

Abstract
Family studies have established that the heritability of blood pressure is significant and genome-wide association studies (GWAS) have identified numerous susceptibility loci, including one within the non-coding part of Rho GTPase-activating protein 42 gene (ARHGAP42) on chromosome 11q22.1. Arhgap42-deficient mice have significantly elevated blood pressure, but the phenotypic effects of human variants in the coding part of the gene are unknown. In a Danish cohort of carriers with apparently balanced chromosomal rearrangements, we identified a family where a reciprocal translocation t(11;18)(q22.1;q12.2) segregated with hypertension and obesity. Clinical re-examination revealed that four carriers (age 50–77 years) have had hypertension for several years along with an increased body mass index (34–43 kg/m2). A younger carrier (age 23 years) had normal blood pressure and body mass index. Mapping of the chromosomal breakpoints with mate-pair and Sanger sequencing revealed truncation of ARHGAP42. A decreased expression level of ARHGAP42 mRNA in the blood was found in the translocation carriers relative to controls and allele-specific expression analysis showed monoallelic expression in the translocation carriers, confirming that the truncated allele of ARHGAP42 was not expressed. These findings support that haploinsufficiency of ARHGAP42 leads to an age-dependent hypertension. The other breakpoint truncated a regulatory domain of the CUGBP Elav-like family member 4 (CELF4) gene on chromosome 18q12.2 that harbours several GWAS signals for obesity. We thereby provide additional support for an obesity locus in the CELF4 domain.
Subject terms: Genetics research, Cytogenetics, Next-generation sequencing, Hypertension
Introduction
Hypertension is a chronic disease defined by a long-term elevated blood pressure (BP), specifically BP ≥ 140 mmHg systolic or ≥90 mmHg diastolic. It is a major public health challenge estimated to affect more than one billion adults worldwide [1, 2], contributing to morbidity and mortality with hypertension-related complications such as an increased risk of stroke, heart failure, kidney damage, and atherosclerosis [2, 3]. Despite this, the pathogenesis of hypertension remains largely unknown, in part, due to the complexity of the trait involving interactions between genes and environmental factors. Family studies have established that the heritability of BP is 30–50%, suggesting that unravelling the genetics of hypertension may be key to understanding its pathogenesis, as well as identifying therapeutic targets [2–4]. This is complicated by the fact that BP variation, in the majority of patients, is determined by many genes that each have only a small effect [3].
Currently, genome-wide association studies (GWAS) have identified in excess of 120 loci, which contribute to hypertension and BP regulation in general [4–9]. One such locus was identified on chromosome 11 within the Rho GTPase-activating protein 42 gene (ARHGAP42), where the BP-associated allele is defined by single-nucleotide polymorphisms (SNPs) located in the non-coding part of the gene [9–14]. The minor allele at this locus is protective and ARHGAP42 mRNA is threefold higher in individuals homozygous for the minor allele, as compared with individuals homozygous for the major allele [13, 14]. The minor T allele at the proposed functional SNP at this locus, rs604723:C > T, binds serum response factor more readily than the major C allele, increasing ARHGAP42 expression and decreasing Ras homologue family member A (RhoA)-dependent vascular smooth muscle cell contractility, thereby modifying hypertension risk [14–16]. Moreover, genotyping of a cohort of 346 human patients with untreated borderline hypertension revealed a 5 mmHg decrease in BP in individuals homozygous for the minor allele compared with individuals homozygous for the major allele [13].
Variants within the coding part of ARHGAP42 have not been described in humans but Arhgap42gt/gt mice, homozygous for a gene-trap-mediated reduction in Arhgap42 mRNA levels, exhibit significant hypertension (+ 20–30 mmHg) and enhanced arterial contraction and BP in response to angiotensin II and endothelin-1, with no other manifestations. Pre-treatment with the RhoA kinase inhibitor, Y27632, completely abrogates this response [15, 16]. A dose-dependent relationship between ARHGAP42 expression and BP is supported by heterozygous Arhgap42+/gt mice having half the amount of vascular Arhgap42 mRNA, when compared with their wild-type counterparts [15]. These heterozygous mice also exhibit significant hypertension (+15 mmHg) [15, 16]. To our knowledge, no human studies have previously described variants in the coding part of ARHGAP42 and the consequences on human BP have therefore been unknown.
We report a Danish family where hypertension and high body mass index (BMI) segregate with a balanced reciprocal translocation that truncates the ARHGAP42 gene. Balanced chromosomal rearrangements, such as reciprocal translocations, associated with phenotypic abnormalities are a valuable resource to increase our understanding of the human genome, as mapping of the breakpoints can provide the link between a disease and the disease-causing gene or mechanism [17]. Herein, we describe the phenotype of the family, the mapping results, and the change in ARHGAP42 mRNA expression, with the aim of reporting the clinical consequences of a loss-of-function variant in this gene. In addition, we describe a potential association between obesity and the other breakpoint truncating a cluster of three obesity-associated SNPs that are located in the topological associated domain of the CUGBP Elav-like family member 4 (CELF4) gene.
Materials and methods
Patients
A nationwide cohort of carriers of a balanced reciprocal translocation or inversion has previously been re-examined by questionnaires [18] and among these families we identified a family where a t(11;18) translocation segregated with hypertension. This reciprocal translocation was originally detected in 1990 by chromosome analysis of the proband, because she had recurrent spontaneous abortions (Fig. 1a). Subsequent cytogenetic analyses of relatives showed that her mother, two brothers, and two of the brothers’ children were all carriers of the translocation (Fig. 1b), whereas the two brothers of the mother had normal karyotypes. When the family participated in the re-examination in 2002, four of the translocation carriers reported that they had hypertension: the proband’s mother was diagnosed with hypertension at the age of 35 years and with ventricular extra systoles at the age of 73 years, the proband has been hypertensive since she was 35–38 years and her two brothers since the age of 48 and 45 years, respectively. One of them had atrial fibrillation since the age of 53 years. The mother’s brothers with normal karyotypes are both reported to be healthy, without hypertension, and with normal weight. The proband’s maternal grandfather and grandmother are both deceased without knowledge about hypertension.
Fig. 1.

a Pedigree of the family with the proband (III.5, indicated with an arrow), her mother (II.2), two brothers (III.2 and III.4), niece (IV.4) and nephew (IV.3) carrying a t(11;18) reciprocal translocation. Individuals with normal karyotypes are indicated in grey, whereas individuals not analysed are indicated in white. Phenotypes are as annotated with hypertension indicated by a white dot and body mass index (BMI) written below. Underneath each translocation carrier, except individuals IV.3 and IV.4, who were unavailable for investigation, the SNP haplotypes from Sanger sequencing of genomic DNA are shown. b Partial karyogram of the t(11;18)(q22.1;q12.2) translocation, showing the normal and derivative (der) chromosomes 11 and 18. The approximate breakpoint positions are indicated by arrows
The family was invited to a medical examination including an oral glucose tolerance test at the Novo Nordisk Foundation Center for Basic Metabolic Research in 2017 and five t(11;18) translocation carriers attended. Blood samples for various clinical analyses were obtained from all five carriers. In addition, samples for DNA and RNA analyses were obtained from four carriers whereas one carrier only contributed with a blood sample for DNA analysis. The study has been approved by the National Scientific Ethics Committee (H-KF-2006-5901) and the Danish Data Protection Agency (2012-54-0053). Written consent was obtained from all participating carriers.
Mapping of the chromosomal breakpoints
Genomic DNA was extracted from the blood samples and mate-pair sequencing (MPS) libraries were prepared as described elsewhere [19]. The MPS libraries were sequenced as 75 bp paired-end reads on the Illumina NextSeq and alignment of reads was done as previously described [19], except the samples were aligned to GRCh38/hg38. The chromosomal breakpoints were confirmed by Sanger sequencing of the breakpoint-spanning fragments (please see Supplementary Table 1 for primer sequences and conditions). The mapping results and the phenotypes have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/, accession number: SCV000864009).
Evaluation of the truncated genomic regions
The genomic regions truncated by the balanced reciprocal translocation were evaluated using public available databases including UCSC Genome browser (https://genome.ucsc.edu/) [20], Ensembl (https://www.ensembl.org) [21], ExAC (http://exac.broadinstitute.org/) and gnomAD (http://gnomad.broadinstitute.org/) [22], Clinical Genome Resource’s (https://clinicalgenome.org/) [23], Genotype-Tissue Expression project (https://www.gtexportal.org/home/) [24], and the 3D genome browser (http://promoter.bx.psu.edu/hi-c/) [25].
Quantitative real-time PCR and allele-specific expression
RNA was isolated and complimentary DNA was prepared from the translocation carriers and six unrelated control individuals using random hexamer primers. Two primer pairs were selected, one proximal (ARHGAP42E1) and one distal (ARHGAP42E21) to the 11q22.1 translocation breakpoint (see Fig. 2a for primer localization and Supplementary Table 2 for primer sequences and conditions). All samples were run in triplicates and all analyses of the same gene were run on the same plate. Relative gene expression was determined using the ΔΔCt approach as detailed elsewhere [26]. Data were normalized with a geometric mean normalization factor (geNorm calculation) as previously published [27] using four reference genes (GAPDH, COX4A, B2M, and HPRT) and data are presented as mean rescaled normalized relative quantities ± SD. Statistical analysis and Grubb’s test for outliers was performed and normality was assessed visually with Q–Q plots. Statistical significance was determined using an unpaired Student’s t-test with Welch’s correction and a p-value of ≤ 0.05 was considered significant.
Fig. 2.

a The chromosome 11 breakpoint truncates ARHGAP42 between exons 4 and 5 as illustrated in the GRCh38/hg38 UCSC Genome Browser [20] view of the 11q22.1 breakpoint and indicated with a black arrow. The positions of the SNPs known to be associated with hypertension and discussed in the main text are illustrated below the gene. The position of the qPCR primers are indicated with arrows: the black arrows show the position of the ARHGAP42E1 qPCR primers and the grey arrows show the position of the ARHGAP42E21 qPCR primers. b, c ARHGAP42 mRNA expression in blood from translocation carriers (grey) and unrelated controls (white) using the two different primer sets: b proximal primer pair (ARHGAP42E1), p-value = 0.0828 and c distal primer pair (ARHGAP42E21), p-value = 0.0219. For the experiments, four translocation carriers (II.2, III.2, III.4, and III.5 in Fig. 1A) and six unrelated controls were investigated, and values represent the mean rescaled normalized relative quantities ± SD. Results are depicted as fold change relative to the minimum ARHAGP42 expression level
Allele-specific expression analysis of ARHGAP42 was performed by Sanger sequencing of a synonymous SNP in exon 10 (rs543146, hg38 chr11:g.100943839 A > G) of ARHGAP42 (see Fig. 2a for SNP localization and Supplementary Table 3 for primer sequences and conditions).
Results
Clinical findings
At the general medical examination in 2017, the four carriers with hypertension (II:2, III:2, III:4, and III:5, Fig. 1a) were all treated by medical antihypertensive treatment. These four carriers were also found to have a BMI > 30 with a body fat percentage exceeding the recommended of whom one (II:2) had type 2 diabetes based on fasting plasma glucose of 7.5 mmol/L, whereas the three others had prediabetes with fasting plasma glucose > 6 mmol/L. Three individuals (II:2, III:2, and III:4) also had borderline high cholesterol level. The proband’s 23 years old niece, a translocation carrier, was found to be normotensive and with normal BMI. The results of the medical examination are presented in Table 1 together with the medicine taken by the carriers. For the proband, the first prescribed antihypertensive medicine was a thiazide diuretic (Bendroflumethiazide, age 35–38 years) and this was subsequently supplemented with a calcium channel blocker (Felodipine, age 38 years), but because of insufficient effect an angiotensin II receptor blocker (Losartan, age 40 years) was added. She used this combination of medicine from the age of 40–50 years (as listed in Table 1). However, 6 months ago she stopped using the thiazide diuretic, reduced the dosage of the calcium channel blocker (from 10 to 5 mg daily), while continuing the angiotensin II receptor blocker on the same dosage without increasing her BP.
Table 1.
Results from the clinical examination of the five translocation carriers
| II.2 | III.2 | III.4 | III.5 (Proband) | IV.4 | |
|---|---|---|---|---|---|
| Gender | Female | Male | Male | Female | Female |
| Age (years) | 77 | 57 | 54 | 50 | 23 |
| BMI (kg/m2) | 36.1 | 34.3 | 35.5 | 42.6 | 20.8 |
| Body fat percentage (%) | 47.2 | 33.8 | 32.6 | 49.6 | 30.1 |
| Fasting p-glukose (mmol/L) | 7.6 | 6.0 | 6.0 | 6.2 | 4.8 |
| Oral glucose tolerance test, p-glucose at 120 min (mmol/L) | 11.0 | 5.9 | 3.9 | 6.7 | 5.4 |
| Fasting s-insulin (pmol/L) | 125 | 141 | 62 | 137 | 43 |
| Oral glucose tolerance test, s-insulin at 120 min (pmol/L) | 940 | 361 | 111 | 373 | 173 |
| HbA1c (mmol/L) | 42 | 37 | 38 | 38 | N/A |
| Plasma cholesterol (mmol/L) | 5.1 | 6.0 | 5.1 | 4.2 | 4.8 |
| Urinary test | Blank | Blank | Blank | Blank | Blank |
| ECG | Normal | Normal | Atrial fibrillation | Normal | Normal |
| Blood pressure (systolic/diastolic mmHg) | 126/73a | 130/90a | 134/87a | 134/95a | 121/64 |
| Antihypertensive treatment | From 35 years | From 48 years | From 45 years | From 35–38 years | |
| Medicine in 2017 | Losartan-Hydrochlorothiazide 100 mg/12.5 mg × 1; Metoprolol 25 mg × 1; Acetylsalicyl acid 75 mg × 1 | Enalapril 10 mg × 1 | Captopril 25 mg × 3; Felodipine 5 mg × 1; Warfarin 2.5 mg × 3 Bendroflumethiazide/potassium chloride 2.5 mg/573 mg × 1 | Losartan 100 mg × 1; Felodipine 10 mg × 1; Bendroflumethiazide/potassium chloride 2.5 mg/573 mg × 1 | None |
| Other diseases | Psoriasis | Cervical dysplasia |
aBlood pressure when treated with antihypertensive medicine
BMI body mass index, ECG electrocardiography, p plasma, s serum
Values exceeding the recommended thresholds are indicated in bold
Identification and evaluation of the t(11;18) translocation breakpoints
The karyotype of the balanced reciprocal translocation was revised to t(11;18)(q22.1;q12.2) by MPS. Both chromosomal breakpoints were subsequently validated by Sanger sequencing revealing a 2 bp duplication (AG) at the der(11) breakpoint and a 1 bp deletion (T) at the der(18) breakpoint (see Supplementary Table 4). These indels (insertions and deletions) are described according to the Human Genome Variation Society nomenclature [28] as g.100863853_100863854dupAG and g.39025190delT (hg38), respectively. Microhomology was observed at the der(11) breakpoint, whereas the der(18) breakpoint was located within a repetitive sequence. Furthermore, a 1 bp insertion (A) was observed at the der(11) junction. The 11q22.1 breakpoint was within intron 4 of ARHGAP42, truncating the canonical transcript of the gene (Fig. 2a) according to Ensembl release 93. The 18q12.2 breakpoint did not disrupt any known genes but was found to truncate the putative regulatory domain of CELF4 (Fig. 3). No fusion transcripts were deduced from the sequence. The karyotype according to ISCN 2016 [29] of the proband was found to be 46,XX,t(11;18)(q23.3;q21).seq[GRCh38] t(11;18)(q22.1;q12.2) with the genomic positions: g.[chr11:pter_cen_100863854::AGA::chr18:39025191_qter] g.[chr18:pter_cen_39025189::chr11:100863853_qter].
Fig. 3.
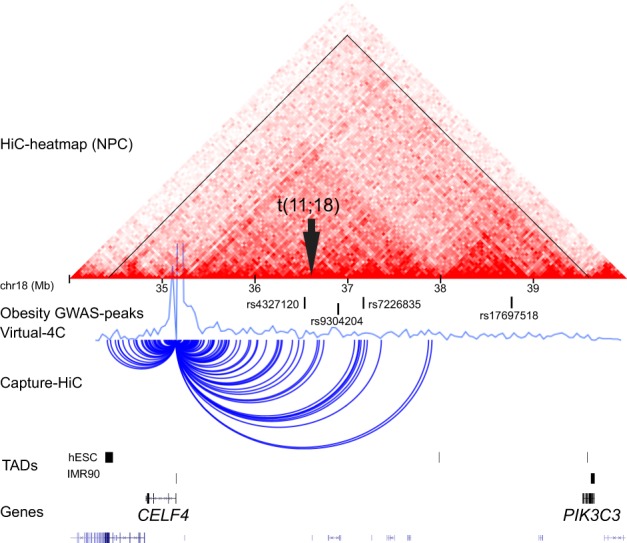
The chromosome 18 breakpoint (black arrow) truncates the putative regulatory domain of the CUGBP Elav-Like Family Member 4 (CELF4) gene. The HiC-heatmap of neural progenitor cells (NPC) and the Virtual 4C-profile with CELF4 as anchor point [25] show that CELF4 has cis-interactions within a > 5 megabase (Mb) region on 18q12.2 (tented region), within topological associating domain (TAD) boundaries in embryonic (hESC) and IMR90 fibroblasts. Four obesity-associated genome-wide association study (GWAS), signals are located within the CELF4 -domain, including three single-nucleotide polymorphisms (SNPs; rs4327120:T > C, rs9304204:G > A, rs7226835:C > A) surrounding the translocation breakpoint. Moreover, Capture-HiC from GM12878 cells show the interactions of CELF4, where both the translocation breakpoints and the cluster of three obesity-associated SNPs are located within the CELF4 interaction range
ARHGAP42 expression analysis
To elucidate the effect of truncating ARHGAP42 on the expression of the gene, we investigated the ARHGAP42 gene expression in the four available translocation carriers and in six non-related control individuals as illustrated in Fig. 2b, c. Quantitative PCR (qPCR) analysis with the distal primer pair (ARHGAP42E21) revealed a statistically significant decrease (about half) in expression in the translocation carriers relative to the controls (p-value = 0.0219, Fig. 2c), whereas qPCR analysis with the proximal primer pair (ARHGAP42E1) revealed a nonsignificant decrease in ARHGAP42 expression (p-value = 0.0828, Fig. 2b).
To investigate whether disruption of ARHGAP42 attenuates expression from the truncated allele specifically, we performed allele-specific expression analysis of ARHGAP42 using a synonymous SNP in exon 10 (rs543146:A > G) of ARHGAP42 (Fig. 2a). The G allele of SNP rs543146:A > G segregated with the derivative chromosome 18 in the family (Fig. 1a), allowing us to use it as a cis marker to test allele-specific expression of ARHGAP42 at the locus. Translocation carrier III.2 and control individual 1, who were both investigated with qPCR (Fig. 2b, c), were found to be heterozygous at the rs543146:A > G SNP, as illustrated by Sanger sequencing of rs543146:A > G in genomic DNA (Supplementary Figure 1). However, Sanger sequencing of the same SNP in cDNA revealed that although control individual 1, as expected, is still heterozygous, translocation carrier III.2 only express mRNA from the non-truncated ‘A’ allele (Supplementary Figure 1, bottom panel). Thus, translocation carrier III.2 and control individual 1 showed monoallelic and bi-allelic expression of ARHGAP42 mRNA, respectively, confirming that the decrease in the overall ARHGAP42 expression seen in qPCR was due to the truncated allele of the gene being specifically silenced.
Discussion
In this study, we describe for the first time the consequence of a heterozygous variant causing loss of function of ARHGAP42 in humans. A search of the ExAC and gnomAD databases revealed ARHGAP42 to be consistently poorly represented, with no information about whether or not the gene is a loss-of-function constraint gene [22]. Furthermore, there is a surprising lack of copy number loss of this specific gene in The Clinical Genome Resources [30]. The low sequencing coverage of ARHGAP42 is most likely due to the high number of pseudogenes, as evident from the high degree of similarity of the ARHGAP42 mRNA sequence to other places in the genome. Thus, the finding of no, or very few, other patients with variants in the coding part of ARHGAP42 may be explained by the low sequencing coverage of this gene, e.g., when using whole-exome and -genome sequencing techniques. In contrast, the detection of a truncated gene by a reciprocal translocation is not limited by low sequencing coverage of a gene as seen in this family, illustrating that mapping of these balanced chromosomal rearrangements is a valuable approach to reveal disease-associated variants, disease genes, and genetic mechanisms [18].
The loss-of-function variant in ARHGAP42 segregated in the current family with both hypertension and high BMI from the age of 35 years. This is in accordance with reports that hypertension and high BMI tend to rise with age in general [31, 32]. The carriers were found to have lower ARHGAP42 mRNA expression, due to allele-specific expression of ARHGAP42, presumably due to non-sense-mediated decay or silencing of the truncated allele. The role of ARHGAP42 in BP homoeostasis has been studied in Arhgap42-deficient mice where ARHGAP42 expression and BP modify hypertension in a dose-dependent manner [15, 16]. We propose that the loss-of-function variant of one copy of ARHGAP42 results in haploinsufficiency and leads to age-dependent hypertension in humans. Previous studies in humans have exclusively reported non-coding variants within ARHGAP42, with individuals homozygous for the minor allele having lower BP and threefold higher ARHGAP42 mRNA levels, when compared with individuals homozygous for the major allele [9–14]. These observations are in accordance with the reduced amount of ARHGAP42 mRNA found in the translocation carriers and their elevated BP.
Only about half of treated patients achieve reasonable control of their BP [14] and a promising area for personalized medicine is therefore to select antihypertensive drugs based on individual genetic testing [33]. Thus, ours and similar studies can have implications for the clinical management when the identified genetic aetiology can be used to choose the optimal antihypertensive medicine for the specific patients. It has been reported that decreased ARHGAP42 expression leads to increased RhoA-dependent SMC contraction [14, 16] and it has therefore been proposed that hypertension patients with variants in ARHGAP42 could be more responsive to antihypertensive therapies that target vascular smooth muscle cell contractility and vessel tone directly including antihypertensive drugs that target angiotensin II, angiotensin-converting-enzyme inhibitors, and angiotensin receptor blockers [16]. We suggest that this is also the case for patients with a loss-of-function variant of ARHGAP42, because the four t(11;18)(q22.1;q12.2) translocation carriers with hypertension are all well treated by antihypertensive medicine that target the angiotensin II pathway. It is noteworthy that they are all treated with dosages within the normal range, and that the angiotensin II receptor blocker seems to be the most important antihypertensive treatment for the proband, because she could stop using most other medicine. It could be interesting to study if this is also the case for hypertensive individuals who are homozygous for the ARHGAP42 major allele or have other variants in the coding part of the gene.
There is no evidence from GWAS studies or mouse models that ARHGAP42 is associated with obesity or diabetes. In contrast, the breakpoint on chromosome 18 truncates a cluster of obesity-associated SNPs that are located in the topological associated domain [34] of CELF4, a brain-specific RNA-binding protein (Fig. 3). Haploinsufficiency of CELF4 has been associated with intellectual disability, autism, and obesity [35], and apart from a complex seizure disorder, hyperphagia-associated weight gain is one of the features in heterozygotic Celf4+/− mice [36, 37]. We cannot assess CELF4 expression in the translocation carriers, because it is exclusively expressed in the central nervous system in adults [36, 38, 39]. However, based on the circumstantial genetic data about the CELF4 domain and obesity, we speculate that the high BMI in the adult translocation carriers could be explained by a change of CELF4 gene regulation by a long-range positional effect.
Weight gain predisposes to hypertension in general by shifting the BP frequency distribution towards higher levels [31, 40]. The translocation carriers might be even more susceptible, because they only have half the amount of ARHGAP42 mRNA compared with unrelated controls and as ARHGAP42 levels are important for modulating vessel tone in response to signals that increase BP [14]. Thus, the translocation carriers may be unable to counteract the cascade of events, initiated by excess adipose tissue, and it might thereby contribute to a higher BP [31]. This theory is in agreement with the notion that progression from a normotensive to hypertensive phenotype results from the complex combination of genetic, environmental, behavioural, and dietary factors [31, 41].
Altogether, our data support the conclusion that haploinsufficiency of ARHGAP42 is involved in the aetiology of age-dependent hypertension and also implicate that genetic variants in the regulatory domain of CELF4 may be associated with obesity.
Supplementary information
Acknowledgements
We thank the family who has participated in the clinical study. We also thank Annemette F. Mikkelsen, Rabab Nima, Lillian E. Rasmussen, Linda B. Dalsgaard, and Bjarke Thomsen for technical assistance in the laboratory, and Annemette Forman and the Clinical Test Center at Novo Nordisk Foundation Center for Basic Metabolic Research for clinical evaluation of the family. This work was supported by Hartmann Fonden (A29354) and the Danish Council for Independent Research (4183-00482B).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41431-019-0382-9) contains supplementary material, which is available to authorized users.
References
- 1.Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365:217–23. doi: 10.1016/S0140-6736(05)17741-1. [DOI] [PubMed] [Google Scholar]
- 2.Widmaier EP, Raff H, Strang KT, Vander AJ. Vander’s human physiology: the mechanisms of body function. 13th ed. New York, NY: McGraw-Hill; 2014. [Google Scholar]
- 3.Coffman TM. Under pressure: the search for the essential mechanisms of hypertension. Nat Med. 2011;17:1402–9. doi: 10.1038/nm.2541. [DOI] [PubMed] [Google Scholar]
- 4.Padmanabhan S, Caulfield M, Dominiczak AF. Genetic and molecular aspects of hypertension. Circ Res. 2015;116:937–59. doi: 10.1161/CIRCRESAHA.116.303647. [DOI] [PubMed] [Google Scholar]
- 5.Franceschini N, Chasman DI, Cooper-DeHoff RM, Arnett DK. Genetics, ancestry, and hypertension: Implications for targeted antihypertensive therapies. Curr Hypertens Rep. 2014;16:461. doi: 10.1007/s11906-014-0461-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41:666–76. doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41:677–87. doi: 10.1038/ng.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warren HR, Evangelou E, Cabrera CP, Gao H, Ren M, Mifsud B, et al. Genome-wide association analysis identifies novel blood pressure loci and offers biological insights into cardiovascular risk. Nat Genet. 2017;49:403–15. doi: 10.1038/ng.3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478:103–9. doi: 10.1038/nature10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wain LV, Verwoert GC, O’Reilly PF, Shi G, Johnson T, Johnson AD, et al. Genome-wide association study identifies six new loci influencing pulse pressure and mean arterial pressure. Nat Genet. 2011;43:1005–11. doi: 10.1038/ng.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kato N, Loh M, Takeuchi F, Verweij N, Wang X, Zhang W, et al. Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nat Genet. 2015;47:1282–93. doi: 10.1038/ng.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–4. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bai X, Mangum KD, Dee RA, Stouffer GA, Lee CR, Oni-Orisan A, et al. Blood pressure – associated polymorphism controls ARHGAP42 expression via serum response factor DNA binding. J Clin Invest. 2017;127:670–80. doi: 10.1172/JCI88899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bai X, Mangum K, Kakoki M, Smithies O, Mack CP, Taylor JM. GRAF3 serves as a blood volume-sensitive rheostat to control smooth muscle contractility and blood pressure. Small GTPases. 2017;1248:1–10. doi: 10.1080/21541248.2017.1375602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bai X, Lenhart KC, Bird KE, Suen AA, Rojas M, Kakoki M, et al. The smooth muscle-selective RhoGAP GRAF3 is a critical regulator of vascular tone and hypertension. Nat Commun. 2013;4:2910. doi: 10.1038/ncomms3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bai X, Dee R, Mangum KD, Mack CP, Taylor JM. RhoA signaling and blood pressure: the consequence of failing to “Tone it Down”. World J Hypertens. 2016;6:18–35. doi: 10.5494/wjh.v6.i1.18. [DOI] [Google Scholar]
- 17.Talkowski ME, Ernst C, Heilbut A, Chiang C, Hanscom C, Lindgren A, et al. Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research. Am J Hum Genet. 2011;88:469–81. doi: 10.1016/j.ajhg.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bache I, Hjorth M, Bugge M, Holstebroe S, Hilden J, Schmidt L, et al. Systematic re-examination of carriers of balanced reciprocal translocations: a strategy to search for candidate regions for common and complex diseases. Eur J Hum Genet. 2006;14:410–7. doi: 10.1038/sj.ejhg.5201592. [DOI] [PubMed] [Google Scholar]
- 19.Aristidou C, Koufaris C, Theodosiou A, Bak M, Mehrjouy MM, Behjati F, et al. Accurate breakpoint mapping in apparently balanced translocation families with discordant phenotypes using whole genome mate-pair sequencing. PLoS One. 2017;12:e0169935. doi: 10.1371/journal.pone.0169935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The Human Genome Browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, et al. Ensembl 2018. Nucleic Acids Res. 2018;46:D754–61. doi: 10.1093/nar/gkx1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, et al. ClinGen—the clinical genome resource. N Engl J Med. 2015;372:2235–42. doi: 10.1056/NEJMsr1406261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–5. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Zhang B, Zhang L, An L, Xu J, Li D et al. The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Biorxiv 2017. 10.1101/112268. [DOI] [PMC free article] [PubMed]
- 26.Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8:R19. doi: 10.1186/gb-2007-8-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:34. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, et al. HGVS recommendations for the description of sequence variants: 2016 Update. Hum Mutat. 2016;37:564–9. doi: 10.1002/humu.22981. [DOI] [PubMed] [Google Scholar]
- 29.Mcgowan-jordan J, Simons A, Schmid M. ISCN: an international system for human cytogenomic nomenclature. S. Karger; Basel, Switzerland, 2016.
- 30.Kirkpatrick BE, Riggs ER, Azzariti DR, Miller VR, Ledbetter DH, Miller DT, et al. GenomeConnect: matchmaking between patients, clinical laboratories, and researchers to improve genomic knowledge. Hum Mutat. 2015;36:974–8. doi: 10.1002/humu.22838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leggio M, Lombardi M, Caldarone E, Severi P, D’Emidio S, Armeni M, et al. The relationship between obesity and hypertension: an updated comprehensive overview on vicious twins. Hypertens Res. 2017;40:947–63. doi: 10.1038/hr.2017.75. [DOI] [PubMed] [Google Scholar]
- 32.Litt JS, Lambert JR, Glueck DH. Gardening and age-related weight gain: Results from a cross-sectional survey of Denver residents. Prev Med Rep. 2017;8:221–5. doi: 10.1016/j.pmedr.2017.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rossi GP, Ceolotto G, Caroccia B, Lenzini L. Genetic screening in arterial hypertension. Nat Rev Endocrinol. 2017;13:289–98. doi: 10.1038/nrendo.2016.196. [DOI] [PubMed] [Google Scholar]
- 34.Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518:331–6. doi: 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Halgren C, Bache I, Bak M, Myatt MW, Anderson CM, Brøndum-Nielsen K, et al. Haploinsufficiency of CELF4 at 18q12.2 is associated with developmental and behavioral disorders, seizures, eye manifestations, and obesity. Eur J Hum Genet. 2012;20:1315–9. doi: 10.1038/ejhg.2012.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wagnon JL, Mahaffey CL, Sun W, Yang Y, Chao HT, Frankel WN. Etiology of a genetically complex seizure disorder in Celf4 mutant mice. Genes, Brain Behav. 2011;10:765–77. doi: 10.1111/j.1601-183X.2011.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Y, Mahaffey CL, Bérubé N, Maddatu TP, Cox GA, Frankel WN. Complex seizure disorder caused by Brunol4 deficiency in mice. PLoS Genet. 2007;3:e124. doi: 10.1371/journal.pgen.0030124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kislinger T, Cox B, Kannan A, Chung C, Hu P, Ignatchenko A, et al. Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell. 2006;125:173–86. doi: 10.1016/j.cell.2006.01.044. [DOI] [PubMed] [Google Scholar]
- 39.Melé M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, et al. Human genomics. The human transcriptome across tissues and individuals. Science. 2015;348:660–5. doi: 10.1126/science.aaa0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hall JE, Do Carmo JM, Da Silva AA, Wang Z, Hall ME. Obesity-induced hypertension: interaction of neurohumoral and renal mechanisms. Circ Res. 2015;116:991–1006. doi: 10.1161/CIRCRESAHA.116.305697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeMarco VG, Aroor AR, Sowers JR. The pathophysiology of hypertension in patients with obesity. Nat Rev Endocrinol. 2014;10:364–76. doi: 10.1038/nrendo.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.