

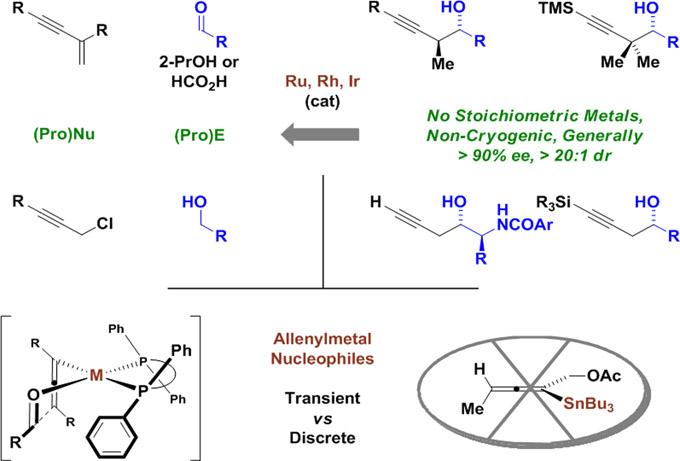
Graphical Abstract
Chiral metal complexes catalyze enantioselective carbonyl propargylation via reductive coupling or as hydrogen auto-transfer processes, in which reactant alcohols serve dually as reductant and carbonyl proelectrophile. Unlike classical propargylation protocols, which rely on allenylmetal reagents or metallic reductants (e.g. NHK reactions), reductive protocols for carbonyl propargylation can occur in the absence of stoichiometric metals, precluding generation of metallic byproducts. Propargylations of this type exploit both enyne and propargyl halide pronucleophiles.

Keywords: Propargylation, Iridium, ruthenium, rhodium, enantioselective, dehydrogenation, hydrogen transfer
I. Introduction and Historical Perspective
Carbonyl propargylations represent a distinctive and useful subset of carbonyl addition reactions.1 In the first reported carbonyl propargylations described by Prévost (1950) and Wotiz (1950), Grignard reagents derived from propargyl bromide were observed to form mixtures of β-acetylenic and α-allenic carbinols or carboxylic acids upon reaction with acetaldehyde or carbon dioxide, respectively.2a,b Chodkiewicz (1969) later demonstrated relative stereocontrol in aldehyde additions of this type.2c In a major advance, preformed allenylmetal reagents based on boron, tin and silicon were developed by Favre and Gaudemar (1966),2d,e Lequam and Guillerm (1973)2f and Danheiser (1980),2g respectively, and were shown to provide homopropargyl alcohols upon reaction with carbonyl electrophiles. Carbonyl propargylations through propargyl halide reductive coupling under Nozaki-Hiyama conditions were reported by Place, Delbecq and Gore (1978).2h Shortly thereafter, Mukaiyama (1981) showed that stannous chloride promoted aldehyde-propargyl halide reductive coupling.2i Finally, using a tartrate-modified allenylboron reagent, the first enantioselective carbonyl propargylations were reported by Yamamoto (1982).3a
Following Yamamoto’s seminal contribution,3a numerous methods for enantioselective carbonyl propargylation were developed (Scheme 1).3–12 The majority of these methods exploit chiral allenylmetal reagents based on boron.3 However, related reagents based on aluminum,5 tin,6 silicon8 and zinc10 also have been developed. While effective, these methods require stoichiometric chiral modifiers. Chiral catalysts for the asymmetric addition of achiral allenyl- or propargylmetal reagents (B,4 Sn,7 Si9) to carbonyl electrophiles overcome this limitation, but these protocols still require hydrolytically unstable premetalated reagents that are prepared through multi-step sequences. A conceptually more appealing strategy for carbonyl propargylation involves the direct catalytic enantioselective reductive coupling of propargyl halides with carbonyl compounds, as realized under Nozaki-Hiyama-Kishi conditions.11 However, the use of stoichiometric chromium(II) chloride as terminal reductant is far from ideal. More recently, in a significant departure from prior art, propargyl halide reductive couplings were accomplished using 2-propanol as terminal reductant and 1,3-enynes were introduced as pronucleophiles.12 Furthermore, catalytic enantioselective propargylations have been achieved using alcohols dually as reductant and carbonyl proelectrophile, that is, via hydrogen auto-transfer.12,13 Notably, the coupling of alcohols with enyne pronucleophiles12a-c,f enables completely atom-efficient carbonyl propargylation. In this monograph, we provide a concise overview of this emergent class of asymmetric alcohol-mediated carbonyl additions.
Scheme 1.

Selected milestones in enantioselective carbonyl propargylation.
I. Enyne Pronucleophiles
The reductive coupling of carbonyl compounds with conjugated enynes using rhodium14,15 and nickel catalysts16 occurs at the acetylenic terminus of the enyne to provide products of carbonyl dienylation. In these processes, alkyne-carbonyl oxidative coupling to form an oxa-metalacyclopentene intermediate is the regio-determining event.17 In 2008, ruthenium complexes were found to catalyze 2-propanol-mediated enyne-carbonyl reductive couplings.12a Remarkably, an alternate regioselectivity was observed wherein C-C coupling occurs at the interior vinylic position of the enyne. Furthermore, for the ruthenium-catalyzed reactions, carbonyl addition could be conducted from the alcohol oxidation level of the reactant via hydrogen auto-transfer. Mechanistic studies corroborate pathways involving enyne hydroruthenation to form nucleophilic allenylruthenium intermediates.18 These processes, which represent the first examples of transfer hydrogenative carbonyl propargylation, set the stage for the development of diverse diastereo- and enantioselective transformations of this type (Scheme 2).12
Scheme 2.

Divergent regioselectivity in enyne-carbonyl reductive coupling and discovery of enyne-mediated carbonyl propargylation.a
aDPPF = 1,1’-bis(diphenylphosphino)ferrocene.
Enforcing relative and absolute stereocontrol in alcohol-mediated carbonyl propargylations catalyzed by ruthenium initially proved to be challenging.12b For 1,3-enynes with unsubstituted vinyl moieties (RC≡CCH=CH2), the transient allenylruthenium nucleophiles that arise upon enyne hydroruthenation are axially chiral. Hence, to achieve high levels of enantiomeric enrichment, the transfer of chirality in both enyne hydroruthenation and carbonyl addition events must occur with high levels of fidelity. In contrast, the commercially available 1,3-enyne, TMSC≡CC(Me)=CH2, delivers an allenylruthenium intermediate that is not axially chiral, mitigating erosion of asymmetry in the transmission of chirality from catalyst to product. Thus, through the action of the chiral ruthenium complex formed in situ from (TFA)2Ru(CO)(PPh3)2 and (R)-BINAP, enyne-mediated carbonyl propargylation is achieved with uniformly high levels of enantioselectivity from aliphatic, allylic and benzylic alcohol reactants (Scheme 3).12f In the presence of 2-propanol under otherwise identical conditions, corresponding reductive couplings of aldehyde reactants are conducted with roughly equivalent yields and enantioselectivities (not shown).
Scheme 3.

Enantioselective ruthenium catalyzed carbonyl propargylation via hydrogen auto-transfer using enyne pronucleophiles.a
aBINAP = 2,2′-bis-(diphenylphosphino)-1,1′-binaphthalene. TFA = trifluoroacetate. TMS = trimethylsilyl. Yields are of material isolated by silica gel chromatography. Enantioselectivities determined by stationary phase HPLC analysis.b95 °C.c120 °C.
The catalytic cycle for enyne-mediated propargylation is initiated by substitution of trifluoroacetate with benzyl alcohol to form the ruthenium alkoxide I (Scheme 4). β-Hydride elimination delivers the ruthenium hydride II with concomitant release of benzaldehyde.19 Enyne hydrometalation provides the σ-propargyl complex III, which isomerizes to the σ-allenyl complex IV.18 Aldehyde coordination, as in complex V, followed by carbonyl addition forms the homopropargyl ruthenium alkoxide VI. Trifluoroacetic acid-mediated protonolysis of alkoxide VI releases the homopropargyl alcohol and regenerates the catalyst to close the catalytic cycle. Alternatively, product may be released through direct exchange of alkoxide VI with benzyl alcohol to regenerate ruthenium alkoxide I. This interpretation of the catalytic mechanism is consistent with the results of deuterium labeling. Deuterium is mostly retained at the carbinol position (Hc = 87%2H), suggesting hydrogen transfer between the enyne and primary alcohol shows little if any reversibility, and that the homopropargyl alcohol is resistant to dehydrogenation, which would diminish enantioselectivity. Adventitious water likely contributes to incomplete transfer of deuterium.20
Scheme 4.

General catalytic mechanism and deuterium labeling studies.
To illustrate the utility of the products obtained upon ruthenium-catalyzed enyne-mediated propargylation, the indicated homopropargyl alcohol derived from p-bromobenzyl alcohol was converted to a range of value-added products (eq. 1–3). Hydrozirconation of the homopropargyl alcohol followed by exposure to iodine provides the trans-vinyl iodide in good yield (eq. 1).21 Conversion of the homopropargyl alcohol to the acetylenic iodide22 followed by diimide reduction using 2-nitrobenzenesulfonylhydrazide (NBSH)23 delivers the isomeric cis-vinyl iodide (eq. 2). Lastly, exposure of the homopropargyl alcohol to 3-buten-2-ol in the presence of a cationic Cp-ruthenium(II) catalyst (Cp = cyclopentadienyl) results in formation of the indicated γ,δ-unsaturated ketone (eq. 3).24
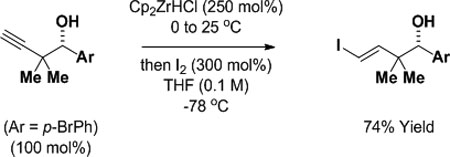 |
(eq. 1) |
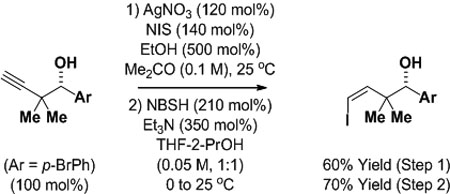 |
(eq. 2) |
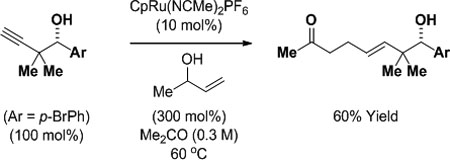 |
(eq. 3) |
In parallel studies, it was found that iridium complexes are also competent catalysts for enyne-mediated propargylation (Scheme 5).12c In the presence of an iridium catalyst modified by (R)-DM-SEGPHOS or (R)-SEGPHOS, the indicated enyne derived from 2-methyl-3-butyn-2-ol (47 USD/Kg) undergoes formic acid-mediated hydrometalation to form aldehyde-allenyliridium pairs, which deliver products of carbonyl anti-(α-methyl)propargylation. Aromatic aldehydes, enals and aliphatic aldehydes all participate in propargylation with uniformly high levels of anti-diastereo- and enantioselectivity. In the absence of formic acid but under otherwise identical conditions, benzylic, allylic and aliphatic alcohols undergo propargylation via hydrogen auto-transfer. For reactions conducted from the alcohol oxidation level, lower yields were evident in certain cases but anti-diastereo- and enantioselectivities were consistently higher. The indicated stereochemical model, which invokes intervention of a square planar allenyliridium species, is inspired by the structure of a related η1-allenylplatinum(II) complex, trans-Pt(PPh3)2(Br)(η1-CH=C=CH2), for which the allenyl moiety lies roughly perpendicular to the square coordination plane.25 Finally, the products of propargylation are easily deprotected to reveal the terminal alkyne in a “one-pot” procedure (eq. 4). Specifically, the propargylation products are initially exposed to TBAF (tetra-n-butylammonium fluoride) in THF at room temperature to cleave the silyl ether. Toluene and NaOH are subsequently added and the reaction mixture is heated to promote elimination of acetone.
Scheme 5.

anti-Diastereo- and enantioselective iridium-catalyzed carbonyl propargylation via formic acid-mediated reductive coupling and hydrogen auto-transfer using enyne pronucleophiles.a
aDM-SEGPHOS = 5,5′−bis-[di(3,5-xylyl)phosphino]-4,4′-bi-1,3-benzodioxole. SEGPHOS = 5,5′-bis-(diphenylphosphino)-4,4′-bi-1,3-benzodioxole. cod = cycloocta-1,5-diene. TIPS = triisopropylsilyl. Yields are of material isolated by silica gel chromatography. Enantioselectivities determined by stationary phase HPLC analysis.bMeCN (200 mol%).cNa2SO4 (100 mol%).dPhCF3 (1 M) was used as solvent, HCO2H (50 mol%).e[Ir(cod)Cl]2 (5 mol%), (R)-SEGPHOS (10 mol%).
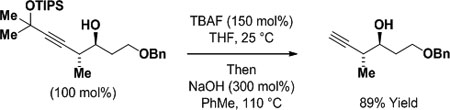 |
(eq. 4) |
In a significant extension of these earlier studies, Buchwald recently developed enantioselective copper(I)-catalyzed enyne-carbonyl reductive couplings mediated by silane that are applicable to ketones (Scheme 6).26 Using a copper complex modified by (S,S)-Ph-BPE, a wide range of ketones were converted to tertiary homopropargyl alcohols with high levels of syn-diastereo- and enantioselectivity. The authors demonstrate that the reaction could be run efficiently on 50 mmol scale with catalyst loadings as low as 0.2 mol% (not shown). Corresponding ketone propargylations from the alcohol oxidative level via hydrogen auto-transfer remain an unmet challenge that would bypass use of exogenous silane reductant.
Scheme 6.

syn-Diastereo- and enantioselective copper-catalyzed carbonyl propargylation via silane-mediated reductive coupling and hydrogen auto-transfer using enyne pronucleophiles.a
a(S,S)-Ph-BPE = 1,2-Bis[(2S,5S)-2,5-diphenylphospholano]ethane. Yields are of material isolated by silica gel chromatography. Enantioselectivities determined by stationary phase HPLC analysis.aDM-SEGPHOS = 5,5′−bis-[di(3,5-xylyl)phosphino]-4,4′-bi-1,3-benzodioxole. SEGPHOS = 5,5′-bis-(diphenylphosphino)-4,4′-bi-1,3-benzodioxole. cod = cycloocta-1,5-diene. TIPS = triisopropylsilyl. Yields are of material isolated by silica gel chromatography. Enantioselectivities determined by stationary phase HPLC analysis.
II. Propargyl Chlorides as Pronucleophiles
The ability to balance alcohol dehydrogenation with C-X (X = OAc, halide, etc.) reductive cleavage13 suggested the feasibility of catalytic propargylations wherein alcohol oxidation is used to generate allenylmetal intermediates from propargyl chlorides – a process akin to NHK-type propargylations,11 but using alcohols rather than chromium(II) chloride as terminal reductant. In an initial step toward this objective, it was found that chiral iridium complexes modified by (R)-SEGPHOS catalyze enantioselective 2-propanol mediated propargylations of aryl aldehydes and related hydrogen auto-transfer processes of benzylic alcohols (Scheme 7).12d Trialkylsilyl substitution was required at the acetylenic terminus. The use of 3-hexyne as an additive improved conversion by mitigating over-reduction of the resulting homopropargyl alcohol products. Attempted propargylation of aliphatic aldehydes were significantly less efficient under these conditions. Remarkably, ether products obtained via O-propargylation were not observed.
Scheme 7.

Enantioselective iridium-catalyzed carbonyl propargylation via 2-propanol mediated reductive coupling and hydrogen auto-transfer using propargyl chloride pronucleophiles.a
aSEGPHOS = 5,5′-bis-(diphenylphosphino)-4,4′-bi-1,3-benzodioxole. OTf = trifluoromethanesulfonate. Yields are of material isolated by silica gel chromatography. Enantioselectivities determined by stationary phase HPLC analysis.
Efforts toward the development of conditions enabling direct use of the unsubstituted propargyl chloride, HC≡CCH2Cl, in alcohol-mediated carbonyl propargylation were sought. Eventually, it was discovered that neutral rhodium catalysts modified by rac-BINAP were effective in such transformations (Scheme 8). Diverse benzylic, allylic and aliphatic alcohols were converted to the homopropargyl alcohols. These transformations could also be conducted from the aldehyde oxidation level under the conditions of 2-propanol-mediated reductive coupling (not shown). Because the use of optically pure BINAP only led to moderate levels of enantiomeric enrichment (40–55% ee), match-mismatch effects in the C-propargylation of chiral α-stereogenic amino alcohols were explored. As we hoped, products of asymmetric C-propargylation could be formed with high levels of stereocontrol (Scheme 8).12e Diastereoselectivities were found to increase with increasing size of the α-substituent. The collective data suggest intervention of an intramolecular NH-O hydrogen bond at the stage of the transient α-amino aldehyde accentuates diastereofacial bias in the carbonyl addition event. Again, ether products obtained via O-propargylation were not observed.
Scheme 8.

Rhodium catalyzed propargylation of alcohols via hydrogen auto-transfer using propargyl chloride.a
aYields are of material isolated by silica gel chromatography.b2-PrOH was omitted.cToluene (1.0 M).
III. Summary and Outlook
Carbonyl addition ranks among the most fundamental methods for C-C bond formation and has played a central role in the field of chemical synthesis since its very inception. Classical methods for carbonyl addition rely on the use of premetalated reagents, which can be hazardous and give rise to stoichiometric quantities of metallic byproducts. Here, as shown in the specific context of carbonyl propargylation, metal catalyzed reductive coupling offers an alternative to preformed carbanions, allowing carbonyl addition to be achieved using safe, inexpensive reductants with low molecular weights. More broadly, these studies contribute to a growing body of green catalytic C-C couplings that exploit the native reducing ability of alcohol reactants.13
Acknowledgments
The Welch Foundation (F-0038), the NIH (RO1-GM069445) and the UT Austin Center for Green Chemistry and Catalysis are acknowledged for partial support of this research. Brett Ambler was supported by an American Cancer Society - Joe and Jessie Crump Postdoctoral Fellowship (PF-17–229-01-CDD).
Biographies

Professor Michael J. Krische (B.S. UC Berkeley; PhD Stanford University) holds the Robert A. Welch Chair in Science at the University of Texas at Austin. Professor Krische has pioneered a new class of C-C bond formations that merge the characteristics of carbonyl addition and catalytic hydrogenation. Professor Krische’s work has been recognized by numerous awards, including the NSF-CAREER Award (2000), Cottrell Scholar Award (2002), Eli Lilly Granteeship (2002), Frasch Award in Chemistry (2002), Dreyfus Teacher-Scholar Award (2003), Sloan Fellowship (2003), Johnson & Johnson Focused Giving Award (2005), Solvias Ligand Prize (2006), Presidential Green Chemistry Award (2007), ACS Corey Award (2007), Dowpharma Prize (2007), Novartis Lectureship (2008), Tetrahedron Young Investigator Award (2009), Humboldt Senior Research Award (2009–2011), Mukaiyama Award (2010), Glaxo-Smith-Kline Scholar Award (2011), ACS Cope Scholar Award (2012), and JSPS Fellow (2013), Royal Society of Chemistry, Pedlar Award (2015).

Professor Sang Kook Woo received a B.S. in chemistry from Yeungnam University and a Ph.D. in organic chemistry from Seoul National University under the supervision of Professor Eun Lee. He carried out postdoctoral research at the University of Texas at Austin under the supervision of Professor Michael J. Krische, where he has contributed catalytic enantioselective carbonyl propargylation and total synthesis of polyketide natural products. In 2013, he joined the faculty at University of Ulsan as an assistant professor of chemistry, and was promoted to associate professor in 2018. His research is focused on the development of synthetic methods and the synthesis of biologically active compounds.

Brett Ambler received a B.A. in chemistry from St. Olaf College and a Ph.D. in Medicinal Chemistry from The University of Kansas under the supervision of Professor Ryan Altman. During his doctoral studies, he developed copper-catalyzed decarboxylative trifluoromethylation reactions. Dr. Ambler is presently an ACS postdoctoral fellow in the laboratory of Professor Michael J. Krische, where he has contributed to the development synthetic methods for the construction of antiproliferative polyketide natural products.
FRONTSPIECE
Footnotes
The authors declare no competing financial interest.
Contributor Information
Sang Kook Woo, Email: woosk@ulsan.ac.kr.
Michael J. Krische, Email: mkrische@mail.utexas.edu.
References
- [1].For selected reviews on carbonyl propargylation, see: Marshall JA, Chem. Rev 1996, 96, 31; Ding C-H, Hou X-L, Chem. Rev 2011, 111, 1914; Wisniewska HM, Jarvo ER, J. Org. Chem 2013, 78, 11629; Thaima T, Zamani F, Hyland CJT, Pyne SG, Synthesis 2017, 49, 1461.
- [2].For selected milestones in carbonyl propargylation, see: Prévost C, Gaudemar M, Honigberg J, Compt. Rend 1950, 230, 1186; Wotiz JH, J. Am. Chem. Soc 1950, 72, 1639; Karila M, Capmau ML, Chodkiewicz W, Compt. Rend 1969, 269, 342; Favre E, Gaudemar M, Compt. Rend 1966, 263, 1332; Favre E, Gaudemar M, Compt. Rend 1966, 263, 1543; Lequan M, Guillerm G, J. Organomet. Chem 1973, 54, 153; Danheiser RL, Carini DJ, J. Org. Chem 1980, 45, 3925; Place P, Delbecq F, Gore J, Tetrahedron Lett. 1978, 19, 3801; Mukaiyama T, Harada T, Chem. Lett 1981, 621.
- [3].For enantioselective carbonyl propargylation via chiral allenylboron reagents, see: Haruta R, Ishiguro M, Ikeda N, Yamamoto H, J. Am. Chem. Soc 1982, 104, 7667; Corey EJ, Yu C-M, Lee D-H, J. Am. Chem. Soc 1990, 112, 878; Matsumoto Y, Naito M, Uozumi Y, Hayashi T, J. Chem. Soc., Chem. Commun 1993, 1468; Lai C, Soderquist JA, Org. Lett 2005, 7, 799; Hernandez E, Burgos CH, Alicea E, Soderquist JA, Org. Lett 2006, 8, 4089.
- [4].For catalytic enantioselective carbonyl propargylation via achiral allenyl/propargylboron reagents, see: Shi S-L, Xu L-W, Oisaki K, Kanai M, Shibasaki M, J. Am. Chem. Soc 2010, 132, 6638; Fandrick DR, Fandrick KR, Reeves JT, Tan Z, Tang W, Capacci AG, Rodriguez S, Song JJ, Lee H, Yee NK, Senanayake CH, J. Am. Chem. Soc 2010, 132, 7600; Fandrick KR, Fandrick DR, Reeves JT, Gao J, Ma S, Li W, Lee H, Grinberg N, Lu B, Senanayake CH, J. Am. Chem. Soc 2011, 133, 10332; Barnett DS, Schaus SE, Org. Lett 2011, 13, 4020; Jain P, Wang H, Houk KN, Antilla JC, Angew. Chem. Int. Ed 2012, 51, 1391; Reddy LR, Org. Lett 2012, 14, 1142; Chen M, Roush WR, J. Am. Chem. Soc 2012, 134, 10947; Fandrick KR, Ogikubo J, Fandrick DR, Patel ND, Saha J, Lee H, Ma S, Grinberg N, Busacca CA, Senanayake CH, Org. Lett 2013, 15, 1214; Fandrick DR, Reeves JT, Bakonyi JM, Nyalapatla PR, Tan Z, Niemeier O, Akalay D, Fandrick KR, Wohlleben W, Ollenberger S, Song JJ, Sun X, Qu B, Haddad N, Sanyal S, Shen S, Ma S, Byrne D, Chitroda A, Fuchs V, Narayanan BA, Grinberg N, Lee H, Yee N, Brenner M, Senanayake CH, J. Org. Chem 2013, 78, 3592; Wei X-F, Shimizu Y, Kanai M, ACS Cent. Sci 2016, 2, 21.
- [5].For catalytic enantioselective carbonyl propargylation via achiral allenylaluminum reagents, see: Minowa N, Mukaiyama T, Bull. Chem. Soc. Jpn 1987, 60, 3697.
- [6].For asymmetric carbonyl propargylation via chiral allenyltin reagents, see: Allenyltin Reagents: Marshall JA, Wang X.-j., J. Org. Chem 1991, 56, 3211.
- [7].For catalytic enantioselective carbonyl propargylation via achiral allenyltin reagents, see: Keck GE, Krishnamurthy D, Chen X, Tetrahedron Lett. 1994, 35, 8323–8324; Yu C-M, Yoon S-K, Choi H-S, Baek K, Chem. Commun 1997, 763–764; Yu C-M, Choi H-S, Yoon S-K, Jung W-H, Synlett 1997, 889–890; Yu C-M, Yoon S-K, Baek K, Lee J-Y, Angew. Chem 1998, 110, 2504–2506; Angew. Chem. Int. Ed. 1998, 37, 2392–2395; Denmark SE, Wynn T, J. Am. Chem. Soc 2001, 123, 6199–6200. Konishi S, Hanawa H, Maruoka K, Tetrahedron: Asymmetry 2003, 14, 1603–1605.
- [8].For asymmetric carbonyl propargylation via chiral allenylsilicon reagents, see: Marshall JA, Maxson K, J. Org. Chem 2000, 65, 630; Brawn RA, Panek JS, Org. Lett 2007, 9, 2689.
- [9].For catalytic enantioselective carbonyl propargylation via achiral allenylsilicon reagents, see: Iseki K, Kuroki Y, Kobayashi Y, Tetrahedron: Asymmetry 1998, 9, 2889; Evans DA, Sweeney ZK, Rovis T, Tedrow JS, J. Am. Chem. Soc 2001, 123, 12095; Nakajima M, Saito M, Hashimoto S, Tetrahedron: Asymmetry 2002, 13, 2449; Chen J, Captain B, Takenaka N, Org. Lett 2011, 13, 1654.
- [10].For asymmetric carbonyl propargylation via chiral allenylzinc reagents, see: Marshall JA, Adams ND, J. Org. Chem 1998, 63, 3812; Marshall JA, Adams ND, J. Org. Chem 1999, 64, 5201; Marino JP, McClure MS, Holub DP, Comasseto JV, Tucci FC, J. Am. Chem. Soc 2002, 124, 1664.
- [11].For catalytic enantioselective carbonyl propargylation via Nozaki-Hiyama-Kishi reaction, see: Bandini M, Cozzi PG, Umani-Ronchi A, Polyhedron 2000, 19, 537; Bandini M, Cozzi PG, Melchiorre P, Tino R, Umani-Ronchi A, Tetrahedron: Asymmetry 2001, 12, 1063; Inoue M, Nakada M, Org. Lett 2004, 6, 2977; Naodovic M, Xia G, Yamamoto H, Org. Lett 2008, 10, 4053; Liu S, Kim JT, Dong C-G, Kishi Y, Org. Lett 2009, 11, 4520; Harper KC, Sigman MS, Science 2011, 333, 1875; g) Harper KC, Vilardi SC, Sigman MS, J. Am. Chem. Soc 2013, 135, 2482.
- [12].For catalytic enantioselective carbonyl propargylation via alcohol-mediated hydrogen transfer, see: Patman RL, Williams VM, Bower JF, Krische MJ, Angew. Chem. Int. Ed 2008, 47, 5220; Geary LM, Leung JC, Krische MJ, Chem. Eur. J 2012, 18, 16823; Geary LM, Woo SK, Leung JC, Krische MJ, Angew. Chem. Int. Ed 2012, 51, 2972; Woo SK, Geary LM, Krische MJ, Angew. Chem. Int. Ed 2012, 51, 7830; Liang T, Woo SK, Krische MJ, Angew. Chem. Int. Ed 2016, 55, 9207; Nguyen KD, Herkommer D, Krische MJ, J. Am. Chem. Soc 2016, 138, 5238.
- [13].For selected reviews on alcohol-mediated carbonyl addition, see: Patman RL, Bower JF, Kim IS, Krische MJ, Aldrichimica Acta 2008, 41, 95; Bower JF, Kim IS, Patman RL, Krische MJ, Angew. Chem. Int. Ed 2009, 48, 34; Hassan A, Krische MJ, Org. Process Res. Dev 2011, 15, 1236; Ketcham JM, Shin I, Montgomery TP, Krische MJ, Angew. Chem. Int. Ed 2014, 53, 9142; Nguyen KD, Park BY, Luong T, Sato H, Garza VJ, Krische MJ, Science 2016, 354, 300; Kim SW, Zhang W, Krische MJ, Acc. Chem. Res 2017, 50, 2371; Holmes M, Schwartz LA, Krische MJ, Chem. Rev 2018, 118, 6026.
- [14].For rhodium-catalyzed enyne-carbonyl reductive coupling, see: Jang H-Y, Huddleston RR, Krische MJ, J. Am. Chem. Soc 2004, 126, 4664; Kong J-R, Ngai M-Y, Krische MJ, J. Am. Chem. Soc 2006, 128, 718; Hong Y-T, Cho C-W, Skucas E, Krische MJ, Org. Lett 2007, 9, 3745; Komanduri V, Krische MJ, J. Am. Chem. Soc 2006, 128, 16448. For a related diyne coupling, see: Huddleston RR, Jang H-Y, Krische MJ, J. Am. Chem. Soc 2003, 125, 11488.
- [15].For rhodium-catalyzed enyne-imine reductive coupling, see: Kong J-R, Cho C-W, Krische MJ, J. Am. Chem. Soc 2005, 127, 11269.
- [16].For nickel-catalyzed enyne-carbonyl reductive coupling, see: Miller KM, Luanphaisarnnont T, Molinaro C, Jamison TF, J. Am. Chem. Soc 2004, 126, 4130; Miller KM, Jamison TF, J. Am. Chem. Soc 2004, 126, 15342; Miller KM, Colby EA, Woodin KS, Jamison TF, Adv. Synth. Catal 2005, 347, 1533; Miller KM, Jamison TF, Org. Lett 2005, 7, 3077; Malik HA, Sormunen GJ, Montgomery J, J. Am. Chem. Soc 2010, 132, 6304.
- [17].For selected theoretical and computational studies on metalacycle formation via oxidative coupling of π-unsaturated reactants, see: Stockis A, Hoffmann R, J. Am. Chem. Soc 1980, 102, 2952; Liu P, McCarren P, Cheong PH-Y, Jamison TF, Houk KN J. Am. Chem. Soc 2010, 132, 2050; Liu P, Krische MJ, Houk KN, Chem. Eur. J 2011, 17, 4021.
- [18].For enyne hydroruthenation to form σ-allenylruthenium complexes, see: Wakatsuki Y, Yamazaki H, Maruyama Y, Shimizu I, J. Chem. Soc., Chem. Commun 1991, 261.
- [19].For alcohol dehydrogenation catalyzed by (TFA)2Ru(CO)(PPh3)2, see: Dobson A, Robinson SD, Inorg. Chem 1977, 16, 137.
- [20].Tse SKS, Xue P, Lin Z, Jia G, Adv. Synth. Catal 2010, 352, 1512. [Google Scholar]
- [21].Hart DW, Blackburn TF, Schwartz J, J. Am. Chem. Soc 1975, 97, 679. [Google Scholar]
- [22].Hofmeister H, Annen K, Laurent H, Wiechert R, Angew. Chem. Int. Ed. Engl 1984, 23, 727. [Google Scholar]
- [23].Myers AG, Zheng B, Movassaghi M, J. Org. Chem 1997, 62, 7507. [DOI] [PubMed] [Google Scholar]
- [24].Trost BM, Toste FD, Pinkerton AB, Chem. Rev 2001, 101, 2067. [DOI] [PubMed] [Google Scholar]
- [25].Huang T-M, Hsu R-H, Yang C-S, Chen J-T, Lee G-H, Wang Y, Organometallics 1994, 13, 3657. [Google Scholar]
- [26].Yang Y, Perry IB, Lu G, Liu P, Buchwald SL, Science 2016, 353, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]