

Abstract
We aimed to investigate the pathogenicity of cardiac ion channel variants previously associated with SIDS. We reviewed SIDS-associated variants previously reported in databases and the literature in three large population-based cohorts; The ExAC database, the Inter99 study, and the UK Biobank (UKBB). Variants were classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines. Of the 92 SIDS-associated variants, 59 (64%) were present in ExAC, 18 (20%) in Inter99, and 24 (26%) in UKBB. Using the Inter99 cohort, we found no difference in J-point amplitude and QTc-interval between carriers and non-carriers for 14/18 variants. There was no difference in the risk of syncope (P = 0.32), malignant ventricular arrhythmia (P = 0.96), and all-cause mortality (P = 0.59) between carriers and non-carriers. The ACMG guidelines reclassified 75% of all variants as variant-of-uncertain significance, likely benign, and benign. We identified ~2/3 of variants previously associated with SIDS and found no significant associations with electrocardiographic traits, syncope, malignant ventricular arrhythmia, or all-cause mortality. These data indicate that many of these variants are not highly penetrant, monogenic causes of SIDS and underline the importance of frequent reappraisal of genetic variants to avoid future misdiagnosis.
Subject terms: Genetics, Genetics research
Introduction
Sudden infant death syndrome (SIDS) is defined as the sudden unexpected death of an infant <1 year of age with onset of the fatal episode apparently occurring during sleep and remaining unexplained despite thorough investigation, which includes a complete autopsy and review of the circumstances of death, and the clinical history [1]. The prevalence of SIDS has fallen dramatically since the American Academy of Pediatrics in 1992 recommended infants to be placed in a non-prone sleeping position [2] following the New Zealand Cot Death Study [3], which resulted in a >50% reduction of SIDS [4]. Nevertheless, SIDS is still one of the major causes of infant mortality [5]. A total of 1563 SIDS cases were accounted for in 2013 in the United States, which corresponds to an incidence of 4 per 10,000 live births [6]. A number of studies have investigated a possible link between SIDS and genetic variants in different physiological pathways [7]. Today, 17 genes encoding cardiac ion channels and ion channel regulators have been associated with SIDS [8, 9].
Postmortem genetic testing of SIDS victims is not routine practice but can be done for family segregation analysis or targeted diagnostic purposes if a primary electrical cardiac disorder is suspected or known in the family, e.g., long QT Syndrome (LQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), and Brugada syndrome (BrS) [10, 11]. Furthermore, guidelines for prevention of sudden cardiac death recommend that first-degree relatives of SIDS victims should undergo cardiac evaluation [12]. In order to provide patients with appropriate genetic counseling and clinical management, the genetic foundation of which decision are made upon must be correct and accurate. In recent years numerous studies have questioned a substantial proportion of variants previously associated with monogenic cardiac traits [13–18]. To facilitate a critical and evidence-based classification of variants, the American College of Medical Genetics and Genomics (ACMG) recently developed new standards and guidelines for classification of variants associated with Mendelian diseases [19].
In this study, we aimed to assess (I) whether variants previously associated with SIDS were present in three large independent genetic databases; (II) whether carrier-status was associated with ECG-traits previously associated with sudden cardiac death; (III) whether carrier-status was associated with an increased risk of syncope, malignant ventricular arrhythmia, and all-cause mortality. Lastly (IV), we aimed to (re)classify identified variants using the recently published ACMG guidelines.
Methods
Identification of SIDS-associated variants
All missense and nonsense variants in ion channel and ion channel regulator genes previously associated with SIDS were identified using the Human Gene Mutation Database (HGMD). The HGMD was queried using the search term “sudden infant death syndrome” and “SIDS”. In addition, variants identified in studies by Andreasen et al. [20] and Wilders et al. [21], and a literature search in PubMed, for any recently published studies were included. Articles that did not report original data, or were not written in English, were excluded.
Study populations
ExAC database
The Exome Aggregation Consortium (ExAC) database contains exome sequencing data from 60,706 unrelated adult individuals from a diverse set of populations. Individuals in ExAC <18 years of age and with severe pediatric diseases were excluded [22]. The ExAC was used to obtain genotype frequencies of variants previously associated with SIDS [22].
The Inter99 study
The Inter99 study constitutes ~6000 individuals randomly drawn from a sex- and age-stratified sample of the population [23] (Table 1). The study is a Danish population-based, randomized intervention study (CT00289237, ClinicalTrials.gov) investigating the effects of lifestyle interventions on cardiovascular death in participants included from the age of 30 years and above. Patients were included from March 1999 to March 2006 and followed until December 2012. The study cohort is of Danish origin and has previously been described in detail [24].
Table 1.
Baseline characteristics for participants in Inter99
| Carrier | Non-carrier | P-value | |
|---|---|---|---|
| Participant information | |||
| Number of subjects | 323 | 5860 | |
| Sex, male; n (%) | 155 (48.0) | 2864 (48.9) | 0.80 |
| Age, years; mean (±SD) | 46.7 (8.1) | 46.2 (7.9) | 0.32 |
| BMI, mean (±SD) | 26.1 (4.1) | 26.3 (4.7) | 0.49 |
| Systolic blood pressure; mean (±SD) | 130.5 (16.6) | 130.7 (17.6) | 0.60 |
| Diastolic blood pressure; mean (±SD) | 82.4 (10.6) | 82.6 (11.4) | 0.51 |
BMI body mass index, n number, SD standard deviation
Electrocardiograms (ECGs) were digitally stored in the MUSE® Cardiology Information System and processed using version 21 of the Marquette 12SL algorithm (GE Healthcare, Wauwatosa, WI). Electronic ECGs were used to assess whether carriers of SIDS variants had longer QTc-intervals, J-point amplitude, and RR-interval. We used Bazett’s formula to obtain rate-corrected values of the QT-interval (QTc). Prolonged QTc was defined as QTc ≥450 ms for men and ≥460 ms for women, according to Schwartz et al. [25]. We report the J-point, as an amplitude in millimeters (1 mm = 0.1 mV). In addition to digitally measured J-point amplitudes, two experienced physicians analyzed all ECGs manually and independently for diagnostic type 1 Brugada pattern [12], and the non-diagnostic type 2 Brugada pattern [26] that may warrant further investigation [27]. The RR-interval was defined as the duration between two successive peaks.
All Danish citizens are assigned a unique personal civil registration number, which enables linkage of data across multiple nationwide healthcare registries. This makes it possible to gather information on death, emigration, any hospital, outpatient clinic, or emergency room discharge diagnosis on an individual level. Malignant ventricular arrhythmia was defined as a composite endpoint of ventricular fibrillation (VF), ventricular tachycardia (VT), and cardiac arrest. Using the Danish National Patient Registry, we identified patients, who had previously been discharged with or who during follow-up had obtained one of the following ICD-10 codes: syncope (R55.9), ventricular tachycardia (I47.2), ventricular fibrillation (I49.0), and cardiac arrest (I46). Data on death during follow-up was accessed using the Danish Causes of Death Registry [28]. Details on genotyping are provided in the Supplementary Methods.
The UK Biobank
The UK Biobank is a large, population-based, prospective cohort study holding phenotype and genotype information on 408,961 ethnically matched European participants, aged 40–69 years at the time of recruitment [29]. Participants were recruited between 2006 and 2010. Using the Michigan PheWeb browser, we investigated whether the interrogated variants were associated with severe cardiac disorders (cardiac arrest [I46], malignant ventricular arrhythmias [VT I47.2; VF I49.0], or cardiomyopathy [I42]) [30]. Details on genotyping are provided in the Supplementary Methods.
ACMG guidelines
The American College of Medical Genetics and Genomics (ACMG) standards and guidelines [19] were used to reevaluate each variant into a five-tier terminology system: pathogenic, likely pathogenic, variant of uncertain significance (VUS), likely benign, and benign. The variant classification builds on different evidence such as functional studies, co-segregation, population data, and computational data. The online tool provided by Kleinberger et al. [31] was used to standardize the process of classifying the variants.
All original articles were searched for available information on functional effect and familial co-segregation of the variants.
Statistical analyses
In Inter99, RR-interval, QTc-interval, and J-point amplitude were compared using a one-way analysis of variance. The QTc-interval was adjusted for gender. Logistic regression was used to estimate the relationship between carrier-status and risk of syncope, malignant ventricular arrhythmia, and all-cause mortality. The model was adjusted for gender and age. Survival probability was assessed using the Kaplan–Meier method, and differences in mortality for carriers and non-carriers was tested with a two-tailed log-rank test.
For the comparison between the proportion of variants identified in ExAC compared with those not identified in ExAC, a two-tailed Exact Binomial Test was employed for each of the five ACMG classification categories.
Minor allele frequencies were obtained by dividing the allele count with the total allele number for each variant. Genotype prevalence was obtained by dividing the total number of heterozygote (one minor allele) and homozygote (two minor alleles) carriers with the total number of individuals. To avoid inflated genotype prevalences, we excluded variants with minor allele frequencies > 3%.
A two-sided P-value < 0.05 was considered statically significant. Genetic analyses were performed using PLINK (version 1.9) [32]. Statistical analyses were performed using the R (version 3.4.3).
Results
We found 92 variants in 17 cardiac ion channel or ion channel regulator genes previously associated with SIDS. Supplementary Table 1 and Supplementary Table 2 summarize variant classification, based on genotypic prevalence in the three interrogated databases, in silico predictions, functional characterizations, and evidence of family history.
ExAC: allele frequencies of SIDS variants
Of the 92 variants previously associated with SIDS, we found 59 (64%) variants in ExAC (Supplementary Table 1). A genotype frequency of > 5% is standalone criteria for benign classification. We found five variants that had allele frequencies higher than this; KCNQ1 p.(P448R) (9.8% in East Asian population), SCN5A p.(H558R) (22% across populations), SCN5A p.(S1103Y) (9.7% in African/African American population), SCN5A p.(R1193Q) (7% in East Asian population), and KCNH2 p.(R1047L) (7% in Finnish population).
Inter99: risk of syncope, malignant ventricular arrhythmia, and all-cause mortality
In the Inter99 cohort, we found 18 of the 92 variants (20%; Supplementary Table 1), in 323 carriers (332 alleles), corresponding to a genotype prevalence of 1:19 (323/6,183). We found no statistically significant difference in RR-intervals, QTc-intervals and J-point amplitude for the majority of the interrogated variants (Table 2). Four of the 18 variants showed significant ECG changes (P < 0.05), these were located in the genes CAV3 (rs72546668), HCN4 (rs62641689), and SCN5A (rs1805124 and rs41261344).
Table 2.
Associations between variants identified in the Inter99 cohort and ECG characteristics
| Genes | Variant | Amino acid | refSNP | Number of individuals | Mean RR-interval | Mean QTcB-interval | Mean STJ V1-amplitude | Mean STJ V2-amplitude | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Carrier | Non-carrier | Carrier, ms (SD) | Non-carrier, ms (SD) | P-value | Carrier, ms (SD) | Non-carrier, ms (SD) | P-value | Carrier, mm (SD) | Non-carrier, mm (SD) | P-value | Carrier, mm (SD) | Non-carrier, mm (SD) | P-value | ||||
| CAV3 | c.216 C > G | p.(Cys72Trp) | rs116840776 | 11 | 6099 | 928.91 (147.16) | 917.85 (152.46) | 0.654 | 422.30 (23.74) | 423.68 (22.82) | 0.563 | 0.01 (0.19) | 0.08 (0.31) | 0.589 | 0.23 (0.40) | 0.39 (0.51) | 0.547 |
| CAV3 | c.233 C > T | p.(Thr78Met) | rs72546668 | 36 | 6074 | 884.00 (145.25) | 918.07 (152.4) | 0.183 | 421.11 (22.95) | 423.69 (22.82) | 0.450 | 0.18 (0.33)a | 0.08 (0.31) | 0.0380 | 0.49 (0.56) | 0.39 (0.51) | 0.177 |
| GPD1L | c.247 G > A | p.(Glu83Lys) | rs72552292 | 1 | 6109 | 1090.00 (NA) | 917.84 (152.43) | 0.149 | 404.20 (NA) | 423.68 (22.82) | 0.135 | 0.34 (NA) | 0.08 (0.31) | 0.359 | 0.92 (NA) | 0.39 (0.51) | 0.100 |
| GPD1L | c.370 A > G | p.(Ile124Val) | rs72552293 | 32 | 6078 | 912.00 (174.82) | 917.90 (152.33) | 0.977 | 426.58 (22.09) | 423.66 (22.83) | 0.680 | 0.06 (0.29) | 0.08 (0.31) | 0.851 | 0.29 (0.52) | 0.39 (0.51) | 0.507 |
| HCN4 | c.2275 G > A | p.(Val759Ile) | rs62641689 | 87 [1] | 6022 | 948.11 (161.79)a [668.00 (NA)] | 917.47 (152.25) | 0.038 [0.076] | 422.02 (21.74) [428.23 (NA)] | 423.70 (22.84) | 0.274 [0.687] | 0.09 (0.30) [0.83 (NA)]a | 0.08 (0.31) | 0.672 [0.024] | 0.38 (0.45) [1.07 (NA)] | 0.39 (0.51) | 0.991 [0.282] |
| KCNH2 | c.442 C > T | p.(Arg148Trp) | rs139544114 | 7 | 6103 | 874.57 (127.14) | 917.92 (152.46) | 0.460 | 428.85 (9.46) | 423.67 (22.83) | 0.551 | 0.11 (0.35) | 0.08 (0.31) | 0.703 | 0.45 (0.49) | 0.39 (0.51) | 0.649 |
| KCNH2 | c.3140 G > T | p.(Arg1047Leu) | rs36210421 | 364 [4] | 5742 | 912.67 (143.86) [863 (131.92)] | 918.23 (152.99) | 0.505 [0.371] | 421.03 (22.41) [430.15 (15.28)] | 423.69 (22.90) | 0.082 [0.649] | 0.07 (0.28) [0.06 (0.29)] | 0.08 (0.31) | 0.510 [0.731] | 0.40 (0.43) [0.49 (0.76)] | 0.39 (0.51) | 0.781 [0.993] |
| KCNQ1 | c.1343 C > G | p.(Pro448Arg) | rs12720449 | 1 | 6109 | 988.00 (NA) | 917.85 (152.45) | 0.682 | 420.53 (NA) | 423.68 (22.82) | 0.938 | 0.48 (NA) | 0.08 (0.31) | 0.293 | 1.02 (NA) | 0.39 (0.51) | 0.304 |
| SCN1B | c.641 G > A | p.(Arg214Gln) | rs66876876 | 71 | 6039 | 886.93 (173.08) | 918.23 (152.16) | 0.083 | 427.48 (24.48) | 423.63 (22.80) | 0.14 | 0.02 (0.29) | 0.08 (0.31) | 0.111 | 0.34 (0.54) | 0.39 (0.51) | 0.388 |
| SCN3B | c.29 T > C | p.(Leu10Pro) | rs121918282 | 1 | 6109 | 1178.00 (NA) | 917.82 (152.41) | 0.117 | 409.08 (NA) | 423.68 (22.82) | 0.714 | 0.39 (NA) | 0.08 (0.31) | 0.398 | 1.07 (NA) | 0.39 (0.51) | 0.304 |
| SCN5A | c.647 C > T | p.(Ser216Leu) | rs41276525 | 5 | 6105 | 855.60 (100.20) | 917.92 (152.47) | 0.357 | 422.43 (18.76) | 423.68 (22.83) | 0.905 | 0.18 (0.25) | 0.08 (0.31) | 0.389 | 0.48 (0.40) | 0.39 (0.51) | 0.595 |
| SCN5A | c.1571 C > A | p.(Ser524Tyr) | rs41313691 | 1 | 6109 | 1142.00 (NA) | 917.83 (152.42) | 0.169 | 413.61 (NA) | 423.68 (22.82) | 0.788 | 0.00 (NA) | 0.08 (0.31) | 0.626 | 0.29 (NA) | 0.39 (0.51) | 0.567 |
| SCN5A | c.1673A > G | p.(His558Arg) | rs1805124 | 2114 [295] | 3701 | 920.91 (157.78) [918.49 (151.53)] | 916.08 (149.38) | 0.324 [0.949] | 423.99 (22.86) [425.72 (21.99)]a | 423.33 (22.86) | 0.130 [0.026] | 0.07 (0.30) [0.08 (0.33)] | 0.08 (0.31) | 0.101 [0.779] | 0.37 (0.51)a [0.41 (0.54)] | 0.40 (0.50) | 0.004 [0.767] |
| SCN5A | c.3578 G > A | p.(Arg1193Gln) | rs41261344 | 18 | 6092 | 968.11 (104.76) | 917.72 (152.54) | 0.170 | 412.53 (17.69)a | 423.71 (22.83) | 0.037 | 0.10 (0.36) | 0.08 (0.31) | 0.792 | 0.45 (0.51) | 0.39 (0.51) | 0.656 |
| SCN5A | c.5851 G > T | p.(Val1951Leu) | rs41315493 | 3 | 6107 | 853.33 (102.01) | 917.90 (152.46) | 0.389 | 429.14 (2.38) | 423.67 (22.83) | 0.509 | −0.05 (0.13) | 0.08 (0.31) | 0.391 | 0.71 (0.64) | 0.39 (0.51) | 0.366 |
| SCN5A | c.6010 T > C | p.(Phe2004Leu) | rs41311117 | 39 | 6071 | 876.92 (142.88) | 918.13 (152.47) | 0.090 | 427.23 (22.63) | 423.65 (22.82) | 0.322 | 0.05 (0.28) | 0.08 (0.31) | 0.512 | 0.35 (0.54) | 0.39 (0.51) | 0.568 |
| SCN5A | c.6016 C > G | p.(Pro2006Ala) | rs45489199 | 10 | 6100 | 887.80 (170.35) | 917.92 (152.42) | 0.418 | 430.97 (25.77) | 423.66 (22.82) | 0.168 | 0.18 (0.35) | 0.08 (0.31) | 0.467 | 0.31 (0.58) | 0.39 (0.51) | 0.297 |
| SNTA1 | c.784 A > C | p.(Thr262Pro) | rs200316080 | 1 | 6109 | 922.00 (NA) | 917.87 (152.45) | 0.978 | 435.32 (NA) | 423.67 (22.82) | 0.548 | 0.00 (NA) | 0.08 (0.31) | 0.591 | 0.34 (NA) | 0.39 (0.51) | 0.683 |
mm millimeter, ms milliseconds, refSNP reference single-nucleotide polymorphism, SD standard deviation
Brackets indicates homozygote minor allele carriers
aCarrier significant different from non-carrier, P-value < 0.05
Two manual raters analyzed the 6110 ECGs and found no Brugada-like ECG patterns in carriers but found one Brugada type 1 pattern in non-carriers.
We found no statistically significant differences in the risk of syncope [odds ratio (OR): 0.64 (95% CI: 0.26–1.57), P = 0.32], malignant ventricular arrhythmia [OR: 1.05 (95% CI: 0.14–7.94), P = 0.96], and all-cause mortality [OR: 1.15 (95% CI: 0.70–1.89), P = 0.59] between carriers and non-carriers. Neither did we observe any difference in overall survival between carriers and non-carriers (P = 0.43; Fig. 1).
Fig. 1.
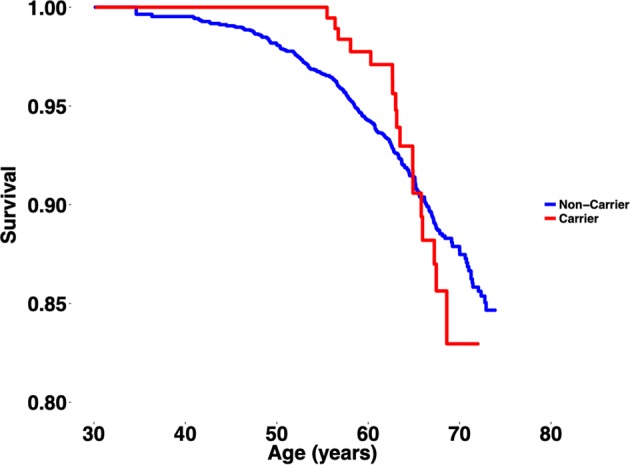
Kaplan–Meier plot displaying the overall survival between carriers and non-carriers of SIDS-associated variants. P-value (log-rank) = 0.43
UK Biobank: associations with cardiac traits
Using the PheWeb browser, we identified 24 (26%) of the 92 SIDS-associated variants in the database. Moreover, six variants showed statistically significant associations with severe cardiac disease (Supplementary Table 1): GPD1L p.(I124V) (I428 other cardiomyopathy, P = 0.018), KCNE2 p.(V14I) (I46 cardiac arrest and VF, P = 0.035; I490 VF, P = 0.002; and 1421 hypertrophic obstructive cardiomyopathy, P = 0.011), SCN3B p.(L10P) (I490 VF, P = 0.017 and I46 cardiac arrest and VF, P = 0.011), SCN5A p.(S524Y) (I490 VF, P = 0.020), SCN5A p.(V1951L) (I472 Paroxysmal VT, P = 0.006 and I517 Cardiomegaly, P = 0.029), and SCN5A p.(P2006A) (I46 cardiac arrest and VF, P = 0.048).
Classification using ACMG guidelines
Using the ACMG guidelines, 9/92 (10%) variants were classified as pathogenic and 14/92 (15%) as likely pathogenic (Supplementary Table 1 and Supplementary Fig. 2). In contrast, 27/92 (29%) variants were reclassified as benign and 7/92 (8%) as likely benign. Variants with conflicting evidence or lack of appropriate information were reclassified as variant of uncertain significance (VUS); corresponding to 35/92 (38%) variants. Of the 18 variants identified in Inter99, 14 were classified as benign, 1 as likely benign, and 3 as VUS.
SIDS victims die within the first year of life, hence the reproductive fitness of pathogenic variants correlated with SIDS should decrease and consequently their frequency is expected to be low [33]. We found that the proportion of SIDS-associated variants in ExAC was significantly higher than those not in ExAC (P = 1.49 × 10−8) for the benign category (Fig. 2). No statistically significant difference was found in the likely benign category (P = 0.289). The SIDS-associated variants classified as VUS were significantly overrepresented in ExAC compared with VUSs not identified in ExAC (P = 0.024). On the other hand, SIDS-associated variants classified as likely pathogenic (P = 0.013) or as pathogenic (P = 0.004) were significantly underrepresented in ExAC compared with variants not identified in ExAC.
Fig. 2.
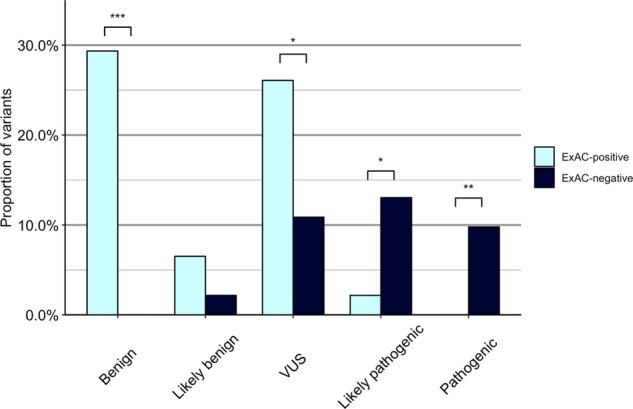
Proportion of variants identified in ExAC and not identified in ExAC, categorized in evidence of pathogenicity. VUS, variant of uncertain significance. Significance: ***P-values < 0.001; **P-values < 0.01; *P < 0.05
Classification in the ClinVar database
We used the online repository ClinVar to evaluate variant curation using their reference SNPs (Supplementary Table 1). Of the 92 variants, 7/92 (8%) were classified as pathogenic or likely pathogenic. In comparison, 9/92 (10%) were classified as benign or likely benign. The remaining 76/92 (83%) variants were classified as either variants with conflicting interpretations of pathogenicity (CIP), variants of unknown significance (VUS), or lacked a classification. Of these 76 variants, we provided new classifications on 44/76 (58%), classifying 28 variants as benign or likely benign and 16 variants as pathogenic or likely pathogenic.
Discussion
The study identified ~2/3 of ion channel variants previously associated with SIDS in the ExAC database (Supplementary Table 1). Moreover, we identified ~1/5 of the variants in Inter99 and ~1/4 of the variants in the UK Biobank, respectively. Overall, we found no significant difference in the risk of syncope, malignant ventricular arrhythmia, and all-cause mortality between carriers and non-carriers.
Variants associated with SIDS must be highly pathogenic and rare due to the low-reproductive fitness in this trait and the low prevalence (1:2500) for SIDS. In this study, we found an unexpectedly high prevalence of SIDS-associated variants in three populations representing the general population. This genotype prevalence was 125 times higher than the expected phenotype prevalence, indicating that the majority of the identified variants are most likely not highly penetrant monogenic causes of SIDS. More importantly, since SIDS is defined as a sudden unexplained death in persons below the age of 1 year, the identification of a majority of SIDS-related variants in three independent middle-aged population cohorts, provides additional evidence that these are at least not the sole cause of the disease. Moreover, ~10% of variants identified in ExAC had a MAF > 5%, a criterion which in itself implies benign classification according to the ACMG guidelines. Carrying a previously reported SIDS variant was not associated with an increased risk of syncope, malignant ventricular arrhythmia or all-cause mortality. Nor did the interrogated variants affect ECG-traits, associated with lethal cardiac arrhythmia.
Notably, other studies on smaller populations have reported similar findings. Consistent with our findings, these studies have suggested that some variants such as p.(R148W) in KCNH2 [20] and p.(R1193Q) in SCN5A [34] may be rare benign variation rather than disease-causing variants while another study found no association in six variants previously reported to be associated with SIDS [35].
We also investigated whether the interrogated variants were associated with cardiac traits in a larger sample size (UK Biobank). We identified 26% of previously SIDS-associated variants in the UK Biobank, of which six variants were associated with severe cardiac traits (Supplementary Table 1). The reason for a lower detection rate in the UK Biobank compared with ExAC could be attributed to the use of different genotyping techniques (GWAS array chip vs. exome sequencing). SIDS has traditionally been categorized as a monogenic disease. Our results show that the majority of variants analyzed in this paper can be identified in the general population. Whether or not SIDS should still be considered a Mendelian disease remains to be further elucidated. Although, the majority of variants were not associated with electrocardiographic traits or adverse outcomes, six variants were associated with other cardiac traits in the UK Biobank, which could indicate that some variants could act as disease modifiers, which in concert with other susceptibility variants could contribute to disease.
Recently, two studies were published using whole exome sequencing to address the role of ultra-rare variants [36, 37]. Despite this ambitious approach, potentially causative variants in genes related to arrhythmia and cardiomyopathy were only found in 25/161 (16%) and 6/71 (8%) of the cases. The relatively low prevalence of variants in cardiac genes, support that variants in these genes only explain a minority of SIDS cases. This further underlines the importance of keeping a multidisciplinary and cautious approach when evaluating the pathogenicity of variants identified in individual SIDS cases. It also indicates the importance of considering pathophysiological mechanisms of putative SIDS-associated variants.
In several of the variants, distinctly different allele frequencies were reported in ExAC compared to data from the UK Biobank and the Inter99 study. As an example, the variant p.(S524Y) of the SCN5A gene has a MAF of 0.5115% in ExAC compared to a MAF of 0.0081% and 0.012% in the data from Inter99 and UK Biobank respectively. When examining the ethnicity for the reported variant, the populations of African descent have a higher reported MAF (4.9%), compared with Latino (0.19%), European (0.14%) and South Asian (0.016%) populations. This exemplifies how the differences in MAF between the datasets may in part be due to differences in ethnicity. More importantly it underlines the importance of using diverse ethnical controls when evaluating the disease-causing potential of a given variant.
When evaluating the genetic causes of SIDS, one must also consider the concept of reduced penetrance, i.e., where an individual harbor a dominantly inherited variant, but do not show any clinical signs of disease [19]. One model incorporating the concept of reduced penetrance is the triple risk model. According to the triple risk model, SIDS may be caused by the presence of three factors, which must all be present; the presence of a vulnerability in the infant, a critical period in development and finally an exogenous stressor. One cannot exclude the possibility that vulnerability, i.e., a genetic variant in this case, which may be lethal for certain infants may not result in any detectable phenotype in individuals outside the critical development period, such as the individuals in the studied cohorts [38]. Variants may also show different degree of pathogenicity. In fact, some studies have suggested that common variants, while not being direct causes of SIDS may instead act as disease modifiers, rather than monogenic causes of disease [39]. Furthermore, recent studies have shown protective variants acting as risk modifiers. These variants rescue the pathogenic effect of the initial variant [40, 41]. This study has not accounted for reduced penetrance of the genetic variants or for protective variants, but we advise the reader to take it under consideration when interpreting the results.
The interpretation of pathogenicity of variants must be based on uniform approaches, i.e., common criteria for variant classification. Therefore, we analyzed the variants according to the ACMG guidelines [19], which include important criteria, such as family segregation, functional characterization, and in silico predictions (see Supplementary Table 2). Previously, variants were classified as disease-causing, when observed in cases, and absent in a few hundred controls, with no or limited supporting information as pathogenic evidence. The standards for variant classification have changed and continue to do so, and we therefore encourage researchers to fully adopt the ACMG guidelines in order to critically assess both novel and old variants [42].
Using the ACMG guidelines, our data provides new insights into the pathogenicity on >50% of the variants previously classified as variants with conflicting interpretations, variants of uncertain significance, or variants lacking a classification in ClinVar. Thus, our data contributes to a more granulated assessment of these variants.
Families experiencing cases of SIDS could be tested for putative variants [10]. In light of cascade screening, any counseling based on erroneously classified variants can have detrimental consequences, and lead to incorrect diagnosis and management of the family. Solid classification is also of utmost importance in genetic counseling in families, who have lost a child due to SIDS, in particular if prenatal genetic testing might be considered. Wrongfully diagnosing an individual with an alleged life-threatening variant may lead to stigmatization and unnecessary intervention and follow-up. Conversely, identifying the erroneous variant might lead to overseeing of the causal variant.
Limitations
First, since age inclusion criteria have been applied in the interrogated cohorts, carriers of highly pathogenic variants could have died before age of inclusion. As discussed above, some variants could result in fatal events in infants and young children, but be without any associated risk in older children and adults who have survived the critical developmental phases without exposure to exogenous stressors, as described in the triple risk model [38].
Secondly, we used genotyping array data in Inter99 and UK Biobank, thus variants not included on the chip were not examined. Third, the syncope analysis included only patients who were hospitalized. Patients, who experienced syncope and pre-syncope and did not seek medical assistance, or were treated in primary care, were not included. When using the ICD-10 code for syncope, syncope cannot be differentiated from arrhythmia and vasovagal causes. Last, associations with cardiac traits found in the UK Biobank were not Bonferroni corrected, and should be interpreted with caution. No Bonferroni correction (P = 0.05/4 = 0.0125) was applied, as we aimed to report lack of association. These associations ought to be subjected to additional research and are included for hypothesis generation purposes.
Conclusion
The study identified ~2/3 of variants previously associated with SIDS in three large population-based cohorts with available genotypic data. For the vast majority of variants, we found no effect on ECG-traits previously associated with sudden cardiac death. Moreover, carriers of SIDS variants were not at increased risk of syncope, malignant ventricular arrhythmia, and all-cause mortality, compared with non-carriers. These findings suggest that a large number of variants, previously implicated with SIDS, are not highly penetrant monogenic causes of the disease and show the importance of temporal re-evaluation of genetic variants, to avoid misapplication in a clinical setting.
Supplementary information
Acknowledgements
We wish to thank the Exome Aggregation Consortium (ExAC) and the contributing projects in creating this resource for biomedical research. ExAC was funded by grants from the National Institutes of Health’s National Institute of General Medical Sciences (R01 GM104371) and National Institute of Diabetes and Digestive and Kidney Diseases R01 (U54 DK105566).
Funding
The study was supported by The John and Birthe Meyer foundation, The Foundation of 17-12-1981, The Research Foundation of the Heart Center Rigshospitalet, The Danish Heart Foundation (grant no. 11-04-R84-A3401-22654), The Danish National Research Foundation Center for Cardiac Arrhythmia, The Arvid Nilsson Foundation, The Aase and Ejnar Danielsen Foundation, Fondsbørsvekselerer Henry Hansen og Hustru Karla Hansen født Westergaards Legat, Direktør Ib Henriksens Fond, and the Hallas Møller Grant of the Novo Nordisk Foundation.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41431-019-0416-3) contains supplementary material, which is available to authorized users.
References
- 1.Krous HF. Sudden infant death syndrome and unclassified sudden infant deaths: a definitional and diagnostic approach. Pediatrics. 2004;114:234–8. doi: 10.1542/peds.114.1.234. [DOI] [PubMed] [Google Scholar]
- 2.American Academy of Pediatrics task force on infant positioning and SIDS. Positioning and SIDS. Pediatrics. 1992;89:1120–6. [PubMed]
- 3.Mitchell EA, Scragg R, Stewart AW, Becroft DM, Taylor BJ, Ford RP, et al. Results from the first year of the New Zealand cot death study. N Z Med J. 1991;104:71–6. [PubMed] [Google Scholar]
- 4.Force on Sudden Infant Death, S. The changing concept of sudden infant death syndrome: diagnostic coding shifts, controversies regarding the sleeping environment, and new variables to consider in reducing risk. Am Acad Pedia Task. 2005;116:1245–55. doi: 10.1542/peds.2005-1499. [DOI] [PubMed] [Google Scholar]
- 5.Goldstein RD, Trachtenberg FL, Sens MA, Harty BJ, Kinney HC. Overall postneonatal mortality and rates of SIDS. Pediatrics. 2016;137:e20152298. doi: 10.1542/peds.2015-2298. [DOI] [PubMed] [Google Scholar]
- 6.Osterman MJ, Kochanek KD, MacDorman MF, Strobino DM, Guyer B. Annual summary of vital statistics: 2012-2013. Pediatrics. 2015;135:1115–25. doi: 10.1542/peds.2015-0434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Norstrand DW, Ackerman MJ. Genomic risk factors in sudden infant death syndrome. Genome Med. 2010;2:86. doi: 10.1186/gm207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arnestad M. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation. 2007;115:361–7. doi: 10.1161/CIRCULATIONAHA.106.658021. [DOI] [PubMed] [Google Scholar]
- 9.Van Norstrand DW, Ackerman MJ. Sudden infant death syndrome: do ion channels play a role? Heart Rhythm. 2009;6:272–8. doi: 10.1016/j.hrthm.2008.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. EP Eur. 2011;13:1077–109. doi: 10.1093/europace/eur245. [DOI] [PubMed] [Google Scholar]
- 11.Gollob MH. Recommendations for the use of genetic testing in the clinical evaluation of inherited cardiac arrhythmias associated with sudden cardiac death: Canadian Cardiovascular Society/Canadian Heart Rhythm Society joint position paper. Can J Cardiol. 2011;27:232–45. doi: 10.1016/j.cjca.2010.12.078. [DOI] [PubMed] [Google Scholar]
- 12.Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J. 2015;36:2793–867. doi: 10.1093/eurheartj/ehv316. [DOI] [PubMed] [Google Scholar]
- 13.Paludan-Müller C, Ahlberg G, Ghouse J, Herfelt C, Svendsen Jh, Haunsø S, et al. Integration of 60,000 exomes and ACMG guidelines question the role of catecholaminergic polymorphic ventricular tachycardia-associated variants. Clin Genet. 2017;91:63–72. doi: 10.1111/cge.12847. [DOI] [PubMed] [Google Scholar]
- 14.Paludan-Müller C, Ahlberg G, Ghouse J, Svendsen JH, Haunsø S, Olesen MS. Analysis of 60 706 exomes questions the role of de novo variants previously implicated in cardiac disease clinical perspective. Circ Cardiovasc Genet. 2017;10:e001878. doi: 10.1161/CIRCGENETICS.117.001878. [DOI] [PubMed] [Google Scholar]
- 15.Ghouse J, Have CT, Weeke P, Nielsen JB, Ahlberg G, Balslev-Harder M, et al. Rare genetic variants previously associated with congenital forms of long QT syndrome have little or no effect on the QT interval. Eur Heart J. 2015;36:2523–9. doi: 10.1093/eurheartj/ehv297. [DOI] [PubMed] [Google Scholar]
- 16.Ghouse J, Have CT, Skov MW, Andreasen L, Ahlberg G, Nielsen JB, et al. Numerous Brugada syndrome-associated genetic variants have no effect on J-point elevation, syncope susceptibility, malignant cardiac arrhythmia, and all-cause mortality. Genet Med. 2017;19;521–28. [DOI] [PubMed]
- 17.Andreasen C, Nielsen JB, Refsgaard L, Holst AG, Christensen AH, Andreasen L, et al. New population-based exome data are questioning the pathogenicity of previously cardiomyopathy-associated genetic variants. Eur J Hum Genet. 2013;21:918–28. doi: 10.1038/ejhg.2012.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manrai AK, Funke BH, Rehm HL, Olesen MS, Maron BA, Szolovits P, et al. Genetic misdiagnoses and the potential for health disparities. N Engl J Med. 2016;375:655–65. doi: 10.1056/NEJMsa1507092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med J Am Coll Med Genet. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andreasen C. Mutations in genes encoding cardiac ion channels previously associated with sudden infant death syndrome (SIDS) are present with high frequency in new exome data. Can J Cardiol. 2013;29:1104–9. doi: 10.1016/j.cjca.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 21.Wilders R. Cardiac ion channelopathies and the sudden infant death syndrome. ISRN Cardiol. 2012;2012:846171. [DOI] [PMC free article] [PubMed]
- 22.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glümer C, Jørgensen T, Borch-Johnsen K. Prevalences of diabetes and impaired glucose regulation in a Danish population: the Inter99 study. Diabetes Care. 2003;26:2335–40. doi: 10.2337/diacare.26.8.2335. [DOI] [PubMed] [Google Scholar]
- 24.Jørgensen T, Borch-Johnsen K, Thomsen TF, Ibsen H, Glümer C, Pisinger C. A randomized non-pharmacological intervention study for prevention of ischaemic heart disease: baseline results Inter99. Eur J Cardiovasc Prev Rehabil J Eur Soc Cardiol Work Groups Epidemiol Prev Card Rehabil Exerc Physiol. 2003;10:377–86. doi: 10.1097/01.hjr.0000096541.30533.82. [DOI] [PubMed] [Google Scholar]
- 25.Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation. 1993;88:782–4. doi: 10.1161/01.CIR.88.2.782. [DOI] [PubMed] [Google Scholar]
- 26.Lohmueller KE, Sparsø T, Li Q, Andersson E, Korneliussen T, Albrechtsen A, et al. Whole-exome sequencing of 2,000 Danish individuals and the role of rare coding variants in type 2 diabetes. Am J Hum Genet. 2013;93:1072–86. doi: 10.1016/j.ajhg.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hedley PL, Jørgensen P, Schlamowitz S, Moolman-Smook J, Kanters JK, Corfield VA, et al. The genetic basis of Brugada syndrome: a mutation update. Hum Mutat. 2009;30:1256–66. doi: 10.1002/humu.21066. [DOI] [PubMed] [Google Scholar]
- 28.Helweg-Larsen K. The Danish register of causes of death. Scand J Public Health. 2011;39(7_suppl):26–9. doi: 10.1177/1403494811399958. [DOI] [PubMed] [Google Scholar]
- 29.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.PheWeb [Internet]. [cited 9 May 2018]. Available from: http://pheweb.sph.umich.edu:5003/
- 31.Kleinberger J, Maloney KA, Pollin TI, Jeng LJB. An openly available online tool for implementing the ACMG/AMP standards and guidelines for the interpretation of sequence variants. Genet Med. 2016;18;1165. [DOI] [PMC free article] [PubMed]
- 32.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–53. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ackerman MJ, Splawski I, Makielski JC, Tester DJ, Will ML, Timothy KW, et al. Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals: implications for arrhythmogenic susceptibility and Brugada/long QT syndrome genetic testing. Heart Rhythm. 2004;1:600–7. doi: 10.1016/j.hrthm.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 35.Yamamoto T, Matsusue A, Umehara T, Kubo SI, Ikematsu K. No association between cardiac ion channel variants and sudden infant death. Pedia Int J Jpn Pedia Soc. 2018;60:483–4. doi: 10.1111/ped.13532. [DOI] [PubMed] [Google Scholar]
- 36.Neubauer J, Lecca MR, Russo G, Bartsch C, Medeiros-Domingo A, Berger W, et al. Post-mortem whole-exome analysis in a large sudden infant death syndrome cohort with a focus on cardiovascular and metabolic genetic diseases. Eur J Hum Genet EJHG. 2017;25:404–9. doi: 10.1038/ejhg.2016.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oshima Y, Yamamoto T, Ishikawa T, Mishima H, Matsusue A, Umehara T, et al. Postmortem genetic analysis of sudden unexpected death in infancy: neonatal genetic screening may enable the prevention of sudden infant death. J Hum Genet. 2017;62:989–95. doi: 10.1038/jhg.2017.79. [DOI] [PubMed] [Google Scholar]
- 38.Filiano JJ, Kinney HC. A perspective on neuropathologic findings in victims of the sudden infant death syndrome: the triple-risk model. Biol Neonate. 1994;65:194–7. doi: 10.1159/000244052. [DOI] [PubMed] [Google Scholar]
- 39.Osawa M, Kimura R, Hasegawa I, Mukasa N, Satoh F. SNP association and sequence analysis of the NOS1AP gene in SIDS. Leg Med Tokyo Jpn. 2009;11(Suppl 1):S307–8. doi: 10.1016/j.legalmed.2009.01.065. [DOI] [PubMed] [Google Scholar]
- 40.Bal NC, Jena N, Sopariwala D, Balaraju T, Shaikh S, Bal C, et al. Probing cationic selectivity of cardiac calsequestrin and its CPVT mutants. Biochem J. 2011;435:391–9. doi: 10.1042/BJ20101771. [DOI] [PubMed] [Google Scholar]
- 41.Klaassen S, Probst S, Oechslin E, Gerull B, Krings G, Schuler P, et al. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation. 2008;117:2893–901. doi: 10.1161/CIRCULATIONAHA.107.746164. [DOI] [PubMed] [Google Scholar]
- 42.Ghouse J, Skov MW, Bigseth RS, Ahlberg G, Kanters JK, Olesen MS. Distinguishing pathogenic mutations from background genetic noise in cardiology: The use of large genome databases for genetic interpretation. Clin Genet. 2018;93:459–66. doi: 10.1111/cge.13066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.