

Abstract
Selection and prioritization of phenotype-centric variants remains a challenging part of variant analysis and interpretation in clinical exome sequencing. Phenotype-driven shortlisting of patient-specific gene lists can avoid missed diagnosis. Here, we analyzed the relevance of using primary Human Phenotype Ontology identifiers (HPO IDs) in prioritizing Mendelian disease genes across 30 in-house, 10 previously reported, and 10 recently published cases using three popular web-based gene prioritization tools (OMIMExplorer, VarElect & Phenolyzer). We assessed partial HPO-based gene prioritization using randomly chosen and top 10%, 30%, and 50% HPO IDs based on information content and found high variance within rank ratios across the former vs the latter. This signified that randomly selected less-specific HPO IDs for a given disease phenotype performed poorly by ranking probe gene farther away from the top rank. In contrast, the use of top 10%, 30%, and 50% HPO IDs individually could rank the probe gene among the top 1% in the ranked list of genes that was equivalent to the results when the full list of HPO IDs were used. Hence, we conclude that use of just the top 10% of HPO IDs chosen based on information content is sufficient for ranking the probe gene at top position. Our findings provide practical guidance for utilizing structured phenotype semantics and web-based gene-ranking tools to aid in identifying known as well unknown candidate gene associations in Mendelian disorders.
Subject terms: Genetic testing, High-throughput screening
Introduction
Whole-exome sequencing has resolved “diagnostic odysseys” for many patients [1, 2] and has changed the course of disease management in some cases [3, 4]. With clinical whole-exome sequencing being increasingly adopted as a diagnostic and discovery tool for rare Mendelian disorders, there still exists a gap in the computational tools bridging variant analysis with clinical phenotyping to resolve cause of the disease [5, 6]. Despite improvements in next-generation sequencing and bioinformatics-based computational platforms, current diagnostic rate in identifying genetic basis of disease in rare disorders has remained between 25 and 40% [7–9]. This has partially been attributed to genetic heterogeneity and overlapping clinical features across specific disease types, making clinical diagnosis extremely challenging in certain cases [10]. It has well been recognized that identification of exact causal mutation is important for providing definitive molecular diagnosis in genetic disorders in supporting clinical management of patients [11–13]. Variant analysis of next-generation sequencing data is a laborious task, involving filtration of thousands of variants based on sequencing metrics, allele frequency, predicted functional impact, conservation scores followed by gene, and phenotype-based variant prioritization [6, 14]. As full experimental and clinical validation of the massive volume of candidate genes retrieved from such platforms is impracticable, there is continuing need for tools that can prioritize a short-list of genes that are more likely to be causal. In addition, gene-based interpretation becomes crucial to avoid missing out valuable information as a gene may have novel undocumented variants not previously associated with any known disease or phenotype [15]. Hence, gene prioritization has an important role in diagnosis using exome sequencing platforms. Recent studies have emphasized the importance of deep phenotyping [16] in increasing the diagnostic capability of clinical exome sequencing [17–19]. As compared with an agnostic approach, the onus of finding a true causal variant lies in efficient phenotype based gene prioritization, which substantially reduces the number of candidates to review. Human Phenotype Ontology (HPO) [20] is one of the most widely used platform for standardized vocabulary of phenotypic descriptions of human disorders in addition to other similar platforms—Medical Subject Headings (MeSH) [21], Unified Medical Language System (UMLS) [22], and SNOMED CT [23]. As of November 2018, HPO comprised of ~ 13,000 descriptive phenotype terms and over 156,000 annotations to hereditary diseases [24]. A recent publication showed that the use of HPO terms could be potentially advantageous over MeSH terms and UMLS concepts in automated text-mining approaches [25]. Semantic similarity of HPO terms to known diseases when used in conjunction with variant prioritization has been shown to improve diagnosis [18, 19], particularly for patients with atypical clinical profiles [17]. Recently, it was also reported that use of specific and large number of relevant HPO terms proved to be beneficial for phenotype based gene-ranking algorithm [19]. With the large number of commercial as well as freely accessible gene prioritization tools available to facilitate this process, the choice of tools still remains ambigous [6, 26–28].
In this context, we sought to assess the utility of full-versus-partial repertoire of patient-specific primary HPO identifiers (HPO ID) in gene prioritization of a varied cohort of Mendelian disorders. Specificity of a given phenotype is defined by how closely associated it was with the causal disease. HPO database contains phenotype terms arranged in a hierarchical manner such that a general phenotype term is sub-divided into specific phenotype terms and each of these terms are described by multiple synonyms. Hence, each specific and general phenotype terms has a specific HPO ID to bypass ambiguity caused by free text descriptions or synonyms. We used these HPO IDs along with the gene-ranking feature of three recently released web-based gene prioritization tools- VarElect [15], Phenolyzer [29], and OMIMExplorer [30], which utilize HPO identifiers as semantics for gene-ranking, on a total of 50 cases.
Materials and methods
Data set generation and HPO standardization
A total of 50 cases with pre-identified causal genes and their associated HPO IDs were used as queries for gene prioritization. Of these cases, 10 were described previously [15, 31, 32] and termed “Known Training set”; another 10 were recently described having novel disease–gene associations [33–38] and referred as “Novel Training set”. At last, 30 cases were derived from in-house exome analysis of neuromuscular disorder cases and termed “In-house”. The novel training set was compiled based on studies published between 25 April and 25 July 2017 and the prioritization analysis exercise in this study was conducted in the week of 25 July 2017. Each clinician provided patient phenotype was translated into primary HPO IDs by entering each description into the HPO browser (http://human-phenotype-ontology.github.io/2016/02/05/browser.html) and retrieving the best fit match. As, clinical symptoms are documented in HPO browser by superclass, primary ID, alternative ID, subclass, and in some cases multiple synonyms of the description, we restricted our selection to primary HPO IDs only, to evade non-recognition or poor specificity of the term for a given disease–gene prioritization exercise. A detailed list of all cases with respective phenotype descriptions, HPO IDs, and disease–gene associations is given in Supplementary Table 1.
Gene prioritization
In all the 50 cases, the disease causing gene had already been resolved and was termed probe gene. The genes obtained from respective exome sequencing accompanied each probe gene and together, this list was referred to as the candidate gene list (Supplementary Table 2). Three web-based gene prioritization tools—VarElect, OMIMExplorer, and Phenolyzer—were used for ranking the candidate genes of all the cases by simultaneously utilizing patient specific HPO IDs. These tools ranked the input genes based on gene–phenotype associations retrieved from tool specific databases. An overview of the gene prioritization workflow is given in Fig. 1.
Fig. 1.
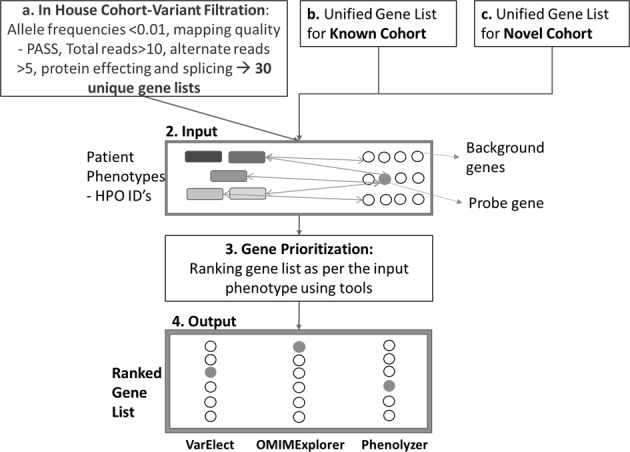
Gene prioritization workflow
Evaluation criteria of gene prioritization analysis
For conducting partial HPO ID-based analysis, 10%, 30%, and 50% of the original HPO ID list were randomly picked using random selection function in excel. Only 26 cases could be assessed for this analysis, which was a modified application of leave one out cross validation (LOOCV) [28], owing to the limitation of choosing at least one HPO ID for the lowest cutoff (10% HPO IDs). Partial phenotype term-based assessment has been previously carried out using up to 40 maximum HPO terms via computational simulations [17–19], whereas all our analysis were based on real patient data. We used this approach of HPO ID selection to bring in uniformity in HPO ID fractions as not all cases had same number of HPO IDs. In addition, we also analyzed these cases using single specific and top 10%, 30%, and 50% HPO IDs based on information content as defined previously [19]. All HPO IDs describing the cases were evaluated for number of genes that they annotate and were ordered in decreasing order of information content from which top 10%, 30%, and 50% HPO IDs were chosen(Supplementary Table 3). To conduct a more robust comparison, we calculated the performance measures by determining the true positive and false-positive rate in a subset of ranked gene list [26]. We used four commonly used thresholds [25, 26, 39] of top 5%, 10%, 30%, and 50% of the respective ranked gene lists per query and the true-positive rate (TPR) was calculated for each threshold. TPR [26] was defined as the fraction of true disease genes ranked in the Top 5%, 10%, 30%, and 50% genes among all the cases by each tool and vice versa. In addition, we determined how well the true positives were ranked in the output list by estimating Median Rank Ratio (MedRR) and Normalized Discounted Gain (NDCG) [39]. MedRR was the median of all rank ratios obtained for each probe gene across all disease–gene prioritizations per tool. The lower the value of MedRR, the higher was the performance of the tool. NDCG penalized the true positives occurring later in the ranked list and showed the importance of a probe gene being detected as higher up in the ranked list. All evaluation criteria used in this study are described in detail in Supplementary Methods.
Results
Gene prioritization using full and partial HPO IDs
In order to assess the impact of using all HPO IDs on gene prioritization, partial HPO IDs in ranking each probe gene, we conducted a threefold validation based on randomized selection of 10%, 30%, and 50% of the HPO IDs for each disease–gene prioritization (Table 1, Supplementary Table 2 and 4). We further randomized each subset (10%, 30%, and 50%) for a total of four times and calculated the MedRR across 26 cases (Supplementary Table 5). This comparison enabled the assessment of the phenotype dependency of each tool with respect to both quantity and specificity of HPO ID chosen for prioritization. The probe gene was ranked poorly in all 26 cases when 10%, and 30% HPO ID list was used across all three tools, which was directly linked to the type and number of HPO IDs that were involved in the gene prioritization (Fig. 2, Table 1). Figure 3 shows the distribution of MedRR across the entire cohort of 26 cases. Significant positive correlation was observed between probe gene-ranking ability across 26 cases for 50% HPO IDs vs 30% HPO IDs (Pearson’s correlation coefficient = 0.88, p value = 2.8 × 10−9) and 50% HPO IDs vs 10% HPO IDs (Pearson’s correlation coefficient = 0.46, p value = 0.017). At last, Phenolyzer and VarElect correlated significantly for all LOOCV’s (p value = 0.0326, 10% HPO; 0.0193, 30% HPO; 0.0002, 50% HPO; and 1.1 × 10−5, 100% HPO).
Table 1.
Performance measures of three HPO-based gene prioritization tools when partial HPO ID’s were used
| Tool | Evaluation criteria | 10% HPO ID’s | 30% HPO ID’s | 50% HPO ID’s | All HPO ID’s |
|---|---|---|---|---|---|
| Omimexplorer | TPR (%) | 84.62 | 84.62 | 84.62 | 84.62 |
| MedRR (%) | 2.65 | 1.67 | 1.06 | 1.06 | |
| Median NDCG | 0.97 | 0.97 | 0.98 | 0.98 | |
| Phenolyzer | TPR (%) | 96.15 | 96.15 | 100 | 100 |
| MedRR (%) | 10.03 | 3.78 | 1.18 | 3.17 | |
| Median NDCG | 0.98 | 0.99 | 0.99 | 0.99 | |
| Varelect | TPR (%) | 100 | 100 | 100 | 100 |
| MedRR (%) | 0.55 | 0.31 | 0.19 | 0.22 | |
| Median NDCG | 0.99 | 0.99 | 0.99 | 0.99 |
The best performance measures per evaluation criteria across the 10%, 30%, 50, and 100% HPO ID’s used are given in bold italics for each tool NDCG-normalized discounted cumulative gain of the probe gene ranks, TPR-true positive rate, MedRR-median rank ratio. This Table represents analysis for 26 cases only
Fig. 2.

Distribution of probe gene rank positions across 26 partial HPO-based queries. Four HPO-based gene query groups (10% HPO, 30% HPO, 50% HPO, and full list of HPO) and their subsequent gene rank positions are summarized for three tools- VarElect, Phenolyzer, and OMIMExplorer. The frequency of probe gene being identified as Rank 1, Rank 2–10, Rank 11–20, Rank 21–100 and > Rank 100 are represented across overall, In-house, known, and novel cohorts
Fig. 3.
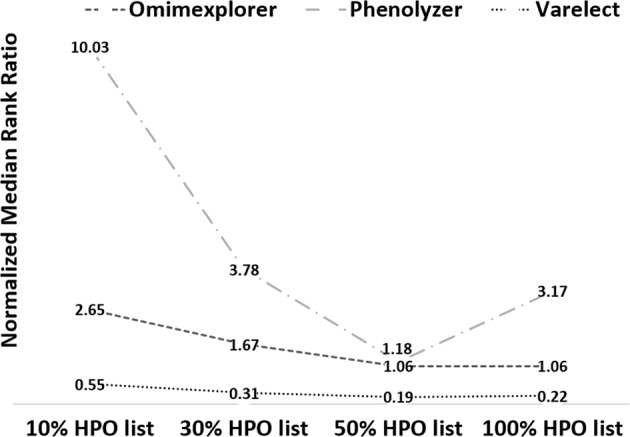
Median rank ratio distribution using partial HPO terms across three gene prioritization tools. The median rank ratio values obtained across all partial HPO-based analysis (10% HPO, 30% HPO, 50% HPO, and full list of HPO) are depicted for each of the three tools studied. The values have been normalized (by 100) for graphical representation
Performance of gene prioritization tools
To assess the impact of using partial gene list in gene prioritization, we utilized three tools across top 5%, 10%, 30%, 50%, and 100% of the ranked gene list across 50 disease–gene associations, which comprised of in-house, known and novel disease–gene associations (Table 2, Supplementary Table 6). VarElect and OMIMExplorer performed well in identifying the probe gene as the number one candidate gene for in-house cohort in > 40% of the cases (Fig. 4a). In addition, VarElect had the lowest MedRR (0.23), followed by OMIMExplorer (1.2) (Supplementary Table 7a). Highest TPR were observed for gene-disease associations in the in-house data set across all three tools when compared with known or novel data set (Supplementary Table 7a). For the known as well as novel data set, VarElect could rank the given probe genes better than the other tools across top 5%, 10%, 30%, and 50% of the gene lists with highest NDCG and lowest MedRR values for the same (Supplementary Table 7b and 7c, Supplementary Fig. 2). When assessing disease inheritance pattern of candidate gene, the best performance measures were obtained for disease with X-linked inheritance, which could be attributed to all but one case being associated with DMD probe gene, hence bringing uniformity to the disease phenotype associations (Supplementary Fig. 2, Supplementary Table 8d, Table 3). Genes with autosomal recessive and unknown inheritance were grouped together and VarElect outperformed the rest two tools across all analyzed performance measures with 100% TPR, lowest MedRR (range from 4.8 to 10.2) and highest NDCG (range from 0.99 to 0.89) (Supplementary Fig. 2, Supplementary Table 8b). We further divided our cohort of 50 cases into those associated with single or multiple (heterogeneous) disorders with autosomal inheritance (Supplementary Fig. 2, Supplementary Table 9). VarElect performed best for the genes related to multiple disorders (TPR: 100–75%, MedRR: 0.32–4.5, Median NDCG: 0.99–0.91) across all LOOCV’s of 100%, 50%, 30%, and 10% gene list (Supplementary Table 9b). OMIMExplorer returned the best receiver-operating characteristics curve overall with area under the curve = 0.823 and sensitivity = 85.4%. Table 4 summarizes the area under the curve values over 10% and 1% false-positive threshold for all gene prioritization tools when all HPO IDs were used.
Table 2.
Overall performance measures of four gene prioritization tools across 50 cases
| Tool | Evaluation criteria | Top 5% | Top 10% | Top 30% | Top 50% | Total gene list |
|---|---|---|---|---|---|---|
| VarElect | TPR (%) | 76 | 78 | 86 | 88 | 100 |
| MedRR (%) | 4.17 | 2.16 | 0.77 | 0.46 | 0.23 | |
| Median NDCG | 0.92 | 0.95 | 0.98 | 0.98 | 0.99 | |
| OMIMExplorer | TPR (%) | 76 | 76 | 86 | 86 | 86 |
| MedRR (%) | 22.01 | 11.01 | 4.02 | 2.41 | 1.2 | |
| Median NDCG | 0.82 | 0.88 | 0.94 | 0.96 | 0.98 | |
| Phenolyzer | TPR (%) | 60 | 64 | 78 | 84 | 96 |
| MedRR (%) | 7.53 | 3.9 | 2.59 | 2.69 | 1.72 | |
| Median NDCG | 0.88 | 0.92 | 0.96 | 0.98 | 0.99 |
NDCG-normalized discounted cumulative gain of the probe gene ranks, TPR-true positive rate, MedRR-median rank ratio. Above performance measures are a summary of all 50 cases used in this study across top 5%, Top 10%, Top 30%, and Top 50% of the prioritized list of genes. All (100%) HPO ID’s per case were used for all the above gene prioritization analysis
Fig. 4.

Distribution of probe gene rank positions across 50 cases by disease cohort. The frequency of probe gene being identified as Rank 1, Rank 2–10, Rank 11–20, Rank 21–100, and > Rank 100 are represented across overall, In-house, known, and novel cohorts for three tools- OMIMExplorer, Phenolyzer, and VarElect
Table 3.
Median rank ratios of probe genes across four tools
| Mode ofinheritance | Genes | VarElect | OMIMExplorer | Phenolyzer |
|---|---|---|---|---|
| AR/unknown | ATXN1L*, THAP11*, AP4B1, GLDN, POC1A, RNASEH2A, SGCB, SPTBN4, SUOX, DYSF, SMN1, CEP290 # , FKRP, CAPN, NPAT # , LAMA2, NEB | 4.83 | 3.17 | 17.51 |
| AD | FGF9, SEC16A, TFAP2A, ELN, GABRB3 # , CACNA1H # | 1.19 | 3.17 | 2.45 |
| AR/AD | LMNA, MYH7, MYBPC1, FBN1 # , RYR1 (3), ENPP1 # , COL6A2, TTN (2) | 0.23 | 1.1 | 1.93 |
| XL | DMD (15), FHL1 | 0.08 | 0.54 | 0.33 |
Values indicate normalized median rank ratios across 50 cases with full candidate gene list used to rank each. Lower value of median rank ratio indicates better ranking ability of the tool as it represents that the probe gene was ranked in top fractions of the gene list. Number of cases with the candidate gene are given in brackets if more than one case had the same candidate gene
AR-Autosomal Recessive, AD-Autosomal Dominant, XL-X linked
* Genes with unknown diseases or inheritance pattern
# Genes associated with disorders of known as well as unknown inheritance patterns
Table 4.
Receiver operator curve characteristics of four gene prioritization tools
| VarElect | OMIMExplorer | Phenolyzer | |
|---|---|---|---|
| 10% FPR threshold | |||
| Accuracy (%) | 58.80% | 71.60% | 60.80% |
| Sensitivity (%) | 64.60% | 78.80% | 70.60% |
| Specificity (%) | 44.10% | 62.80% | 50.80% |
| AUC | 0.575 | 0.738 | 0.632 |
| 1% FPR threshold | |||
| Accuracy (%) | 67.90% | 52.40% | 57.60% |
| Sensitivity (%) | 84.80% | 100% | 87.90% |
| Specificity (%) | 55.10% | 45.70% | 48.40% |
| AUC | 0.745 | 0.832 | 0.731 |
AUC – area under the curve, FPR – false-positive rate
All HPO ID’s per case were used for this analysis
Bold entries indicate highest value for Accuracy, Sensitivity, Specificity and AUC across all three tools for each comparison
Discussion
Candidate gene identification is more challenging for Mendelian disorders with genetic heterogeneity than for those that are associated with unique and well-characterized list of associated phenotype terms. In the former, gene prioritization and variant analysis is compounded by complexities whereby one gene may be associated with multiple disorders or multiple genes may be associated with similar or overlapping phenotypes, leading to multiple molecular diagnosis [10, 40].
The primary aim of our study was to investigate the relevance of specific HPO ID-based semantics rather than mere quantitative reliance of using large inputs of HPO IDs for effective gene prioritization in genetically heterogeneous Mendelian disorders. The rate of diagnostic success has been reported to be lower in cases with less-specific phenotypes [41]. Recent studies have emphasized the use of relevant HPO-descriptive terms in gene prioritization [17–19, 42]. These studies reported efficient disease–gene prioritization when using all HPO terms describing the phenotype as opposed to when incomplete (1–5, 6–10 terms etc.), imprecise (more general HPO term included), or noisy (unrelated terms) HPO terms were used [17–19]. In our study, we followed modified approach of random partial HPO ID selection [18] to simulate a real life scenario of having partial or incomplete phenotypes available for each case, from the clinic. We further randomized the selection of HPO IDs four times per case and observed that the trend observed in Fig. 3 was normalized and best MedRR values were inclined toward 100% HPO IDs followed by 50%, 30%, and 10% HPO IDs for VarElect and Phenolyzer tools (Supplementary Fig. 3). This result could be correlated to imperfect HPO IDs as reported previously [17–19]. In addition, randomly chosen 10% HPO IDs subset had highest variance followed by 30% HPO IDs and 50% HPO IDs for Phenolyzer, while 30% HPO IDs had highest variance followed by 10% HPO IDs and 50% HPO IDs for VarElect (Supplementary Table 10). This highlights the probability of choosing a less-specific phenotype term, which ranks a given probe gene away from the top position, resulting in higher variability in probe gene ranks. We observed that the analysis utilizing single-specific HPO ID with maximum information content yielded favorable probe gene ranks (Supplementary Table 11). In addition, in cases with well-characterized disorders, top 10% HPO IDs ranked the probe gene equally well as that when all HPO IDs were used (Supplementary Fig 3a, Supplementary Table 12). The novel disease–gene associations could not be ranked well, which highlights the dependency of tool on underlying gene-search algorithm and known databases. OMIMExplorer could not be used for information content-based gene prioritization and multiple randomization as it was taken offline owing to hardware issues at the time of these analyses and hence, we substituted this tool with commonly used semantic similarity based tool—Phenomizer and a recently introduced machine learning based tool—Amelie [43]. At last, we carried out a deeper case study of 15 cases with DMD probe gene, whereby each case was described by a different set of HPO IDs based on original clinical symptoms provided by the clinician (Supplementary Table 13). HPO database has annotated Duchenne muscular dystrophy with 23 HPO IDs and Becker muscular dystrophy with 15 HPO IDs. In 46.7% (7/15) of cases for which ≥ 10 specific HPO IDs were used to describe patient phenotype, DMD gene was ranked as number one candidate by all tools. However, when ≤ 4 HPO IDs were used to describe other DMD-related queries, the gene could be ranked as top candidate more frequently by VarElect as compared to the rest of the two tools (Supplementary Table 13). This could be attributed to the use of more generic HPO IDs such as HP:0003236—elevated serum creatine phosphokinase and HP:0003560—muscular dystrophy in latter, whereas former had more disease-specific HPO IDs provided in the list. For cases 13, 14, and 15, the HPO ID for muscular dystrophy—HP:0003560 was used for prioritization which in turn is annotated with 76 genes in HPO and is one of the HPO IDs described for Becker muscular dystrophy. Poor ranking of DMD gene in these cases could be correlated with lower information content of the HPO ID used. For Case 27, Phenolyzer ranked the gene poorly as compared with its counterparts at 10%, 30%, and 50% HPO IDs. This could be owing to underlying variability in gene score generation by each tool. In addition, when we used single specific HPO ID with highest information content–HP:0008981 (Calf muscle hypertrophy with 38 annotated genes in HPO), DMD could be ranked as top candidate gene. Hence, these results suggest, whereas specific phenotyping is key in describing clinical cases, the use of specific HPO IDs leads to effective gene-ranking whereby even top 10% of HPO IDs are sufficient to rank the candidate gene. However, the choice of these HPO IDs has to be based on clinical acumen besides mere computational assessment of number of genes annotated by the corresponding HPO ID. Second, we assessed the diagnostic rate on retrospective, known, and novel disease–gene cohorts using three gene prioritization tools, which use pair-wise gene–phenotype-based similarity as their underlying semantics. Our findings show that OMIMExplorer works best only on disease phenotypes that are well-characterized in OMIM. VarElect has previously been shown to outperform Phenolyzer [15] and in our study, we observed that it performed better than both OMIMExplorer and Phenolyzer. In addition, VarElect remained less affected by use of partial HPO IDs-based gene-ranking. The individual differences between the gene prioritization obtained from these three tools can be attributed to their unique workflow, which is based on their underlying databases and algorithms (Supplementary Table 14). When assessing the use of HPO IDs by each tool, Phenolyzer queries the HPO IDs to create a weighted ranked gene list via logistic regression model, whereas VarElect uses Boolean logic and guilt by association methodology to search rare, unique, and uncharacterized medical terminology matching the input HPO IDs to identify genes sharing same pathways. At last, OMIMExplorer uses semantic similarity to match the HPO ID to the known OMIM diseases and calculates similarity scores of disease genes. We also looked into the gene-ranking ability of all tools based on disease–gene inheritance and found that VarElect performed fairly well as compared with the other two counterparts across all disease inheritance patterns (Supplementary Fig. 2, Supplementary Table 8). In a cohort of 52 known cases, Zemojtel et al. [18] had reported that best rank ratios were returned via PhenIX tool for AD and X-linked disease genes. Our results when analyzed by disease inheritance pattern reflected similar trend with X-linked giving best MedRR followed by AD and then AR and unknown inheritance models. If we compare our gene prioritization approach with an agnostic approach whereby phenotype associations are not considered for gene-ranking, one could focus on sequence ontology and in silico predictive assessment. However, taking the example of case 1 from our retrospective cohort, candidate variants in 2740 genes were identified after removing low quality, synonymous and > 1% minor allele frequency variants. Focusing only on protein-effecting variants without phenotype consideration, further reduced our list to 936 genes (Supplementary Table 6). However, to prioritize candidate variants in these 936 genes in the absence of any gene-related ranking feature would have made the analysis more laborious. Hence, the use of gene prioritization tools helped in reducing this list to a manageable set by ranking each of the 936 genes from rank 1 to 936, whereby only top 10 ranked genes could be further curated.
Drawing conclusions from previous studies [19, 44] on small subsets of specific phenotypes, we analyzed 50 cases of neuromuscular disorder in this study, which suggests that specific phenotyping alone can enhance gene rankings and reduce analysis time in a clinical setting. Although this study was performed mostly on neuromuscular cases, as pointed by these other studies [19, 44], such conclusions could generally be extended to other disorders. We recommend the use of specific HPO ID-based gene prioritization using web-based, and easy to use comprehensive tools. We speculate this approach could be effective toward uncovering not just monogenic but also “blended phenotypes” for cases that portray multiple monogenic disorders and would be an advantageous tool to fill in the gaps in clinician derived gene lists for efficient molecular diagnosis. In addition, it would further provide the user more freedom to choose various gene lists based on different filtering criteria, which in turn could highlight similar genes associated with given phenotypic features. A potential limitation of our study was the small sample size of 26 cases used for assessing partial phenotype based gene prioritization and future work would involve validating the findings of this study in a larger study cohort. Second, quantitative estimation of the specificity of each HPO ID to the given disease–gene association was beyond the scope of this study. In summary, our study shows that effective gene prioritization is linked to the specificity of well-defined phenotypic identifiers to a given disorder rather than the number of HPO IDs. Hence, from a clinical standpoint, it is imperative to provide specific phenotype identifiers, which the clinician deems as most relevant to a given disease, as inputs for efficient gene prioritization in both known and unknown monogenic disease associations. Lastly, the availability of more user-friendly and comprehensive web-based gene-ranking tools with fast processing time would facilitate candidate gene identification in clinical sequencing.
Supplementary information
Acknowledgements
This work was performed with grant funding support from National Medical Research Council of Singapore. We also thank Arthi Shanmugasundaram for help with the gene prioritization work and Grace Tan for assistance in filtering the in-house exome data.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41431-019-0412-7) contains supplementary material, which is available to authorized users.
References
- 1.Tetreault M, Bareke E, Nadaf J, Alirezaie N, Majewski J. Whole-exome sequencing as a diagnostic tool: current challenges and future opportunities. Expert Rev Mol Diagn. 2015;15:1–12. doi: 10.1586/14737159.2015.1039516. [DOI] [PubMed] [Google Scholar]
- 2.Valencia CA, Husami A, Holle J, Johnson JA, Qian Y, Mathur A et al. Clinical impact and cost-effectiveness of whole exome sequencing as a diagnostic tool: a pediatric center’s experience. Front Pediatr 2015;3:67. [DOI] [PMC free article] [PubMed]
- 3.Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6:265ra168. doi: 10.1126/scitranslmed.3010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Srivastava S, Cohen JS, Vernon H, Barañano K, McClellan R, Jamal L, et al. Clinical whole-exome sequencing in child neurology practice. Ann Neurol. 2014;76:473–83. doi: 10.1002/ana.24251. [DOI] [PubMed] [Google Scholar]
- 5.Boycott KM, Rath A, Chong JX, Hartley T, Alkuraya FS, Baynam G et al. International cooperation to enable the diagnosis of all rare genetic diseases. 2017;100:695–705. [DOI] [PMC free article] [PubMed]
- 6.Eilbeck K, Quinlan A, Yandell M. Settling the score: variant prioritization and Mendelian disease. Nat Rev Genet. 2017;18:599–12. [DOI] [PMC free article] [PubMed]
- 7.Seco CZ, Wesdorp M, Feenstra I, Pfundt R, Hehir-Kwa JY, Lelieveld SH et al. The diagnostic yield of whole-exome sequencing targeting a gene panel for hearing impairment in The Netherlands. 2017;25:308–14. [DOI] [PMC free article] [PubMed]
- 8.Tan TY, Dillon OJ, Stark Z, Schofield D, Alam K, Shrestha R, et al. Diagnostic impact and cost-effectiveness of whole-exome sequencing for ambulant children with suspected monogenic conditions. JAMA Pediatr. 2017;171:855–62. doi: 10.1001/jamapediatrics.2017.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, et al. Clinical exome sequencing for genetic identification of rare mendelian disorders. JAMA. 2014;312:1880. doi: 10.1001/jama.2014.14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Donnell-Luria AH, Miller DT. A Clinician’s perspective on clinical exome sequencing. Hum Genet. 2016;135:643–54. doi: 10.1007/s00439-016-1662-x. [DOI] [PubMed] [Google Scholar]
- 11.Tomar S, Sethi R, Sundar G, Quah TC, Quah BL, Lai PS. Mutation spectrum of RB1 mutations in retinoblastoma cases from Singapore with implications for genetic management and counselling. PLoS ONE. 2017;12:e0178776. doi: 10.1371/journal.pone.0178776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Travaglini L, Aiello C, Stregapede F, D’Amico A, Alesi V, Ciolfi A et al. The impact of next-generation sequencing on the diagnosis of pediatric-onset hereditary spastic paraplegias: new genotype-phenotype correlations for rare HSP-related genes. Neurogenetics 2018;19:111–21. [DOI] [PubMed]
- 13.Tranebjærg L, Strenzke N, Lindholm S, Rendtorff ND, Poulsen H, Khandelia H, et al. The CAPOS mutation in ATP1A3 alters Na/K-ATPase function and results in auditory neuropathy which has implications for management. Hum Genet. 2018;137:111–27. doi: 10.1007/s00439-017-1862-z. [DOI] [PubMed] [Google Scholar]
- 14.Seaby EG, Pengelly RJ, Ennis S. Exome sequencing explained: a practical guide to its clinical application. Brief Funct Genomics. 2016;15:374–84. doi: 10.1093/bfgp/elv054. [DOI] [PubMed] [Google Scholar]
- 15.Stelzer G, Plaschkes I, Oz-Levi D, Alkelai A, Olender T, Zimmerman S, et al. VarElect: the phenotype-based variation prioritizer of the GeneCards Suite. BMC Genomics. 2016;17:444. doi: 10.1186/s12864-016-2722-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson PN. Deep phenotyping for precision medicine. Hum Mutat. 2012;33:777–80. doi: 10.1002/humu.22080. [DOI] [PubMed] [Google Scholar]
- 17.Bone WP, Washington NL, Buske OJ, Adams DR, Davis J, Draper D, et al. Computational evaluation of exome sequence data using human and model organism phenotypes improves diagnostic efficiency. Genet Med. 2016;18:608–17. doi: 10.1038/gim.2015.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zemojtel T, Kohler S, Mackenroth L, Jäger M, Hecht J, Krawitz P, et al. Effective diagnosis of genetic disease by computational phenotype analysis of the disease-associated genome. Sci Transl Med. 2014;6:252ra123–252ra123. doi: 10.1126/scitranslmed.3009262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masino AJ, Dechene ET, Dulik MC, Wilkens A, Spinner NB, Krantz ID, et al. Clinical phenotype-based gene prioritization: an initial study using semantic similarity and the human phenotype ontology. BMC Bioinformatics. 2014;15:248. doi: 10.1186/1471-2105-15-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinson PN, Köhler S, Bauer S, Seelow D, Horn D, Mundlos S. The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am J Hum Genet. 2008;83:610–5. doi: 10.1016/j.ajhg.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheung WA, Ouellette BFF, Wasserman WW. Compensating for literature annotation bias when predicting novel drug-disease relationships through Medical Subject Heading Over-representation Profile (MeSHOP) similarity. BMC Med Genomics. 2013;6:S3. doi: 10.1186/1755-8794-6-S2-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bodenreider O. The Unified Medical Language System (UMLS): integrating biomedical terminology. Nucleic Acids Res. 2004;32:267D–270. doi: 10.1093/nar/gkh061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rothwell DJ, Cote RA, Cordeau JP, Boisvert MA. Developing a standard data structure for medical language--the SNOMED proposal. Proc Symp Comput Appl Med Care 1993;695–9. [PMC free article] [PubMed]
- 24.Köhler S, Carmody L, Vasilevsky N, Jacobsen JOB, Danis D, Gourdine JP, et al. Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res. 2019;47:D1018–D1027. doi: 10.1093/nar/gky1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J, Lin X, Teng Y, Qi S, Xiao D, Zhang J, et al. A comprehensive evaluation of disease phenotype networks for gene prioritization. PLoS ONE. 2016;11:1–18. doi: 10.1371/journal.pone.0159457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Börnigen D, Tranchevent LC, Bonachela-Capdevila F, Devriendt K, De Moor B, De Causmaecker P, et al. An unbiased evaluation of gene prioritization tools. Bioinformatics. 2012;28:3081–8. doi: 10.1093/bioinformatics/bts581. [DOI] [PubMed] [Google Scholar]
- 27.Tranchevent LC, Capdevila FB, Nitsch D, de Moor B, de Causmaecker P, Moreau Y. A guide to web tools to prioritize candidate genes. Brief Bioinformatics. 2011;12:22–32. doi: 10.1093/bib/bbq007. [DOI] [PubMed] [Google Scholar]
- 28.Masoudi-Nejad A, Meshkin A. Gene prioritization: rationale, methodologies and algorithms. 66 (Publisher Springer International Publishing AG, Cham, Switzerland, 2014).
- 29.Yang H, Robinson PN, Wang K. Phenolyzer: phenotype-based prioritization of candidate genes for human diseases. Nat Methods. 2015;12:841–3. doi: 10.1038/nmeth.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.James RA, Campbell IM, Chen ES, Boone PM, Rao MA, Bainbridge MN, et al. A visual and curatorial approach to clinical variant prioritization and disease gene discovery in genome-wide diagnostics. Genome Med. 2016;8:13. doi: 10.1186/s13073-016-0261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koparir A, Karatas OF, Yuceturk B, Yuksel B, Bayrak AO, Gerdan OF, et al. Novel POC1A mutation in primordial dwarfism reveals new insights for centriole biogenesis. Hum Mol Genet. 2015;24:5378–87. doi: 10.1093/hmg/ddv261. [DOI] [PubMed] [Google Scholar]
- 32.Izumi R, Niihori T, Takahashi T, Suzuki N, Tateyama M, Watanabe C, et al. Genetic profile for suspected dysferlinopathy identified by targeted next-generation sequencing. Neurol Genet. 2015;1:e36. doi: 10.1212/NXG.0000000000000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knierim E, Gill E, Seifert F, von Moers A, Schuelke M. A recessive mutation in beta-IV-spectrin (SPTBN4) associates with congenital myopathy, neuropathy, and central deafness. Hum Genet. 2017;136:903–10. doi: 10.1007/s00439-017-1814-7. [DOI] [PubMed] [Google Scholar]
- 34.Quintana AM, Yu HC, Brebner A, Pupavac M, Geiger EA, Watson A, et al. Mutations in THAP11 cause an inborn error of cobalamin metabolism and developmental abnormalities. Hum Mol Genet. 2017;26:2838–49. doi: 10.1093/hmg/ddx157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Monies D, Abouelhoda M, AlSayed M, Alhassnan Z, Alotaibi M, Kayyali H, et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum Genet. 2017;136:921–39. doi: 10.1007/s00439-017-1821-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriguez-Zabala M, Aza-Carmona M, Rivera-Pedroza CI, Belinchón A, Guerrero-Zapata I, Barraza-García J. et al. FGF9 mutation causes craniosynostosis along with multiple synostoses. Hum Mutat 2017;38:1471–76. [DOI] [PubMed]
- 37.Wambach JA, Stettner GM, Haack TB, Writzl K, Škofljanec A, Maver A et al. Survival among children with “Lethal” congenital contracture syndrome 11 caused by novel mutations in the gliomedin gene (GLDN). Hum Mutat 2017;38:1477–84. [DOI] [PMC free article] [PubMed]
- 38.Le SV, Le PHT, Van LeTK, Kieu Huynh TT, Hang Do TT. A mutation in GABRB3 associated with Dravet syndrome. Am J Med Genet Part A. 2017;173:2126–31. doi: 10.1002/ajmg.a.38282. [DOI] [PubMed] [Google Scholar]
- 39.Guala D, Sonnhammer ELL. A large-scale benchmark of gene prioritization methods. Sci Rep. 2017;7:46598. doi: 10.1038/srep46598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med. 2017;376:21–31. doi: 10.1056/NEJMoa1516767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baynam G, Walters M, Claes P, Kung S, LeSouef P, Dawkins H, et al. Phenotyping: targeting genotype’s rich cousin for diagnosis. J Paediatr Child Health. 2015;51:381–6. doi: 10.1111/jpc.12705. [DOI] [PubMed] [Google Scholar]
- 42.Requena T, Gallego-Martinez A, Lopez-Escamez JA. A pipeline combining multiple strategies for prioritizing heterozygous variants for the identification of candidate genes in exome datasets. Hum Genomics. 2017;11:11. doi: 10.1186/s40246-017-0107-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johannes Birgmeier A, Haeussler M, Deisseroth CA, Jagadeesh KA, Ratner AJ, Guturu H et al. AMELIE accelerates Mendelian patient diagnosis directly from the primary literature. bioRxiv 2017;1–23.
- 44.Köhler S, Schulz MH, Krawitz P, Bauer S, Dölken S, Ott CE, et al. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am J Hum Genet. 2009;85:457–64. doi: 10.1016/j.ajhg.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.