vesicle exocytosis | 9 | 1.70×10−10 |
| GO:0005886 (cellular component) | Plasma membrane | 94 | 8.92×10−10 |
| GO:0030054 (cellular component) | Cell junction | 51 | 1.60×10−32 |
| GO:0005887 (cellular component) | Integral component of plasma membrane | 32 | 1.80×10−3 |
| GO:0048786 (cellular component) | Presynaptic active zone | 11 | 1.40×10−12 |
| GO:0005524 (molecular function) | ATP binding | 28 | 3.50×10−2 |
| GO:0005509 (molecular function) | Calcium ion binding | 24 | 3.60×10−5 |
| GO:0005516 (molecular function) | Calmodulin binding | 13 | 4.90×10−6 |
| Downregulated | |||
| GO:0007155 (biological process) | Cell adhesion | 10 | 7.76×10−3 |
| GO:0051301 (biological process) | Cell division | 9 | 4.90×10−3 |
| GO:0010628 (biological process) | Positive regulation of gene expression | 8 | 3.60×10−3 |
| GO:0005737 (cellular component) | Cytoplasm | 52 | 8.35×10−3 |
| GO:0070062 (cellular component) | Extracellular exosome | 42 | 4.50×10−6 |
| GO:0016020 (cellular component) | Membrane | 26 | 4.50×10−6 |
| GO:0005515 (molecular function) | protein binding | 89 | 2.10×10−5 |
| GO:0005509 (molecular function) | Calcium ion binding | 15 | 8.60×10−4 |
| GO:0005102 (molecular function) | Receptor binding | 8 | 1.60×10−2 |
GO, Gene Ontology.
Table III.
Significantly enriched GO terms that contain the largest number of differentially expressed genes in glioblastoma.
| Term | Description | Count | P-value |
|---|---|---|---|
| A, Upregulated | |||
| GO:0007268 (biological process) | Chemical synaptic transmission | 28 | 4.30×10−18 |
| GO:0005524 (molecular function) | ATP binding | 28 | 3.50×10−2 |
| GO:0005886 (cellular component) | Plasma membrane | 94 | 8.92×10−10 |
| B, Downregulated | |||
| GO:0007155 (biological process) | Cell adhesion | 10 | 7.76×10−3 |
| GO:0005515 (molecular function) | Protein binding | 89 | 2.10×10−5 |
| GO:0005737 (cellular component) | Cytoplasm | 52 | 8.35×10−3 |
GO, Gene Ontology.
Figure 3.

Top 30 GO terms of DEGs associated with glioblastomas GBM based on their functions. GO, Gene Ontology.
Signaling pathway enrichment analysis
DAVID tools were used to perform DEGs functional and signaling pathway enrichment analyses. The DEGs including both upregulated and downregulated genes associated with glioblastoma were significantly enriched in signaling pathways involing retrograde endocannabinoid signaling, synaptic vesicle cycle, and dopaminergic synapse (Fig. 4). In particular, as shown in Table IV, the upregulated genes were mainly enriched in retrograde endocannabinoid signaling and calcium signaling pathways, whilst downregulated genes were mainly associated with pathways in cancer and the phosphatidylinositol 4,5-bisphosphate 3-kinase/RAC-α serine/threonine-protein kinase signaling pathway.
Figure 4.

Top 30 significantly enriched Kyoto Encyclopedia of Genes and Genomes pathways terms of DEGs associated with glioblastomas using Database for Annotation, Visualization and Integrated Discovery software.
Table IV.
Signaling pathway enrichment analysis of DEGs function in glioblastoma.
| Pathway | Name | Gene count | P-value | Genes |
|---|---|---|---|---|
| A, Upregulated DEG | ||||
| hsa04723 | Retrograde endocannabinoid signaling | 15 | 4.65×10−11 | GABRG1, GABRA1, GABRB2, ADCY5, GABRA5, RIMS1, GRM1, KCNJ3, ITPR1, PRKCB, GRM5, SLC17A7, KCNJ9, GNG3, CACNA1B |
| hsa04020 | Calcium signaling pathway | 14 | 6.51×10−7 | SLC8A2, GRIN2A, GRM1, ITPR1, PRKCB, GRM5, GNAL, ATP2B2, ATP2B3, PDE1A, RYR2, CACNA1E, CAMK2A, CACNA1B |
| hsa04728 | Dopaminergic synapse | 13 | 1.13×10−7 | GNAL, KCNJ9, KIF5A, ADCY5, KIF5C, GRIN2A, GNG3, CAMK2A, KCNJ3, PPP2R2C, ITPR1, PRKCB, CACNA1B |
| hsa04721 | Synaptic vesicle cycle | 12 | 4.98×10−1° | SLC17A7, RAB3A, SYT1, CPLX2, CPLX1, STXBP1, VAMP2, UNC13C, RIMS1, SNAP25, UNC13A, CACNA1B |
| hsa04724 | Glutamatergic synapse | 11 | 2.97×10−7 | SLC17A7, GRM5, GRM3, ADCY5, DLG4, GRIN2A, GNG3, GRM1, KCNJ3, ITPR1, SHANK3, PRKCB |
| hsa04921 | Oxytocin signaling pathway | 12 | 7.64×10−6 | KCNJ9, CACNG8, ADCY5, RYR2, CACNG3, CACNA2D2, CAMK2A, KCNJ3, ITPR1, PRKCB, CAMKK2, CAMK1D |
| hsa05032 | Morphine addiction | 11 | 3.13×10−7 | GABRG1, GABRA1, KCNJ9, GABRB2, ADCY5, PDE1A, GABRA5, GNG3, KCNJ3, PRKCB, CACNA1B |
| hsa04261 | Adrenergic signaling in cardiomyocytes | 11 | 2.35×10−5 | ATP2B2, ATP2B3, CACNG8, ADCY5, PPP1R1A, RYR2, CACNG3, RAPGEF4, CACNA2D2, CAMK2A, PPP2R2C |
| hsa04080 | Neuroactive ligand-receptor interaction | 11 | 3.89×10−3 | GRM5, GABRG1, GRM3, GABRA1, RXFP1, GABRB2, GABRA5, GRIN2A, VIPR1, NTSR2, GRM1 |
| B, Downregulated DEG | ||||
| hsa05200 | Pathways in cancer | 10 | 2.36×10−3 | GNAI3, COL4A1, TGFBR1, CKS2, LAMC1, GNG12, MMP2, CDK2, FN1, F2R |
| hsa04151 | PI3K-Akt signaling pathway | 7 | 4.36×10−2 | COL4A1, TNC, LAMC1, GNG12, CDK2, FN1, F2R |
| hsa05222 | Small cell lung cancer | 5 | 3.68×10−3 | COL4A1, CKS2, LAMC1, CDK2, FN1 |
| hsa04512 | Extracellular matrix-receptor interaction | 5 | 4.01×10−3 | COL4A1, CD44, TNC, LAMC1, FN1 |
| hsa05161 | Hepatitis B | 5 | 2.31×10−2 | DDX58, TGFBR1, MAP3K1, PCNA, CDK2 |
| hsa02010 | ATP-binding cassette transporters | 3 | 4.30×10−2 | TAP1, ABCA1, ABCB7 |
DEGs, differentially expressed genes; PI3K-Akt, phosphatidylinositol 4,5-bisphosphate 3-kinase/RAC-α serine/threonine-protein kinase.
Identification of key candidate genes and pathways using DEGs PPI network and module analysis
Using Cytoscape software and the STRING database (34,35), 245 DEGs, including 92 downregulated genes and 153 upregulated genes out of 378, were filtered into the DEGs PPI network complex containing 245 nodes and 741 edges (Fig. 5A). Another 133 genes out of the 378 commonly altered DEGs failed to fall into the DEGs PPI network. The identification of 35 hub genes among 245 nodes was successfully achieved by filtering nodes with >11 degrees (also known as interactions or connections; Table I). Additionally, the 10 most vital nodes with >11 degrees were DLG4, SYT1, SNAP251, VAMP2, CACNA1B, SYN1, GNG3, GNG12, CD44 and GNAI3. Based on the degree of importance, two significant modules in the PPI network complex were collected for further analysis using the MCODE plugin. GO and pathway enrichment analyses revealed that 30 nodes and 152 edges existed in Module 1 (Fig. 5B and Table V), which were mainly associated with neurotransmitter secretion, plasma membrane, syntaxin-1 binding and the synaptic vesicle cycle. In addition, Module 2 included 27 nodes and 86 edges (Fig. 5C and Table V), mainly associated with extracellular matrix (ECM) organization, cytoplasm, protein binding and ECM-receptor interaction.
Figure 5.

DEGs PPI network complex and modular analysis. (A) A total of 245 DEGs (153 upregulated genes and 92 downregulated genes) were filtered into the DEGs PPI network complex using the Search Tool for the Retrieval of Interacting Genes/Proteins database. (B) Module 1 was composed of 30 nodes and 152 edges. (C) Module 2 was composed of 27 nodes and 86 edges. Nodes in red signified upregulation and nodes in green signified downregulation. DEGs, differentially expressed genes; PPI, protein-protein interaction.
Table V.
GO and pathway enrichment analysis of Module 1 and Module 2 gene functions.
| Term | Description | Count | P-value |
|---|---|---|---|
| A, Module 1 | |||
| GO:0007269 (biological process) | Neurotransmitter secretion | 11 | 4.99×10−19 |
| GO:0005886 (cellular component) | Plasma membrane | 19 | 5.79×10−6 |
| GO:0017075 (molecular function) | Syntaxin-1 binding | 6 | 8.77×10−11 |
| hsa04721 (KEGG pathway) | Synaptic vesicle cycle | 10 | 3.74×10−14 |
| B, Module 2 | |||
| GO:0030198 (biological process) | ECM organization | 5 | 1.88×10−4 |
| GO:0005737 (cellular component) | Cytoplasm | 17 | 4.80×10−4 |
| GO:0005515 (molecular function) | Protein binding | 20 | 2.38×10−3 |
| hsa04512 (KEGG pathway) | ECM-receptor interaction | 3 | 6.60×10−3 |
GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; ECM, extracellular matrix.
Survival analysis of hub genes
Based on the OncoLnc database, it was demonstrated that high calcium-binding protein 1 (CABP1) expression was negatively associated with the RFS of patients with GBM (Fig. 6).
Figure 6.

High CABP1 expression negatively associated with RFS for GBM patients. The RFS of patients with GBM who have low- and high-expression of CABP1 was analyzed with a Kaplan-Meier plot. RFS, relapse-free survival; CABP1, calcium-binding protein 1; GBM, glioblastomas.
Validation of the DEGs in the TCGA dataset
To confirm the reliability of the identified DEGs from the 3 datasets, the TCGA GBM dataset was downloaded and analyzed using the same strategy used in the current study. A total of 195 of the 240 upregulated genes identified in the present study were also overexpressed in the TCGA GBM dataset, whilst 123 of the 138 downregulated genes identified in the current study were also downregulated in the TCGA GBM dataset (data not shown). The total consistency of the up- and downregulated genes was 84.13%, suggesting that the results of the identified candidate genes are reliable.
Discussion
In the current study, a total of 378 genes were identified, including 240 upregulated genes and 138 downregulated genes. Almost all the DEGs obtained in the training set were verified by the validation set. The GO biological process analysis revealed that the upregulated DEGs were significantly associated with chemical synaptic transmission and nervous system development, while downregulated DEGs were significantly associated with cell adhesion, cell division, positive regulation of gene expression and the innate immune response. The onset and progression of GBM are associated with a complex network of cellular components, molecular functions and biological processes (38,39). For chemical synaptic transmission, available data demonstrated that neurotransmitters can influence the proliferation, quiescence and differentiation status of central nervous system-resident cells (40). Furthermore, the disruption of cell adhesion and the cell cycle were demonstrated to often be involved in the development of GBM (41). A study also revealed that the expression of pro-inflammatory and suppressive cytokines associated with the innate immune response changed significantly (42).
Among the DEGs, two co-expression modules comprising 30 and 27 genes, respectively, and 35 hub genes were identified using Cytoscape MCODE. Module 1 was associated with neurotransmitter secretion, plasma membrane, syntaxin-1 binding and the synaptic vesicle cycle, while Module 2 was associated with ECM organization, cytoplasm, protein binding and ECM-receptor interaction. Following identification of the hub genes, Kaplan-Meier analysis of hub genes revealed that increased expression of CABP1 was negatively associated with RFS. Therefore, CABP1 may be a key gene, and its associated biological processes may be a crucial mechanism of GBM progression. Additionally, all enriched GO terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways may participate in mechanisms underlying GBM occurrence and progression, thus further study is required.
CABP1 is a protein-coding gene located on chromosome 12q24.31 (43). The protein encoded by this gene can regulate the gating of L-type Ca2+ channels by inhibiting calcium-dependent inactivation via the voltage-dependent L-type calcium channel subunit-α1C to control neurotransmitter release, gene expression, muscle contraction, apoptosis, and disease processes (44). In addition, CABP1 can regulate the calcium-dependent activity of inositol 1,4,5-triphosphate receptors, P/Q-type voltage-gated calcium channels and the transient receptor potential channel, short transient receptor potential channel 5 (43). A study identified that CABP1 is involved in drug refractory epilepsy due to mesial temporal sclerosis, providing important insight into the understanding of the genomic basis of the condition (45). CABP1 was observed to be significantly upregulated during P. berghei infection (46). One study also demonstrated the importance of CABP1 in modulating stimulus-secretion coupling in excitable cells, and the ability of CABP1 to inhibit Ca2+ currents and exocytosis in bovine chromaffin cells (47). Thus, it is speculated that CABP1 works as a Ca2+-dependent regulator in GBM and may help to predict the prognosis of patients with GBM. However, CABP1 has rarely been studied in the field of oncology. Certain studies have demonstrated that aberrant CABP1 expression was associated with adenoma risk, particularly for multiple/advanced adenomas (44,48,49). In addition, Zhang et al (50) previously reported that CABP1 is upregulated in glioblastoma tissues compared with normal brain tissues. Using Cox regression analysis and log-rank test, CABP1 was identified to be one of the top six DEGs associated with GBM prognosis, where high CABP1 expression is negatively correlated with overall patient survival. However, the molecular underlying the role of CABP1 in the development and progression of GBM remains unknown. To the best of our knowledge, no previous study has investigated CABP1 in GBM.
In the present study, three datasets generated from Turkish, Swiss and American patients were used to represent three subsets of worldwide populations. Certain genes and small non-coding RNA molecules exhibit abnormal expression patterns in GBM (6). However, genes and biomarkers from different races vary widely (51), which may account for the low consistency of the DEGs from the three datasets.
To conclude, the current study identified a total of 378 DEGs. Among them, two co-expression modules and 35 hub genes were identified. The enriched GO terms and KEGG pathways may be closely associated with GBM occurrence. CABP1 may be a key gene associated with the prognosis of GBM. Altogether, these data introduced CABP1 as a good candidate for experimental studies into GBM. However, clinical experiments are urgently needed to evaluate the molecular role of CABP1 in GBM progression, as well as its specificity and sensitivity as a biomarker of GBM. Therefore, the molecular mechanisms and clinical applications of these genes and pathways require exploration in future studies.
Acknowledgements
The authors would like to thank Dr Sajan Pandey, Department of Neurosurgery, Shanghai Tenth People's Hospital, Tongji University School of Medicine (Shanghai, China) for his technical help and writing assistance.
Glossary
Abbreviations
- GBM
glioblastoma
- DEGs
differentially expressed genes
- PPI
protein-protein interaction network
- HGGs
high-grade gliomas
- DIPG
diffuse intrinsic pontine glioma
- GO
Gene Ontology
- RFS
relapse-free survival
Funding
The current study was supported by a grant given to Professor Liang Gao from the Health and Family Planning Commission (grant no. 03.02.16.001).
Availability of data and material
The datasets generated and/or analyzed during the current study are available in the NCBI-Gene Expression Omnibus (NCBI-GEO; http://www.ncbi.nlm.nih.gov/geo) database.
Authors' contributions
DC and LG conceived and designed the study. LL, XM, XD, TJ, PH, RW and HQ collected the data and performed data analysis. DC, XL and LL interpreted the result. LL and XL were major contributors in writing the manuscript. LG and XL revised the paper. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The local ethics committee of Shanghai Tenth People's Hospital provided the ethical approval for the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Wen WS, Hu SL, Ai Z, Mou L, Lu JM, Li S. Methylated of genes behaving as potential biomarkers in evaluating malignant degree of glioblastoma. J Cell Physiol. 2017;232:3622–3630. doi: 10.1002/jcp.25831. [DOI] [PubMed] [Google Scholar]
- 2.Yano S, Miwa S, Kishimoto H, Toneri M, Hiroshima Y, Yamamoto M, Bouvet M, Urata Y, Tazawa H, Kagawa S, et al. Experimental curative fluorescence-guided surgery of highly invasive glioblastoma multiforme selectively labeled with a killer-reporter adenovirus. Mol Ther. 2015;23:1182–1188. doi: 10.1038/mt.2015.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, Toms S, Idbaih A, Ahluwalia MS, Fink K, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA. 2017;318:2306–2316. doi: 10.1001/jama.2017.18718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 5.Ostrom QT, Bauchet L, Davis FG, Deltour I, Fisher JL, Langer CE, Pekmezci M, Schwartzbaum JA, Turner MC, Walsh KM, et al. The epidemiology of glioma in adults: A ‘state of the science’ review. Neuro Oncol. 2014;16:896–913. doi: 10.1093/neuonc/nou087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeng T, Li L, Zhou Y, Gao L. Exploring long noncoding RNAs in glioblastoma: Regulatory mechanisms and clinical potentials. Int J Genomics. 2018;2018:2895958. doi: 10.1155/2018/2895958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Filbin MG, Suvà ML. Gliomas genomics and epigenomics: Arriving at the start and knowing it for the first time. Annu Rev Pathol. 2016;11:497–521. doi: 10.1146/annurev-pathol-012615-044208. [DOI] [PubMed] [Google Scholar]
- 8.Stupp R, Hegi ME, Gilbert MR, Chakravarti A. Chemoradiotherapy in malignant glioma: Standard of care and future directions. J Clin Oncol. 2007;25:4127–4136. doi: 10.1200/JCO.2007.11.8554. [DOI] [PubMed] [Google Scholar]
- 9.Delgado-López PD, Corrales-García EM. Survival in glioblastoma: A review on the impact of treatment modalities. Clin Transl Oncol. 2016;18:1062–1071. doi: 10.1007/s12094-016-1497-x. [DOI] [PubMed] [Google Scholar]
- 10.Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15:422–442. doi: 10.1038/s41571-018-0003-5. [DOI] [PubMed] [Google Scholar]
- 11.Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370:709–722. doi: 10.1056/NEJMoa1308345. [DOI] [PubMed] [Google Scholar]
- 12.Furnari FB, Cloughesy TF, Cavenee WK, Mischel PS. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat Rev Cancer. 2015;15:302–310. doi: 10.1038/nrc3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ludwig K, Kornblum HI. Molecular markers in glioma. J Neurooncol. 2017;134:505–512. doi: 10.1007/s11060-017-2379-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan Y, Xu Z, Li Z, Sun L, Gong Z. An insight into the increasing role of LncRNAs in the pathogenesis of gliomas. Front Mol Neurosci. 2017;10:53. doi: 10.3389/fnmol.2017.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sturm D, Bender S, Jones DT, Lichter P, Grill J, Becher O, Hawkins C, Majewski J, Jones C, Costello JF, et al. Paediatric and adult glioblastoma: Multiform (epi)genomic culprits emerge. Nat Rev Cancer. 2014;14:92–107. doi: 10.1038/nrc3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long H, Liang C, Zhang X, Fang L, Wang G, Qi S, Huo H, Song Y. Prediction and analysis of key genes in glioblastoma based on bioinformatics. Biomed Res Int. 2017;2017:7653101. doi: 10.1155/2017/7653101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang SW, Ali ND, Zhong L, Shi J. MicroRNAs as biomarkers for human glioblastoma: Progress and potential. Acta Pharmacol Sin. 2018;39:1405–1413. doi: 10.1038/aps.2017.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan SY, Sandanaraj E, Tang C, Ang BT. Biobanking: An important resource for precision medicine in glioblastoma. Adv Exp Med Biol. 2016;951:47–56. doi: 10.1007/978-3-319-45457-3_4. [DOI] [PubMed] [Google Scholar]
- 20.Guo Y, Bao Y, Ma M, Yang W. Identification of key candidate genes and pathways in colorectal cancer by integrated bioinformatical analysis. Int J Mol Sci. 2017;18:E722. doi: 10.3390/ijms18040722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanwar MK, Gilbert MR, Holland EC. Gene expression microarray analysis reveals YKL-40 to be a potential serum marker for malignant character in human glioma. Cancer Res. 2002;62:4364–4368. [PubMed] [Google Scholar]
- 22.Kunkle B, Yoo C, Roy D. Discovering gene-environment interactions in glioblastoma through a comprehensive data integration bioinformatics method. Neurotoxicology. 2013;35:1–14. doi: 10.1016/j.neuro.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Li W, Li K, Zhao L, Zou H. Bioinformatics analysis reveals disturbance mechanism of MAPK signaling pathway and cell cycle in Glioblastoma multiforme. Gene. 2014;547:346–350. doi: 10.1016/j.gene.2014.06.042. [DOI] [PubMed] [Google Scholar]
- 24.Griesinger AM, Birks DK, Donson AM, Amani V, Hoffman LM, Waziri A, Wang M, Handler MH, Foreman NK. Characterization of distinct immunophenotypes across pediatric brain tumor types. J Immunol. 2013;191:4880–4888. doi: 10.4049/jimmunol.1301966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gulluoglu S, Tuysuz EC, Sahin M, Kuskucu A, Kaan Yaltirik C, Ture U, Kucukkaraduman B, Akbar MW, Gure AO, Bayrak OF, Dalan AB. Simultaneous miRNA and mRNA transcriptome profiling of glioblastoma samples reveals a novel set of OncomiR candidates and their target genes. Brain Res. 2018;1700:199–210. doi: 10.1016/j.brainres.2018.08.035. [DOI] [PubMed] [Google Scholar]
- 26.Bady P, Diserens AC, Castella V, Kalt S, Heinimann K, Hamou MF, Delorenzi M, Hegi ME. DNA fingerprinting of glioma cell lines and considerations on similarity measurements. Neuro Oncol. 2012;14:701–711. doi: 10.1093/neuonc/nos072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sciuscio D, Diserens AC, van Dommelen K, Martinet D, Jones G, Janzer RC, Pollo C, Hamou MF, Kaina B, Stupp R, et al. Extent and patterns of MGMT promoter methylation in glioblastoma- and respective glioblastoma-derived spheres. Clin Cancer Res. 2011;17:255–266. doi: 10.1158/1078-0432.CCR-10-1931. [DOI] [PubMed] [Google Scholar]
- 28.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. NCBI GEO: Archive for functional genomics data sets-update. Nucleic Acids Res. 2013;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He WQ, Gu JW, Li CY, Kuang YQ, Kong B, Cheng L, Zhang JH, Cheng JM, Ma Y. The PPI network and clusters analysis in glioblastoma. Eur Rev Med Pharmacol Sci. 2015;19:4784–4790. [PubMed] [Google Scholar]
- 30.Li Y, Min W, Li M, Han G, Dai D, Zhang L, Chen X, Wang X, Zhang Y, Yue Z, Liu J. Identification of hub genes and regulatory factors of glioblastoma multiforme subgroups by RNA-seq data analysis. Int J Mol Med. 2016;38:1170–1178. doi: 10.3892/ijmm.2016.2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei B, Wang L, Du C, Hu G, Wang L, Jin Y, Kong D. Identification of differentially expressed genes regulated by transcription factors in glioblastomas by bioinformatics analysis. Mol Med Rep. 2015;11:2548–2554. doi: 10.3892/mmr.2014.3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 33.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen WJ, Tang RX, He RQ, Li DY, Liang L, Zeng JH, Hu XH, Ma J, Li SK, Chen G. Clinical roles of the aberrantly expressed lncRNAs in lung squamous cell carcinoma: A study based on RNA-sequencing and microarray data mining. Oncotarget. 2017;8:61282–61304. doi: 10.18632/oncotarget.18058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scardoni G, Petterlini M, Laudanna C. Analyzing biological network parameters with CentiScaPe. Bioinformatics. 2009;25:2857–2859. doi: 10.1093/bioinformatics/btp517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anaya J. OncoLnc: Linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. Peer J Computer Sci. 2016;2:e67. doi: 10.7717/peerj-cs.67. [DOI] [Google Scholar]
- 38.Iser IC, Pereira MB, Lenz G, Wink MR. The epithelial- to-mesenchymal transition-like process in glioblastoma: An updated systematic review and in silico investigation. Med Res Rev. 2017;37:271–313. doi: 10.1002/med.21408. [DOI] [PubMed] [Google Scholar]
- 39.Alshabi AM, Vastrad B, Shaikh IA, Vastrad C. Identification of crucial candidate genes and pathways in glioblastoma multiform by bioinformatics analysis. Biomolecules. 2019;9:E201. doi: 10.3390/biom9050201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caragher SP, Hall RR, Ahsan R, Ahmed AU. Monoamines in glioblastoma: Complex biology with therapeutic potential. Neuro Oncol. 2018;20:1014–1025. doi: 10.1093/neuonc/nox210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng YC, Tsai WC, Sung YC, Chang HH, Chen Y. Interference with PSMB4 expression exerts an anti-tumor effect by decreasing the invasion and proliferation of human glioblastoma cells. Cell Physiol Biochem. 2018;45:819–831. doi: 10.1159/000487174. [DOI] [PubMed] [Google Scholar]
- 42.Turkowski K, Brandenburg S, Mueller A, Kremenetskaia I, Bungert AD, Blank A, Felsenstein M, Vajkoczy P. VEGF as a modulator of the innate immune response in glioblastoma. Glia. 2018;66:161–174. doi: 10.1002/glia.23234. [DOI] [PubMed] [Google Scholar]
- 43.Li C, Chan J, Haeseleer F, Mikoshiba K, Palczewski K, Ikura M, Ames JB. Structural insights into Ca2+-dependent regulation of inositol 1,4,5-trisphosphate receptors by CaBP1. J Biol Chem. 2009;284:2472–2481. doi: 10.1074/jbc.M806513200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oz S, Tsemakhovich V, Christel CJ, Lee A, Dascal N. CaBP1 regulates voltage-dependent inactivation and activation of Ca(V)1.2 (L-type) calcium channels. J Biol Chem. 2011;286:13945–13953. doi: 10.1074/jbc.M110.198424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dixit AB, Banerjee J, Srivastava A, Tripathi M, Sarkar C, Kakkar A, Jain M, Chandra PS. RNA-seq analysis of hippocampal tissues reveals novel candidate genes for drug refractory epilepsy in patients with MTLE-HS. Genomics. 2016;107:178–188. doi: 10.1016/j.ygeno.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 46.Mubaraki MA, Hafiz TA, Al-Quraishy S, Dkhil MA. Oxidative stress and genes regulation of cerebral malaria upon Zizyphus spina-christi treatment in a murine model. Microb Pathog. 2017;107:69–74. doi: 10.1016/j.micpath.2017.03.017. [DOI] [PubMed] [Google Scholar]
- 47.Chen ML, Chen YC, Peng IW, Kang RL, Wu MP, Cheng PW, Shih PY, Lu LL, Yang CC, Pan CY. Ca2+ binding protein-1 inhibits Ca2+ currents and exocytosis in bovine chromaffin cells. J Biomed Sci. 2008;15:169–181. doi: 10.1007/s11373-007-9217-8. [DOI] [PubMed] [Google Scholar]
- 48.Zhao J, Zhu X, Shrubsole MJ, Ness RM, Hibler EA, Cai Q, Long J, Chen Z, Jiang M, Kabagambe EK, et al. Interactions between calcium intake and polymorphisms in genes essential for calcium reabsorption and risk of colorectal neoplasia in a two-phase study. Mol Carcinog. 2017;56:2258–2266. doi: 10.1002/mc.22678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun P, Shrubsole MJ, Ness RM, Cai Q, Long J, Edwards T, Chen Z, Zhu X, Deng X, Luo J, et al. Calcium intake, CABP1 polymorphisms, and the risk of colorectal adenoma: Results from Tennessee Colorectal Polyp Study. Cancer Res. 2011;71(Suppl 8):S3763. [Google Scholar]
- 50.Zhang Y, Xu J, Zhu X. A 63 signature genes prediction system is effective for glioblastoma prognosis. Int J Mol Med. 2018;41:2070–2078. doi: 10.3892/ijmm.2018.3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Delfino KR, Serão NV, Southey BR, Rodriguez-Zas SL. Therapy-, gender- and race-specific microRNA markers, target genes and networks related to glioblastoma recurrence and survival. Cancer Genomics Proteomics. 2011;8:173–183. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and/or analyzed during the current study are available in the NCBI-Gene Expression Omnibus (NCBI-GEO; http://www.ncbi.nlm.nih.gov/geo) database.