

Abstract
The DOCK3 gene encodes the Dedicator of cytokinesis 3 (DOCK3) protein, which belongs to the family of guanine nucleotide exchange factors and is expressed almost exclusively in the brain and spinal cord. We used whole exome sequencing (WES) to investigate the molecular cause of developmental delay and hypotonia in three unrelated probands. WES identified truncating and splice site variants in Patient 1 and compound heterozygous and homozygous missense variants in Patients 2 and 3, respectively. We studied the effect of the three missense variants in vitro by using site-directed mutagenesis and pull-down assay and show that the induction of Rac1 activation was significantly lower in DOCK3 mutant cells compared with wild type human DOCK3 (P < 0.05). We generated a protein model to further examine the effect of the two missense variants within or adjacent to the DHR-2 domain in DOCK3 and this model supports pathogenicity. Our results support a loss of function mechanism but the data on the patients with missense variants should be cautiously interpreted because of the variability of the phenotypes and limited number of cases. Prior studies have described DOCK3 bi-allelic loss of function variants in two families with ataxia, hypotonia, and developmental delay. Here, we report on three patients with DOCK3-related developmental delay, wide-based or uncoordinated gait, and hypotonia, further supporting DOCK3’s role in a neurodevelopmental syndrome and expanding the spectrum of phenotypic and genotypic variability.
Subject terms: Disease genetics, Genetics of the nervous system
Introduction
Global developmental delay (GDD) and intellectual disability (ID) affect 1–3% of children in the US [1, 2]. Whole exome sequencing (WES) can facilitate the diagnostic process in patients with GDD/ID and can lead to the discovery of novel genetic disorders. In addition, the identification of genetic variants that affect the function of proteins through WES in patients with developmental delay (DD) has increased the understanding of the molecular and cellular mechanisms leading to these conditions and provided targets for possible therapeutic intervention. We demonstrate the use of WES to identify variants in DOCK3 that are predicted to affect function in three unrelated probands with GDD, wide-based or uncoordinated gait, and hypotonia.
The DOCK3 gene (MIM 603123) is located on 3p21.2 and has 53 exons that encode the 2030 amino acid Dedicator of cytokinesis 3 (DOCK3) protein, also known as Modifier of Cell Adhesion (MOCA) and presenilin-binding protein (PBP), which is expressed specifically in the brain and spinal cord [3, 4]. DOCK3 belongs to the family of guanine nucleotide exchange factors (GEFs) that activate GTPases by exchanging bound GDP for free GTP [5, 6].
DOCK3 has four conserved domains that each play unique roles in its function as a mediator of cell adhesion, axonal regeneration and outgrowth, and neuroprotection of cells. The SH3 (src Homology-3) domain is located at the N-terminus and functions as an auto-inhibitor of DOCK3 by binding to the DHR-2 (Dock Homology Region-2) domain and nearby proline-rich C-terminus. The DHR-1 domain binds phosphatidylinositol-3,4,5-triphosphate in the WAVE protein (WASP family verprolin-homologous protein) and the DOCK3/WAVE complex localizes to the cell periphery, where the complex is involved in stimulating DHR-2 to activate Rho-GTPase protein Rac1 from Rac-GDP (inactive form) to Rac-GTP (active form). The dissociation of Rac-GTP and WAVE from the phosphorylated DOCK3 stimulates actin reorganization and axonal outgrowth [6, 7]. The last conserved domain is the proline-rich C terminus, which has been demonstrated to bind the GluN2B and GluN2D subunits of the NMDA receptor and this interaction increases the internalization and degradation of the NMDA receptor [7, 8].
Three prior studies have described DOCK3 variants in children with GDD or ID [3, 9, 10]. A pericentric inversion of chromosome 3, inv(3)(p14:q21), involving DOCK3 has been described in a family with attention deficit-hyperactivity disorder (ADHD) and ID [3]. However, the association of this inversion to the phenotypic features in this family is not certain, particularly since the inversion is affecting at least one other potential gene involved in ADHD or ID. More recently, compound heterozygous loss of function variants in DOCK3 have been reported in two siblings with DD, ataxia, and hypotonia [9]. An additional boy with biallelic DOCK3 deletion and GDD, hypotonia, and ataxia was also recently described [10].
Prior to its association with developmental disorders, DOCK3 was implicated in neurodegenerative disease. It was first discovered to play a role in the regulation of tau phosphorylation and the formation of neurofibrillary tangles in Alzheimer disease [11, 12]. DOCK3 has also been shown to stimulate axon regeneration in retinal ganglion cells after optic nerve transection [6]. Increased expression of DOCK3 has also been implicated in epileptogenesis, and when overexpressed in oligodendrocytes, DOCK3 was shown to protect against demyelination [13, 14].
In this study, we present detailed clinical and molecular findings of three unrelated patients with GDD and hypotonia. WES identified variants in DOCK3. The results of in silico analysis and in vitro studies support loss of function mechanism although data on the patients with missense variants should be cautiously interpreted because of the phenotypic differences and limited number of patients. We compare the phenotypic and genetic data of our cohort with previously reported families with DOCK3 variants.
Methods
Patients
Clinical data were obtained by retrospective chart review and direct patient interaction and interview of patients’ parents.
Whole exome sequencing
The Institutional Review Board of Washington University School of Medicine approved the study protocol used for Patients 1 and 2 and the Mayo Clinic Institutional Review Board approved the study protocol used for Patient 3. Informed consents were obtained from the parents for publication of genetic results and photographs. WES of Patient 1 and his mother was performed on DNA collected from peripheral blood by GeneDx (Gaithersburg, MD) using the Agilent Clinical Research Exome kit and NextGen sequencing with an Illumina HiSeq system with 100 bp paired-end reads. Mean depth of coverage was 128× and quality threshold 97.2%. Reported variants were confirmed with Sanger sequencing. Sequencing methodology and variant interpretation protocol has previously been described [15].
Genomic DNA of Patient 2 and his parents was isolated from peripheral blood leukocytes and exome-coding DNA was captured with the Agilent Sure Select Clinical Research Exome (CRE) kit (v1). Sequencing was performed on an Illumina HiSeq platform with 101 bp paired-end reads. Reads were aligned to hg19 using BWA (http://bio-bwa.sourceforge.net) and variants were called using the GATK (http://www.broadinstitute.org/gatk/). Detected variants were annotated, filtered, and prioritized using the Bench lab NGS platform (Agilent Technologies). Initially, only genes known to be involved in ID were analyzed, followed by a full exome analysis.
Trio WES of Patient 3 was performed utilizing a custom reagent developed by Mayo Clinic and Agilent Technologies. Sequencing was performed on an Illumina HiSeq 2500 Next Generation sequencing instrument, using HapMap Sample NA12878 as an internal control. Paired-end 101 base-pair reads were aligned to a modified human reference genome (GRCh37/hg19) using Novoalign (Novocraft Technologies, Malaysia). Approximately 97% of the exome is sequenced to a depth of at least 20×. In our reference sample NA12878 (Genome in a Bottle Consortium), greater than 99% of single nucleotide variants and 95% of small insertions and/or deletions up to 30 bases were accurately identified.
Plasmids
Full-length human DOCK3 cDNA was purchased from transOMIC Technologies (Huntsville, AL) and was subcloned into the expression vector pCMV (TAKARA, Kyoto, Japan). Three DOCK3 mutants [p.(Arg392Gln), p.(Lys1298Arg), and p.(Met1647Leu)] were prepared from full-length DOCK3 cDNA by site-directed mutagenesis PCR. WT Rac1 cDNA was also subcloned into the expression vector pCMV (TAKARA).
Measurement of Dock3 GEF activity
The activity of Rac1 was measured by a pull-down assay as described previously [6]. Briefly, glutathione-S-transferase (GST)-fused CRIB (Cdc42/Rac interactive binding motif) was purified from bacterial lysates using glutathione-agarose (Thermo Fisher Scientific, Rockford, IL). Cos7 cells were transfected with plasmids encoding WT DOCK3 or mutant DOCK3. Cos7 cells were cultured in normal conditions for 24 h after transfection, and additionally in serum-free medium for 3 h. Cos7 cells were washed twice with cold PBS and lysed with a lysis buffer (1% Triton X-100, 50 mM Tris (pH 7.4), 150 mM NaCl, 10% glycerol, and 10 mM MgCl2 containing a protease inhibitor mixture), and then centrifuged for 15 min at 10,000 × g at 4 °C. The resulting supernatants were incubated with glutathione agarose beads for 45 min at 4 °C with gentle agitation for binding active Rac1. The beads were washed three times with lysis buffer.
The bound active Rac1 was subjected to SDS-PAGE, and immunoblot analysis was performed using anti-Rac1 (1:1000, BD Bioscience, Franklin Lakes, NJ) and anti-DOCK3 (1:1000, Cosmo Bio, Tokyo, Japan) antibodies. Quantitative analysis of the signal bands was carried out using ImageJ software version 1.46r (US National Institutes of Health).
DHR2 domain model
We used homology-based methods to generate a protein structure model for the DHR-2 domain of human DOCK3 from the available experimental structure of the DHR-2 domain of human DOCK2 [16] (PDB 3b13; 44% identical [17]); amino acids 1214 through 1635. The amino acids from the C-terminal to the DHR-2 domain lack homology to any known structure and are enriched for aromatic residues and methionine. We used ab initio prediction tools, QUARK [18] and RaptorX [19], to generate models for residues 1600–1719. We considered the consistency of structural features among ab initio predictions in model quality assessment. We visualized protein structures using PyMOL [The PyMOL Molecular Graphics System, Version 1.9 Schrödinger, LLC].
Results
Photographs of the patients are shown in Fig. 1, and Table 1 summarizes the clinical and molecular findings in the probands described in this article in comparison with three previously reported patients [9, 10].
Fig. 1.

Facial images of 3 patients with DOCK3 variants. From left to right: Patient 1 (a, b), Patient 2 (c, d), and Patient 3 (e, f)
Table 1.
Clinical and molecular findings in patients with DOCK3 mutations
| Patient 1 | Patient 2 | Patient 3 | Helbig et al. Female |
Helbig et al. Male |
Iwata-Otsubo et al. | |
|---|---|---|---|---|---|---|
| DOCK3 mutations | ||||||
| Allele 1 | c.1038-2A>G: IVS12-2A>G | c.1175G>A: p. (Arg392Gln) | c.5020A>T: p.(Met1674Leu) | c.382C>G:p.(Gln128*) | c.382C>G: p.(Gln128*) | 170 kb deletion exons 6–12 |
| Allele 2 | c.3107-3110delACTT: p.(Tyr1036Leufs*8) | c.3887A>G: p.(Lys1296Arg) | c.5020A>T: p.(Met1674Leu) | 458 kb heterozygous deletion on 3p21.2 | 458 kb heterozygous deletion on 3p21.2 | 170 kb deletion including exons 6–12 |
| Sex | Male | Male | Female | Female | Male | Male |
| Ethnicity | Caucasian | Dutch | Caucasian | Ashkenazi and Yemeni Jewish | Ashkenazi and Yemeni Jewish | Caucasian |
| Pregnancy duration (weeks) | 37 | 36 | 35 | 42 | 41 | Unknown |
| Growth (last evaluation) | ||||||
| OFC (%) | 45 | 15 | >97 | 75 | 30 | 14 |
| Weight (%) | 99 | <3 | >97 | 25 | 40 | 4 |
| Height (%) | 70 | 3 | 84 | 15 | 8 | 5 |
| Developmental delay | ||||||
| Age of 1st walking (months) | 36 | 48 | 18 | 60 | 30 | 22 |
| Age of 1st words (months) | 48 | 84 | 15 | Nonverbal | 48 (single word vocab at 11 years of age) | 28 |
| Behavioral abnormalities | No | Auto-mutilation, autism spectrum disorder | Autism spectrum disorder | Not reported | Not reported | Not reported |
| Brain MRI | Day of life 1: shallow sulci, dysmorphic gyri, underdeveloped white matter, and a cavum septum pellucidum that were not evident on repeat brain MRI at 3 years of age; T2 hyperintensity in the right periventricular corona radiata and putamen | Diminished white matter in bilateral occipital lobe, slightly enlarged Virchow Robin spaces, slightly hypoplastic splenium of corpus callosum | Resolved Chiari malformation, enlarged lateral and third ventricles | Normal | Not reported | Slightly bulky and dysmorphic corpus callosum |
| Spine MRI | Syrinx C3-L2 | Not done | History of spina bifida, mild diffuse atrophy of thoracic spinal cord, elongated conus medullaris ending in soft tissue placode at L5 | Not done | Not reported | Not reported |
| EEG | Excessive discontinuity, no seizures | Not done, no clinical seizures | Left temporal slowing with left temporal spikes | Normal | Not reported | Not reported |
| Hypotonia | Yes | Yes | Yes | Yes | Yes | Yes |
| Coordination or gait abnormalities | Mild wide-based gait | Wide-based gait | Poor coordination | Ataxia | Ataxia | Wide-based gait |
| Dysmorphic features | Prominent forehead | Brachycephaly, prominent philtrum | No | Relative macrocephaly, prominent chin, high arched palate, malocclusion, long fingers | Downslanting palpebral fissures, long face, pointed chin | Bilateral epicanthal folds, upturned nasal tip/anteverted nares, prominent cheeks |
| Other congenital abnormalities | TE fistula, vertebral anomalies, thoracolumbar scoliosis, single right kidney with megaureter; myopia and astigmatism | Phimosis | High arched palate, Dental abnormalities | |||
Clinical reports
Patient 1
Patient 1 is a 5-year-old Caucasian male who was born via cesarean section at 37 weeks estimated gestational age to a 24-year-old Caucasian mother. Pregnancy was complicated by polyhydramnios and maternal hypoglycemia and hypothyroidism. The APGAR scores were 7 and 9 at 1 and 5 min, respectively. The birth weight was 3.4 kg (25–50th centile), length 48 cm (10–25th centile), and occipitofrontal circumference (OFC) 36.2 cm (50–75th centile). During the newborn period, he was noted to have multiple congenital anomalies suggesting VACTERL association, including tracheoesophageal fistula with esophageal atresia, vertebral and rib anomalies, and a single right kidney with hydronephrosis and right hydroureter.
The patient was noted to have DD during the 1st year of life. He started walking at 36 months old. At 5 years old, he is not able to run or jump and continues to wear ankle braces. He also exhibited delay of his fine motor skills and started using utensils at 5 years of age. He began to consistently use 5–10 words at the age of 5 years but does not combine two words. He is not toilet trained at the age of 5 years. He has myopia and astigmatism, requiring corrective lenses. During neuropsychological testing at 5 years of age, he had increased distractibility and hyperactivity, as well as deficits in verbal and nonverbal reasoning, executive function, memory, receptive and expressive language, visual motor integration, fine motor skills, and early academic skills.
On physical examination at 5 years, his weight was 31.9 kg (>99th centile), height 113.5 cm (66th centile), and OFC 53.5 cm (85th centile). He was noted to have a broad forehead with deep-set and hooded eyes (Fig. 1).
His previous workup included normal chromosome microarray (CMA), fragile X syndrome testing, and extensive metabolic workup. His brain MRI on day of life 1 showed shallow sulci, dysmorphic gyri, underdeveloped white matter, and a cavum septum pellucidum which suggested a developmental brain anomaly. His repeat brain MRI at 3 years of age showed no evidence of myelination or cerebral cortex abnormality but T2 hyperintensity in the right periventricular corona radiata and putamen, which may represent sequela of prior infarct. His spine MRI showed a syrinx extending from approximately C3 to the conus at L1–L2. He had an EEG during the 2nd week of life with excessive discontinuity during sleep, indicating mild generalized cerebral dysfunction.
Patient 2
Patient 2 is a 6-year-old Caucasian male who was born via spontaneous vaginal delivery at 36 weeks estimated gestational age to a 27-year-old Caucasian mother. Pregnancy was complicated by premature contractions starting at 23 weeks gestational age, maternal bicornuate uterus, maternal use of fluoxetine and seroquel, and maternal smoking of 10–15 cigarettes per day. APGAR scores were 1 and 9 at 1 and 5 min, respectively. Birth weight was 2 kg (10th centile). Birth length and OFC are unavailable. He required tube feeding for 2 weeks after birth. He currently eats by mouth but is very selective in his food choices.
The patient has GDD. He started sitting independently at the age of 30 months and walking at 48 months. At age 6 years, he has not developed speech and language, has been diagnosed with autism spectrum disorder, and exhibits self-injurious behaviors. At age 7 years, he was able to say 4 words and exhibited wide-based gait.
The patient also had phimosis, which required surgical correction and exhibits myopia, diagnosed at age 3 years.
On physical examination at 5.5 years, his weight was 15 kg (50th percentile), height was 103.2 cm (25th percentile), and his OFC was 49.2 cm (7th percentile). He had brachycephaly, plagiocephaly, and a prominent philtrum (Fig. 1). He had marked generalized hypotonia, and displayed fits of laughter.
Previous workup included normal CMA, fragile X syndrome testing, methylation studies for Angelman syndrome, sequencing of UBE3A and the MECP2 genes, thyroid function studies, and extensive metabolic workup. Brain MRI showed diminished white matter in bilateral occipital lobe, slightly enlarged Virchow Robin spaces, slightly hypoplastic splenium of corpus callosum.
Patient 3
Patient 3 is a 3-year-old Caucasian female born at 35 weeks of gestation via planned C-section because of an in utero diagnosis of spina bifida corrected by microsurgery at 24 weeks of gestation. She was the first child born to a 25-year-old mother and her birth weight was 2.6 kg (65th centile) at delivery. Birth length and OFC are unavailable. She had an 18-day hospital stay post-delivery for respiratory support and feeding difficulties. She exhibited early DD: she walked at 18 months and said her first words at 15 months, but later lost all speech. The patient exhibited socialization difficulties and poor eye contact and was diagnosed with ASD. She also had a history of cyclic vomiting.
On physical examination at 2 years of age, the patient’s weight was 20.9 kg, height was 95.6 cm, body mass index was 22.87 kg/m2, and OFC was 54.5 cm; all above the 97% centile. She exhibited macrocephaly, frontal bossing, deep-set eyes which were somewhat widely set, and a high arched palate (Fig. 1).
Her diagnostic workup included normal CMA, fragile X syndrome testing, and PTEN gene sequencing, thyroid hormone level, plasma amino acids, urine organic acids, acylcarnitine profile, lead levels. Brain MRI at 21 months showed moderately enlarged lateral and third ventricles and resolution of the Chiari malformation seen previously in utero. EEG showed left temporal slowing with left temporal spikes.
Molecular results
Exome sequencing results
Figure 2 illustrates the locations of the DOCK3 variants found in Patients 1–3 in relation to the conserved domains in DOCK3. The reference transcript used for all exonic variants is NM_004947.4 and the genomic reference for the intronic variant is NG_028012.1. Exons are numbered like in NG_028012.1.
Fig. 2.
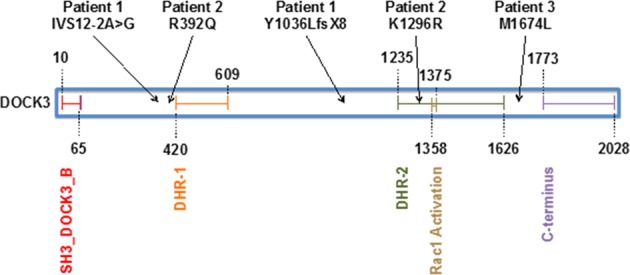
DOCK3 protein domains (UniProt: Q8IZD9) and location of variants in Patients 1, 2, and 3. Numbers refer to amino acids. The SH3_DOCK3_B domain (amino acids 10–65) is shown in red. The DHR-1 domain (amino acids 420–609) is orange. The DHR-2 domain (amino acids 1235–1626) is green and contains the catalytic activity for Rho/Rac GTPases. Amino acids 1358–1375 are conserved amino acids required for Rac1 activation, which is outlined in brown. The last conserved domain is the proline-rich C terminus (amino acids 1773–2028) in purple, which is involved in the internalization and degradation of the NMDA receptor. The variants described in this paper are shown above the protein diagram
WES of Patient 1 revealed two variants in the DOCK3 gene. The first variant is c.1038-2A>G:IVS12-2A>G, which is predicted to destroy the canonical splice acceptor site in intron 12. The destruction of the splice acceptor site is expected to cause abnormal gene splicing, which either leads to nonsense-mediated decay or to an abnormal protein product. This variant was not detected in the mother. The second variant is a maternally inherited frameshift in exon 30, c.3107_3110delACTT: p.(Tyr1036Leufs*8). The frameshift causes a premature stop codon at position 8 of the new reading frame, which is predicted to cause loss of normal protein function through either protein truncation or nonsense mediated mRNA decay. In silico analysis using Mutation Taster predicts the variant is disease-causing. These variants were not found in large population cohorts [20].
Patient 2 also has compound heterozygous variants, c.[1175G>A];[3887A>G] in DOCK3. The variant c.1175G>A is maternally inherited and predicted to cause a missense substitution, p.(Arg392Gln), that has an allele frequency of 1.0E−03 among Europeans in the gnomAD database (http://gnomad.broadinstitute.org/variant/3-51251601-G-A; accessed 11/14/2018). It was also reported in homozygous state in 3 individuals of South Asian descent. This variant was predicted to be probably damaging by Polyphen and disease-causing by Mutation Taster and its Combined Annotation Dependent Depletion (CADD) score is 29.5. The c.3887A>G variant is paternally inherited and causes a single amino acid substitution, p.(Lys1296Arg). Its frequency among Europeans is 5.00E−04 in gnomAD (http://gnomad.broadinstitute.org/variant/3-51378788-A-G; accessed 11/14/2018). There are no homozygotes in gnomAD for this missense variant. It is predicted to be possibly damaging by Polyphen and disease-causing by Mutation Taster and its CADD Score is 25.3.
Patient 3 has a homozygous variant, c.[5020A>T];[5020A>T], which is predicted to cause a single amino acid substitution, p.(Met1674Leu), that has an allele frequency of 1.75E−03 among Europeans in the gnomAD database [20] (http://gnomad.broadinstitute.org/variant/3-51399303-A-T; accessed 11/14/2018). There are no homozygotes in gnomAD for this missense variant. Both parents were confirmed to be heterozygous of this variant. In silico predictions for this variant were conflicting. Polyphen and SIFT predicted that the variant was benign and tolerated, respectively, while Mutation Taster predicted the variant as disease causing and its CADD score was 15.03.
The variants in Patient 1 were submitted to ClinVar: https://www.ncbi.nlm.nih.gov/clinvar/variation/599269/ and https://www.ncbi.nlm.nih.gov/clinvar/variation/599268/. The variants c.[1175G>A];[3887A>G] in Patient 2 were submitted to the Leiden Open Variation Database and can be found at http://www.lovd.nl/DOCK3 (Patient ID: #00218378). The variant c.5020A>T in Patient 3 was submitted to ClinVar: https://www.ncbi.nlm.nih.gov/clinvar/variation/547977/.
The effect of missense variants on Dock3 GEF activity
The Rac1 activity was strongly increased by WT human DOCK3 in Cos7 cells (5.00-fold) (Fig. 3). Although the three DOCK3 mutants [p.(Arg392Gln), p.(Lys1298Arg), and p.(Met1647Leu)] induced Rac1 activation, the levels of Rac1 activities were significantly lower compared with WT human DOCK3 (3.41-fold, 3.98-fold, and 3.64-fold for the three mutants, respectively) (Fig. 3).
Fig. 3.
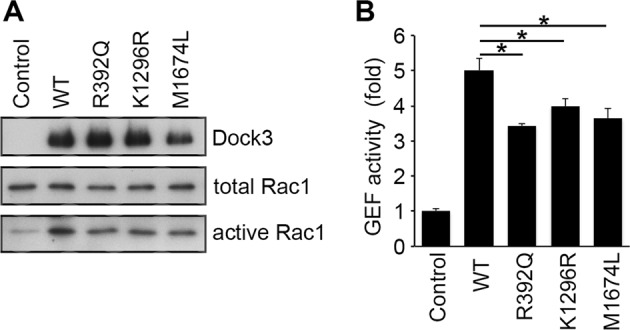
Dock3 GEF activity. Lysates from Cos-7 cells transfected with the indicated plasmids were subjected to the pull-down assay for measurement of Rac1 activity using the GST-CRIB fusion protein. Expression of Dock3, total Rac1, and activated Rac1 were detected by immunoblot analysis (left panel). The level of activated Rac1 was quantified and shown in the histogram (right panel). The data are presented as means ± S.E.M. of four samples. *P < 0.05
While DOCK3 is a large protein with 2030 amino acids, it is intolerant of missense variation; the MisZ score is 3.56 standard deviations above the average human gene [20]. To further examine the potential effect of the missense variants in DOCK3, we generated a protein model of the full DHR-2 domain of DOCK3, using a combination of homology-based and ab initio methods (Fig. 4). Ab initio methods consistently predict a helical structure from residues 1606 through 1629 and two short helices from 1688 through 1692 and 1707 through 1714. Models were consistent with a small pair of strands alternating with these two short helices. Inspection of this model showed that much of the hydrophobic core consisted of methionine and a few aromatic residues. The electrostatic surface of DOCK3 shows that Lys1296Arg is within a large positive patch, surrounded by a triad of negative patches.
Fig. 4.

The DOCK3 DHR-2 domain structure is shown in cartoon representation colored by protein and secondary structure. The sites altered by genetic variation are shown in thick sticks. a Our protein model shows that neither of the missense variant positions, p.(Lys1296) and p.(Met1674), are at the Rac1 interface. b Focusing on the surface around p.(Lys1296), it is within a large positively charged patch (blue semi-transparent surface) surrounded by a triad of negative patches (red semi-transparent surface). c p.(Met1674) is within the hydrophobic core of the methionine-rich region of the catalytic domain. Additional methionine and aromatic residues within the region are shown in sticks representation
Discussion
We describe three unrelated patients with variants in DOCK3 associated with GDD, hypotonia, and wide-based or uncoordinated gait. This is consistent with previous descriptions of GDD, gait abnormalities, and hypotonia in children with DOCK3 variants [9, 10]. There did not seem to be a different or milder phenotype in patients with missense variants versus patients with truncating variants. All of the patients in our study exhibited brain abnormalities and Patients 1 and 3 also had spine abnormalities (Table 1). However, the brain abnormality was different in all of the patients and again there did not seem to be a difference between the patients with truncating and missense variants. Of note, Iwata-Otsubo et al. reported a patient with DOCK3 variants with a slightly bulky and dysmorphic corpus callosum [10] and brain MRI of Patient 2 in this study showed a hypoplastic splenium of the corpus callosum.
The three patients described in this report also exhibited no major or recognizable dysmorphic features although minor facial abnormalities are found (Table 1). Patient 1 did not have behavioral abnormalities, Patient 2 displayed self-injurious behaviors, and Patients 2 and 3 have been diagnosed with ASD. Growth also varied and no specific pattern was found among the patients with DOCK3 variants, from less than 5th centile to greater than 97th pcentile for weight. Height and OFC were also varied (Table 1). Patient 1 also had congenital anomalies, including TE fistula, vertebral and rib abnormalities and hydroureter, likely related to VACTERL association, and it is unclear whether these findings are related to the DOCK3 variants since no other patients reported with DOCK3 variants have had these birth defects.
The two variants in Patient 1 are predicted to cause loss of function through either protein truncation or nonsense mediated mRNA decay. Most of the domains involved in the major functions of the protein would be affected by these variants. Downstream of the premature stop codon are the DHR-2 domain and C-terminus, which contain the activation site for the Rac1 activity and the amino acids involved in NMDA receptor internalization, respectively. The predicted loss of function of these two variants in Patient 1 is expected to have functional consequences based on Probability of Loss of Function Intolerance (pLI) = 1 in ExAc [20].
To understand the functional consequences of the three missense variants in Patients 2 and 3, we studied the activity of DOCK3 mutants. The DOCK3 mutants, p.(Arg392Gln), p.(Lys1296Arg), and p.(Met1674Leu), induced Rac1 activation, but the levels of Rac1 activities were significantly lower in the DOCK3 mutants compared with the wild type, 3.41-fold, 3.98-fold, and 3.64-fold for the three mutants, respectively, as compared to 5.00-fold in controls (Fig. 3). These results suggest that the three variants have functional consequences and likely affect DOCK3/Rac1-mediated function in neurons.
The mechanism for the loss of function in DOCK3/Rac1 activity may be extrapolated from the location of the variant within the protein. The c.1175G>A:p.(Arg392Gln) missense variant exists in front of the DHR-1 domain in the amino acid sequence, the c.3887A>G:p.(Lys1296Arg) variant exists within the DHR-2 domain of human DOCK3, and the c.5020A>T:p.(Met1674Leu) missense variant exists near the DHR2 domain. Since the DHR-1 domain is necessary for the recruitment of DOCK3 to the plasma membrane, which is required for the full activation of Rac1, the c.1175G>A:p.(Arg392Gln) missense variant may affect DOCK3’s ability to re-localize to the plasma membrane. On the other hand, DHR2 is the GEF enzymatic domain, and so the c.3887A>G:p.(Lys1296Arg) missense variant may reduce DOCK3 GEF activity directly. Moreover, the c.5020A>T:p.(Met1674Leu) missense variant may also affect DOCK3 GEF activity directly based on its location, which is close to DHR2. Future studies could identify the details of how these variants alter DOCK3 function. Circular dichroism spectroscopy can assess changes to the overall conformation of DOCK3, for each genomic variant. DOCK3 immunostaining combined with confocal microscopy or other imaging techniques can identify altered localization. Gel-shift assays can identify differential binding by functionally related proteins such as WASF1 (WAVE). These experimental techniques combined with additional molecular modeling may not only identify which function is altered, but the details as to how.
Each of the observed missense variants in the DHR-2 domain has a moderate effect on the biochemical properties of each amino acid—c.5020A>T:p.(Met1674Leu) maintains a small hydrophobic sidechain and c.3887A>G:p.(Lys1296Arg) maintains a positive charge. The methionine-aromatic structural motif has been previously studied and shown to be common in proteins [21]. Thus, even though the c.5020A>T:p.(Met1674Lys) variant maintains a hydrophobic sidechain, the biochemistry of this region of the protein may depend on methionine specifically. The electrostatic surface of DOCK3 shows that c.3887A>G:p.(Lys1296Arg) is within a large positive patch, surrounded by a triad of negative patches, which implies that the likely effect of c.3887A>G:p.(Lys1296Arg) is subtle, driven by the larger sidechain and potentially affecting a different molecular interaction. It may be that variants with more significant changes to amino acid properties would be incompatible with life; however the observation of nonsense variants would seemingly contradict this hypothesis. The subtler changes we have observed could represent a balance between affecting DOCK3 DHR-2 domain function, but not triggering pathway-level compensation.
There are three prior reports implicating DOCK3 in DD [3, 9, 10] (Table 1). Two siblings with severe DD, hypotonia, and ataxic gait were recently reported with compound heterozygous variants in DOCK3 [9]. The two affected siblings had a paternally-inherited 458 kb heterozygous deletion in chromosomal region 3p21.2, which includes part of the DOCK3 gene, and a c.382C>G:p.(Gln128*) DOCK3 variant that is predicted to introduce a premature stop codon within exon 6 [9]. Iwata-Otsubo et al. also reported on a boy with a 170 kb homozygous deletion that included exons 6–12 of the DOCK3 gene with unsteady gait, hypotonia, and DD [10]. The patients reported in these studies with variants in DOCK3 display very similar phenotypic features to previously reported DOCK3 knockout mice [9, 10]. This led Iwata-Otsubo et al. to conclude that biallelic disease-causing variants of DOCK3 lead to a specific DOCK3-related neurodevelopmental syndrome [10].
DOCK3 knockout mice displayed ataxic gait, limb weakness, and impairment in learning [22]. Mice homozygous for a null DOCK3 variant had sensorimotor impairments and changes associated with impaired axonal transport in several brain regions, including the spinal cord and cerebellum, which supports that loss of DOCK3 leads to central axonal pathology and behavioral abnormalities [22].
Our study was limited by the inclusion of only three patients. All three patients displayed GDD, variable gait abnormalities, and hypotonia, but also had other, unshared clinical findings, such as VACTERL association in Patient 1, self-injurious behaviors in Patient 2, and myelomeningocele in Patient 3. Lastly, it is unclear whether VACTERL-like association in Patient 1 is due to the heterozygous DOCK3 variants or due to other genetic factors somewhere else in the genome.
In summary, we describe three patients with genetic variants in DOCK3 who presented with GDD, hypotonia, and wide-based or uncoordinated gait. Considered in the context of prior studies describing DOCK3 variants in families with GDD, hypotonia, and ataxia, our study of three unrelated patients with DOCK3 variants provides a strong support for DOCK3 loss of function variants leading to DD, hypotonia and a range of coordination problems, including ataxia. Given the location of the missense variants in the patients in this study, the likely mechanism by which the variants may cause GDD/ID is alteration of function of the DHR-1 and DHR-2 domains. Reports of additional patients, especially with missense variants, are needed to clarify the full spectrum of clinical manifestation and potential genotype–phenotype correlation.
Web resources
The URLs for online tools and data presented herein are as follows:
OMIM (http://www.omim/org/).
UCSC (http://genome.ucsc.edu/).
Mutation Nomenclature (http://www.hgvs.org/mutnomen/recs.html).
Exome Aggregation Consortium (http://exac.broadinstitute.org/).
The Genome Aggregation Database (gnomAD) (http://gnomad.broadinstitute.org/).
Acknowledgements
The authors thank the families of the patients for participating in this study.
Compliance with ethical standards
Conflict of interest
RES discloses her employment with GeneDx. The other authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Michelson DJ, Shevell MJ, Sherr EH, Moeschler JB, Gropman AL, Ashwal S. Evidence report: genetic and metabolic testing on children with global developmental delay. Neurology. 2011;77:1629–35. doi: 10.1212/WNL.0b013e3182345896. [DOI] [PubMed] [Google Scholar]
- 2.Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6:265ra168. doi: 10.1126/scitranslmed.3010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Silva MG, Elliott K, Dahl HH, Fitzpatrick E, Wilcox S, Delatycki M, et al. Disruption of a novel member of a sodium/hydrogen exchanger family and DOCK3 is associated with an attention deficit hyperactivity disorder-like phenotype. J Med Genet. 2003;40:733–40. doi: 10.1136/jmg.40.10.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Namekata K, Enokido Y, Iwasawa K, Kimura H. MOCA induces membrane spreading by activating Rac1. J Biol Chem. 2004;279:14331–7. doi: 10.1074/jbc.M311275200. [DOI] [PubMed] [Google Scholar]
- 5.Côté JF, Vuori K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J Cell Sci. 2002;115:4901–13. doi: 10.1242/jcs.00219. [DOI] [PubMed] [Google Scholar]
- 6.Namekata K, Harada C, Taya C, Guo X, Kimura H, Parada LF, et al. Dock3 induces axonal outgrowth by stimulating membrane recruitment of the WAVE complex. Proc Natl Acad Sci USA. 2010;107:7586–91. doi: 10.1073/pnas.0914514107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Namekata K, Kimura A, Kawamura K, Harada C, Harada T. Dock GEFs and their therapeutic potential; Neuroprotection and axon regeneration. Prog Retin Eye Res. 2014;43:1–16. doi: 10.1016/j.preteyeres.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 8.Kimura A, Namekata K, Guo X, Harada C, Harada T. Dock3-NMDA receptor interaction as a target for glaucoma therapy. Histol Histopathol. 2017;32:215–21. doi: 10.14670/HH-11-820. [DOI] [PubMed] [Google Scholar]
- 9.Helbig KL, Mroske C, Moorthy D, Sajan SA, Velinov M. Biallelic loss-of-function variants in DOCK3 cause muscle hypotonia, ataxia, and intellectual disability. Clin Genet. 2017;92:430–3. doi: 10.1111/cge.12995. [DOI] [PubMed] [Google Scholar]
- 10.Iwata-Otsubo A, Ritter A, Weckselbatt B, Ryan NR, Burgess D, Conlin LR, et al. DOCK3-related neurodevelopmental syndrome: biallelic intragenic deletion of DOCK3 in a boy with developmental delay and hypotonia. Am J Med Genet A. 2018;176A:241–5. doi: 10.1002/ajmg.a.38517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kashiwa A, Yoshida H, Lee S, Paladino T, Liu Y, Chen Q, et al. Isolation and characterization of novel presenilin binding protein. J Neurochem. 2000;75:109–16. doi: 10.1046/j.1471-4159.2000.0750109.x. [DOI] [PubMed] [Google Scholar]
- 12.Chen Q, Yoshida H, Schubert D, Hamer P, Mallory M, Masliah E. Presenilin binding protein is associated with neurofibrillary alterations in Alzheimer’s disease and stimulates tau phosphorylation. Am J Pathol. 2001;159:1597–602. doi: 10.1016/S0002-9440(10)63005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Mi X, Chen L, Jiang G, Wang N, Zhang Y, et al. Dock3 participate in epileptogenesis through rac1 pathway in animal models. Mol Neurobiol. 2016;53:2715–25. doi: 10.1007/s12035-015-9406-9. [DOI] [PubMed] [Google Scholar]
- 14.Namekata K, Kimura A, Harada C, Yoshida H, Matsumoto Y, Harada T. Dock3 protects myelin in the cuprizone model for demyelination. Cell Death Dis. 2014;5:e1395. doi: 10.1038/cddis.2014.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanaka AJ, Cho MT, Millan F, Juusola J, Retterer K, Joshi C, et al. Mutations in SPATA5 are associated with microcephaly, intellectual disability, seizures, and hearing loss. Am J Hum Genet. 2015;97:457–64. doi: 10.1016/j.ajhg.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pieper U, Webb BM, Dong GQ, Schneidman-Duhovny D, Fan H, Kim SJ, et al. ModBase, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 2014;42:D336–46. doi: 10.1093/nar/gkt1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanawa-Suetsugu K, Kukimoto-Nino M, Mishima-Tsumagari C, Akasaka R, Ohsawa N, Sekine S, et al. Structural basis for mutual relief of the Rac guanine nucleotide exchange factor DOCK2 and its partner ELMO1 from their autoinhibited forms. Proc Natl Acad Sci USA. 2012;109:3305–10. doi: 10.1073/pnas.1113512109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu D, Zhang Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins. 2012;80:1715–35. doi: 10.1002/prot.24105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morten Källberg M, Margaryan G, Wang S, Ma J, Xu J. RaptorX server: a resource for template-based protein structure modeling. Methods Mol Biol. 2014;1137:17–27. doi: 10.1007/978-1-4939-0366-5_2. [DOI] [PubMed] [Google Scholar]
- 20.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valley CC, Cembran A, Perlmutter JD, Lewis AK, Labello NP, Gao J, et al. The methionine-aromatic motif plays a unique role in stabilizing protein structure. J Biol Chem. 2012;287:34979–91. doi: 10.1074/jbc.M112.374504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Q, Peto C, Shelton GD, Mizisin A, Sawchenoko PE, Schubert D. Loss of modifier of cell adhesion reveals a pathway leading to axonal degeneration. J Neurosci. 2009;21:118–30. doi: 10.1523/JNEUROSCI.3985-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]