

Abstract
The neuropeptide FF (NPFF) system has been implicated in a number of physiological processes including modulating the pharmacological activity of opioid analgesics and several other classes of drugs of abuse. In this study, we report the discovery of a novel proline scaffold with antagonistic activity at the NPFF receptors through a high throughput screening campaign using a functional calcium mobilization assay. Focused structure-activity relationship studies on the initial hit 1 have resulted in several analogs with calcium mobilization potencies in the submicromolar range and modest selectivity for the NPFF1 receptor. Affinities and potencies of these compounds were confirmed in radioligand binding and functional cAMP assays. Two compounds 16 and 33 had good solubility and blood-brain barrier permeability that fall within the range of CNS permeant candidates without the liability of being a P-glycoprotein substrate. Finally, both compounds reversed fentanyl-induced hyperalgesia in rats when administered intraperitoneally. Together, these results point to the potential of these proline analogs as promising NPFF receptor antagonists.
Keywords: proline, neuropeptide FF, antagonist, structure-activity relationship
Graphical Abstract
Introduction
Neuropeptide FF (NPFF, FLFQPQRF-amide) belongs to a family of neuropeptides called RFamide peptides, members of which all contain an Arg-Phe-NH2 (RF-amide) motif at the C terminus.1–3 NPFF is an endogenous peptide that binds to and activates two G protein-coupled receptors (GPCRs), NPFF1 and NPFF2. These receptors are members of the rhodopsin family and predominantly couple to Gαi/o proteins. Originally isolated from bovine brain,4 NPFF and its receptors have recently been identified in the central nervous system (CNS) of various animal species. Ligand binding studies performed on rodents confirmed that both subtypes are widely expressed in the brain, whereas only NPFF2 receptors are expressed in the spine at detectable levels.5–7 The NPFF system has been implicated in the regulation of a variety of physiological processes, such as insulin release, food intake, memory, blood pressure, electrolyte balance, and neural regeneration.8, 9 Most interestingly, the NPFF system has also been shown to play an important role in modulating the effects of opioids and several other classes of drugs of abuse.10–12
It is well documented that NPFF, having no affinity for the opioid receptors, is an important modulator of opioid receptor function. Several studies have shown that the effects of NPFF on opioid modulation are dependent on the route of administration. For example, intracerebroventricular (i.c.v.) administration of NPFF in rats attenuated morphine-induced analgesia and locomotion, and precipitated opioid withdrawal syndromes in morphine-dependent rats, whereas intrathecal (i.t.) administration produced opioid-induced analgesia and also prolonged morphine-induced analgesia.4, 13, 14 Ventricular injection of NPFF antiserum restored the analgesic response to morphine in morphine-tolerant rats but did not affect opiate-naïve rats.15 RF9, a dipeptide NPFF½ receptor antagonist, dose-dependently blocked the long-lasting hyperalgesia produced by either acute fentanyl or chronic morphine administration and prevented the development of associated tolerance.16, 17 This effect appears to be mainly mediated by the NPFF1 receptor, as the selective NPFF1 antagonist AC-262620 also reduced opioid tolerance.18 Together, these results confirm the opioid-modulating properties of the NPFF system and suggest that co-administration of an NPFF antagonist with opioids may alleviate hyperalgesia and tolerance, two main side effects associated with chronic opioid use.
The finding that NPFF precipitates a nicotine withdrawal syndrome also suggests that NPFF participates in the processes of dependence and addiction to other drugs.19 Chronic administration of NPFF into the lateral ventricle potentiated the behavioral sensitization to amphetamine.20 More recently, it was demonstrated that stimulation of NPFF receptors decreased the expression of amphetamine-induced condition-placed preference, while inhibition of NPFF receptors decreased amphetamine withdrawal anxiety.21 Moreover, it was suggested that NPFF is involved in the mechanism of expression of sensitization to cocaine hyperlocomotion, although this effect could be non-specific.22 Consistent with these observations, there is evidence that NPFF binding sites are abundant in the ventral tegmental area (VTA), while NPFF-like immunoreactivity was detected in the nucleus accumbens (NAc), two brain regions belonging to the mesolimbic dopamine DA projections which are known to be involved in drug addiction.23 Together, these findings suggest that the NPFF system is a viable target for the treatment of drug addiction.
In addition to NPFF, several other neuropeptides from the RFamide family activate one or both NPFF receptors, including NPSF, NPAF and NPVF. A number of peptidomimetic NPFF ligands have also been reported, which retained the guanidine functional group (Figure 1), including the putative NPFF½ antagonist RF9.24 In these ligands, acylation of the N-terminus of the last two amino acid residues (RF) have been reported to be critical for NPFF activities (e.g. BIBP3226 and RF9).5, 17 Subsequent modification of RF9 led to the discovery of the potent NPFF1 selective dipeptide biphenyl-RF and peptidomimetic RF313.25–27 These peptides or peptidomimetics were effective in preventing fentanyl-induced hyperalgesia in rats by subcutaneous or oral administration, acting as antagonists (Figure 1).17, 25–27 However, other studies suggest that RF9 is not an NPFF receptor antagonist in some assays, as it did not reverse phosphorylation of MAPK/ERK½ and anorectic effects in fasting mice induced by [Tyr1]NPFF, but rather exhibited anorectic effects itself after i.c.v or subcutaneous administration.28
Figure 1.

Reported peptidic, peptidomimetics and small molecule NPFF ligands.
Several classes of non-peptidic NPFF ligands have recently been disclosed. Quinazolino-, quinolino-, pyrimidine-, and thiazole-guanidines were reported in several patents as NPFF ligands.29–33 A series of N-benzylpiperidines with mixed activities as agonists/antagonists at NPFF1 and antagonists at NPFF2 were recently reported.34 These series still contained the guanidine functionality, which is often associated with high plasma-protein binding and limited BBB penetration.35 Despite this, some of these guanidine-containing ligands have been reported to enter the CNS, although to a relatively small extent.32 So far, only two series of small molecule NPFF ligands, fused imidazole-pyrazines and indole derivatives (Figure 1),24 which do not possess the guanidine functionality have been reported in patents; however, only limited biological data were presented on these compounds.36, 37 Together, these results underscore the importance of discovering novel NPFF ligands with improved pharmacological profiles (agonist or antagonist) and properties, particularly small non-peptidic molecules that are not subject to peptidolytic degradation and thus are more favorable tools to explore the biological roles of the NPFF receptors.
In an effort to develop novel small molecule NPFF ligands, we performed a high throughput screen (HTS) of a GPCR-oriented compound library. Proline 1 emerged as a promising hit with moderate activities at the two NPFF subtypes with reasonable physiochemical properties (Figure 2). We have thus developed a synthetic route for this scaffold and prepared a focused library of proline analogs to explore the structure-activity relationships (SARs) at the carboxamide region, some of which displayed improved potency at the NPFF receptors when characterized in the functional calcium mobilization assays. Several active compounds were further evaluated for their effects in modulating cellular levels of cyclic adenosine monophosphate (cAMP) and their binding affinities to the two NPFF receptors. The drug-like properties such as solubility and blood-brain barrier permeability were then examined. Finally, the effects of these compounds in reversing fentanyl-induced hyperalgesia were investigated. Herein, we describe the discovery and characterization of these proline-based novel NPFF antagonists.
Figure 2.
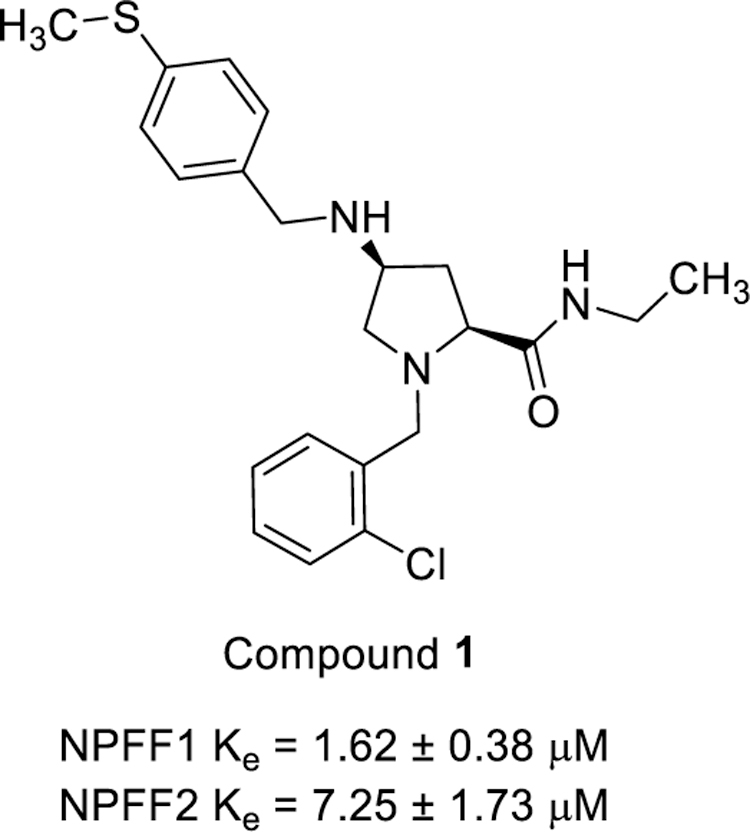
Hit compound 1 in calcium mobilization assays
Results and Discussion
NPFF Calcium Mobilization Assay Development.
Traditionally, NPFF activity has been examined using assays such as radioligand binding, GTP-γ-S or cAMP assays. To establish a platform allowing for low-cost HTS, we developed functional calcium mobilization assays using CHO cells simultaneously over-expressing the promiscuous Gα16 protein and either human NPFF1 or NPFF2 receptors. The NPFF1 and NPFF2 stable cell lines were created by transfecting the expression plasmids into RD-HGA16 CHO cells (Molecular Devices), selecting for positive clones using antibiotic resistance, and testing for functional expression of NPFF1 and 2 following procedures previously disclosed by our group for other receptor systems.38–40 Clones were first screened against 10 μM NPFF to identify clones that had functional NPFF receptors. Eight clones with the highest maximal NPFF response were further evaluated with NPFF concentration-response curves; the clone with the most potent and efficacious NPFF response was chosen as the working clone. Figure 3 shows the data (N=3) obtained when our stable NPFF1 and NPFF2 cell lines are treated with NPFF in 96-well format with a 1% DMSO final concentration. In the NPFF1-RD-HGA16 cells, NPFF has an EC50 value of 64 ± 1 nM and the signal window is 17,900 ± 1,300 relative fluorescent units (RFUs). In the NPFF2-RD-HGA16 cells, NPFF has an EC50 value of 49 ± 3 nM and the signal window is 9,100 ± 230 RFUs. These NPFF EC50 values are consistent with results from cAMP and GTP-γ-S assays.18, 41, 42 In parental RD-HGA-16 CHO cells, there was no response to NPFF, confirming its signaling through NPFF receptors. Both NPFF functional assays tolerated DMSO up to a final concentration of 1%.
Figure 3.

NPFF EC50 in functional calcium mobilization assays. (A) NPFF response in stable NPFF1-RD-HGA16 cells (●) and parental RD-HGA16 CHO cells (○). (B) NPFF response in stable NPFF2-RD-HGA16 cells (■) and parental RD-HGA16 CHO cells (○). Representative data from one experiment is shown and each data point is the mean ± SD of duplicate determinations.
HTS of 22,000 Diverse Compound Libary.
The NPFF1 FLIPR-based calcium mobilization assay was used to screen our 22,000 ChemBridge Premium Diversity Library, which is a subset of both the ChemBridge EXPRESS-PICK and CORE library stock collections. The library is composed of highly diverse scaffolds designed to provide lead compounds for new targets as well as enable in-house scaffold hopping. The content and properties of the diversity set were critically appraised for screening against GPCRs based on a number of factors including maximum diversity, optimal ADME parameters, structural novelty (minimal overlap with scaffolds found in known GPCR ligands and drugs), and pharmacophoric compliance with characteristics prototypical of GPCR ligands.
Because the NPFF1 assay was originally developed in 96-well format, we miniaturized the assay to 384-well format and determined the cell density that provided a confluent cell monolayer and NPFF potency/efficacy consistent with the 96-well assay. We then tested the calcium mobilization assay using our automated liquid handling equipment which includes a Biotek cell dispenser, Beckman Coulter Biomek NX, and FLIPR Tetra to determine whether automation resulted in adverse effects; no changes in assay performance were observed.43 Using the automated and miniaturized assay, we generated a Z’-factor by testing positive (10 μM final NPFF) and negative controls (1% DMSO/assay buffer) on three individual days.44 The Z’-factor (N=3) was determined to be 0.75 with a standard deviation of 0.06; a Z’-factor greater than 0.5 is considered appropriate for HTS.
Library compounds were screened at 10 μM final concentration for both agonist and antagonist activities as part of a 3-addition protocol our laboratory developed. This protocol allows for evaluation of both modes of activity with a single assay plate thereby reducing the overall time and cost of the screen. Concentration-response curves were run with 37 cherry picked antagonists (27 with >65% inhibition and 10 with > 85% inhibition) resulting in the confirmed activities of these compounds including proline 1.
Chemistry.
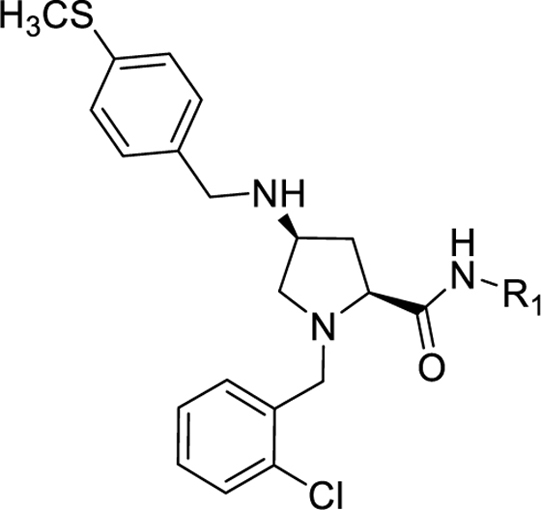
Compounds 1, 10 - 47 were synthesized following procedures depicted in Scheme 1. trans-4-Hydroxy-L-proline methyl ester (2) underwent reductive amination by sodium triacetoxyborohydride in 1,2-dichloroethane to give the intermediate 3 which was then converted to a tosylated derivative 4 in the presence of pyridine in dichloromethane. Substitution of the tosyl group with sodium azide provided azide 5, which subsequently underwent Staudinger reduction with triphenylphosphine to give amine 6. A second reductive amination with either 4-methoxybenzaldehyde or 4-(methylthio)benzaldehyde provided intermediates 7a and 7b, respectively. This three-step conversion from the tosylate 4 to amines 7a-b gave higher yields (overall three-step yield 32%) than the direct displacement with 4-methoxybenzylamine or 4-(methylthio)benzylamine (<10%).45 While the low yields of this direct conversion could be slightly improved by the employment of a large excess of the amines under elevated reaction temperatures in a polar solvent, the unreacted amines greatly complicated the purification of the product. Furthermore, the use of excess amines is not amenable to scale-up synthesis of this common intermediate. The secondary amines 7a-b were then protected with a Boc group and the resulting intermediates 8a-b were subsequently hydrolyzed under basic conditions to give acids 9a-b. HBTU-assisted amide coupling between 9a-b and corresponding amines followed by cleavage of the Boc protective group provided the final products (1, 10 - 47). All target compounds were characterized by 1H and 13C NMR, HRMS, and HPLC.
Scheme 1.

Synthesis of compounds 10 – 47.
Reagents and conditions: (a) 2-chlorobenzaldehyde, Na(OAc)3BH, 1,2-DCE, rt, 24 h (b) TsCl, pyridine, DCM, rt, 24 h (c) NaN3, DMF, 70 oC, 16 h (d) PPh3, THF, H2O, reflux, 16 h (e) 4-MeOPhCHO or 4-MeSPhCHO, Na(OAc)3BH, 1,2-DCE, rt, 24 h (f) Boc2O, aq. Na2CO3, 1,4-dioxane, rt, 16 h (g) aq. LiOH, MeOH, rt, 16 h (h) corresponding amine, HBTU, DIEA, DMF, rt, 24 h (i) TFA, DCM, rt, 1 h.
Pharmacological Evaluations.
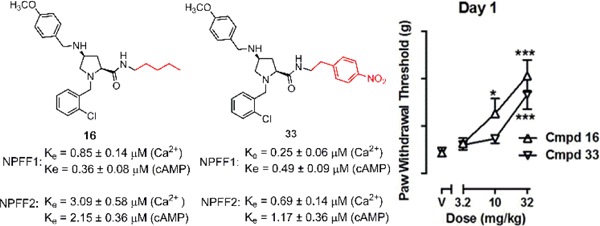
All the synthesized compounds were first characterized in NPFF1 and NPFF2 calcium mobilization assays for their ability to antagonize NPFF stimulated calcium influx. Since the NPFF receptors natively couple to Gαi/o proteins and inhibit adenylate cyclase, we chose active compounds with Ke values of ≤ 1 μM in NPFF1 or NPFF2 calcium assays, and further evaluated them in PerkinElmer’s Lance cAMP assays. Figure 4 displays the concentration-response curve of a representative compound (16) in the NPFF1 and NPFF2 calcium mobilization assays. The concentration-response curve of compounds 16 and 33 in NPFF1 and NPFF2 cAMP assays are shown in Figure 5. In the cAMP assay, the NPFF + 16 curve had a Hill slope of 1.3 in both NPFF1 and FF2 cells while the NPFF + 33 curve had a Hill slope of 1.4 and 1 in NPFF1 and FF2 cells, respectively, thus indicating that the activity of these lead compounds is not due to the formation of aggregates.46, 47 These results correlate with those from the calcium mobilization assays where the NPFF + 16 curve had a Hill slope of 1.7 and 1.1 in NPFF1 and FF2 cells, respectively, and the NPFF + 33 curve had a Hill slope of 1.4 in both cell lines.
Figure 4.

Antagonist activity of compound 16 in NPFF1 (A) and NPFF2 (B) calcium mobilization functional Ke assays. (A) Concentration-response curves of NPFF alone (○) and NPFF + 5 μM final 16 (□) in stable NPFF1-RD-HGA16 cells. (B) Concentration-response curves of NPFF alone (○) and NPFF + 10 μM final 16 (□) in stable NPFF2-RD-HGA16 cells. The right shift of the NPFF curve in the presence of test compound was used to calculate Ke values as described in the Methods. Representative data from one experiment are shown and each data point is mean ± SD of duplicate determinations.
Figure 5.

Antagonist activity of compounds 16 and 33 in NPFF1 (A) and FF2 (B) cAMP functional Ke assays. (A) Concentration-response curves of NPFF alone (○), NPFF + 4 μM final 16 (□), and NPFF + 2 μM final 33 (◊) in stable NPFF1-CHO cells. (B) Concentration-response curves of NPFF alone (○), NPFF + 10 μM final 16 (□), and NPFF + 10 μM final 33 (◊) in stable NPFF2-CHO cells. The right shift of the NPFF curve in the presence of test compound was used to calculcate Ke values as described in the Methods. Each data point is mean ± SEM of at least N=3 conducted in duplicate.
In both functional assays, the apparent dissociation constant Ke of the test compounds was calculated from compound-mediated inhibition of NPFF activity, where the EC50 curves of the agonist NPFF were obtained alone and together with the test compound and the right-shift of the agonist curve was measured, as previously described by our group.48, 49 The selected compounds were also characterized in radioligand binding assays to measure affinity and confirm that the proline scaffold is a bona fide template as an NPFF ligand. All compounds were also tested for agonist activity using the calcium mobilization assay; none showed any significant agonist activity at either the NPFF1 or NPFF2 receptor (< 20% of NPFF Emax at 10 µM final concentration).
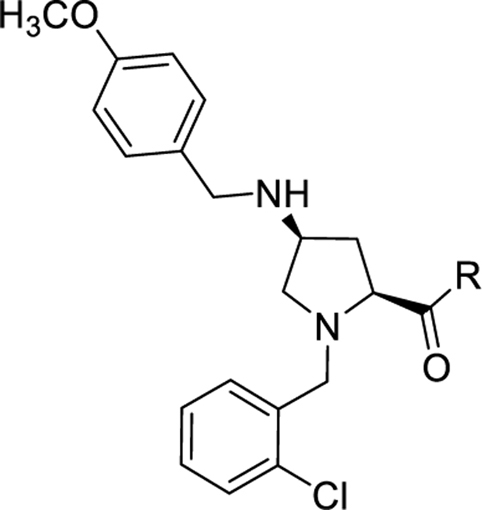
Analogs with a 4-methoxybenzyl group at the 4-position of the proline core, instead of the 4-(methylthio)benzyl group in the hit compound 1, were synthesized in the SAR studies at the carboxamide region because the methoxy moiety is known to have better metabolic stability. The corresponding starting material, 4-methoxybenzaldehyde is also more readily available compared to the methylthio counterpart. As can be seen from Table 1, the NPFF1 antagonist activity of this series was sensitive to the length of the side chain R of the carboxamide functionality. Methyl and ethyl analogs (10 and 11) were inactive at both NPFF receptors. The antagonist activities at the NPFF1 receptor increased from n-propyl to n-pentyl (12, 13, 16), then decreased slightly with n-hexyl (18) and was completely abolished with n-decyl chain (19). Among the three butyl isomers (13-15), the NPFF1 antagonist activity slightly decreased in the order of n-butyl, s-butyl and t-butyl. In contrast, the NPFF2 antagonist activity increased significantly in the same order, indicating that a linear chain is preferred for NPFF1 selectivity. A similar trend was observed with n-pentyl and i-pentyl isomers (16 and 17). Between these two isomers, n-pentyl is the more potent and selective NPFF1 antagonist (16, NPFF1 Ke = 0.72 µM, NPFF2 Ke = 3.09 µM). Compounds 20-21 have aliphatic side chains with a bulkier moiety at the terminal end. The cyclohexyl analog 21 (NPFF1 Ke = 1.12 µM) is slightly more potent than the more hydrophilic morpholinyl counterpart 20 (NPFF1 Ke = 1.96 µM). The flexible N,N-diethylaminoethyl side chain (22) was inactive at both receptors. These results suggest that introduction of a heteroatom is not well tolerated in this region. Most of the aliphatic analogs did not show significant activities at the NPFF2 receptor except the t-Bu (15), i-pentyl (17), n-hexyl (18), and (cyclohexyl)ethyl (21) analogs which displayed modest antagonist activities in the micromolar range.
Table 1.
SAR at the carboxamide region.
 | |||||
|---|---|---|---|---|---|
| Compound | R | Ca2+ | cAMP | ||
| NPFF1 Ke (μM)a | NPFF2 Ke (μM)a | NPFF1 Ke (μM)a | NPFF2 Ke (μM)a | ||
| 10 | NHMe | >10b | >10b | N.D. | N.D. |
| 11 | NHEt | >10b | >10b | N.D. | N.D. |
| 12 | NH(n-Pr) | 2.60 ± 0.15 | >10b | N.D. | N.D. |
| 13 | NH(n-Bu) | 1.24 ± 0.14 | d | N.D. | N.D. |
| 14 | NH(s-Bu) | 1.50 ± 0.06 | 3.68 ± 0.35b | N.D. | N.D. |
| 15 | NH(t-Bu) | 1.88 ± 0.21 | 1.26 ± 0.8c | N.D. | N.D. |
| 16 | NH(n-Pentyl) | 0.72 ± 0.01 | 3.09 ± 0.58b | 0.36 ± 0.08 | 2.15 ± 0.36 |
| 17 | NH(i-Pentyl) | 1.32 ± 0.09 | 1.54 ± 0.13c | N.D. | N.D. |
| 18 | NH(n-hexyl) | 0.82 ± 0.08 | 1.49 ± 0.20c | 1.98 ± 0.28 | 2.48 ± 0.27 |
| 19 | NH(n-decyl) | >10b,c | d | N.D. | N.D. |
| 20 | 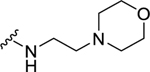 |
1.96 ± 0.07 | d | N.D. | N.D. |
| 21 | 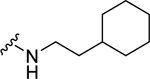 |
1.12 ± 0.23 | 1.22 ± 0.36c | N.D. | N.D. |
| 22 | 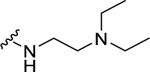 |
>10b | d | N.D. | N.D. |
| 23 | 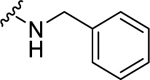 |
0.78 ± 0.08c | 2.01 ± 0.44c | 1.72 ± 0.32 | 3.90 ± 1.1 |
| 24 | 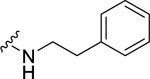 |
0.85 ± 0.14 | 2.34 ± 0.31c | 0.77 ± 0.12 | 3.79 ± 0.45 |
| 25 | 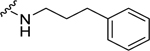 |
> 10f | 2.60 ± 0.28c | N.D. | N.D. |
| 26 | 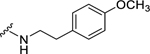 |
0.67 ± 0.06 | 1.75 ± 0.11c | 0.57 ± 0.15c | 2.56 ± 0.20 |
| 27 | 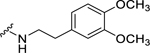 |
1.03 ± 0.05 | d | 1.52 ± 0.11 | > 10b |
| 28 | 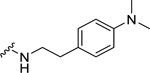 |
1.20 ± 0.25 | 2.08 ± 0.38c | N.D. | N.D. |
| 29 | 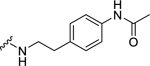 |
2.51 ± 0.08 | >10b | N.D. | N.D. |
| 30 | 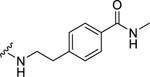 |
>10b | >10b | N.D. | N.D. |
| 31 | 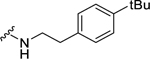 |
e | 2.42 ± 0.26c | N.D. | N.D. |
| 32 | 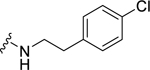 |
0.99 ± 0.22c | 1.83 ± 0.09c | 1.49 ± 0.10 | 2.53 ± 0.36 |
| 33 | 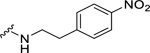 |
0.25 ± 0.06c | 0.69 ± 0.14c | 0.49 ± 0.09 | 1.17 ± 0.36 |
| 34 | 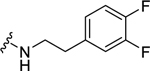 |
0.61 ± 0.13 | 3.49 ± 1.4b,c | 0.57 ± 0.17 | 2.16 ± 0.30 |
| 35 | 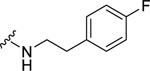 |
1.13 ± 0.11 | >10b | 1.41 ± 0.18 | >10b |
| 36 | 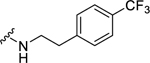 |
1.67 ± 0.22c | d | N.D. | N.D. |
| 37 | 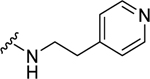 |
1.82 ± 0.20 | d | N.D. | N.D. |
| 38 | 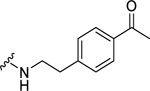 |
>10b | >10b | N.D. | N.D. |
| 39 | 1.88 ± 0.25 | d | N.D. | N.D. | |
| 40 | NEt2 | 2.90 ± 0.15 | 4.93 ± 0.02b | N.D. | N.D. |
| 41 | N(n-Pr)2 | 0.88 ± 0.12c | 1.73 ± 0.27c | 2.31 ± 0.42 | >10b |
| 42 | 1.33 ± 0.29c | 2.08 ± 0.30c | N.D. | N.D. | |
Values are the mean ± SEM of at least three independent experiments in duplicate
Values are the mean ± SEM of two independent experiments in duplicate
Pre-incubation of antagonist and test compound was 45 min or 1 hr
Compound was inactive in antagonist screen at 10 µM final (N=2).
Compound appeared cytotoxic in the assay and potency was not determined.
Value is IC50 because compound was noncompetitive in Ke assay. N.D., Not determined.
When the cyclohexyl (21) was replaced by a phenyl group (24), the NPFF1 activity was slightly improved while retaining selectivity over the NPFF2 receptor. The shorter benzyl group (23, NPFF1 Ke = 0.78 µM) has equivalent NPFF1 antagonist activity to the phenethyl analog (24, NPFF1 Ke = 0.85 µM). However, lengthening the side by another methylene group as in phenylpropyl (25) completely abolished the activity at the NPFF1 receptor. While displaying similar antagonist activity at the NPFF1 receptor, the phenethyl analog seemed to have better selectivity at the NPFF1 receptor over the NPFF2 receptor. As 24 emerged as a potent, selective NPFF1 antagonist, it is warranted to study the substituent effect on the phenyl ring of compound 24.
Among electron-donating substituents at the para position of the phenyl ring (26-31), 4-methoxy (26, NPFF1 Ke = 0.67 µM) was the most potent NPFF1 antagonist. 3,4-Dimethoxy (27), and 4-dimethylamino (28) were slightly less potent. Bulky groups such as 4-acetamido (29), 4-(methylamino)carbonyl (30), and t-butyl (31) were not well tolerated, in agreement with the previous observation that there is limited space at the binding pocket. Turning to electron-withdrawing substituents, 4-nitro (33, NPFF1 Ke = 0.25 µM) demonstrated the best NPFF1 antagonist potency among all proline analogs. Our results indicate that strong electron-withdrawing groups are favored for good NPFF1 antagonist activity as 3,4-difluoro (34, NPFF1 Ke = 0.61 µM) was more potent than 4-chloro (32, NPFF1 Ke = 0.99 µM), 4-fluoro (35, NPFF1 Ke = 1.13 µM) and 4-trifluoromethyl (36, NPFF1 Ke = 1.67 µM). 4-Pyridinyl (37) which is commonly used as an isosteric replacement of 4-nitrophenyl,50 only displayed moderate NPFF1 activity (Ke = 1.82 µM). Finally, the NPFF1 antagonist activity of the two bulky electron-withdrawing groups, acetyl (38) and 4-methylsulfonyl (39) was significantly dampened compared to the 4-nitro analog. These data imply that small, strong electron-withdrawing substituents were preferred whereas bulky groups proved to be deleterious for NPFF1 antagonist activity. Similar to the aliphatic series, these phenethyl analogs were not potent at the NPFF2 receptor except 4-methoxy (26), diethylamino (28), 4-chloro (32), 4-nitro (33), and 3,4-difluoro (34).
Next, the effect of disubstituted amides at this region was also investigated. Diethylamino and dipropylamino analogs (40, NPFF1 Ke = 2.90 µM and 41, NPFF1 Ke = 0.88 µM) were more potent at the NPFF1 receptor compared to their monosubstituted amide counterparts (11, NPFF1 Ke >10 µM and 12, NPFF1 Ke = 2.60 µM). Compound 42 (NPFF1 Ke = 1.33 µM) with a rigid spacer between the phenyl ring and the amide was less active at the NPFF1 receptor compared to 24 (NPFF1 Ke = 0.85 µM) with a flexible ethylene linker.
Since the initial hit 1 has a 4-methylthio at the 4-benzyl group on the proline scaffold, after exploring various substituents at the carboxamide region with a 4-(4-methoxybenzyl) substitution of the proline scaffold, we selected several of the more potent 4-methoxybenzyl analogs and examined the effects of the 4-methylthio substitution (Table 2). The pentyl analog at the carboxamide of the (methylthio)benzyl series was a slightly more potent NPFF1 antagonist than its methoxy counterpart in the calcium mobilization assay (43 Ke = 0.36 µM vs. 16 Ke = 0.72 µM). Similarly, phenethyl (44) and 3,4-difluorophenethyl (47) analogs demonstrated slightly better NPFF1 activities in the thioether series than their methoxy counterparts (44 Ke = 0.53 µM vs. 24 Ke = 0.85 µM; 47 Ke = 0.47 µM vs. 34 Ke = 0.61 µM). On the other hand, the 4-methoxy (45) and 4-nitro (46) analogs were similar in potency as their methoxy equivalents (45 Ke = 0.87 µM vs. 26 Ke = 0.67 µM, 46 Ke = 0.37 µM vs. 33 Ke = 0.25 µM). At the NPFF2 receptor, this series appeared to be more active and thus, less selective for the NPFF1 receptor compared the 4-(4-methoxybenzyl)amino analogs except for 45 and 46.
Table 2.
SAR at the carboxamide region of the 4-(4-methylthio)benzylamino series.
 | |||||
|---|---|---|---|---|---|
| Ca2+ | cAMP | ||||
| Compound | R1 | NPFF1 Ke (μM)a | NPFF2 Ke (μM)a | NPFF1 Ke (μM)a | NPFF2 Ke (μM)a |
| 43 | n-Pentyl | 0.36 ± 0.05 | 0.97 ± 0.03c | 1.07 ± 0.20 | 2.74 ± 0.21 |
| 44 |  |
0.53 ± 0.08 | 3.70 ± 0.94 | 1.30 ± 0.14 | >10b |
| 45 |  |
0.87 ± 0.10 | 2.30 ± 0.28c | 1.15 ± 0.12 | >10b |
| 46 |  |
0.37 ± 0.07c | 1.35 ± 0.20 | 0.89 ± 0.16 | 0.75 ± 0.12 |
| 47 |  |
0.47 ± 0.06 | 0.88 ± 0.16c | 1.07 ± 0.27 | >10b |
Values are the mean ± SEM of at least three independent experiments in duplicate
Values are the mean ± SEM of two independent experiments in duplicate
Pre-incubation of antagonist and test compound was 1 hr.
Throughout the course of these studies, most of the compounds we tested displayed competitive antagonism in the curve-shift assays; however, several compounds (23, 32, 33, 41, 42, 45) showed evidence of insurmountable antagonism by shifting the curve to the right and also depressing the maximal NPFF signal. While allosteric modulators commonly produce such a response, this type of antagonism can also be observed with competitive orthosteric antagonists with slow dissociation rates. Our group has previously reported that by performing the curve-shift assays with longer antagonist-receptor incubation periods, the system reaches equilibrium, and hence the compounds produce a typical competitive antagonist profile.51 Indeed, when we applied the longer incubations to the NPFF assays, the compounds displayed the typical competitive antagonist activity profile.
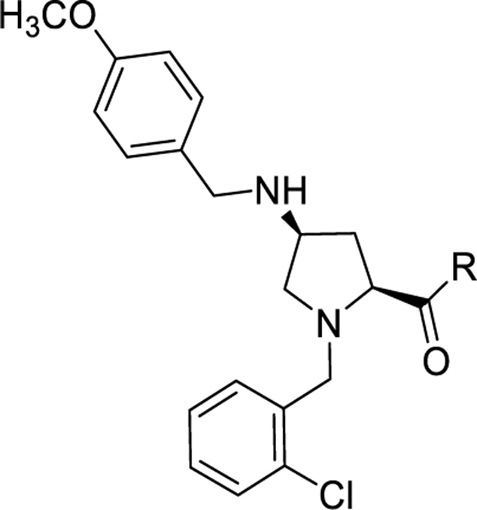
Several compounds were further characterized in radioligand binding and cAMP assays (Table 3). In general, the data obtained from all three assays are in good concordance with each other. Compounds that were potent antagonists in the calcium mobilization assays also showed good antagonist potency in the secondary cAMP assay that measures native G-protein coupling (Fig. 5). The binding assays confirm that all compounds bind to the NPFF receptors and that potent compounds (33 and 34) in the NPFF1 functional assays have potent binding affinities as well.
Table 3.
Results from the cAMP, radioligand binding and calcium mobilization assays of representative compounds
 | |||||||
|---|---|---|---|---|---|---|---|
| # | R1 | Radioligand bindinga | cAMP assayb | Calcium mobilization assayb | |||
| Ki NPFF1 (μM) | Ki NPFF2 (μM) | Ke NPFF1 (nM) | Ke NPFF2 (nM) | Ke NPFF1 (nM) | Ke NPFF2 (nM) | ||
| 16 | NH(n-Pentyl) | 0.89 ± 0.05 | 1.44 ± 0.13 | 0.36 ± 0.08 | 2.15 ± 0.36 | 0.72 ± 0.01 | 3.09 ± 0.58 |
| 24 | 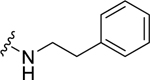 |
0.71 ± 0.02 | 1.23 ± 0.27 | 0.77 ± 0.12 | 3.79 ± 0.45 | 0.85 ± 0.14 | 2.34 ± 0.31 |
| 26 | 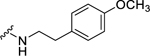 |
0.92 ± 0.03 | 1.46 ± 0.05 | 0.57 ± 0.15 | 2.56 ± 0.20 | 0.67 ± 0.06 | 1.75 ± 0.11 |
| 33 | 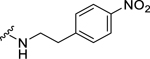 |
0.61 ± 0.03 | 1.67 ± 0.06 | 0.49 ± 0.09 | 1.17 ± 0.36 | 0.25 ± 0.06c | 0.69 ± 0.14 |
| 34 | 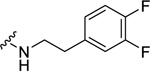 |
0.56 ± 0.04 | 1.49 ± 0.27 | 0.57 ± 0.17 | 2.16 ± 0.30 | 0.61 ± 0.13 | 3.49 ± 1.4 |
Values are the mean ± SEM of at least two independent experiments in duplicate
Values are the mean ± SEM of at least three independent experiments in duplicate.
Collectively, these results highlight the importance of the substituent size, a preference for lipophilicity, and some flexibility at the NPFF binding pocket around the carboxamide region. Several ligands with moderate activity against NPFF1 and no/weak NPFF2 activity were identified. The most potent NPFF1 compounds of the series were the pentyl (16) and 4-nitro (33) analog based on their potency and affinity in all three assays. These compounds were then further evaluated for the preliminary ADME properties and in vivo activities in a hyperalgesia model.
Physicochemical Properties and Preliminary ADME Studies.
The proline scaffold has favorable physicochemical properties with a balance between lipophilic and hydrophilic groups (Table 4, calculated in ACD Labs), which would normally lead to good solubility and blood-brain barrier (BBB) permeability required for successful CNS drugs.52, 53 Therefore, we tested two potent NPFF1 antagonists, 16 and 33, as free base forms in the kinetic solubility and bidirectional MDCK-MDR1 permeability assays. Both assays were performed by Paraza Pharma Inc (Montreal, Canada) according to their standard protocols.
Table 4.
Physicochemical and Preliminary ADME Properties of Compounds 16 and 33
| Desired value | 1 | 16 | 33 | |
|---|---|---|---|---|
| MW | < 500 | 418 | 444 | 523 |
| ClogP | 1 – 4 | 3.57 | 4.59 | 4.37 |
| PSA | < 70 | 69. 7 | 53.6 | 54.4 |
| pKa | < 8 | 8.6, 5.0 |
9.1, 4.8 |
9.1, 4.8 |
| HBD | < 3 | 2 | 2 | 2 |
| HBA | < 7 | 4 | 5 | 8 |
| Solubility (δM) | > 60 | N.D. | 146.8 ± 6.9 | 45.9 ± 7.7 |
| Papp (10−6 cm/sec) A-to-B |
> 2 | N.D. | 7.6 | 2.7 |
| Papp (10−6 cm/sec) B-to-A |
N.D. | 6.7 | 3.6 | |
| Efflux ratio | < 2.5 | N.D. | 0.9 | 1.3 |
HBD: H-bond donor; HBA: H-bond acceptor. N.D.: Not determined.
As seen from table 4, 16 was soluble in aqueous solutions, displaying a kinetic solubility of 146.8 ± 6.9 µM (Mean ± %CV) which falls in the range of compounds with good solubility.54 33 has a lower solubility of 45.9 ± 7.7 µM, which is expected for a bigger molecule with higher molecular weight. Moreover, these compounds contain multiple protonable nitrogen atoms which can be converted to salt forms to enhance solubility and bioavailability.
One of the major challenges for CNS drugs is their ability to cross the blood-brain barrier (BBB) and reach the CNS. For the majority of drugs, the BBB permeability is affected by two factors: the ability to permeate through the BBB passively and the avoidance of being effluxed out by the transport proteins such as the P-glycoprotein. 16 and 33 were evaluated in the bidirectional transport assay using MDCK-MDR1 cells which are stably transfected with human MDR1 cDNA and express a higher level of the P-glycoprotein (Pgp) compared to the wild type. Table 4 showed that 16 traversed the cell barrier from the apical (A) to basolateral (B) at a rate of 7.6 × 10−6 cm/s, and the reverse direction B to A at a rate of 6.7 × 10−6 cm/s, demonstrating a moderate BBB permeability (within the range of 3–6 × 10−6 cm/s).55 33 could also penetrate the BBB albeit at lower permeability compared to 16. Both compounds were not Pgp substrates as indicated by the efflux ratio (PB→A/PA→B).54 Together, the data demonstrated the encouraging potential of these compounds as CNS positive ligands.
Anti-hyperalgesia effects of 16 and 33 in rats
The NPFF1/2 antagonist RF9 has been shown to block the long-lasting hyperalgesia induced by heroin and fetanyl.16, 17 Several other NPFF antagonists have also been demonstrated to show the same effects.26 We therefore tested 16 and 33 in a fentanyl-induced hyperalgesia model in rats.
Prior to fentanyl treatment, rats displayed a mean paw withdrawal threshold (PWT) of 24.2 ± 1.8 g, which decreased to 5.7 ± 0.8 g on day 1 after fentanyl administration. One-way repeated measures ANOVA, with time entered as the repeated measure factor, revealed a significant main effect of fentanyl treatment on paw withdrawal threshold (F(6, 35) = 23.42, p < 0.0001). Bonferroni’s post hoc tests revealed significant differences on days 1–3 as compared to day 0.
Both 16 and 33 dose-dependently increased PWT over a dose range of 3.2–32 mg/kg when tested on day 1 (Fig. 6). Treatment with 16 produced a significant main effect as determined by one-way repeated measures ANOVA, with treatment entered as the within subject factor: F(3, 20) = 15.10, p < 0.0001. Additionally, Bonferroni’s post hoc tests revealed significant differences at 10 and 32 mg/kg of 16 as compared to vehicle. Similarly, treatment with 33 produced a significant main effect (F(3, 20) = 12.45, p < 0.001) and Bonferroni’s post hoc tests revealed significant differences at 32 mg/kg of 33 as compared to vehicle.
Figure 6.

(A) Fentanyl-induced mechanical hyperalgesia; (B): anti-hyperalgesic effects of compounds 16 and 33. (N=6 per group). Abscissa: time. Ordinate: paw withdrawal threshold (gram). P < 0.05 compared to pre-fentanyl treatment (Day 0) (left) or compared to “V” (vehicle) treatment.
Conclusions
The NPFF system has been implicated in a number of important physiological functions, particularly the modulation of opioid analgesia. Opioids remain the most effective analgesics for many pain conditions, particularly for chronic pain; however, the adverse effects related to opioid use such as physical dependence, hyperalgesia and tolerance preclude adequate dosing and effective pain control in a large population of pain sufferers. Combination therapy, which combines opioids with another drug that may increase the efficacy of opioids and/or reduce the untoward effects, offers a promising alternative strategy for pain management. Given these unmet challenges associated with opioids in the treatment of pain, the NPFF system represents a promising therapeutic target for developing add-on therapies for pain management.56
Several classes of NPFF-like compounds have been reported, most of which were either peptide or peptidomimetics retaining the guanidine functionality. We have conducted a HTS of a GPCR-focused compound library and identified a novel NPFF receptor antagonist hit containing a proline scaffold. The present study explores the potential of these prolines as a promising novel scaffold for the design of NPFF antagonists. The initial SAR investigation focused on the carboxamide region, and revealed substitution at this position influenced NPFF receptor antagonism and subtype selectivity. Specifically, the carboxamide region prefers substituents such as n-pentyl, n-hexyl, and phenethyl groups. Several compounds with submicromolar NPFF1 potencies were identified as well. Compounds 16 with the pentyl group and 33 with a 4-nitrophenethyl substituent emerged as the most potent analogs at NPFF1 and showed modest preference over NPFF2. Results from the secondary cAMP assays further confirmed the NPFF antagonistic activities of these compounds. Radioligand binding assays demonstrated that the compounds bind to the NPFF receptors with moderate affinity. Both 16 and 33 possess good solubility and BBB permeability, demonstrating the proline scaffold as a potential potent template with favorable drug-like properties for further investigation of antagonist activity at the NPFF receptors. Finally, both compounds reversed opioid-induced hyperalgesia in rats when i.p. administrated, consistent with previously reported NPFF antagonists. Optimization of other regions of the proline core is in progress and will be reported in the future.
Experimental section
Chemistry.
All solvents and chemicals were reagent grade. Unless otherwise mentioned, all reagents and solvents were purchased from commercial vendors and used as received. Flash column chromatography was carried out on a Teledyne ISCO CombiFlash Rf system using prepacked columns. Solvents used include hexane, ethyl acetate (EtOAc), dichloromethane, methanol, and chloroform/methanol/ammonium hydroxide (80:18:2) (CMA-80). Purity and characterization of compounds were established by a combination of HPLC, TLC, mass spectrometry, and NMR analyses. 1H and 13C NMR spectra were recorded on a Bruker Avance DPX-300 (300 MHz) spectrometer and were determined in CDCl3, DMSO-d6, or CD3OD with tetramethylsilane (TMS) (0.00 ppm) or solvent peaks as the internal reference. Chemical shifts are reported in ppm relative to the reference signal, and coupling constant (J) values are reported in hertz (Hz). Thin layer chromatography (TLC) was performed on EMD precoated silica gel 60 F254 plates, and spots were visualized with UV light or iodine staining. Nominal mass spectra were obtained using a Waters Alliance HT/Micromass ZQ system (ESI). High resolution mass spectra were obtained using Agilent 1290 Infinity UHPLC-6230 TOP system (ESI). All final compounds were greater than 95% pure as determined by HPLC on an Agilent 1100 system using an Agilent Zorbax SB-Phenyl, 2.1 mm × 150 mm, 5 μm column using a 15 minute gradient elution of 5–95% solvent B at 1 mL/min followed by 10 minutes at 95% solvent B (solvent A, water with 0.1% TFA; solvent B, acetonitrile with 0.1% TFA and 5% water; absorbance monitored at 220 and 280 nm).
Methyl (2S,4R)-1-[(2-chlorophenyl)methyl]-4-hydroxypyrrolidine-2-carboxylate (3).
To a solution of methyl trans-4-hydroxy-L-proline (16.5 mmol, 3.00 g) and 2-chlorobenzaldehyde (21.5 mmol, 2.1 ml) in 1,2-dichloroethane (55 ml) was added acetic acid (0.9 ml) and sodium triacetoxyborohydride (24.8 mmol, 5.26 g). The reaction was stirred at room temperature for 24 h. After quenching with saturated solution of sodium bicarbonate, the reaction mixture was extracted three times with dichloromethane. The combined organic layers were washed sequentially with water and brine, dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo. The residue was purified by column chromatography to give the desired product as yellow liquid (4.00 g, 90%). 1H NMR (300 MHz, CDCl3) δ 7.44 (dd, J = 1.98, 7.44 Hz, 1H), 7.34 (m, 1H), 7.20 (m, 2H), 4.43 (m, 1H), 3.92 (m, 2H), 3.63 – 3.74 (m, 4H), 3.35 (dd, J = 5.56, 10.08 Hz, 1H), 2.52 (dd, J = 3.58, 10.17 Hz, 1H), 2.24 (m, 1H), 2.12 (m, 1H). MS (ESI) m/z calcl. for C13H17ClNO3 [M+H]+ 270.1, m/z found 270.2.
Methyl (2S,4R)-1-[(2-chlorophenyl)methyl]-4-[(4-methylbenzenesulfonyl)oxy]pyrrolidine-2-carboxylate (4).
To a solution of 3 (14.8 mmol, 4.00 g) in pyridine (11.4 ml) and anhydrous dichloromethane (11.4 ml) at 0 oC was added dropwise tosyl chloride (17.8 mmol, 3.39 g). The reaction was refluxed for 24 h. After removal of the solvent in vacuo, the residue was dissolved in dichloromethane and washed with saturated copper sulfate, water, and brine. The combined organic layers were dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo. The residue was purified by column chromatography (silica gel, ethyl acetate/hexanes) to provide the desired product as colorless liquid (3.77 g, 60%). 1H NMR (300 MHz, CDCl3) δ 7.74 – 7.79 (m, 2H), 7.37 – 7.46 (m, 1H), 7.28 – 7.35 (m, 3H), 7.17 – 7.23 (m, 2H), 5.01 (d, J = 5.46 Hz, 1H), 3.74 – 4.04 (m, 2H), 3.69 (s, 1H), 3.66 (s, 3H), 3.29 (dd, J = 6.03, 11.11 Hz, 1H), 2.67 – 2.73 (m, 1H), 2.44 (s, 3H), 2.28 (dd, J = 5.46, 7.54 Hz, 2H). MS (ESI) m/z calcd. for C20H23ClNO5S [M+H]+ 424.1, m/z found 424.2.
Methyl (2S,4S)-4-azido-1-[(2-chlorophenyl)methyl]pyrrolidine-2-carboxylate (5).
To a solution of 4 (6.87 mmol, 2.91 g) in DMF (40 ml) was added sodium azide (13.74 mmol, 0.89 g). After stirring at 65 oC for 16 h, the reaction mixture was diluted with water, and extracted three times with ethyl acetate. The combined organic layers were dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo. The residue was purified by column chromatography (SiO2, hexanes/ethyl acetate) to give the desired product as yellow liquid (1.48 g, 73%). 1H NMR (300 MHz, CDCl3) δ 7.54 (dd, J = 1.70, 7.54 Hz, 1H), 7.34 (dd, J = 1.51, 7.72 Hz, 1H), 7.16 – 7.29 (m, 2H), 4.02 – 4.09 (m, 1H), 3.90 – 3.98 (m, 1H), 3.81 – 3.87 (m, 1H), 3.72 (s, 3H), 3.45 (dd, J = 6.03, 9.23 Hz, 1H), 3.13 (dd, J = 1.51, 10.36 Hz, 1H), 2.71 (dd, J = 5.75, 10.27 Hz, 1H), 2.54 (ddd, J = 7.72, 9.23, 14.13 Hz, 1H), 2.12 – 2.21 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 173.2, 135.7, 133.9, 130.7, 129.4, 128.4, 126.7, 63.7, 59.2, 57.8, 54.2, 51.8, 35.7. MS (ESI) m/z calcd. for C13H16ClN4O2 [M+H]+ 295.1, m/z found 295.1.
Methyl (2S,4S)-4-amino-1-[(2-chlorophenyl)methyl]pyrrolidine-2-carboxylate (6).
To a solution of azide 5 (4.3 mmol, 1.27 g) in THF (19 ml) under nitrogen was added PPh3 (8.6 mmol, 2.26 g) and water (0.2 ml). The reaction mixture was refluxed with stirring for 6 h. After the solvent was removed, the residue was dissolved in diethyl ether, treated with 0.1 N HCl for 5 min, and then extracted twice with diethyl ether. The aqueous layer was then treated with 1 N NaOH until pH 10, and then extracted with dichloromethane. The combined dichloromethane fractions were dried over anhydrous MgSO4, concentrated in vacuo to afford the desired product as yellow liquid (1.08 g, 93%). 1H NMR (300 MHz, CDCl3) δ 7.50 (dd, J = 1.9, 7.54 Hz, 1H), 7.33 (m, 1H), 7.16 – 7.20 (m, 2H), 3.95 (d, J = 14.7 Hz, 1H), 3.81 (d, J = 13.9 Hz, 1H), 3.66 (s, 3H), 3.45 (m, 1H), 3.39 (dd, J = 5.5, 9.6 Hz, 1H), 2.85 (m, 1H), 2.68 (dd, J = 5.5, 9.4 Hz, 1H), 2.48 (m, 1H), 1.79 (m, 1H), 1.74 (br, 2H). 13C NMR (75 MHz, CDCl3) δ 174.2, 135.9, 133.6, 130.7, 129.1, 128.1, 126.5, 64.2, 62.2, 54.5, 51.5, 50.6, 39.6. MS (ESI) m/z calcd. for C13H18ClN2O2 [M+H]+ 269.1, m/z found 269.3.
Methyl (2S,4S)-1-[(2-chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino} pyrrolidine-2-carboxylate (7a).
To a solution of amine 6 (3.72 mmol, 1 g) in 1,2-dichloroethane (12.4 ml) was added 4-methoxybenzaldehyde (3.72 mmol, 0.45 ml), sodium triacetoxyborohydride (5.58 mmol, 1.18 g) and glacial acetic acid (3.72 mmol, 0.21 ml). After stirring at room temperature for 16 h, the reaction was quenched with saturated sodium bicarbonate, extracted three times with dichloromethane. The combined organic layers were dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo. The residue was purified by column chromatography (SiO2, dichloromethane/methanol) to give the desired product as colorless liquid (0.68 g, 47%). 1H NMR (300 MHz, CDCl3) δ 7.49 (dd, J = 1.79, 7.44 Hz, 1H), 7.33 (m, 1H), 7.16 – 7.25 (m, 4H), 6.84 (m, 2H), 3.95 (d, J = 14.30 Hz, 1H), 3.78 – 3.84 (m, 4H), 3.67 (d, J = 3.20 Hz, 5H), 3.39 (dd, J = 6.03, 9.04 Hz, 1H), 3.28 (m, 1H), 3.03 (dd, J = 2.54, 9.51 Hz, 1H), 2.61 (dd, J = 6.12, 9.51 Hz, 1H), 2.40 (m, 1H), 1.93 (m, 1H). MS (ESI) m/z calcd. for C21H26ClN2O3 [M+H]+ 389.2, m/z found: 389.4.
Methyl (2S,4S)-1-[(2-chlorophenyl)methyl]-4-{[(4-(methylsulfanyl)phenyl)methyl]amino} pyrrolidine-2-carboxylate (7b) was synthesized from 6 and 4-(methylthio)benzylaldehyde according to the procedure for the synthesis of 7a as yellow liquid (45% yield). 1H NMR (300 MHz, CDCl3) δ 7.42 (br. s., 1H), 7.32 (d, J = 6.78 Hz, 1H), 7.13 – 7.24 (m, 6H), 3.90 (d, J = 7.91 Hz, 1H), 3.66 (s, 3H), 3.64 (br. s., 1H), 3.22 – 3.30 (m, 1H), 2.87 – 2.97 (m, 1H), 2.47 – 2.50 (m, 2H), 2.46 (s, 3H), 2.02 – 2.12 (m, 1H). MS (ESI) m/z calcd. for C21H26ClN2O2S [M+H]+ 405.1, m/z found 405.3.
Methyl (2S,4S)-4-{[(tert-butoxy)carbonyl][(4-methoxyphenyl)methyl]amino}-1-[(2-chlorophenyl)methyl]pyrrolidine-2-carboxylate (8a).
To a solution of amine 7a (1.75 mmol, 0.68 g) in 1,4-dioxane (10 ml) was added 10% w/v Na2CO3 solution (2ml) and Boc2O (1.92 mmol, 0.42 g). After stirring at room temperature after 16 h, 1,4-dioxane was removed under reduced pressure. The remaining aqueous solution was extracted three times with dichloromethane. The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated in vacuo and the crude was used for the next step without further purification. 1H NMR (300 MHz, CDCl3) δ 7.30 – 7.38 (m, 2H), 7.16 – 7.22 (m, 2H), 7.06 (d, J = 8.67 Hz, 2H), 6.80 (d, J = 8.67 Hz, 2H), 4.47 – 4.63 (m, 1H), 4.26 – 4.44 (m, 2H), 3.83 (d, J = 6.97 Hz, 2H), 3.78 (s, 3H), 3.65 (s, 3H), 3.61 (d, J = 6.97 Hz, 1H), 3.06 (t, J = 8.29 Hz, 1H), 2.67 (dd, J = 7.44, 8.95 Hz, 1H), 2.13 – 2.25 (m, 2H), 1.53 (s, 9H). MS (ESI) m/z calcd. for C26H34ClN2O5 [M+H]+ 489.2, m/z found 489.6.
Methyl (2S,4S)-4-{[(tert-butoxy)carbonyl][(4-(methylsulfanyl)phenyl)methyl]amino}-1-[(2-chlorophenyl)methyl]pyrrolidine-2-carboxylate (8b) was synthesized from 7b according to the procedure for the synthesis of 8a as yellow liquid (quant. yield). 1H NMR (300 MHz, CDCl3) δ 7.34 (d, J = 6.22 Hz, 1H), 7.26 – 7.32 (m, 1H), 7.08 – 7.19 (m, 4H), 7.01 – 7.06 (m, 2H), 4.79 – 5.03 (m, 1H), 4.58 (s, 2H), 3.91 (d, J = 13.75 Hz, 1H), 3.64 (s, 3H), 3.58 (d, J = 13.94 Hz, 1H), 3.27 (t, J = 8.29 Hz, 1H), 2.87 – 2.96 (m, 1H), 2.49 – 2.60 (m, 2H), 2.45 (s, 3H), 1.87 – 1.99 (m, 1H), 1.34 (s, 9H). MS (ESI) m/z calcd. for C26H33ClN2O4S [M+H]+ 505.2, m/z found 505.6.
(2S,4S)-4-{[(tert-butoxy)carbonyl][(4-methoxyphenyl)methyl]amino}-1-[(2-chlorophenyl)methyl]pyrrolidine-2-carboxylic acid (9a).
To a solution of ester 8a (1.75 mmol, 0.86 g) in methanol (16 ml) and water (16 ml) was added LiOH (8.75 mmol, 0.21 g). After stirring at room temperature for 3 h, methanol was removed under reduced pressure. The remaining solution was diluted in water and acidified with 1N HCl to pH 5. The mixture was then extracted with ethyl acetate. The combined organic layers were washed with water and brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo to provide the desired product as white solid (0.83 g, quantitative yield). 1H NMR (300 MHz, CDCl3) δ 7.35 (d, J = 8.10 Hz, 2H), 7.17 – 7.24 (m, 2H), 7.02 (d, J = 8.48 Hz, 2H), 6.77 (d, J = 8.48 Hz, 2H), 4.26 – 4.55 (m, 3H), 4.06 – 4.17 (m, 1H), 3.71 – 3.82 (m, 4H), 3.40 – 3.52 (m, 1H), 3.08 (d, J = 6.40 Hz, 1H), 2.70 – 2.82 (m, 1H), 2.44 – 2.58 (m, 1H), 2.07 – 2.17 (m, 1H), 1.40 (s, 9H). MS (ESI) m/z calcd. for C25H32ClN2O5 [M+H]+ 473.2, m/z found 473.5.
(2S,4S)-4-{[(tert-butoxy)carbonyl][(4-(methylsulfanyl)phenyl)methyl]amino}-1-[(2-chlorophenyl)methyl]pyrrolidine-2-carboxylic acid (9b) was synthesized from 8b according to the procedure for the synthesis of 9a as white solid (quant. yield). 1H NMR (300 MHz, CDCl3) δ 7.42 (br. s., 1H), 7.34 (d, J = 7.72 Hz, 1H), 7.16 – 7.26 (m, 2H), 7.13 (d, J = 8.10 Hz, 2H), 7.02 (d, J = 8.10 Hz, 2H), 4.47 – 4.61 (m, 1H), 4.31 – 4.46 (m, 2H), 4.23 (d, J = 10.93 Hz, 1H), 3.77 – 3.88 (m, 1H), 3.58 (d, J = 10.74 Hz, 1H), 3.02 – 3.13 (m, 1H), 2.81 (d, J = 10.36 Hz, 1H), 2.52 – 2.66 (m, 1H), 2.44 (s, 3H), 2.06 – 2.17 (m, 1H), 1.38 (s, 8H). MS (ESI) m/z calcd. for C25H32ClN2O4S [M-H]− 489.2, m/z found 489.5.
General procedure A:
To a solution of acid 9 (0.2 mmol, 1 eq) in DMF (0.6 ml, 0.3 M) was added HBTU (0.22 mmol, 1.1 eq), corresponding amine (0.22 mmol, 1.1 eq), DIEA (0.66 mmol, 3 eq). After stirring at room temperature for 16 h, the reaction mixture was diluted with water, extracted three times with ethyl acetate. The combined organic fractions were dried over anhydrous magnesium sulfate and concentrated in vacuo. The residue was dissolved in 20% v/v trifluoroacetic acid (1 ml) in dichloromethane and stirred at room temperature for 16 h. The reaction mixture was concentrated under reduced pressure and the residue was purified on column chromatography (SiO2, methanol/dichloromethane) to afford the desired product.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino}-N-methylpyrrolidine-2-carboxamide (10) was prepared according to the general procedure A as yellow oil (9%). 1H NMR (300 MHz, CDCl3) δ 7.43 (dd, J = 1.79, 7.44 Hz, 1H), 7.28 – 7.34 (m, 2H), 7.12 – 7.25 (m, 3H), 6.87 (s, 2H), 3.90 – 3.96 (m, 2H), 3.75 (s, 3H), 3.58 – 3.69 (m, 2H), 3.28 (s, 2H), 3.09 (dd, J = 4.14, 7.54 Hz, 1H), 2.68 (d, J = 4.90 Hz, 3H), 2.43 – 2.59 (m, 1H), 2.03 – 2.14 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 173.3, 160.5, 134.4, 133.9, 131.4, 131.0, 129.6, 129.0, 127.1, 114.6, 66.2, 55.6, 55.2, 54.9, 54.8, 53.6, 42.0, 33.5, 11.8. HRMS (ESI) m/z calcd. for C21H27ClN3O2 [M+H]+ 388.1791, m/z found 388.1789.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-N-ethyl-4-{[(4-methoxyphenyl)methyl]amino} pyrrolidine-2-carboxamide (11) was prepared according to the general procedure A as yellow oil (32%). 1H NMR (300 MHz, CDCl3) δ 7.41 (br. s., 1H), 7.30 – 7.38 (m, 2H), 7.14 – 7.25 (m, 4H), 6.84 (d, J = 8.67 Hz, 1H), 3.83 – 3.91 (m, 1H), 3.79 (s, 3H), 3.58 – 3.73 (m, 3H), 3.33 (td, J = 2.80, 5.89 Hz, 1H), 3.19 – 3.28 (m, 1H), 3.10 – 3.19 (m, 2H), 3.00 (d, J = 9.98 Hz, 1H), 2.62 (dd, J = 5.56, 10.08 Hz, 1H), 2.49 (ddd, J = 6.40, 9.84, 13.70 Hz, 1H), 1.87 – 1.97 (m, 1H), 1.02 (t, J = 14.70 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 173.9, 158.9, 135.7, 134.3, 131.1, 129.7, 129.5, 128.9, 126.9, 113.9, 67.1, 59.5, 57.4, 56.2, 55.3, 51.1, 37.0, 33.7, 14.6. HRMS (ESI) m/z calcd. for C22H29ClN3O2 [M+H]+ 402.1948, m/z found 402.1930.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino}-N-propylpyrrolidine-2-carboxamide (12) was prepared according to the general procedure A as yellow oil (48%). 1H NMR (300 MHz, CDCl3) δ 7.82 (t, J = 5.27 Hz, 1H), 7.29 – 7.50 (m, 4H), 7.24 (d, J = 8.67 Hz, 2H), 6.85 (d, J = 8.67 Hz, 2H), 4.26 – 4.46 (m, 3H), 4.14 (d, J = 13.00 Hz, 2H), 3.94 – 4.03 (m, 1H), 3.78 (s, 3H), 3.46 (d, J = 5.09 Hz, 2H), 3.07 – 3.22 (m, 2H), 2.88 – 3.02 (m, 1H), 2.27 – 2.40 (m, 1H), 1.42 – 1.54 (m, 2H), 0.87 (t, J = 7.44 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 174.0, 158.9, 135.7, 134.1, 131.0, 129.7, 129.6, 128.8, 126.9, 113.9, 67.1, 59.2, 57.2, 56.1, 55.3, 51.0, 40.7, 36.8, 22.7, 11.4. HRMS (ESI) m/z calcd. for C23H31ClN3O2 [M+H]+ 416.2105, m/z found 416.2091.
(2S,4S)-N-Butyl-1-[(2-chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino} pyrrolidine-2-carboxamide (13) was prepared according to the general procedure A as yellow oil (12%). 1H NMR (300 MHz, CDCl3) δ 7.74 (br. s., 1H), 7.47 (d, J = 7.72 Hz, 1H), 7.39 – 7.45 (m, 2H), 7.34 (dd, J = 2.07, 7.16 Hz, 1H), 7.24 (d, J = 8.67 Hz, 2H), 6.87 (d, J = 8.29 Hz, 2H), 4.34 – 4.56 (m, 3H), 4.19 (d, J = 13.37 Hz, 2H), 3.99 – 4.07 (m, 1H), 3.78 (s, 3H), 3.56 (d, J = 3.20 Hz, 2H), 3.13 – 3.27 (m, 2H), 2.94 – 3.08 (m, 1H), 2.33 – 2.46 (m, 1H), 1.38 – 1.50 (m, 2H), 1.29 (dd, J = 6.97, 14.88 Hz, 2H), 0.90 (t, J = 7.25 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 173.2, 159.8, 135.1, 133.9, 130.8, 130.6, 129.6, 128.8, 127.0, 114.3, 66.7, 57.2, 55.8, 55.5, 55.2, 49.6, 39.0, 34.8, 31.4, 20.0, 13.6. HRMS (ESI) m/z calcd. for C24H33ClN3O2 [M+H]+ 430.2261, m/z found 430.2253.
(2S,4S)-N-(Butan-2-yl)-1-[(2-chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino} pyrrolidine-2-carboxamide (14) was prepared according to the general procedure A as colorless oil (74%). 1H NMR (300 MHz, CDCl3) δ 7.29 – 7.39 (m, 3H), 7.16 – 7.25 (m, 4H), 6.83 (d, J = 8.67 Hz, 1H), 3.87 – 3.94 (m, 1H), 3.74 – 3.84 (m, 5H), 3.61 – 3.72 (m, 3H), 3.20 – 3.33 (m, 2H), 2.97 (td, J = 1.62, 10.13 Hz, 1H), 2.60 (ddd, J = 3.01, 5.60, 10.03 Hz, 1H), 2.45 – 2.54 (m, 1H), 1.87 – 1.96 (m, 1H), 1.76 (br. s., 1H), 1.29 – 1.39 (m, 2H), 1.02 (d, J = 6.59 Hz, 2H), 0.94 (d, J = 6.59 Hz, 2H), 0.74 – 0.85 (m, 5H). 13C NMR (75 MHz, CDCl3) δ 173.4, 173.4, 158.7, 158.7, 135.9, 134.3, 134.2, 131.9, 131.2, 130.9, 129.8, 129.8, 129.4, 128.9, 128.8, 126.9, 126.9, 113.8, 113.8, 67.3, 67.2, 59.7, 59.6, 57.7, 57.5, 56.2, 56.1, 55.3, 51.3, 46.0, 45.8, 37.4, 37.2, 29.6, 20.1, 10.5, 10.4. HRMS (ESI) m/z calcd. for C24H33ClN3O2 [M+H]+ 430.2261, m/z found 430.2277.
(2S,4S)-N-tert-Butyl-1-[(2-chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino} pyrrolidine-2-carboxamide (15) was prepared according to the general procedure A as colorless oil (88%). 1H NMR (300 MHz, CDCl3) δ 7.30 – 7.39 (m, 2H), 7.15 – 7.25 (m, 5H), 6.82 – 6.87 (m, 2H), 3.76 – 3.82 (m, 5H), 3.60 – 3.75 (m, 2H), 3.28 – 3.36 (m, 1H), 3.14 (dd, J = 5.27, 9.80 Hz, 1H), 3.06 (d, J = 10.17 Hz, 1H), 2.64 (dd, J = 5.65, 9.98 Hz, 1H), 2.47 (ddd, J = 6.40, 9.80, 13.56 Hz, 1H), 1.89 (td, J = 3.67, 13.56 Hz, 1H), 1.18 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 173.5, 158.8, 136.0, 134.4, 131.0, 129.7, 129.4, 128.9, 127.0, 113.9, 67.8, 59.9, 57.6, 56.2, 55.3, 51.2, 50.2, 37.2, 28.5. HRMS (ESI) m/z calcd. for C24H33ClN3O2 [M+H]+ 430.2261, m/z found 430.2242.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino}-N-pentylpyrrolidine-2-carboxamide (16) was prepared according to the general procedure A as yellow oil (25%). 1H NMR (300 MHz, CDCl3) δ 7.32 – 7.43 (m, 3H), 7.17 – 7.24 (m, 4H), 6.81 – 6.86 (m, 2H), 3.84 – 3.91 (m, 1H), 3.78 (s, 3H), 3.64 – 3.74 (m, 3H), 3.31 – 3.38 (m, 1H), 3.25 (dd, J = 5.65, 9.80 Hz, 1H), 2.99 – 3.16 (m, 3H), 2.61 (dd, J = 5.56, 10.27 Hz, 1H), 2.48 (dd, J = 2.83, 6.78 Hz, 1H), 1.92 (d, J = 18.08 Hz, 1H), 1.31 – 1.43 (m, 2H), 1.16 – 1.30 (m, 4H), 0.84 (t, J = 6.97 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 173.9, 158.9, 135.7, 134.1, 130.9, 129.7, 129.6, 128.8, 126.9, 113.9, 67.1, 59.2, 57.2, 56.1, 55.3, 51.0, 38.9, 36.8, 29.1, 29.1, 22.3, 13.9. HRMS (ESI) m/z calcd. for C25H35ClN3O2 [M+H]+ 444.2418, m/z found 444.2411.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino}-N-(3-methylbutyl)pyrrolidine-2-carboxamide (17) was prepared according to the general procedure A as colorless oil (71%). 1H NMR (300 MHz, CDCl3) δ 7.23 – 7.34 (m, 3H), 7.10 – 7.18 (m, 4H), 6.76 (d, J = 8.67 Hz, 2H), 3.77 – 3.83 (m, 1H), 3.71 (s, 3H), 3.57 – 3.67 (m, 3H), 3.27 (td, J = 2.87, 5.93 Hz, 1H), 3.17 (dd, J = 5.46, 9.80 Hz, 1H), 3.05 (dt, J = 7.72, 12.90 Hz, 2H), 2.92 – 2.98 (m, 1H), 2.54 (dd, J = 5.65, 10.17 Hz, 1H), 2.42 (ddd, J = 6.59, 9.80, 13.75 Hz, 1H), 1.80 – 1.89 (m, 1H), 1.44 (td, J = 6.69, 13.37 Hz, 1H), 1.16 – 1.23 (m, 2H), 0.77 (dd, J = 3.39, 6.59 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 172.9, 157.9, 134.7, 133.1, 130.0, 128.7, 128.6, 127.8, 125.9, 112.9, 66.1, 58.2, 56.2, 55.1, 54.2, 49.9, 37.3, 36.2, 35.8, 24.8, 21.4. HRMS (ESI) m/z calcd. for C25H35ClN3O2 [M+H]+ 444.2418, m/z found 444.2419.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino}-N-hexylpyrrolidine-2-carboxamide (18) was prepared according to the general procedure A as colorless oil (49%). 1H NMR (300 MHz, CDCl3) δ 7.45 (t, J = 5.65 Hz, 1H), 7.31 – 7.38 (m, 2H), 7.16 – 7.24 (m, 4H), 6.81 – 6.85 (m, 2H), 3.84 – 3.91 (m, 1H), 3.79 (s, 3H), 3.58 – 3.72 (m, 3H), 3.31 (td, J = 2.76, 5.98 Hz, 1H), 3.25 (dd, J = 5.56, 9.89 Hz, 1H), 3.11 (dt, J = 7.06, 12.86 Hz, 2H), 3.00 (d, J = 10.17 Hz, 1H), 2.60 (dd, J = 5.56, 10.08 Hz, 1H), 2.49 (ddd, J = 6.50, 9.84, 13.61 Hz, 1H), 1.90 (td, J = 3.84, 13.61 Hz, 2H), 1.31 – 1.41 (m, 2H), 1.17 – 1.29 (m, 6H), 0.85 (t, J = 13.40 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 174.0, 158.8, 135.8, 134.2, 131.8, 131.0, 129.7, 129.4, 128.8, 126.9, 113.9, 67.2, 59.6, 57.4, 56.2, 55.2, 51.2, 38.9, 37.2, 31.5, 29.4, 26.6, 22.5, 14.0. HRMS (ESI) m/z calcd. for C26H37ClN3O2 [M+H]+ 458.2574, m/z found 458.2583.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-N-decyl-4-{[(4-methoxyphenyl)methyl]amino} pyrrolidine-2-carboxamide (19) was prepared according to the general procedure A as colorless oil (58%). 1H NMR (300 MHz, CDCl3) δ 7.42 (t, J = 5.75 Hz, 1H), 7.33 – 7.37 (m, 2H), 7.16 – 7.25 (m, 5H), 6.83 (d, J = 8.67 Hz, 1H), 3.84 – 3.91 (m, 1H), 3.78 (s, 3H), 3.62 – 3.75 (m, 4H), 3.33 – 3.40 (m, 1H), 3.25 (dd, J = 5.65, 9.61 Hz, 1H), 2.90 – 3.16 (m, 6H), 2.61 (dd, J = 5.65, 10.17 Hz, 1H), 2.49 (ddd, J = 6.59, 9.61, 13.75 Hz, 1H), 1.89 – 1.97 (m, 1H), 1.16 – 1.42 (m, 18H), 0.85 – 0.90 (m, 3H). 13C NMR (75 MHz, CDCl3) δ 173.9, 159.0, 135.7, 134.1, 130.9, 129.7, 129.7, 128.8, 127.0, 114.0, 67.1, 59.1, 57.1, 56.1, 55.3, 50.9, 39.0, 36.7, 31.9, 29.6, 29.5, 29.5, 29.3, 27.0, 22.7, 14.1. HRMS (ESI) m/z calcd. for C30H45ClN3O2 [M+H]+ 514.3200, m/z found 514.3190.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino}-N-[2-(morpholin-4-yl)ethyl]pyrrolidine-2-carboxamide (20) was prepared according to the general procedure A as colorless oil (18%). 1H NMR (300 MHz, CDCl3) δ 7.52 (t, J = 5.27 Hz, 1H), 7.34 – 7.38 (m, 2H), 7.18 – 7.25 (m, 4H), 6.84 (d, J = 8.67 Hz, 2H), 3.84 – 3.91 (m, 1H), 3.74 – 3.78 (m, 4H), 3.68 – 3.73 (m, 2H), 3.59 – 3.65 (m, 4H), 3.42 (t, J = 7.72 Hz, 1H), 3.22 – 3.37 (m, 4H), 2.35 – 2.45 (m, 7H), 2.13 – 2.21 (m, J = 1.00, 1.00 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 173.5, 159.2, 135.6, 134.2, 130.9, 129.8, 129.7, 128.9, 126.9, 114.0, 66.8, 66.4, 57.4, 57.3, 55.4, 55.2, 53.4, 51.4, 36.7, 35.3, 30.9. HRMS (ESI) m/z calcd. for C26H36ClN4O3 [M+H]+ 487.2476, m/z found 487.2460.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-N-(2-cyclohexylethyl)-4-{[(4-methoxyphenyl)methyl] amino}pyrrolidine-2-carboxamide (21) was prepared according to the general procedure A as colorless oil (53%). 1H NMR (300 MHz, CDCl3) δ 7.32 – 7.41 (m, 3H), 7.15 – 7.25 (m, 4H), 6.84 (d, J = 8.67 Hz, 2H), 3.83 – 3.90 (m, 1H), 3.79 (s, 3H), 3.59 – 3.73 (m, 3H), 3.32 (td, J = 2.80, 5.89 Hz, 1H), 3.25 (dd, J = 5.56, 9.70 Hz, 1H), 3.08 – 3.19 (m, 2H), 3.02 (d, J = 10.17 Hz, 1H), 2.60 (dd, J = 5.65, 9.98 Hz, 1H), 2.49 (ddd, J = 6.50, 9.80, 13.66 Hz, 1H), 1.86 – 1.95 (m, 1H), 1.56 – 1.70 (m, 5H), 1.08 – 1.30 (m, 6H), 0.76 – 0.93 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 173.9, 158.8, 135.8, 134.1, 130.9, 129.7, 129.5, 128.8, 126.9, 113.9, 67.2, 59.4, 57.3, 56.2, 55.2, 51.1, 37.0, 36.8, 36.7, 35.3, 33.1, 33.1, 30.9, 26.5, 26.2. HRMS (ESI) m/z calcd. for C28H39ClN3O2 [M+H]+ 484.2731, m/z found 484.2731.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-N,N-diethyl-4-{[(4-methoxyphenyl)methyl]amino} pyrrolidine-2-carboxamide (22) was prepared according to the general procedure A as colorless oil (11%). 1H NMR (300 MHz, CDCl3) δ 7.78 (br. s., 1H), 7.46 (dd, J = 2.26, 6.97 Hz, 1H), 7.33 – 7.38 (m, 1H), 7.16 – 7.25 (m, 4H), 6.80 – 6.88 (m, 2H), 3.83 – 3.91 (m, 1H), 3.79 (s, 3H), 3.64 – 3.76 (m, 3H), 3.16 – 3.36 (m, 4H), 3.03 (d, J = 9.61 Hz, 1H), 2.43 – 2.61 (m, 7H), 1.79 – 2.05 (m, 3H), 0.96 (t, J = 7.06 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 174.2, 158.8, 135.8, 134.0, 130.8, 129.5, 129.4, 128.6, 126.9, 113.9, 67.2, 56.8, 56.1, 55.3, 51.5, 51.2, 46.6, 37.3, 36.2, 11.1. HRMS (ESI) m/z calcd. for C26H38ClN4O2 [M+H]+ 473.2683, m/z found 473.2687.
(2S,4S)-N-Benzyl-1-[(2-chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino} pyrrolidine-2-carboxamide (23) was prepared according to the general procedure A as yellow oil (44%). 1H NMR (300 MHz, CDCl3) δ 7.82 (t, J = 5.75 Hz, 1H), 7.22 – 7.27 (m, 5H), 7.10 – 7.21 (m, 6H), 6.77 – 6.83 (m, 2H), 4.32 (dd, J = 5.93, 8.95 Hz, 2H), 3.87 (d, J = 13.19 Hz, 1H), 3.77 (s, 3H), 3.58 – 3.71 (m, 3H), 3.34 – 3.41 (m, 1H), 3.31 (dd, J = 5.56, 9.70 Hz, 1H), 2.98 (d, J = 10.36 Hz, 1H), 2.60 (dd, J = 5.46, 10.36 Hz, 1H), 2.49 (ddd, J = 6.78, 9.70, 13.85 Hz, 1H), 1.92 – 2.02 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 173.9, 159.1, 138.3, 135.4, 134.1, 131.0, 129.7, 129.7, 128.8, 128.6, 127.8, 127.3, 126.9, 114.0, 67.0, 58.7, 56.9, 56.0, 55.3, 50.7, 43.1, 36.3. HRMS (ESI) m/z calcd. for C27H31ClN3O2 [M+H]+ 464.2105, m/z found 464.2104.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino}-N-(2-phenylethyl)pyrrolidine-2-carboxamide (24) was prepared according to the general procedure A as yellow oil (59%). 1H NMR (300 MHz, CDCl3) δ 7.52 (t, J = 5.84 Hz, 1H), 7.32 – 7.37 (m, 1H), 7.09 – 7.24 (m, 11H), 6.83 (d, J = 8.67 Hz, 1H), 3.73 – 3.81 (m, 4H), 3.63 – 3.67 (m, 1H), 3.56 – 3.62 (m, 2H), 3.43 – 3.55 (m, 1H), 3.30 – 3.42 (m, 1H), 3.19 – 3.28 (m, 2H), 2.92 (d, J = 10.17 Hz, 1H), 2.67 – 2.76 (m, 2H), 2.42 – 2.55 (m, 2H), 1.96 (br. s., 1H), 1.81 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 174.1, 158.7, 138.9, 135.8, 134.1, 132.0, 130.7, 129.6, 129.4, 128.7, 128.6, 128.5, 126.9, 126.4, 113.9, 67.3, 59.5, 57.2, 56.0, 55.3, 51.2, 39.8, 37.3, 35.5. HRMS (ESI) m/z calcd. for C28H33ClN3O2 [M+H]+ 478.2261, m/z found 478.2264.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino}-N-(3-phenylpropyl)pyrrolidine-2-carboxamide (25) was prepared according to the general procedure A as colorless oil (70%). 1H NMR (300 MHz, CDCl3) δ 7.44 (t, J = 5.37 Hz, 1H), 7.13 – 7.36 (m, 10H), 7.09 (d, J = 6.78 Hz, 1H), 6.80 (d, J = 8.67 Hz, 2H), 3.83 – 3.90 (m, 1H), 3.75 (s, 3H), 3.66 – 3.73 (m, 3H), 3.34 – 3.41 (m, 1H), 3.25 (dd, J = 5.56, 9.70 Hz, 1H), 3.11 – 3.20 (m, 2H), 3.05 (d, J = 10.55 Hz, 1H), 2.43 – 2.64 (m, 4H), 1.96 (d, J = 3.20 Hz, 1H), 1.65 – 1.77 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 173.9, 159.1, 141.5, 135.6, 134.1, 130.9, 129.8, 129.7, 128.9, 128.4, 128.3, 127.0, 125.9, 114.0, 67.0, 58.9, 56.9, 56.0, 55.2, 50.8, 38.6, 36.5, 33.2, 31.1. HRMS (ESI) m/z calcd. for C29H35ClN3O2 [M+H]+ 492.2418, m/z found 492.2411.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-N-[2-(4-methoxyphenyl)ethyl]-4-{[(4-methoxyphenyl) methyl]amino}pyrrolidine-2-carboxamide (26) was prepared according to the general procedure A as colorless oil (20%). 1H NMR (300 MHz, CDCl3) δ 7.40 (t, J = 5.65 Hz, 1H), 7.32 – 7.36 (m, 1H), 7.15 – 7.22 (m, 4H), 7.10 – 7.14 (m, 1H), 7.05 (d, J = 8.67 Hz, 2H), 6.85 (d, J = 8.67 Hz, 2H), 6.77 (d, J = 8.48 Hz, 2H), 3.78 (s, 3H), 3.75 (s, 3H), 3.70 – 3.73 (m, 1H), 3.65 (d, J = 9.04 Hz, 2H), 3.42 – 3.52 (m, 1H), 3.37 (dd, J = 5.65, 9.61 Hz, 1H), 3.23 – 3.32 (m, 1H), 3.10 – 3.22 (m, 2H), 2.67 (dt, J = 3.30, 7.02 Hz, 2H), 2.25 (t, J = 8.01 Hz, 1H), 2.01 – 2.16 (m, 2H), 1.40 – 1.68 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 173.8, 158.8, 158.2, 135.7, 134.2, 132.1, 131.0, 130.9, 129.6, 129.6, 129.2, 128.8, 126.9, 114.0, 113.9, 66.7, 60.1, 57.8, 56.1, 55.3, 55.2, 52.1, 40.0, 37.9, 34.8, 30.9. HRMS (ESI) m/z calcd. for C29H35ClN3O3 [M+H]+ 508.2367, m/z found 508.2357.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-N-[2-(3,4-dimethoxyphenyl)ethyl]-4-{[(4-methoxyphenyl)methyl]amino}pyrrolidine-2-carboxamide (27) was prepared according to the general procedure A as white solid (61%). 1H NMR (300 MHz, CDCl3) δ 7.29 – 7.35 (m, 1H), 7.18 – 7.25 (m, 3H), 7.09 – 7.15 (m, 2H), 6.82 (d, J = 8.67 Hz, 2H), 6.68 (s, 3H), 3.63 – 3.88 (m, 13H), 3.45 – 3.60 (m, 3H), 3.28 – 3.41 (m, 1H), 3.12 – 3.20 (m, 2H), 2.70 (qd, J = 7.21, 14.93 Hz, 2H), 2.43 – 2.58 (m, 2H), 1.99 (ddd, J = 3.49, 6.26, 13.99 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 172.6, 160.3, 148.9, 147.6, 134.6, 133.6, 131.2, 131.1, 130.4, 129.5, 128.7, 126.9, 120.6, 114.5, 111.8, 111.1, 66.8, 56.0, 55.8, 55.2, 55.0, 54.9, 48.9, 40.1, 34.8, 33.8. HRMS (ESI) m/z calcd. for C30H37ClN3O4 [M+H]+ 538.2473, m/z found 538.2457.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-N-{2-[4-(dimethylamino)phenyl]ethyl}-4-{[(4-methoxyphenyl)methyl]amino}pyrrolidine-2-carboxamide (28) was prepared according to the general procedure A as yellow oil (25%). 1H NMR (300 MHz, CDCl3) δ 7.30 – 7.34 (m, 1H), 7.15 – 7.25 (m, 5H), 6.98 (d, J = 8.67 Hz, 2H), 6.84 (d, J = 8.67 Hz, 2H), 6.59 (d, J = 8.48 Hz, 2H), 3.62 – 3.81 (m, 7H), 3.33 – 3.45 (m, 3H), 3.20 (dd, J = 5.46, 9.42 Hz, 1H), 3.05 (d, J = 10.36 Hz, 1H), 2.86 (s, 6H), 2.53 – 2.69 (m, 3H), 2.40 – 2.51 (m, 1H), 1.91 (d, J = 13.56 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 173.7, 159.3, 149.4, 135.4, 133.9, 130.8, 130.1, 129.5, 129.2, 128.6, 126.9, 126.5, 114.1, 112.9, 66.8, 58.2, 56.1, 55.8, 55.3, 50.3, 40.7, 40.3, 35.8, 34.4. HRMS (ESI) m/z calcd. for C30H38ClN4O2 [M+H]+ 521.2683, m/z found 521.2661.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-N-[2-(4-acetamidophenyl)ethyl]-4-{[(4-methoxyphenyl)methyl]amino}pyrrolidine-2-carboxamide (29) was prepared according to the general procedure A as colorless liquid (50%). 1H NMR (300 MHz, CDCl3) δ 8.44 (br. s., 1H), 7.67 (t, J = 5.75 Hz, 1H), 7.37 (dd, J = 1.60, 7.44 Hz, 1H), 7.23 – 7.31 (m, 6H), 7.11 – 7.21 (m, 2H), 7.03 (d, J = 8.29 Hz, 2H), 6.86 (d, J = 8.67 Hz, 2H), 3.84 (s, 2H), 3.76 (s, 3H), 3.62 – 3.74 (m, 2H), 3.39 – 3.48 (m, 2H), 3.34 (td, J = 6.31, 12.62 Hz, 1H), 3.11 – 3.23 (m, 2H), 2.66 – 2.75 (m, 2H), 2.54 (dd, J = 5.56, 11.02 Hz, 1H), 2.28 – 2.41 (m, 1H), 2.07 (s, 3H), 1.76 (d, J = 14.32 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 173.1, 168.9, 160.1, 136.1, 134.5, 134.5, 133.5, 130.9, 130.5, 129.2, 129.0, 128.5, 126.8, 120.6, 114.3, 65.6, 55.0, 54.8, 54.4, 53.5, 48.7, 39.8, 34.4, 33.7, 23.9. HRMS (ESI) m/z calcd. for C30H36ClN4O3 [M+H]+ 535.2476, m/z found 535.2470.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino}-N-{2-[4-(methylcarbamoyl)phenyl]ethyl}pyrrolidine-2-carboxamide (30) was prepared according to the general procedure A as colorless oil (28%). 1H NMR (300 MHz, CDCl3) δ 7.75 (t, J = 5.84 Hz, 1H), 7.53 (d, J = 8.10 Hz, 2H), 7.37 (dd, J = 2.17, 6.88 Hz, 1H), 7.22 (d, J = 8.67 Hz, 2H), 7.09 – 7.18 (m, 4H), 6.82 (d, J = 8.48 Hz, 3H), 3.86 (d, J = 2.45 Hz, 2H), 3.71 – 3.77 (m, 3H), 3.57 – 3.68 (m, 2H), 3.33 – 3.54 (m, 3H), 3.12 – 3.23 (m, 2H), 3.06 (q, J = 7.41 Hz, 1H), 2.89 (d, J = 4.71 Hz, 3H), 2.74 – 2.83 (m, 2H), 2.38 – 2.55 (m, 2H), 1.94 (dd, J = 3.58, 14.13 Hz, 1H), 1.34 – 1.45 (m, 5H). 13C NMR (75 MHz, CDCl3) δ 173.0, 168.3, 160.4, 142.5, 134.7, 133.6, 132.7, 131.3, 130.5, 129.5, 128.8, 128.7, 127.1, 114.5, 66.5, 55.2, 54.9, 54.7, 53.7, 48.9, 42.0, 39.8, 35.0, 33.8, 26.7, 11.8. HRMS (ESI) m/z calcd. for C30H35ClN4O3 [M+H]+ 535.2476, m/z found 535.2458.
(2S,4S)-N-[2-(4-tert-Butylphenyl)ethyl]-1-[(2-chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino}pyrrolidine-2-carboxamide (31) was prepared according to the general procedure A as colorless oil (36%). 1H NMR (300 MHz, CDCl3) δ 7.51 (br. s., 1H), 7.31 – 7.36 (m, 1H), 7.14 – 7.25 (m, 8H), 7.05 (d, J = 8.29 Hz, 2H), 6.83 (d, J = 8.67 Hz, 2H), 3.75 – 3.83 (m, 5H), 3.57 – 3.67 (m, 3H), 3.40 – 3.50 (m, 1H), 3.30 – 3.39 (m, 1H), 3.20 – 3.26 (m, 2H), 2.92 (d, J = 10.17 Hz, 1H), 2.67 (q, J = 7.03 Hz, 2H), 2.41 – 2.56 (m, 2H), 1.80 – 1.89 (m, 1H), 1.26 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 174.1, 158.7, 149.2, 135.8, 135.8, 134.1, 132.1, 130.9, 129.6, 129.3, 128.7, 128.3, 126.9, 125.4, 113.8, 67.2, 59.6, 57.2, 56.1, 55.2, 51.2, 39.9, 37.3, 35.1, 34.3, 31.3. HRMS (ESI) m/z calcd. for C32H41ClN3O2 [M+H]+ 534.2887, m/z found 534.2887.
(2S,4S)-N-[2-(4-Chlorophenyl)ethyl]-1-[(2-chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino}pyrrolidine-2-carboxamide (32) was prepared according to the general procedure A as colorless oil (55%). 1H NMR (300 MHz, CDCl3) δ 7.46 (t, J = 5.93 Hz, 1H), 7.27 – 7.31 (m, 1H), 7.08 – 7.25 (m, 7H), 7.00 – 7.04 (m, 2H), 6.81 – 6.86 (m, 2H), 3.75 – 3.80 (m, 0H), 3.73 (s, 3H), 3.68 (d, J = 13.19 Hz, 2H), 3.53 – 3.60 (m, 1H), 3.31 – 3.50 (m, 3H), 3.18 (dd, J = 6.78, 9.04 Hz, 1H), 3.08 (d, J = 10.93 Hz, 1H), 2.69 (q, J = 7.03 Hz, 2H), 2.42 – 2.55 (m, 2H), 1.92 (dt, J = 3.20, 7.06 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 173.1, 159.8, 137.2, 134.9, 133.7, 132.1, 130.6, 130.2, 130.0, 129.6, 128.7, 128.5, 126.9, 114.3, 67.1, 57.3, 55.8, 55.2, 55.2, 49.7, 39.7, 34.9, 34.7. HRMS (ESI) m/z for C28H32Cl2N3O2 [M+H]+ 512.1872, m/z found 512.1868.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-N-[2-(4-methoxyphenyl)ethyl]-4-{[(4-nitrophenyl) methyl]amino}pyrrolidine-2-carboxamide (33) was prepared according to the general procedure A as yellow oil (22%). 1H NMR (300 MHz, CDCl3) δ 8.06 (d, J = 8.67 Hz, 2H), 7.45 – 7.51 (m, 1H), 7.31 – 7.36 (m, 1H), 7.27 (s, 1H), 7.13 – 7.26 (m, 6H), 6.82 – 6.89 (m, 2H), 3.79 (s, 3H), 3.62 – 3.74 (m, 4H), 3.34 – 3.53 (m, 3H), 3.11 – 3.22 (m, 2H), 2.83 (dt, J = 2.92, 6.92 Hz, 2H), 2.22 – 2.33 (m, 1H), 2.08 (t, J = 7.54 Hz, 2H), 1.46 – 1.72 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 174.1, 158.9, 146.7, 146.7, 135.5, 134.1, 130.9, 129.7, 129.5, 129.2, 129.0, 128.8, 127.0, 123.7, 113.9, 66.6, 60.1, 58.1, 55.9, 55.3, 52.0, 39.2, 37.8, 35.6. HRMS (ESI) m/z calcd. for C28H32ClN4O4 [M+H]+ 523.2112, m/z found 523.2103.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-N-[2-(3,4-difluorophenyl)ethyl]-4-{[(4-methoxyphenyl)methyl]amino}pyrrolidine-2-carboxamide (34) was prepared according to the general procedure A as white solid (62%). 1H NMR (300 MHz, CDCl3) δ 7.55 (t, J = 6.03 Hz, 1H), 7.29 (d, J = 2.26 Hz, 1H), 7.11 – 7.25 (m, 5H), 6.88 – 6.96 (m, 2H), 6.75 – 6.86 (m, 3H), 3.72 – 3.79 (m, 4H), 3.54 – 3.71 (m, 3H), 3.30 – 3.47 (m, 3H), 3.19 (dd, J = 6.69, 9.14 Hz, 1H), 3.07 (d, J = 10.93 Hz, 1H), 2.67 (q, J = 6.91 Hz, 2H), 2.43 – 2.56 (m, 2H), 1.94 (dd, J = 3.11, 6.50 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 173.2, 159.7, 135.8, 135.0, 133.7, 130.5, 130.2, 129.6, 128.7, 126.9, 124.5, 124.5, 124.4, 124.4, 117.5, 117.2, 117.1, 116.9, 114.2, 67.2, 57.6, 56.0, 55.3, 55.2, 49.8, 39.6, 38.6, 35.1, 34.5. HRMS (ESI) m/z calcd. for C28H30ClF2N3O2 [M+H]+ 514.2073, m/z found 514.2065.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-N-[2-(4-fluorophenyl)ethyl]-4-{[(4-methoxyphenyl)methyl]amino}pyrrolidine-2-carboxamide (35) was prepared according to the general procedure A as colorless oil (74%). 1H NMR (300 MHz, CDCl3) δ 7.38 (t, J = 5.84 Hz, 1H), 7.16 – 7.26 (m, 4H), 7.01 – 7.12 (m, 4H), 6.77 – 6.86 (m, 4H), 3.73 – 3.87 (m, 2H), 3.70 (s, 3H), 3.60 – 3.67 (m, 1H), 3.47 – 3.58 (m, 3H), 3.24 – 3.37 (m, 1H), 3.10 – 3.21 (m, 2H), 2.63 – 2.81 (m, 2H), 2.44 – 2.56 (m, 2H), 1.93 – 2.04 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 172.3, 160.5, 134.4, 134.3, 134.3, 133.5, 131.4, 130.0, 129.9, 129.5, 128.6, 126.9, 121.8, 115.3, 115.0, 114.5, 67.1, 55.8, 55.2, 54.9, 54.6, 48.7, 40.1, 34.4, 33.4. HRMS (ESI) m/z calcd. for C28H31ClFN3O2 [M+H]+ 496.2167; m/z found 496.2168.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino}-N-{2-[4-(trifluoromethyl)phenyl]ethyl}pyrrolidine-2-carboxamide (36) was prepared according to the general procedure A as yellow oil (69%). 1H NMR (300 MHz, CDCl3) δ 7.54 (t, J = 5.84 Hz, 1H), 7.43 (d, J = 8.10 Hz, 2H), 7.32 (dd, J = 1.88, 7.35 Hz, 1H), 7.14 – 7.24 (m, 7H), 6.81 – 6.86 (m, 2H), 3.73 – 3.80 (m, 4H), 3.57 – 3.70 (m, 3H), 3.38 – 3.48 (m, 2H), 3.29 – 3.36 (m, 1H), 3.23 (dd, J = 5.84, 9.80 Hz, 1H), 3.00 (s, 1H), 2.71 – 2.81 (m, 2H), 2.42 – 2.58 (m, 2H), 1.80 – 1.90 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 174.0, 159.0, 143.0, 135.5, 134.0, 130.6, 129.6, 129.0, 128.8, 126.9, 125.4, 125.3, 114.0, 67.1, 59.0, 57.0, 56.0, 55.2, 50.9, 39.5, 36.7, 35.4. HRMS (ESI) m/z calcd. for C29H32ClF3N3O2 [M+H]+ 546.2135, m/z found 546.2140.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino}-N-[2-(pyridin-4-yl)ethyl]pyrrolidine-2-carboxamide (37) was prepared according to the general procedure A as white solid (73%). 1H NMR (300 MHz, CDCl3) δ 8.34 (d, J = 5.84 Hz, 2H), 7.65 (t, J = 5.93 Hz, 1H), 7.24 – 7.33 (m, 3H), 7.20 (d, J = 8.67 Hz, 2H), 7.10 – 7.15 (m, 2H), 7.05 (d, J = 6.03 Hz, 2H), 6.82 (d, J = 8.67 Hz, 2H), 3.75 – 3.86 (m, 2H), 3.67 (br. s., 5H), 3.50 (quin, J = 6.83 Hz, 2H), 3.35 – 3.44 (m, 1H), 3.10 – 3.20 (m, 2H), 2.76 (dt, J = 2.83, 7.06 Hz, 2H), 2.44 – 2.57 (m, 2H), 1.93 (ddd, J = 2.73, 7.16, 14.22 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 172.7, 160.3, 149.4, 148.3, 134.5, 133.6, 131.2, 130.2, 129.6, 128.7, 126.9, 124.2, 114.5, 67.1, 56.2, 55.2, 54.8, 49.1, 38.9, 34.6, 34.0. HRMS (ESI) m/z calcd. for C27H32ClN4O2 [M+H]+ 479.2214, m/z found 479.2212.
(2S,4S)-N-[2-(4-Acetylphenyl)ethyl]-1-[(2-chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino}pyrrolidine-2-carboxamide (38) was prepared according to the general procedure A as colorless liquid (35%). 1H NMR (300 MHz, CDCl3) δ 7.65 – 7.72 (m, 2H), 7.35 – 7.42 (m, 1H), 7.18 – 7.24 (m, 3H), 7.05 – 7.16 (m, 4H), 6.83 (d, J = 8.67 Hz, 2H), 3.78 – 3.89 (m, 2H), 3.66 – 3.74 (m, 4H), 3.48 – 3.61 (m, 3H), 3.37 (d, J = 5.65 Hz, 1H), 3.18 (d, J = 8.10 Hz, 2H), 2.74 – 2.87 (m, 2H), 2.43 – 2.60 (m, 2H), 2.18 (d, J = 5.84 Hz, 3H), 1.93 – 2.06 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 172.4, 160.5, 157.6, 140.4, 136.6, 134.4, 133.7, 131.5, 130.5, 129.5, 128.7, 128.6, 127.0, 126.7, 121.7, 114.6, 66.9, 55.5, 55.2, 54.7, 54.2, 48.6, 40.0, 35.1, 33.4, 14.8. HRMS (ESI) m/z calcd. for C30H35ClN3O3 [M+H]+ 520.2367, m/z found 520.2407.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-N-[2-(4-methanesulfonylphenyl)ethyl]-4-{[(4-methoxyphenyl)methyl]amino}pyrrolidine-2-carboxamide (39) was prepared according to the general procedure A as colorless oil (55%). 1H NMR (300 MHz, CDCl3) δ 7.75 (d, J = 8.29 Hz, 2H), 7.64 (t, J = 5.93 Hz, 1H), 7.25 – 7.35 (m, 5H), 7.16 – 7.22 (m, 4H), 6.81 – 6.86 (m, 2H), 3.76 (s, 3H), 3.59 – 3.74 (m, 4H), 3.42 (q, J = 6.97 Hz, 2H), 3.32 – 3.37 (m, 1H), 3.23 (dd, J = 5.84, 9.61 Hz, 1H), 3.01 (d, J = 1.70 Hz, 1H), 2.98 (s, 3H), 2.79 (qd, J = 6.99, 14.46 Hz, 2H), 2.55 (dd, J = 5.65, 10.55 Hz, 1H), 2.41 – 2.50 (m, 1H), 1.81 – 1.91 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 174.0, 159.2, 145.6, 138.7, 135.4, 133.9, 130.7, 129.9, 129.7, 129.6, 128.9, 127.5, 127.0, 114.1, 67.0, 58.6, 56.7, 55.9, 55.3, 50.7, 44.5, 39.5, 36.3, 35.5. HRMS (ESI) m/z calcd. for C29H35ClN3O4S [M+H]+ 556.2037, m/z found 556.2035.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-N,N-diethyl-4-{[(4-methoxyphenyl)methyl]amino} pyrrolidine-2-carboxamide (40) was prepared according to the general procedure A as yellow oil (54%). 1H NMR (300 MHz, CDCl3) δ 7.37 – 7.44 (m, 3H), 7.31 – 7.35 (m, 1H), 7.18 – 7.23 (m, 2H), 6.87 – 6.93 (m, 2H), 4.09 (d, J = 2.07 Hz, 2H), 3.91 – 4.00 (m, 3H), 3.82 – 3.86 (m, 1H), 3.81 (s, 3H), 3.39 – 3.51 (m, 2H), 3.18 – 3.29 (m, 1H), 2.88 – 3.08 (m, 3H), 2.36 – 2.48 (m, 1H), 2.14 (d, J = 14.69 Hz, 1H), 1.04 (t, J = 7.25 Hz, 3H), 0.97 (t, J = 7.16 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 174.0, 160.3, 135.1, 133.6, 131.3, 130.7, 129.5, 128.8, 127.1, 123.1, 114.6, 59.0, 56.7, 56.4, 55.3, 54.3, 48.5, 42.4, 41.6, 33.7, 15.0, 12.8. HRMS (ESI) m/z calcd. for C24H33ClN3O2 [M+H]+ 430.2261, m/z found 430.2239.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-4-{[(4-methoxyphenyl)methyl]amino}-N,N-dipropylpyrrolidine-2-carboxamide (41) was prepared according to the general procedure A as colorless oil (63%). 1H NMR (300 MHz, CDCl3) δ 7.55 (dd, J = 1.70, 7.54 Hz, 1H), 7.13 – 7.34 (m, 6H), 6.85 (d, J = 8.48 Hz, 2H), 3.73 – 3.92 (m, 7H), 3.70 (dd, J = 4.52, 8.85 Hz, 1H), 3.44 (d, J = 5.84 Hz, 1H), 2.99 – 3.35 (m, 6H), 2.81 (dd, J = 6.03, 9.61 Hz, 1H), 2.40 (td, J = 8.34, 13.28 Hz, 1H), 1.80 – 1.89 (m, 1H), 1.43 – 1.57 (m, 4H), 0.86 (dt, J = 4.52, 7.44 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 173.4, 158.9, 136.4, 133.6, 130.8, 129.7, 129.2, 128.1, 126.8, 113.9, 60.9, 58.3, 56.3, 55.3, 53.9, 50.7, 49.4, 48.3, 36.3, 22.8, 20.9, 11.4, 11.2. HRMS (ESI) m/z calcd. for C26H37ClN3O2 [M+H]+ 458.2574, m/z found 458.2563.
(3S,5S)-1-[(2-Chlorophenyl)methyl]-N-[(4-methoxyphenyl)methyl]-5-(4-phenylpiperazine-1-carbonyl)pyrrolidin-3-amine (42) was prepared according to the general procedure A as colorless oil (23%). 1H NMR (300 MHz, CDCl3) δ 7.45 (dd, J = 1.79, 7.44 Hz, 1H), 7.27 – 7.39 (m, 5H), 7.14 – 7.25 (m, 2H), 6.84 – 6.95 (m, 5H), 3.90 – 3.99 (m, 4H), 3.85 (dd, J = 3.49, 9.14 Hz, 1H), 3.80 (s, 3H), 3.77 (s, 1H), 3.54 – 3.71 (m, 3H), 3.46 (br. s., 1H), 3.35 (d, J = 10.55 Hz, 1H), 2.88 – 3.10 (m, 5H), 2.37 – 2.49 (m, 1H), 2.06 (d, J = 14.13 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 172.6, 159.7, 150.7, 135.4, 133.7, 130.9, 130.6, 129.6, 129.3, 128.8, 127.0, 120.7, 116.6, 114.3, 57.5, 56.4, 55.3, 54.6, 49.7, 49.6, 49.3, 45.6, 42.2, 34.7. HRMS (ESI) m/z calcd. for C30H36ClN4O2 [M+H]+ 519.2527, m/z found 519.2511.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-N-ethyl-4-{[(4-(methylthio)phenyl)methyl]amino} pyrrolidine-2-carboxamide (1) was prepared according to the general procedure A as colorless oil (52%). 1H NMR (300 MHz, CDCl3) δ 7.45 (br. s., 1H), 7.37 (dd, J = 3.67, 5.56 Hz, 1H), 7.29 – 7.33 (m, 1H), 7.23 (dd, J = 3.58, 5.84 Hz, 2H), 7.16 – 7.21 (m, 4H), 3.83 – 3.90 (m, 1H), 3.63 – 3.73 (m, 3H), 3.29 (d, J = 5.84 Hz, 1H), 3.24 (dd, J = 5.46, 9.98 Hz, 1H), 3.10 – 3.19 (m, 2H), 2.98 (d, J = 9.98 Hz, 1H), 2.61 (dd, J = 5.56, 10.08 Hz, 1H), 2.49 – 2.54 (m, 1H), 2.46 (s, 3H), 1.91 (dd, J = 2.73, 8.76 Hz, 3H), 1.02 (t, J = 7.35 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 173.5, 136.6, 136.3, 135.3, 133.8, 130.7, 129.3, 128.5, 128.3, 126.5, 126.5, 66.6, 59.2, 57.1, 55.8, 50.8, 36.7, 33.2, 15.6, 14.1. HRMS (ESI) m/z calcd. for C22H29ClN3OS [M+H]+ 418.1720, m/z found 418.1707.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-4-{[(4-(methylthio)phenyl)methyl]amino}-N-pentylpyrrolidine-2-carboxamide (43) was prepared according to the general procedure A as colorless oil (62%). 1H NMR (300 MHz, CDCl3) δ 7.47 (br. s., 1H), 7.31 – 7.39 (m, 2H), 7.21 – 7.26 (m, 2H), 7.17 – 7.20 (m, 4H), 3.84 – 3.91 (m, 1H), 3.59 – 3.75 (m, 3H), 3.30 (br. s., 1H), 3.25 (dd, J = 5.46, 9.80 Hz, 1H), 3.10 (dt, J = 6.97, 12.72 Hz, 2H), 2.99 (d, J = 10.17 Hz, 1H), 2.60 (dd, J = 5.46, 9.98 Hz, 1H), 2.48 – 2.54 (m, 1H), 2.46 (s, 3H), 1.91 (d, J = 10.36 Hz, 2H), 1.32 – 1.43 (m, 2H), 1.17 – 1.29 (m, 4H), 0.84 (t, J = 6.78 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 174.0, 137.1, 136.7, 135.8, 134.2, 131.0, 129.7, 128.9, 128.7, 126.9, 67.1, 59.6, 57.5, 56.3, 51.3, 38.9, 37.1, 29.2, 29.1, 22.3, 16.1, 13.9. HRMS (ESI) m/z calcd. for C25H35ClN3OS [M+H]+ 460.2189, m/z found 460.2172.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-4-{[(4-(methylthio)phenyl)methyl]amino}-N-(2-phenylethyl)pyrrolidine-2-carboxamide (44) was prepared according to the general procedure A as colorless oil (46%). 1H NMR (300 MHz, CDCl3) δ 7.40 (br. s., 1H), 7.33 (dd, J = 2.92, 4.43 Hz, 1H), 7.09 – 7.24 (m, 12H), 3.60 – 3.79 (m, 4H), 3.37 – 3.50 (m, 2H), 3.34 (d, J = 6.03 Hz, 1H), 3.21 (dd, J = 5.75, 9.51 Hz, 1H), 2.98 (d, J = 10.36 Hz, 1H), 2.67 – 2.76 (m, 2H), 2.47 – 2.58 (m, 2H), 2.45 (s, 3H), 1.86 (d, J = 12.62 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 173.8, 138.8, 137.8, 135.5, 134.7, 134.0, 130.7, 129.6, 129.1, 128.7, 128.6, 128.5, 126.9, 126.8, 126.4, 67.0, 58.6, 56.6, 55.9, 50.7, 39.9, 36.4, 35.4, 15.9. HRMS (ESI) m/z calcd. for C28H33ClN3OS [M+H]+ 494.2033, m/z found 494.2022.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-N-[2-(4-(methylthio)phenyl)ethyl]-4-{[(4-methoxyphenyl) methyl]amino}pyrrolidine-2-carboxamide (45) was prepared according to the general procedure A as colorless oil (50%). 1H NMR (300 MHz, CDCl3) δ 7.47 (br. s., 1H), 7.32 – 7.38 (m, 1H), 7.15 – 7.24 (m, 7H), 7.02 (d, J = 8.48 Hz, 2H), 6.73 (d, J = 8.67 Hz, 2H), 3.71 – 3.80 (m, 4H), 3.59 – 3.69 (m, 3H), 3.42 (dd, J = 6.69, 13.28 Hz, 1H), 3.32 – 3.38 (m, 1H), 3.26 (d, J = 4.14 Hz, 1H), 3.19 – 3.24 (m, 1H), 2.93 (d, J = 9.98 Hz, 1H), 2.61 – 2.71 (m, 2H), 2.51 (dd, J = 5.37, 10.08 Hz, 2H), 2.45 (s, 3H), 2.04 (br. s., 2H), 1.78 – 1.88 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 174.0, 158.2, 137.2, 136.5, 135.7, 134.0, 130.8, 130.7, 129.6, 129.5, 128.8, 128.7, 126.9, 113.9, 67.2, 59.3, 57.1, 56.1, 55.2, 51.2, 40.0, 37.1, 34.6, 16.0. HRMS (ESI) m/z calcd. for C29H35ClN3O2S [M+H]+ 524.2138, m/z found 524.2128.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-N-[2-(4-(methylthio)phenyl)ethyl]-4-{[(4-nitrophenyl) methyl]amino}pyrrolidine-2-carboxamide (46) was prepared according to the general procedure A as colorless oil (44%). 1H NMR (300 MHz, CDCl3) δ 7.93 (d, J = 8.67 Hz, 2H), 7.53 (t, J = 6.03 Hz, 1H), 7.23 – 7.28 (m, 1H), 7.06 – 7.18 (m, 8H), 3.51 – 3.70 (m, 4H), 3.37 (q, J = 6.78 Hz, 2H), 3.22 (d, J = 3.20 Hz, 1H), 3.17 (dd, J = 5.65, 9.80 Hz, 1H), 2.88 (d, J = 10.17 Hz, 1H), 2.72 (qd, J = 7.10, 14.13 Hz, 2H), 2.40 – 2.49 (m, 2H), 2.39 (s, 3H), 1.85 (br. s., 1H), 1.70 – 1.79 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 173.3, 145.7, 145.6, 136.4, 135.4, 134.6, 132.9, 129.4, 128.7, 128.4, 127.8, 127.7, 125.9, 125.8, 122.6, 66.1, 58.4, 56.3, 55.3, 50.3, 38.2, 36.1, 34.5, 14.9. HRMS (ESI) m/z calcd. for C28H32ClN4O3S [M+H]+ 539.1884, m/z found 539.1858.
(2S,4S)-1-[(2-Chlorophenyl)methyl]-N-[2-(3,4-difluorophenyl)ethyl]-4-{[(4-(methylthio)phenyl)methyl]amino}pyrrolidine-2-carboxamide (47) was prepared according to the general procedure A as colorless liquid (56%). 1H NMR (300 MHz, CDCl3) δ 7.58 (br. s., 1H), 7.34 (d, J = 7.35 Hz, 1H), 7.15 – 7.25 (m, 7H), 6.88 – 7.00 (m, 2H), 6.75 – 6.83 (m, 1H), 3.70 – 3.80 (m, 1H), 3.59 – 3.69 (m, 3H), 3.35 – 3.45 (m, 1H), 3.33 (d, J = 6.97 Hz, 2H), 3.24 (dd, J = 5.65, 9.61 Hz, 1H), 2.96 (d, J = 9.80 Hz, 2H), 2.60 – 2.70 (m, 2H), 2.54 (dd, J = 5.56, 10.46 Hz, 2H), 2.41 – 2.49 (m, 4H), 1.85 (d, J = 13.00 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 174.1, 137.5, 135.9, 135.6, 134.0, 130.6, 129.7, 128.8, 128.8, 126.9, 126.9, 124.5, 124.5, 124.4, 124.4, 117.5, 117.3, 117.2, 117.0, 67.1, 59.2, 57.1, 56.1, 51.2, 39.6, 36.9, 34.7, 15.9. HRMS (ESI) m/z calcd. for C28H31ClF2N3OS [M+H]+ 530.1844, m/z found 530.1824.
96-well calcium mobilization assay.
Two individual stable cell lines were created by over-expressing human NPFFR1 and NPFFR2 receptors in CHO-RD-HGA16 (Molecular Devices) cells. The day before the assay, cells were plated into 96-well black-walled assay plates at 30,000 cells/well (100 µL volume) in Ham’s F12 supplemented with 10% fetal bovine serum, 100 units of penicillin/streptomycin, and 100 μg/mL normocin™. The cells were incubated overnight at 37°C, 5% CO2. Prior to the assay, Calcium 5 dye (Molecular Devices) was reconstituted according to the manufacturer’s instructions. The reconstituted dye was diluted 1:40 in warm assay buffer (1X HBSS, 20 mM HEPES, 2.5 mM probenecid, pH 7.4 at 37°C). Growth medium was removed and the cells were gently washed with 100 μL of warm assay buffer. The cells were incubated for 45 minutes at 37°C, 5% CO2 in 200 μL of the diluted Calcium 5 dye. A single concentration of each test compound was prepared at 10x the desired final concentration in 2.25% BSA/8% DMSO/assay buffer. Serial dilutions of NPFF were prepared at 10x the desired final concentration in 0.25% BSA/1% DMSO/assay buffer, aliquoted into 96-well polypropylene plates, and warmed to 37°C. After the dye-loading incubation period, the cells were pretreated with 25 µL of the test compounds and incubated for 15 min at 37°C. After the pretreatment incubation period, the plate was read with a FlexStation II (Molecular Devices). Calcium-mediated changes in fluorescence were monitored every 1.52 seconds over a 60 second time period, with the FlexStation II adding 25 μL of the NPFF serial dilutions at the 19 second time point (excitation at 485 nm, detection at 525 nm). Peak kinetic reduction (SoftMax, Molecular Devices) relative fluorescent units (RFU) were plotted against the log of compound concentration. Nonlinear regression analysis (3-parameter) was used to generate EC50 values (GraphPad Prism, GraphPad Software, Inc., San Diego, CA). Apparent Ke values were calculated using the equation Ke = [L]/((EC50+/EC50−) – 1) where [L] is the concentration of test compound, EC50+ is the EC50 of NPFF with test compound, and EC50− is the EC50 of NPFF alone. Ke values were considered valid when the EC50+/EC50− ratio was at least 4.
384-well HTS.
Stable human NPFFR1 CHO-RD-HGA16 cells were plated in 30 μL/well volume at 5,000 cells/well in Ham’s F12 medium supplemented with 1% FBS and 100 units penicillin/streptomycin in 384-well Greiner µClear® black walled microplates using a MicroFlo™ Select dispenser fitted with a 5 µL cassette (BioTek). The plated cells were incubated overnight at 37 °C, 5% CO2, 95% relative humidity. The next day, compound test plates were prepared by diluting previously replicated library daughter plates with assay buffer to achieve a 100 μM (10X desired final concentration) working solution and filling columns 1, 2, 23, and 24 with positive and negative controls. An additional compound test plate containing the NPFF EC60 concentration (250 nM prepared at 10x the desired final concentration in 1% DMSO/assay buffer) was prepared for the antagonist portion of the screen. Calcium 5 dye (Bulk Kit, Molecular Devices), reconstituted according to the manufacturer’s instructions, was diluted 1:20 in pre-warmed (37°C) assay buffer (1X HBSS, 20 mM HEPES, 2.5 mM probenecid, pH 7.4 at 37 °C) and 30 µL was added to the plate with the Biomek NX, which was then incubated for 45 minutes at 37 °C, 5% CO2, 95% relative humidity. Using the Biomek NX, the dye loaded plate was pretreated with 8.5 µL of 8% DMSO/assay buffer and incubated for 15 minutes at 37 °C, 5% CO2, 95% relative humidity. After this incubation period, the plate was read with the Tetra to evaluate agonist activity. Calcium-mediated changes in fluorescence were monitored every 1 second over a 60 second time period, with the Tetra adding 8.5 µL from the compound plate at the 10 second time point (excitation at 470–495 nm, detection at 515–575 nm). The cell plate was then incubated for another 15 minutes at 37 °C, 5% CO2, 95% relative humidity after which it was read with the Tetra to evaluate antagonist activity. Calcium-mediated changes in fluorescence were monitored every 1 second over a 60 second time period, with the Tetra adding 8.5 µL from the NPFF EC60 plate at the 10 second time point (excitation at 470–495 nm, detection at 515–575 nm). Data was exported from ScreenWorks (Molecular Devices) using the response over baseline (ROB) statistic which presents data as a fold-response compared with the baseline sample. Percent inhibition was calculated using the equation (1 – (cmpd ROB / NPFF EC60 ROB)) × 100.
cAMP assay.
Stable human NPFFR1-CHO (ES-491-C) and NPFFR2-CHO (ES-490-C) cell lines were purchased from PerkinElmer and used with the Lance Ultra kit (TRF0262) to detect cAMP accumulation in low volume 96-well plates. Stimulation buffer containing 1X HBSS, 5 mM HEPES (pH 7.4), 0.1% BSA stabilizer, and 0.5 mM IBMX was prepared. Serial dilutions of the agonist control NPFF were prepared at 8X the desired final concentration in 2% DMSO/stimulation buffer and 2.5 µL was added to the assay plate. A single concentration of each test compound was prepared at 4X the desired final concentration in 2% DMSO/stimulation buffer and 5 µL was added to the assay plate. The EC80 concentration of forskolin (1 µM) was prepared at 8X in 2% DMSO/stimulation buffer and 2.5 µL was added to the assay plate. Cells were lifted from flasks with versene and spun at 270g for 10 minutes. The cell pellet was resuspended in stimulation buffer and 4,000 cells (10 µL) were added to each well. After incubating for 30 min at RT, Eu-cAMP tracer and uLIGHT-anti-cAMP working solutions were added per the manufacturer’s instructions. After 1 hour, the TR-FRET signal (ex 337 nm) was read on a CLARIOstar multimode plate reader (BMG Biotech, Cary NC). Fluorescence values at 665 nm were plotted against the log of compound concentration and nonlinear regression analysis (3-parameter) was used to generate EC50 values (GraphPad Prism, GraphPad Software, Inc., San Diego CA). Ke values were calculated using the same equation as described in the 96-well calcium mobilization methods.
Radioligand binding assay.
Binding assays were performed according to PerkinElmer’s protocol in a final volume of 500 µL of assay buffer (50 mM Tris-HCl, 1 mM MgCl2, 60 mM NaCl, 0.5% BSA, pH 7.4). For NPFF1, the assay mixture contained 25 µL of 0.065 nM [125I]NPFF (PerkinElmer, KD = 0.11 nM, NEX381), 25 µL of test compounds (prepared at 8X the final desired concentration in 8% DMSO/assay buffer), and 150 µL of CHO-hNPFFR1 membranes (1 µg protein/well, PerkinElmer, RBHNF1M400UA). Specific binding was defined as the difference between [125I]NPFF binding in the absence and presence of 100 nM final nonradiolabeled NPFF. After incubating at 27°C for 120 minutes, the binding assay was terminated by vacuum filtration onto Unifilter GF/C glass-fiber filters (pre-soaked in 0.1% PEI) using a Brandel (Gaithersburg, MD, USA) 96-well harvester, followed by three washes with ice-cold wash buffer (50 mM Tris-HCl, 0.1% BSA, pH 7.4). The filter plate was dried for 1 hr at 55°C. Microscint 20 (50 μL) was added to each well and filter-bound radioactivity was counted on a Packard TopCount NXT microplate scintillation and luminescence counter. Percentage of specific [125I]NPFF binding was plotted against the log of compound concentration. Data were fit to a one site (fit Ki) competitive binding model, logEC50=log(10^logKi*(1+RadioligandNM/HotKdNM)), to generate Ki values for the test compounds using GraphPad Prism (GraphPad Software, Inc., San Diego CA). NPFF2 binding assays were conducted with the same protocol, except that 0.1 nM [125I]NPFF (PerkinElmer, KD = 0.15 nM, NEX381) and CHO-hNPFFR2 membranes (1 µg protein/well, PerkinElmer, RBHNF2M400UA) were used.
Kinetic solubility assay.
10 μL of test compound stock solution (10 mM DMSO) was combined with 490 μL of PBS (potassium phosphate monobasic 1 mM, sodium phosphate dibasic 3 mM and sodium chloride 155 mM buffer). The solution was agitated on a VX-2500 multi-tube vortexer (VWR) for 2 hours at room temperature. Following agitation, the sample was filtrated on a glass-fiber filter (1 μm) and the eluate was diluted 200-fold with a mixture of acetonitrile: water (1:1). On each experimental occasion, nicardipine and imipramine were assessed as reference compounds for low and high solubility, respectively. All samples were assessed in triplicate and analyzed by LC-MS/MS using electrospray ionization against standards prepared in the same matrix.
Bidirectional MDCK-MDR1 permeability assay.
MDCK-mdr1 cells at passage 5 were seeded onto permeable polycarbonate supports in 12-well Costar Transwell plates and allowed to grow and differentiate for 3 days. On day 3, culture medium (DMEM supplemented with 10% FBS) was removed from both sides of the transwell inserts and cells were rinsed with warm HBSS. After the rinse step, the chambers were filled with warm transport buffer (HBSS containing 10 mM HEPES, 0.25% BSA, pH 7.4) and the plates were incubated at 37 oC for 30 min prior to TEER (Trans Epithelial Electric Resistance) measurements.
The buffer in the donor chamber (apical side for A-to-B assay, basolateral side for B-to-A assay) was removed and replaced with the working solution (10 μM test article in transport buffer). The plates were then placed at 37 oC under light agitation. At designated time points (30, 60 and 90 min), an aliquot of transport buffer from the receiver chamber was removed and replenished with fresh transport buffer. Samples were quenched with ice-cold ACN containing internal standard and then centrifuged to pellet protein. Resulting supernatants are further diluted with 50/50 ACN/H2O (H2O only for Atenolol) and submitted for LC-MS/MS analysis. Reported apparent permeability (Papp) values were calculated from single determination. Atenolol and propranolol were tested as low and moderate permeability references. Bidirectional transport of digoxin was assessed to demonstrate Pgp activity/expression.
The apparent permeability (Papp, measured in cm/s) of a compound is determined according to the following formula:
dQ/dt is the net rate of appearance in the receiver compartment
A is the area of the Transwell measured in cm2 (1.12 cm2)
Ci is the initial concentration of compound added to the donor chamber
60 is the conversion factor for minute to second
In vivo Pharmacology
Animals.
Adult male (n = 18) Sprague-Dawley rats (Harlan, Indianapolis, IN) weighing 225–300 g were individually housed on a 12/12-hour light/dark cycle with behavioral experiments conducted during the light period. Rats had free access to food and water except during test sessions, and were maintained and experiments were conducted in accordance with guidelines of the International Association for the Study of Pain 57 and with the 2011 Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources on Life Sciences, National Research Council, National Academy of Sciences, Washington, DC), and were approved by the Institutional Animal Care and Use Committee, University at Buffalo, the State University of New York (Buffalo, NY).
Drugs.
Fentanyl was purchased from Sigma-Aldrich (St. Louis, MO), dissolved in 0.9% saline, and injected subcutaneously in a volume of 1 ml/kg. Compounds 16 and 33 were dissolved in a vehicle of 20% dimethyl sulfoxide in saline and injected intraperitoneally in a volume of 1 ml/kg.
Fentanyl-induced hyperalgesia.
Nociceptive thresholds were measured using calibrated von Frey filaments (1.4–26 g; North Coast Medical, Morgan Hill, CA). Rats (n = 6 per group) were placed in elevated plastic chambers with a wire mesh floor (IITC Life Science Inc., Woodland Hills, CA) and allowed to habituate prior to testing. Filaments were applied perpendicularly to the medial plantar surface of the hind paw from below the mesh floor in an ascending order of filament force, beginning with the lowest filament. Filaments were applied until buckling occurred for approximately two seconds. Mechanical paw withdrawal thresholds (PWTs) correspond to the lowest force that elicited a withdrawal of the hind paw in at least two out of three applications. Forces larger than 26 g would physically elevate the non-CFA-treated paw and did not reflect pain-like behavior.
After a nociceptive baseline was established for each rat on day prior to and the day of fentanyl treatment (D-1 and D0), four subcutaneous injections of 0.06 mg/kg fentanyl each were injected at 15 min intervals for a total dose of 0.24 mg/kg.58, 59 In one group of rats, PWT measurements were taken on days 1–5 to monitor the onset of and recovery from the fentanyl-induced hyperalgesia. In the other two groups of rats, antinociceptive dose-effect curves for compounds 16 and 33 were established on day 1 using a multi-cycle cumulative dosing procedure, in which measurements were taken immediately prior to drug administration then 30 min after drug administration before the next injection, and continued for doses ranging from 3.2–32 mg/kg.
Data analysis.
PWTs were averaged within each group and plotted as a function of dose. Repeated measures one-way ANOVAs, with time or treatment entered as the within-subject factor, followed by Bonferroni’s post-hoc test were used to determine the statistical significances. P < 0.05 was considered statistically significant for all tests.
Acknowledgments
Funding Sources
This work was supported by Research Triangle Institute and National Institute on Drug Abuse, National Institutes of Health, U.S. (Grants DA040693 and DA032837 to Y.Z.).
ABBREVIATIONS
- DCM
dichloromethane
- 1,2-DCE
1,2-dichloroethane
- DME
1,2-dimethoxyethane
- FLIPR
fluorometric imaging plate reader
- GPCR
G-protein-coupled receptor
- HPLC
high performance liquid chromatography
- EC50
half-maximum effective concentration
- IC50
half-maximum inhibitory concentration
- MS
mass spectrometry
- NPFF
neuropeptide FF
- SAR
structure−activity relationship
- TLC
thin-layer chromatography
Footnotes
ASSOCIATED CONTENT
Supporting Information
HPLC analysis of target compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Fukusumi S, Fujii R, and Hinuma S (2006) Recent advances in mammalian RFamide peptides: the discovery and functional analyses of PrRP, RFRPs and QRFP, Peptides 27, 1073–1086. [DOI] [PubMed] [Google Scholar]
- (2).Elphick MR, and Mirabeau O (2014) The Evolution and Variety of RFamide-Type Neuropeptides: Insights from Deuterostomian Invertebrates, Front. Endocrinol 5, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Quillet R, Ayachi S, Bihel F, Elhabazi K, Ilien B, and Simonin F (2016) RF-amide neuropeptides and their receptors in Mammals: Pharmacological properties, drug development and main physiological functions, Pharmacol. Ther 160, 84–132. [DOI] [PubMed] [Google Scholar]
- (4).Yang HY, Fratta W, Majane EA, and Costa E (1985) Isolation, sequencing, synthesis, and pharmacological characterization of two brain neuropeptides that modulate the action of morphine, Proc. Natl. Acad. Sci. U. S. A 82, 7757–7761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bonini JA, Jones KA, Adham N, Forray C, Artymyshyn R, Durkin MM, Smith KE, Tamm JA, Boteju LW, Lakhlani PP, Raddatz R, Yao WJ, Ogozalek KL, Boyle N, Kouranova EV, Quan Y, Vaysse PJ, Wetzel JM, Branchek TA, Gerald C, and Borowsky B (2000) Identification and characterization of two G protein-coupled receptors for neuropeptide FF, J. Biol. Chem 275, 39324–39331. [DOI] [PubMed] [Google Scholar]
- (6).Liu Q, Guan XM, Martin WJ, McDonald TP, Clements MK, Jiang Q, Zeng Z, Jacobson M, Williams DL Jr., Yu H, Bomford D, Figueroa D, Mallee J, Wang R, Evans J, Gould R, and Austin CP (2001) Identification and characterization of novel mammalian neuropeptide FF-like peptides that attenuate morphine-induced antinociception, J. Biol. Chem 276, 36961–36969. [DOI] [PubMed] [Google Scholar]
- (7).Yang HY, and Iadarola MJ (2006) Modulatory roles of the NPFF system in pain mechanisms at the spinal level, Peptides 27, 943–952. [DOI] [PubMed] [Google Scholar]
- (8).Simonin F (2006) Neuropeptide FF receptors as therapeutic targets, Drugs Future 31, 603–609. [Google Scholar]
- (9).Yu HP, Zhang N, Zhang T, Wang ZL, Li N, Tang HH, Zhang R, Zhang MN, Xu B, Fang Q, and Wang R (2016) Activation of NPFF2 receptor stimulates neurite outgrowth in Neuro 2A cells through activation of ERK signaling pathway, Peptides 86, 24–32. [DOI] [PubMed] [Google Scholar]
- (10).Yang HY, Tao T, and Iadarola MJ (2008) Modulatory role of neuropeptide FF system in nociception and opiate analgesia, Neuropeptides 42, 1–18. [DOI] [PubMed] [Google Scholar]
- (11).Mouledous L, Mollereau C, and Zajac JM (2010) Opioid-modulating properties of the neuropeptide FF system, Biofactors 36, 423–429. [DOI] [PubMed] [Google Scholar]
- (12).Mollereau C, Roumy M, and Zajac JM (2005) Opioid-modulating peptides: mechanisms of action, Curr. Top. Med. Chem 5, 341–355. [DOI] [PubMed] [Google Scholar]
- (13).Malin DH, Lake JR, Fowler DE, Hammond MV, Brown SL, Leyva JE, Prasco PE, and Dougherty TM (1990) FMRF-NH2-like mammalian peptide precipitates opiate-withdrawal syndrome in the rat, Peptides 11, 277–280. [DOI] [PubMed] [Google Scholar]
- (14).Gouarderes C, Sutak M, Zajac JM, and Jhamandas K (1993) Antinociceptive effects of intrathecally administered F8Famide and FMRFamide in the rat, Eur. J. Pharmacol 237, 73–81. [DOI] [PubMed] [Google Scholar]
- (15).Lake JR, Hammond MV, Shaddox RC, Hunsicker LM, Yang HY, and Malin DH (1991) IgG from neuropeptide FF antiserum reverses morphine tolerance in the rat, Neurosci. Lett 132, 29–32. [DOI] [PubMed] [Google Scholar]
- (16).Elhabazi K, Trigo JM, Mollereau C, Mouledous L, Zajac JM, Bihel F, Schmitt M, Bourguignon JJ, Meziane H, Petit-demouliere B, Bockel F, Maldonado R, and Simonin F (2012) Involvement of neuropeptide FF receptors in neuroadaptive responses to acute and chronic opiate treatments, Br. J. Pharmacol 165, 424–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Simonin F, Schmitt M, Laulin JP, Laboureyras E, Jhamandas JH, MacTavish D, Matifas A, Mollereau C, Laurent P, Parmentier M, Kieffer BL, Bourguignon JJ, and Simonnet G (2006) RF9, a potent and selective neuropeptide FF receptor antagonist, prevents opioid-induced tolerance associated with hyperalgesia, Proc. Natl. Acad. Sci. U. S. A 103, 466–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lameh J, Bertozzi F, Kelly N, Jacobi PM, Nguyen D, Bajpai A, Gaubert G, Olsson R, and Gardell LR (2010) Neuropeptide FF receptors have opposing modulatory effects on nociception, J. Pharmacol. Exp. Ther 334, 244–254. [DOI] [PubMed] [Google Scholar]
- (19).Malin DH, Lake JR, Short PE, Blossman JB, Lawless BA, Schopen CK, Sailer EE, Burgess K, and Wilson OB (1996) Nicotine abstinence syndrome precipitated by an analog of neuropeptide FF, Pharmacol. Biochem. Behav 54, 581–585. [DOI] [PubMed] [Google Scholar]
- (20).Chen JC, Li JY, Liang KW, and Huang YK (1999) Neuropeptide FF potentiates the behavioral sensitization to amphetamine and alters the levels of neurotransmitters in the medial prefrontal cortex, Brain Res 816, 220–224. [DOI] [PubMed] [Google Scholar]
- (21).Kotlinska JH, Gibula-Bruzda E, Koltunowska D, Raoof H, Suder P, and Silberring J (2012) Modulation of neuropeptide FF (NPFF) receptors influences the expression of amphetamine-induced conditioned place preference and amphetamine withdrawal anxiety-like behavior in rats, Peptides 33, 156–163. [DOI] [PubMed] [Google Scholar]
- (22).Kotlinska J, Pachuta A, and Silberring J (2008) Neuropeptide FF (NPFF) reduces the expression of cocaine-induced conditioned place preference and cocaine-induced sensitization in animals, Peptides 29, 933–939. [DOI] [PubMed] [Google Scholar]
- (23).Wu CH, Tao PL, and Huang EY (2010) Distribution of neuropeptide FF (NPFF) receptors in correlation with morphine-induced reward in the rat brain, Peptides 31, 1374–1382. [DOI] [PubMed] [Google Scholar]
- (24).Mankus JV, and McCurdy CR (2012) Nonpeptide ligands of neuropeptide FF: current status and structural insights, Future Med. Chem 4, 1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Gealageas R, Schneider S, Humbert JP, Bertin I, Schmitt M, Laboureyras E, Dugave C, Mollereau C, Simonnet G, Bourguignon JJ, Simonin F, and Bihel F (2012) Development of sub-nanomolar dipeptidic ligands of neuropeptide FF receptors, Bioorg. Med. Chem. Lett 22, 7471–7474. [DOI] [PubMed] [Google Scholar]
- (26).Bihel F, Humbert JP, Schneider S, Bertin I, Wagner P, Schmitt M, Laboureyras E, Petit-Demouliere B, Schneider E, Mollereau C, Simonnet G, Simonin F, and Bourguignon JJ (2015) Development of a peptidomimetic antagonist of neuropeptide FF receptors for the prevention of opioid-induced hyperalgesia, ACS Chem. Neurosci 6, 438–445. [DOI] [PubMed] [Google Scholar]
- (27).Elhabazi K, Humbert JP, Bertin I, Quillet R, Utard V, Schneider S, Schmitt M, Bourguignon JJ, Laboureyras E, Ben Boujema M, Simonnet G, Ancel C, Simonneaux V, Beltramo M, Bucher B, Sorg T, Meziane H, Schneider E, Petit-Demouliere B, Ilien B, Bihel F, and Simonin F (2017) RF313, an orally bioavailable neuropeptide FF receptor antagonist, opposes effects of RF-amide-related peptide-3 and opioid-induced hyperalgesia in rodents, Neuropharmacol 118, 188–198. [DOI] [PubMed] [Google Scholar]
- (28).Maletinska L, Ticha A, Nagelova V, Spolcova A, Blechova M, Elbert T, and Zelezna B (2013) Neuropeptide FF analog RF9 is not an antagonist of NPFF receptor and decreases food intake in mice after its central and peripheral administration, Brain Res 1498, 33–40. [DOI] [PubMed] [Google Scholar]
- (29).Kawakami JK, Konkel MJ, Boteju LW, Wetzel JM, Noble SA, and Wan H (2003) Quinazolino- and quinolino- guanidines as ligands for the neurop eptide ff (npff) receptors, Synaptic Pharmaceutical Corporation, France, WO 200302667 A1. [Google Scholar]
- (30).Scully AL, Davis RE, Vanover KE, Gardell LR, Lameh J, Kelly NM, Bertozzi F, and Sherbukhin V (2005) Treating neuropathic pain with neuropeptide FF receptor 2 agonists, Acadia Pharmaceuticals Inc, US, WO 2005/031000 A2. [Google Scholar]
- (31).Fecher A, Fretz H, Hilpert K, Breu V, Giller T, and Valdenaire O (2005) Guanidine derivatives, Actelion Pharmaceuticals Ltd., Switzerland, WO 2005023781 A1. [Google Scholar]
- (32).Gaubert G, Bertozzi F, Kelly NM, Pawlas J, Scully AL, Nash NR, Gardell LR, Lameh J, and Olsson R (2009) Discovery of selective nonpeptidergic neuropeptide FF2 receptor agonists, J. Med. Chem 52, 6511–6514. [DOI] [PubMed] [Google Scholar]
- (33).Caroff E, Steger M, Valdenaire O, Fecher A, Breu V, Hilpert K, Fretz H, and Giller T (2004) Guanidine derivatives and use thereof as neuropeptide FF receptor antagonists, Actelion Pharmaceuticals Ltd, Switzerland, WO 2004/083218 A1. [Google Scholar]
- (34).Journigan VB, Mesangeau C, Vyas N, Eans SO, Cutler SJ, McLaughlin JP, Mollereau C, and McCurdy CR (2014) Nonpeptide small molecule agonist and antagonist original leads for neuropeptide FF1 and FF2 receptors, J. Med. Chem 57, 8903–8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Mahar Doan KM, Lakhman SS, and Boje KM (2000) Blood-brain barrier transport studies of organic guanidino cations using an in situ brain perfusion technique, Brain Res 876, 141–147. [DOI] [PubMed] [Google Scholar]
- (36).Osakada N, Shinoda K, Kunori S, Shirai T, Toki S, Ichikawa S, Kosaka N, Ichimura M, and Shimada J (2004) Neuropeptide FF receptor antagonist, Kyowa Hakko Kogyo Co. Ltd., Japan, WO 2004080965 A1. [Google Scholar]
- (37).Nakamura T, Saito S, Yoshinaga M, Ohta H, Kawamura M, and Ishizaka T (2009) Heterocyclic compound, Taisho Pharmaceuticals, Japan, WO 2009038012 A1. [Google Scholar]
- (38).Zhang Y, Gilliam A, Maitra R, Damaj MI, Tajuba JM, Seltzman HH, and Thomas BF (2010) Synthesis and biological evaluation of bivalent ligands for the cannabinoid 1 receptor, J. Med. Chem 53, 7048–7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).German N, Decker AM, Gilmour BP, Gay EA, Wiley JL, Thomas BF, and Zhang Y (2014) Diarylureas as allosteric modulators of the cannabinoid CB1 receptor: structure-activity relationship studies on 1-(4-chlorophenyl)-3-{3-[6-(pyrrolidin-1-yl)pyridin-2-yl]phenyl}urea (PSNCBAM-1), J. Med. Chem 57, 7758–7769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Nguyen T, German N, Decker AM, Li JX, Wiley JL, Thomas BF, Kenakin TP, and Zhang Y (2015) Structure-activity relationships of substituted 1H-indole-2-carboxamides as CB1 receptor allosteric modulators, Bioorg. Med. Chem 23, 2195–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Gouarderes C, Mazarguil H, Mollereau C, Chartrel N, Leprince J, Vaudry H, and Zajac JM (2007) Functional differences between NPFF1 and NPFF2 receptor coupling: high intrinsic activities of RFamide-related peptides on stimulation of [35S]GTPgammaS binding, Neuropharmacol 52, 376–386. [DOI] [PubMed] [Google Scholar]
- (42).Vyas N, Mollereau C, Cheve G, and McCurdy CR (2006) Structure-activity relationships of neuropeptide FF and related peptidic and non-peptidic derivatives, Peptides 27, 990–996. [DOI] [PubMed] [Google Scholar]
- (43).Decker AM, Gay EA, Mathews KM, Rosa TC, Langston TL, Maitra R, and Jin C (2017) Development and validation of a high-throughput calcium mobilization assay for the orphan receptor GPR88, J. Biomed. Sci 24, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Zhang JH, Chung TD, and Oldenburg KR (1999) A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays, J. Biomol. Screen 4, 67–73. [DOI] [PubMed] [Google Scholar]
- (45).Cui BQ, Yu J, Yu FC, Li YM, Chang KJ, and Shen YH (2015) Synthesis of (1R,4R)-2,5-diazabicyclo[2.2.1]heptane derivatives by an epimerization-lactamization cascade reaction, Rsc. Adv 5, 10386–10392. [Google Scholar]
- (46).Feng BY, Simeonov A, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, and Austin CP (2007) A high-throughput screen for aggregation-based inhibition in a large compound library, J. Med. Chem 50, 2385–2390. [DOI] [PubMed] [Google Scholar]
- (47).Sassano MF, Doak AK, Roth BL, and Shoichet BK (2013) Colloidal aggregation causes inhibition of G protein-coupled receptors, J. Med. Chem 56, 2406–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Perrey DA, German NA, Gilmour BP, Li JX, Harris DL, Thomas BF, and Zhang Y (2013) Substituted tetrahydroisoquinolines as selective antagonists for the orexin 1 receptor, J. Med. Chem 56, 6901–6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Perrey DA, German NA, Decker AM, Thorn D, Li JX, Gilmour BP, Thomas BF, Harris DL, Runyon SP, and Zhang Y (2015) Effect of 1-substitution on tetrahydroisoquinolines as selective antagonists for the orexin-1 receptor, ACS Chem. Neurosci 6, 599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Gnewuch CT, and Friedman HL (1972) Pyridine isosteres of the -adrenergic antagonists, 2-(p-nitrophenyl)-1-isopropylamino-2-ethanol and 3-(p-nitrophenoxy)-1-isopropylamino-2-propanol, J. Med. Chem 15, 1321–1324. [DOI] [PubMed] [Google Scholar]
- (51).Perrey DA, Decker AM, Li JX, Gilmour BP, Thomas BF, Harris DL, Runyon SP, and Zhang Y (2015) The importance of the 6- and 7-positions of tetrahydroisoquinolines as selective antagonists for the orexin 1 receptor, Bioorg. Med. Chem 23, 5709–5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Rankovic Z (2015) CNS drug design: balancing physicochemical properties for optimal brain exposure, J. Med. Chem 58, 2584–2608. [DOI] [PubMed] [Google Scholar]
- (53).Wenlock MC, Austin RP, Barton P, Davis AM, and Leeson PD (2003) A comparison of physiochemical property profiles of development and marketed oral drugs, J. Med. Chem 46, 1250–1256. [DOI] [PubMed] [Google Scholar]
- (54).Di L, and Kerns E (2016) Solubility, In Drug-Like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization 2nd ed., pp 56–85, Academic Press, New York. [Google Scholar]
- (55).Wang Q, Rager JD, Weinstein K, Kardos PS, Dobson GL, Li J, and Hidalgo IJ (2005) Evaluation of the MDR-MDCK cell line as a permeability screen for the blood-brain barrier, Int. J. Pharm 288, 349–359. [DOI] [PubMed] [Google Scholar]
- (56).Bihel F (2016) Opioid adjuvant strategy: improving opioid effectiveness, Future Med. Chem 8, 339–354. [DOI] [PubMed] [Google Scholar]
- (57).Zimmermann M (1983) Ethical guidelines for investigations of experimental pain in conscious animals, Pain 16, 109–110. [DOI] [PubMed] [Google Scholar]
- (58).Celerier E, Rivat C, Jun Y, Laulin JP, Larcher A, Reynier P, and Simonnet G (2000) Long-lasting hyperalgesia induced by fentanyl in rats: preventive effect of ketamine, Anesthesiol 92, 465–472. [DOI] [PubMed] [Google Scholar]
- (59).Laboureyras E, Aubrun F, Monsaingeon M, Corcuff JB, Laulin JP, and Simonnet G (2014) Exogenous and endogenous opioid-induced pain hypersensitivity in different rat strains, Pain Res. Manag 19, 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]