

Abstract
Hexokinase 1 (HK1) phosphorylates glucose to glucose-6-phosphate, the first rate-limiting step in glycolysis. Homozygous and heterozygous variants in HK1 have been shown to cause autosomal recessive non-spherocytic hemolytic anemia, autosomal recessive Russe type hereditary motor and sensory neuropathy, and autosomal dominant retinitis pigmentosa (adRP). We report seven patients from six unrelated families with a neurodevelopmental disorder associated with developmental delay, intellectual disability, structural brain abnormality, and visual impairments in whom we identified four novel, de novo missense variants in the N-terminal half of HK1. Hexokinase activity in red blood cells of two patients was normal, suggesting that the disease mechanism is not due to loss of hexokinase enzymatic activity.
Subject terms: Neurodevelopmental disorders, Medical genetics, Next-generation sequencing, Genetics research
Introduction
Neurodevelopmental disorders affect 1–3% of children and encompass a wide range in severity and associated behavioral differences [1]. Identifying the etiology of neurodevelopmental disorders has been challenging given the diversity of genetic and non-genetic causes. Exome sequencing (ES) is an effective tool to diagnose patients with phenotypically similar and etiologically heterogeneous neurodevelopmental disorders and to discover new genetic etiologies. Many of these conditions affect reproductive fitness and arise from de novo variants in genes with a critical role in brain development and/or function [2].
Hexokinases catalyze the first rate-limiting step of glycolysis; phosphorylation of glucose to produce glucose-6-phosphate (G6P). There are four hexokinases (I-IV), each of which has a specific tissue expression pattern. HK1 is ubiquitously expressed but is abundant in the brain and is known as the ‘brain-type hexokinase’. HK1 consists of N-terminal regulatory and C-terminal catalytic domains. Bi-allelic variants affecting the catalytic active site in the C-terminus and in the 5’ UTR cause non-spherocytic hemolytic anemia (OMIM #235700) and hereditary motor and sensory neuropathy, Russe type (OMIM #605285), respectively [3–6]. In addition, a rare heterozygous variant close to the C-terminal end, c.2539G>A (p.(Glu847Lys)), has recently been shown to cause autosomal dominant retinitis pigmentosa 79 (OMIM #617460) [7–9]. In this study, we report four novel, de novo variants in HK1 in seven patients from six unrelated families with neurodevelopmental problems, structural brain abnormalities, and visual impairment, expanding the phenotype of variants in HK1.
Subjects and methods
This study was approved by the Institutional Review Board of Columbia University. Informed consent was obtained from all participants. Genomic DNA was extracted from whole blood from the affected children and their biological parents. ES was performed in 12,289 (5132 females; 7157 males) individuals with mainly developmental delay/intellectual disabilities (DD/ID) with or without accompanying abnormalities in multiple organ systems at GeneDx with exon targets captured using the Agilent SureSelect Human All Exon V4 (50 Mb) kit or the Clinical Research Exome (Agilent Technologies, Santa Clara, CA). The sequencing methodology and variant interpretation protocol have been previously described [10]. One additional patient with one of the same variants we identified in the original series was identified through personal communication with an external clinic and laboratory. All HK1 variants were confirmed by Sanger sequencing.
Results
Seven patients from six unrelated families were found to carry four different, de novo heterozygous missense variants in HK1 (NM_000188.2; NP_000179.2): c.1241G>A (p.(Gly414Glu)) and c.1252A>G (p.(Lys418Glu)) in exon 9 and c.1334C>T (p.(Ser445Leu)) and c.1370C>T (p.(Thr457Met)) in exon 10 (Exons are numbered like in NG_012077.1). Predictions of variant pathogenicity are given in Table 1. Two of the variants, c.1370C>T (p.(Thr457Met)) and c.1334C>T (p.(Ser445Leu)), were each observed recurrently in two unrelated families. In addition, the c.1370C>T (p.(Thr457Met)) variant was identified in two affected siblings in one family, and parental analyses did not show parental mosaicism in the blood. We calculated the probability of identifying de novo variants in HK1 with the same phenotype. Assuming a mutation rate of 1e-8 per base pair per meiosis, and considering the six independent meiosis identified from a single laboratory (assuming parental origin for the two that were recurrent in the same family), the probability of our findings is p = 6.68e-5 using a Poisson test and p = 0.0016 with the full-likelihood VarPrism approach [11]. Identified variants have been deposited in ClinVar under the following accession numbers: SCV000570679.3, SCV000570071.3, SCV000854591, and SCV000491089.1.
Table 1.
Predicted pathogenicity of de novo HK1 (NM_000188.2) variants
| Variant | Chr10 Coordinates (GRHc38) | Polyphen-2 | MetaSVM | SIFT | Mutation Taster | PROVEAN | CADD Phred | REVEL | M-CAP | MPC |
|---|---|---|---|---|---|---|---|---|---|---|
| c.1241G>A p.(Gly414Glu) | 69380071 | 0.9711 (Damaging) | 0.93853 (D) | 0.90636 (Damaging) | 0.70825 (Disease Causing) | 0.90248 (Deleterious) | 31 | 0.738 | 0.424 | 2.15 |
| c.1252A>G p.(Lys418Glu) | 69380082 | 0.54346 (Possibly Damaging) | 0.26258 (T) | 0.17625 (Tolerated) | 0.70825 (Disease Causing) | 0.32326 (Neutral) | 25.2 | 0.354 | 0.022 | 1.6 |
| c.1334C>T p.(Ser445Leu)a | 69382555 | 0.9711 (Damaging) | 0.99239 (D) | 0.90636 (Damaging) | 0.70825 (Disease Causing) | 0.80328 (Deleterious) | 21.7 | 0.924 | 0.664 | 1.8 |
| c.1370C>T p.(Thr457Met)b | 69382591 | 0.9711 (Damaging) | 0.58994 (T) | 0.90636 (Damaging) | 0.70825 (Disease Causing) | 0.77077 (Deleterious) | 22.1 | 0.358 | 0.049 | 1.94 |
aThis variant is seen in two individuals from two unrelated families
bThis variant is seen in three individuals from two unrelated families
The four missense variants are all located in highly conserved residues (Fig. 1a). None of the variants were observed in 1000 Genomes [12], the NHLBI GO Exome Sequencing Project (Exome Variant Server, http://evs.gs.washington.edu/EVS), ExAC (exac.broadinstitute.org), gnomAD (gnomad.broadinstitute.org), or in our own local (GeneDx) database of >100,000 exomes consisting of individuals affected with various largely pediatric phenotypes and their healthy first degree relatives. GnomAD v2.1 gene constraint metrics for HK1 indicate the gene is under constraint for both loss-of-function (pLI = 0.91) and missense (Z-score = 3.34) variation in the general population. Regional missense constraint scores (MPC) for each variant are high and range from 1.6 to 2.15 (Table 1) [13].
Fig. 1.

a Sequence alignment of HK1 across different species. Mutated residues in our patients are shown in bold red. b 3D crystal structure of HK1 obtained from Protein Data Bank (PDB ID: 1HKC) and variants localizations. Our variants are localized in the vicinity of the glucose-6-phosphate binding site near the end of N-terminus and in the interdomain α-helix
Clinical findings of our patients are summarized in Table 2. Of note, patients 6 and 7 were similarly affected siblings, and they each died around the age of 1 year due to respiratory infections. Prenatal and neonatal histories were largely unremarkable except a resolved cystic brain lesion in one patient and prematurity in another. Features common to the majority of individuals with the HK1 variants include global developmental delay (7/7), intellectual disability (5/5), optic atrophy and/or retinitis pigmentosa (7/7), structural brain anomalies on MRI including cerebral and cerebellar atrophy and thin corpus callosum (6/7), hypotonia/hypertonia (5/7), speech problems (4/5), and ataxia (3/4). Three patients have feeding difficulties and two patients have musculoskeletal abnormalities (torticollis, scoliosis, hip dislocation, and pes planus). Two patients have nonspecific mildly dysmorphic facial features (Fig. 2). The deceased siblings were noted to have laryngotracheomalacia at birth. There were several features observed in only a single patient including unilateral facial weakness, cerebellar atrophy, and hearing loss.
Table 2.
Detailed clinical findings of patients with novel, de novo HK1 (NM_000188.2) variants
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | |
|---|---|---|---|---|---|---|---|
| Age (years) | 34 | 9 | 14 | 44 | 8 | 1a | 1a |
| Gender | Female | Female | Male | Male | Male | Male | Female |
| Variant | c.1241G>A p.(Gly414Glu) | c.1252A>G p.(Lys418Glu) | c.1334C>T p.(Ser445Leu) | c.1334C>T p.(Ser445Leu) | c.1370C>T p.(Thr457Met) | c.1370C>T p.(Thr457Met) | c.1370C>T p.(Thr457Met) |
| Prenatal | Unremarkable | Cystic brain lesions found at 7mos gestation but later resolved | Unremarkable | Premature, born at 8 mos gestation | Unremarkable | Unremarkable | Unremarkable |
| DD | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Intellectual Disability | Yes (IQ is ~ 70) | Yes | Yes | Yes | Yes | n/a | n/a |
| Microcephaly | No | No | No | No | No | No | No |
| Short Stature | Yes | Yes | No | No | No | No | No |
| Age at Sitting | Delayed | 2 years | 8 mos | 8 mos | n/a | Not achieved | Not achieved |
| Age at Walking | 27 mos | Cannot walk unassisted | 18 mos | 16 mos | 12 mos | n/a | n/a |
| Age at Talking & Current speech | Delayed | First words at 14 months & Significant limited expressive language now, only a few words | 2 years & Speaks in sentences and has word finding difficulties | 18 mos | Delayed & Dysarthric speech | n/a | n/a |
| Dysmorphic features | 5th finger clinodactyly | Right earlobe crease, frontal bossing, epicanthal folds, anteverted nose, bulbous nasal tip, preauricular pit, thin upper lip | No | No | Flat occiput, slight synophrys (father), widely spaced teeth | No | No |
| Neurologic & Behavioral Problems | Ataxia Anxiety | Truncal hypotonia Limb hypertonia (lower > upper) Brisk DTRs Some contractures in hamstrings, No self-care skills | Ataxia, Staring spells, Tingling in legs (EMG normal), Hypotonia Will hug and kiss strangers | No | Progressive neurologic decline, abnormal tone, mild wide spaced gait, bulbar weakness (Mild drooling), Swallowing dysfunction Recent onset right-sided facial weakness | Seizures Infantile spasms Limb hypertonia | Seizures Infantile spasms Hypertonia |
| EEG | n/a | Normal | Normal | n/a | Mild diffuse slowing | Consistent with early myoclonic epileptic encephalopathy | Abnormal (verbal disclosure) |
| Brain MRI | Abnormal signaling in caudate pathway and putamen | Cerebral & cerebellar atrophy, Thin corpus callosum, Periventricular leukomalacia, Possible gray matter heterotropia | Cystic lesion on brain | n/a | Lesions in brainstem (concerning for a demyelinating disease vs autoimmune process vs Guillain Barre syndrome) | Abnormal autopsy findings Volume loss along the white matter adjacent to the atria and frontal horns of the ventricle, atrophy of pons and brainstem, bilateral subdural fluid collection | Ventricular dilatation predominantly involving the occipital and temporal horns of the lateral ventricles with prominence of the subarachnoid space raising the possibility of underlying atrophy and a migrational anomaly |
| Visual | Retinitis Pigmentosa Peripheral vision loss and gaze abnormality | Cortical visual impairment, Strabismus, Astigmatism | Retinitis pigmentosa, Cone-rod dystrophy, Optic atrophy, Photophobia, slight exotropia, bilateral strabismus | Retinitis Pigmentosa (Age of diagnosis: 7 years) Optic atrophy | Bilateral optic atrophy | Optic atrophy Nystagmus | Optic atrophy |
| Other | Kyphoscoliosis Pes planus |
Scoliosis, bilateral hip dislocation (Wears AFOs) Torticollis, clonus, and spasticity as infant, Poor weight gain |
FMF (Het MEFV mutation), Gags easily and sometimes chokes on food |
Hypertension |
Full cheeks (steroid usage) Truncal obesity High blood vs zero CSF glutamine, high CSF lactate and pyruvate, high peripheral blood lactate |
Feeding difficulty GERD Small umbilical hernia Laryngotracheomalacia |
Failure to thrive GERD G-tube Hearing loss Laryngotracheomalacia Respiratory distress |
DD developmental delay, IQ intelligence quotient, OFC occipitofrontal circumference; DTR deep tendon reflex, VUS variant of unknown significance
aThese siblings passed away at the age of 1 year
Fig. 2.

Frontal and profile photos of Patient 2 showing mildly dysmorphic facial features of frontal bossing, anteverted nose, bulbous nasal tip, and thin upper lip
We determined the kinetic properties of red blood cell hexokinase in two patients (c.1252A>G, p.(Lys418Glu) and c.1334C>T, p.(Ser445Leu)) using previously described methods [14] and found no difference between control and patient samples in the affinity of mutant HK for glucose or ATP, nor was there any difference in thermal stability of these two variants (Fig. 3).
Fig. 3.
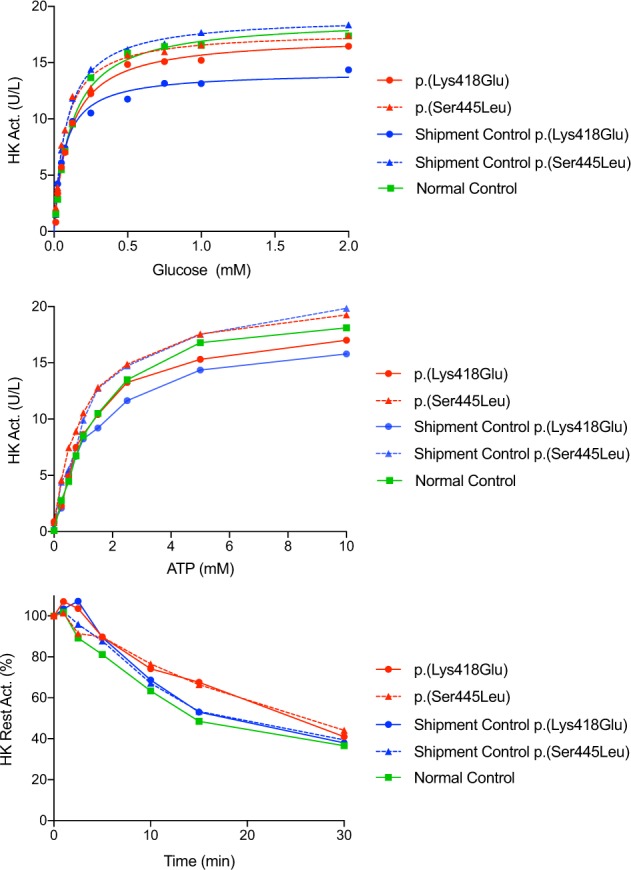
Thermostability, glucose affinity, and ATP affinity measurements on red blood cells from two patients carrying c.1252A>G (p.(Lys418Glu)) and c.1334C>T (p.(Ser445Leu)) variants and controls. In addition to the internal healthy control sample, peripheral blood samples from one unaffected parent of each patient were used to adjust the possible effects of shipment (“shipment control”) from the interpretation
Discussion
We identified seven patients from six independent families with overlapping neurodevelopmental features, structural brain abnormalities including cerebral and cerebellar atrophy and thin corpus callosum, and optic atrophy and/or retinitis pigmentosa and identified four novel, de novo missense variants in highly conserved residues in the N-terminal half of HK1.
There are four hexokinases in humans, each encoded by a different gene that phosphorylates glucose to G6P, the first rate-limiting step in glucose metabolism. HK1 contains an amino-terminal regulatory and carboxy-terminal catalytic domains. Alternative splicing results in different isoforms, which differ only in the first exon and are specific to different cell types such as red blood cells, retina, and spermatogenic cells [7, 15–17]. All of our variants fall into the exons shared across isoforms. The canonical HK1 isoform is expressed ubiquitously; however, due to its abundance in brain, it is known as ‘brain-type hexokinase.’ It is also known, along with HK2, as a mitochondrial hexokinase. It can bind the porin protein (VDAC1) of the outer mitochondrial membrane (OMM) via its N-terminus 12 amino acid hydrophobic sequence and couple oxidative phosphorylation with glycolysis. It has been shown that with this unique feature, HK1 also interacts with the apoptotic pathway genes such as Akt [18–22].
The variants that have been associated with HK1-related clinical phenotypes are shown in Fig. 4. The most common clinical phenotype associated with HK1 variants is non-spherocytic hemolytic anemia (OMIM #235700) caused by bi-allelic variants affecting the hexokinase activity [3, 4, 14, 23, 24]. Homozygous variants in the 5’ UTR of HK1 have been associated with hereditary motor and sensory neuropathy, Russe Type (OMIM #605285) [5, 6, 25]. A rare heterozygous missense variant close to the C-terminal end, c.2539G>A (p.(Glu847Lys)), has been shown to cause autosomal dominant retinitis pigmentosa 79 (OMIM #617460) and other forms of retinal dystrophies [7–9]. Interestingly, Sullivan et al., reported homozygosity of the c.2539G>A (p.(Glu847Lys)) variant in a patient with early onset (at age 4 years old) retinitis pigmentosa but without hemolytic anemia or developmental abnormalities, and Wang et al., reported two asymptomatic adult family members who carry the heterozygous c.2539G>A (p.(Glu847Lys)) variant [7, 8]. In both hereditary motor and sensory neuropathy, Russe Type disease and retinitis pigmentosa 79, no change in hexokinase activity has been reported, and the pathogenic mechanism of those variants is unknown. Additionally, increased expression of HK1 was reported in patients with congenital hyperinsulinism [26], and three non-coding variants were reported in one large family with congenital hyperinsulinism as potentially causative [27].
Fig. 4.

Schematic representation of variants reported in the literature for different phenotypes (brown: autosomal recessive hereditary sensory and motor neuropathy, Russe type [numbered according to NM_033500.2]; green: autosomal recessive non-spherocytic hemolytic anemia [numbered according to NM_033496.2]; blue: autosomal dominant retinitis pigmentosa 79 [numbered according to NM_000188.2]; and red: identified in our cohort [numbered according to NM_000188.2]). Hexokinase activities for red, brown, and blue colored variants were measured normal. § Carrier of p.(Arg94Gln)+/− was also homozygous for glucose-6-phosphate dehydrogenase (G6PDH) variants. *c.−193A>G is reported to be seen in 2 siblings and only one of them exhibited severe hemolysis. † Individual carrying p.(Thr601Met) variant did not have hemolysis although hexokinase activity was diminished. ‡ Patient carrying p.(His868Tyr) had hemolysis but hexokinase activity was measured normal
Glucose-6-phosphate inhibits the kinase activity by binding multiple sites in the N- and C-terminal halves. Binding of the phosphate and pyranose moieties of G6P to the residues 413–415 (Asp-Gly-Ser) and residue 449 (Ser), respectively, results in conformational changes propagated to the C-terminal half and inhibits ATP binding, and hence kinase activity [28, 29]. Orthophosphate (Pi) relieves this inhibitory effect by competing with the phosphate moiety of G6P. The N- and C-terminal halves of HK1 are connected by an interdomain helix required to exert full Pi relief [30]. Since all of our variants fall into the region (Fig. 1b) common to all HK1 isoforms, we measured the hexokinase activity in red blood cells of two patients (c.1334C>T, p.(Ser445Leu) and c.1252A>G, p.(Lys418Glu)) but found no difference in enzyme activity or kinetic properties between controls and patient samples (Fig. 3), consistent with the lack of (hemolytic) anemia in our patients. These results, along with the normal hexokinase activity found in other HK1-related phenotypes without non-spherocytic hemolytic anemia, suggest a different pathogenic mechanism for the dominant missense variants.
All but one of our patients show structural brain abnormalities and neurodevelopmental problems along with optic atrophy and/or retinitis pigmentosa (Table 2). Although neurodevelopmental problems and structural brain abnormalities have been reported in some patients with hexokinase deficiency-related non-spherocytic hemolytic anemia (Table 3) [3, 31–33], whether these findings were direct consequences of HK1 variants, indirect consequences of severe intrauterine anemia due to hexokinase deficiency, or due to different genetic and/or non-genetic factors is not known. In addition, there are individuals with both homozygous and heterozygous variants with decreased hexokinase activity but no phenotypic manifestations including non-spherocytic hemolytic anemia. We hypothesize that heterozygous missense variants identified in this study may result in a gain-of-function which might lead to accumulation of the protein in cells of affected tissues leading to cellular dysfunction, apoptosis, or cell death, adverse effect on mitochondrial function, or conferring new phosphorylation targets for HK1, thereby impacting eye and brain function. It has been shown that the intracellular accumulation of misfolded proteins plays an important role in some forms of retinitis pigmentosa [34]. Accumulation of misfolded proteins, in general, leads to endoplasmic reticulum stress which later results in apoptotic cell death via unfolded protein response [35]. Autophagy and mitophagy are among the well-known pathophysiological mechanisms in optic atrophy and neurological disorders [36]. A gain-of-function mechanism has been hypothesized for some missense variants in OPA1 and has also been proposed for autosomal dominant optic atrophy with extraocular manifestations such as sensorineural hearing loss, ataxia, myopathy, spasticity, and peripheral neuropathy [36, 37]. In addition to the neurodevelopmental problems, we have noted optic atrophy in four patients and retinitis pigmentosa in three patients.
Table 3.
Previous publications in which additional neurodevelopmental findings were also reported in patients with hexokinase deficiency-related non-spherocytic hemolytic anemia
| Publication | Goebel et al. [31] | Gilsanz et al. [32] | Magnani et al. [33] | Kanno et al. [3] | Koralkova et al. [14] |
| Hemolytic anemia | + | + | + | + | + |
| Additional findings | Dysmorphic findings |
Dysmorphic findings Mild MR Short stature Hearing problem |
Hypertonia and opisthotonos DTR↓ Psychomotor retardation HSM |
IUGR Periventricular leucomalacia |
Psychomotor retardation Epilepsy (CNS bleeding?) |
| Hexokinase deficiency | + | + | +a | +a | + |
| HK1 variant & Inheritance | ? | ? | ? |
c.493-481_1028+454del & AR |
c.278 A > G (p.(Arg94Gln)) & AR |
MR mental retardation, DTR deep tendon reflex, HSM hepatosplenomegaly, IUGR intrauterine growth retardation, CNS central nervous system, AR autosomal recessive
Variants are numbered according to NM_033496.2
aThese patients’ heterozygous parents also had diminished hexokinase activity without any phenotypic manifestation
By using protein–protein interaction network analysis, about 30% of the human proteins have been proposed to have multifunctional properties [38, 39] or ‘moonlighting’ activities [40]. Although it has been previously classified in this group along with other glycolytic pathway enzymes such as glucose-6-phosphate isomerase (GPI) [41], HK1 has yet to be classified as multifunctional by network analyses. Several studies have shown that HK1 has an anti-apoptotic function via its mitochondrial role [19, 20, 42–45] and it has been proposed to have a role in some psychiatric [46, 47] and late-onset neurologic disorders [48–50] via its effect on mitochondrial homeostasis. Variants identified in this study could alter HK1 binding to porin protein in the outer mitochondrial membrane and alter the apoptotic pathway or some other aspect of mitochondrial function.
Two of the four variants, c.1334C>T (p.(Ser445Leu)) and c.1370C>T (p.(Thr457Met)), are recurrently observed in unrelated individuals in our study. Furthermore, the c.1370C>T (p.(Thr457Met)) variant is seen in two affected siblings of one family in which parental analyses did not show mosaicism in parental blood. This family also has an unaffected child who does not carry the HK1 variant. These two variants are among the four heterozygous variants detected in four different patients in Deciphering Developmental Disorders study along with another missense variant in C-terminal domain and a canonical splice site variant close to N-terminus [51]. Further phenotypic delineation of those and other patients would contribute to the understanding of the genotype-phenotype relationships, mutational hot spots, mode of action of variants, and disease progress.
In conclusion, we describe de novo heterozygous missense variants in patients with both neurodevelopmental problems, structural brain abnormalities, and optic atrophy and/or retinitis pigmentosa thereby expanding the HK1-associated human disease spectrum. Future clinical and functional studies are needed to elucidate the underlying pathophysiological mechanism leading to the observed phenotypes.
Acknowledgements
We thank families for their generous contribution. This work was supported in part by a grant from the Simons Foundation and the JPB Foundation. This work was supported in part by grants from the National Eye Institute [RO1EY012910 (EAP), R01EY026904 (KMB/EAP) and P30EY014104 (MEEI core support)], the Foundation Fighting Blindness (USA, EAP). Sequencing and analysis of samples from MEEI was provided by the Center for Mendelian Genomics at the Broad Institute of MIT and Harvard and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1 HG008900 to Daniel MacArthur and Heidi Rehm.
Compliance with ethical standards
Conflict of interest
Megan Cho, Aida Telegrafi, Ganka Douglas, Kristin G. Monaghan, Amber Begtrup, and Kyle Retterer are employees of GeneDx.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Soden SE, Saunders CJ, Willig LK, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6:265ra168. doi: 10.1126/scitranslmed.3010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ku CS, Polychronakos C, Tan EK, et al. A new paradigm emerges from the study of de novo mutations in the context of neurodevelopmental disease. Mol Psychiatry. 2013;18:141–53. doi: 10.1038/mp.2012.58. [DOI] [PubMed] [Google Scholar]
- 3.Kanno H, Murakami K, Hariyama Y, Ishikawa K, Miwa S, Fujii H. Homozygous intragenic deletion of type I hexokinase gene causes lethal hemolytic anemia of the affected fetus. Blood. 2002;100:1930. doi: 10.1182/blood-2002-05-1599. [DOI] [PubMed] [Google Scholar]
- 4.van Wijk R, Rijksen G, Huizinga EG, Nieuwenhuis HK, van Solinge WW. HK Utrecht: missense mutation in the active site of human hexokinase associated with hexokinase deficiency and severe nonspherocytic hemolytic anemia. Blood. 2003;101:345–7. doi: 10.1182/blood-2002-06-1851. [DOI] [PubMed] [Google Scholar]
- 5.Hantke J, Chandler D, King R, et al. A mutation in an alternative untranslated exon of hexokinase 1 associated with hereditary motor and sensory neuropathy—Russe (HMSNR) Eur J Hum Genet. 2009;17:1606–14. doi: 10.1038/ejhg.2009.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sevilla T, Martinez-Rubio D, Marquez C, et al. Genetics of the Charcot-Marie-Tooth disease in the Spanish Gypsy population: the hereditary motor and sensory neuropathy-Russe in depth. Clin Genet. 2013;83:565–70. doi: 10.1111/cge.12015. [DOI] [PubMed] [Google Scholar]
- 7.Sullivan LS, Koboldt DC, Bowne SJ, et al. A dominant mutation in hexokinase 1 (HK1) causes retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2014;55:7147–58. doi: 10.1167/iovs.14-15419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang F, Wang Y, Zhang B, et al. A missense mutation in HK1 leads to autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2014;55:7159–64. doi: 10.1167/iovs.14-15520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan Z, Li B, Xu M, et al. The phenotypic variability of HK1-associated retinal dystrophy. Sci Rep. 2017;7:7051. doi: 10.1038/s41598-017-07629-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanaka AJ, Cho MT, Millan F, et al. Mutations in SPATA5 are associated with microcephaly, intellectual disability, seizures, and hearing loss. Am J Hum Genet. 2015;97:457–64. doi: 10.1016/j.ajhg.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu H, Coon H, Li M, Yandell M, Huff CD. VARPRISM: incorporating variant prioritization in tests of de novo mutation association. Genome Med. 2016;8:91. doi: 10.1186/s13073-016-0341-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abecasis GR, Auton A, Brooks LD, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Samocha KE, Kosmicki JA, Karczewski KJ, O'Donnell-Luria AH, Pierce-Hoffman E, MacArthur DG, et al. Regional missense constraint improves variant deleteriousness prediction. bioRxiv. 2017;148353.
- 14.Koralkova P, Mojzikova R, van Oirschot B, et al. Molecular characterization of six new cases of red blood cell hexokinase deficiency yields four novel mutations in HK1. Blood Cells Mol Dis. 2016;59:71–6. doi: 10.1016/j.bcmd.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 15.Murakami K, Blei F, Tilton W, Seaman C, Piomelli S. An isozyme of hexokinase specific for the human red blood cell (HKR) Blood. 1990;75:770–5. [PubMed] [Google Scholar]
- 16.Mori C, Nakamura N, Welch JE, Shiota K, Eddy EM. Testis-specific expression of mRNAs for a unique human type 1 hexokinase lacking the porin-binding domain. Mol Reprod Dev. 1996;44:14–22. doi: 10.1002/(SICI)1098-2795(199605)44:1<14::AID-MRD2>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 17.Mori C, Welch JE, Fulcher KD, O’Brien DA, Eddy EM. Unique hexokinase messenger ribonucleic acids lacking the porin-binding domain are developmentally expressed in mouse spermatogenic cells. Biol Reprod. 1993;49:191–203. doi: 10.1095/biolreprod49.2.191. [DOI] [PubMed] [Google Scholar]
- 18.Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, Thompson CB. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol. 2003;23:7315–28. doi: 10.1128/MCB.23.20.7315-7328.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V. In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem J. 2004;377:347–55. doi: 10.1042/bj20031465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Majewski N, Nogueira V, Bhaskar P, et al. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–30. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 21.Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–18. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Majewski N, Nogueira V, Robey RB, Hay N. Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol Cell Biol. 2004;24:730–40. doi: 10.1128/MCB.24.2.730-740.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Vooght KM, van Solinge WW, van Wesel AC, Kersting S, van Wijk R. First mutation in the red blood cell-specific promoter of hexokinase combined with a novel missense mutation causes hexokinase deficiency and mild chronic hemolysis. Haematologica. 2009;94:1203–10. doi: 10.3324/haematol.2008.002881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bianchi M, Magnani M. Hexokinase mutations that produce nonspherocytic hemolytic anemia. Blood Cells Mol Dis. 1995;21:2–8. doi: 10.1006/bcmd.1995.0002. [DOI] [PubMed] [Google Scholar]
- 25.Dohrn MF, Glockle N, Mulahasanovic L, et al. Frequent genes in rare diseases: panel-based next generation sequencing to disclose causal mutations in hereditary neuropathies. J Neurochem. 2017;143:507–22. doi: 10.1111/jnc.14217. [DOI] [PubMed] [Google Scholar]
- 26.Henquin JC, Sempoux C, Marchandise J, et al. Congenital hyperinsulinism caused by hexokinase I expression or glucokinase-activating mutation in a subset of beta-cells. Diabetes. 2013;62:1689–96. doi: 10.2337/db12-1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pinney SE, Ganapathy K, Bradfield J, et al. Dominant form of congenital hyperinsulinism maps to HK1 region on 10q. Horm Res Paediatr. 2013;80:18–27. doi: 10.1159/000351943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang TY, Alechina O, Aleshin AE, Fromm HJ, Honzatko RB. Identification of a phosphate regulatory site and a low affinity binding site for glucose 6-phosphate in the N-terminal half of human brain hexokinase. J Biol Chem. 1998;273:19548–53. doi: 10.1074/jbc.273.31.19548. [DOI] [PubMed] [Google Scholar]
- 29.Sui D, Wilson JE. Functional interactions between the noncovalently associated N- and C-terminal halves of mammalian Type I hexokinase. Arch Biochem Biophys. 2002;401:21–8. doi: 10.1016/S0003-9861(02)00029-2. [DOI] [PubMed] [Google Scholar]
- 30.Hashimoto M, Wilson JE. Kinetic and regulatory properties of HK I(+), a modified form of the type I isozyme of mammalian hexokinase in which interactions between the N- and C-terminal halves have been disrupted. Arch Biochem Biophys. 2002;399:109–15. doi: 10.1006/abbi.2001.2744. [DOI] [PubMed] [Google Scholar]
- 31.Goebel KM, Gassel WD, Goebel FD, Kaffarnik H. Hemolytic anemia and hexokinase deficiency associated with malformations. Klin Wochenschr. 1972;50:849–51. doi: 10.1007/BF01496345. [DOI] [PubMed] [Google Scholar]
- 32.Gilsanz F, Meyer E, Paglia DE, Valentine WN. Congenital hemolytic anemia due to hexokinase deficiency. Am J Dis Child. 1978;132:636–7. doi: 10.1001/archpedi.1978.02120310100023. [DOI] [PubMed] [Google Scholar]
- 33.Magnani M, Stocchi V, Canestrari F, et al. Human erythrocyte hexokinase deficiency: a new variant with abnormal kinetic properties. Br J Haematol. 1985;61:41–50. doi: 10.1111/j.1365-2141.1985.tb04058.x. [DOI] [PubMed] [Google Scholar]
- 34.Datta R, Waheed A, Bonapace G, Shah GN, Sly WS. Pathogenesis of retinitis pigmentosa associated with apoptosis-inducing mutations in carbonic anhydrase IV. Proc Natl Acad Sci Usa. 2009;106:3437–42. doi: 10.1073/pnas.0813178106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin JH, Li H, Yasumura D, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–9. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kane MS, Alban J, Desquiret-Dumas V, et al. Autophagy controls the pathogenicity of OPA1 mutations in dominant optic atrophy. J Cell Mol Med. 2017;21:2284–97. doi: 10.1111/jcmm.13149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu-Wai-Man P, Griffiths PG, Gorman GS, et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain. 2010;133:771–86. doi: 10.1093/brain/awq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Becker E, Robisson B, Chapple CE, Guenoche A, Brun C. Multifunctional proteins revealed by overlapping clustering in protein interaction network. Bioinformatics. 2012;28:84–90. doi: 10.1093/bioinformatics/btr621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chapple CE, Robisson B, Spinelli L, Guien C, Becker E, Brun C. Extreme multifunctional proteins identified from a human protein interaction network. Nat Commun. 2015;6:7412. doi: 10.1038/ncomms8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Campbell RM, Scanes CG. Endocrine peptides ‘moonlighting’ as immune modulators: roles for somatostatin and GH-releasing factor. J Endocrinol. 1995;147:383–96. doi: 10.1677/joe.0.1470383. [DOI] [PubMed] [Google Scholar]
- 41.Sriram G, Martinez JA, McCabe ER, Liao JC, Dipple KM. Single-gene disorders: what role could moonlighting enzymes play? Am J Hum Genet. 2005;76:911–24. doi: 10.1086/430799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robey RB, Hay N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene. 2006;25:4683–96. doi: 10.1038/sj.onc.1209595. [DOI] [PubMed] [Google Scholar]
- 43.Schindler A, Foley E. A functional RNAi screen identifies hexokinase 1 as a modifier of type II apoptosis. Cell Signal. 2010;22:1330–40. doi: 10.1016/j.cellsig.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 44.Craig TA, Ramachandran PL, Bergen HR, 3rd, Podratz JL, Windebank AJ, Kumar R. The regulation of apoptosis by the downstream regulatory element antagonist modulator/potassium channel interacting protein 3 (DREAM/KChIP3) through interactions with hexokinase I. Biochem Biophys Res Commun. 2013;433:508–12. doi: 10.1016/j.bbrc.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schindler A, Foley E. Hexokinase 1 blocks apoptotic signals at the mitochondria. Cell Signal. 2013;25:2685–92. doi: 10.1016/j.cellsig.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 46.Regenold WT, Pratt M, Nekkalapu S, Shapiro PS, Kristian T, Fiskum G. Mitochondrial detachment of hexokinase 1 in mood and psychotic disorders: implications for brain energy metabolism and neurotrophic signaling. J Psychiatr Res. 2012;46:95–104. doi: 10.1016/j.jpsychires.2011.09.018. [DOI] [PubMed] [Google Scholar]
- 47.Shan D, Mount D, Moore S, Haroutunian V, Meador-Woodruff JH, McCullumsmith RE. Abnormal partitioning of hexokinase 1 suggests disruption of a glutamate transport protein complex in schizophrenia. Schizophr Res. 2014;154:1–13. doi: 10.1016/j.schres.2014.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCoy MK, Kaganovich A, Rudenko IN, Ding J, Cookson MR. Hexokinase activity is required for recruitment of parkin to depolarized mitochondria. Hum Mol Genet. 2014;23:145–56. doi: 10.1093/hmg/ddt407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okatsu K, Iemura S, Koyano F, et al. Mitochondrial hexokinase HKI is a novel substrate of the Parkin ubiquitin ligase. Biochem Biophys Res Commun. 2012;428:197–202. doi: 10.1016/j.bbrc.2012.10.041. [DOI] [PubMed] [Google Scholar]
- 50.Saraiva LM, Seixas da Silva GS, Galina A, et al. Amyloid-beta triggers the release of neuronal hexokinase 1 from mitochondria. PLoS One. 2010;5:e15230. doi: 10.1371/journal.pone.0015230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542:433–8. doi: 10.1038/nature21062. [DOI] [PMC free article] [PubMed] [Google Scholar]