

SUMMARY
The HIV-1 capsid is an ordered protein shell that houses the viral genome during early infection. Its expansive surface consists of an ordered and interfacing array of capsid protein hexamers and pentamers that are recognized by numerous cellular proteins. Many of these proteins recognize specific, assembled capsid interfaces not present in unassembled capsid subunits. We used protein engineering tools to capture diverse capsid assembly intermediates. We built a repertoire of capsid assemblies (ranging from two to 42 capsid protein molecules) that recreate the various surfaces in infectious capsids. These assemblies reveal unique capsid-targeting mechanisms for each of the anti-HIV factors TRIMCyp, MxB, and TRIM5α linked to inhibition of virus uncoating and nuclear entry, as well as the HIV-1 cofactor FEZ1 that facilitates virus intracellular trafficking. This capsid assembly repertoire enables elucidation of capsid recognition modes by known capsid-interacting factors, identification of new capsid-interacting factors, and potentially, development of capsid-targeting therapeutics.
Graphical Abstract
Summers et al. used a series of protein engineering tools to ‘trap’ the naturally self-polymerizing HIV-1 capsid protein in discrete, soluble fragments. These fragments enabled precision analysis of the capsid recognition surfaces and binding modes used by host factors to facilitate or block HIV-1 infection.
INTRODUCTION
HIV-1 packages its genome inside a cone-shaped protein shell, the capsid, which enters the target cell after host-viral membrane fusion. The capsid serves a critical role to simultaneously protect the viral genome from host immune detection and to traffic the genome towards the nucleus for integration into the host genome (reviewed in (Campbell and Hope, 2015)). The capsid is a nearly 40 megadalton protein super-structure composed of roughly 1,500 copies of the 25-kDa capsid protein (CA) (Briggs et al., 2004). A properly assembled capsid is built from approximately 250 CA hexamer subunits and exactly 12 CA pentamer subunits (Ganser et al., 1999; Li et al., 2000). Hexamers and pentamers share a quasi-equivalent structure and form capsids following fullerene cone geometry (Ganser et al., 1999; Li et al., 2000; Mattei et al., 2016; Pornillos et al., 2011). CA contains independently folded N- and C-terminal α-helical domains (termed NTD and CTD, respectively) separated by a flexible linker (Ganser-Pornillos et al., 2007). NTD-NTD and NTD-CTD interfaces form and stabilize rigid hexamers and pentamers. CTD-CTD interfaces, including independent dimerization and trimerization motifs, are responsible for bridging adjacent hexamers and pentamers to form the cone-shaped capsid (Byeon et al., 2009; Ganser-Pornillos et al., 2007; Mattei et al., 2016; Zhao et al., 2013).
While most capsids disassemble immediately after entry, it appears that the subset of capsids that lead to productive infection stay at least partially assembled and are associated with the viral genome until the virus reaches the nuclear pores (Francis and Melikyan, 2018; Mamede et al., 2017). These capsids are exposed to the cellular environment for several hours and provide an expansive surface area for recognition by cellular proteins. In support of this, numerous capsid-interacting host factors have been identified (Yamashita and Engelman, 2017). Many are viral cofactors that are recruited to the capsid surface to aid infection. Others are highly adapted restriction factors that inhibit viral replication after direct capsid recognition. Strikingly, most known capsid-binding host factors have either entirely lacked or displayed only partial recognition of CA monomers. These factors instead must specifically target unique high-order interfaces only found in the assembled CA lattice (Bhattacharya et al., 2014; Biris et al., 2012; Fribourgh et al., 2014; Li et al., 2016; Liu et al., 2016; Morger et al., 2018; Price et al., 2014; Yang et al., 2012). Some of these lattice-targeting factors are cytoplasmic, while others are primarily associated with nuclear pores. These data support the view that the capsid is an expansive protein-docking platform whose finely-tuned structural integrity is critical for viral infection.
Despite the advances towards understanding the apo-capsid structure (reviewed in (Perilla and Gronenborn, 2016)), how host factors, and particularly restriction factors, recognize the assembled capsid remains poorly understood. This is largely due to their special CA-binding modes. As they do not recognize soluble, unassembled capsid building blocks—like native CA dimers or engineered disulfide-crosslinked hexamers (Gamble et al., 1997; Pornillos et al., 2009)—the vast array of solution-based biochemical and structural techniques is not amenable to analyzing their interactions. It is also challenging to use either virion-purified capsids or in vitro assembled CA tubes (Li et al., 2000) in mechanistic or high-resolution structural studies. Both are insoluble and can be highly heterogeneous and unstable (Frank et al., 2015; Mattei et al., 2016; Yu et al., 2013). Additionally, many host factors have low capsid-binding affinity and/or form natural high-order oligomers themselves (Alvarez et al., 2017; Li et al., 2016; Liu et al., 2016), which prevent their uniform distribution along the surface of capsids or CA tubes required for high-resolution studies. To better define host factor-capsid interactions, an experimental balance is needed between insoluble CA polymers and their unassembled CA building blocks.
To address these concerns, we adopted a range of protein engineering techniques to stabilize CA oligomers in soluble, lattice-like fragments that are intermediates between unassembled and fully assembled capsid. Our engineered lattice “assemblies” range in size from 50 kDa to 1 MDa and exhibit every unique capsid lattice interface found in infectious virions. With these tools, solution-based biochemical assays and structural biology techniques can now be applied to many capsid-targeting factors and therapeutics. We used these assemblies to better define the capsid-binding modes of three HIV-1 restriction factors—TRIM5α, TRIMCyp, and MxB (Goujon et al., 2013; Kane et al., 2013; Liu et al., 2013; Sayah et al., 2004; Stremlau et al., 2004). In an accompanying report, we also define the capsid-binding properties of a recently described viral cofactor, FEZ1 (Huang et al., 2019). Each of these four factors demonstrated a unique capsid lattice-sensing ability that could only be defined with our designed capsid assemblies. This work significantly expands our understanding of the diverse array of capsid-sensing motifs and targetable capsid surfaces. Our assemblies support a rapid pipeline between first identification of a new capsid-binding host factor and a thorough analysis of its binding mode. They may also be valuable in searching for additional capsid-binding factors or designing specific capsid-targeting therapeutics to inhibit HIV-1 infectivity.
RESULTS
Existing CA constructs are insufficient to define the diverse capsid lattice-sensing modes of host factors
To investigate how host cell factors recognize the capsid surface during infection, we used previously published capsid oligomers (Figure 1A) to analyze several evolutionarily distinct capsid-binding host factors with poorly understood binding mechanisms. We optimized the recombinant expression and purification of three established HIV-1 restriction factors—rhesus macaque TRIM5α (Stremlau et al., 2004), crab-eating macaque TRIMCyp (Brennan et al., 2008; Dietrich et al., 2011), and human MxB. For each, we produced stable constructs containing at least the minimal domains required for capsid recognition (Figure 1B)(Figure S1A). For TRIM5α and TRIMCyp we used constructs containing: a mutated B-box domain (E120K/R121D to prevent high-order oligomerization) (Diaz-Griffero et al., 2009; Goldstone et al., 2014), the coiled-coil domain, and CA recognition motifs (PRY/SPRY for TRIM5α and CypA for TRIMCyp) (Sayah et al., 2004; Stremlau et al., 2006; Stremlau et al., 2005). We additionally made an artificially trimeric SPRY construct by directly fusing the SPRY domain to the trimeric proliferating cell nuclear antigen protein (PCNA) (termed PCNA-SPRY). For MxB, we fused the reported capsid-binding N-terminal 83 residues to maltose binding protein (MBP) to produce MxB1–83-MBP (Fribourgh et al., 2014; Fricke et al., 2014; Goujon et al., 2014).
Figure 1. Host factors demonstrate diverse CA-binding modes to established capsid assemblies.

(A) Schematics of several reported capsid oligomers. (B) Schematics of host capsid-binding proteins with their potential capsid-binding regions in red. (C-E). Capsid-host factor co-pelleting assays using A14C/E45C disulfide crosslinked CA tubes (top) and SDS-PAGE quantification of the reduction of protein in the soluble fraction (bottom). Error bars represent standard error of the mean of three independent experiments. (F-H) Size-exclusion chromatography co-elution assays between host factors and disulfide crosslinked hexamers (top) or CA monomers/dimers (bottom). A shift of the elution volume (red relative to others) indicates co-elution. (C) A soluble construct of TRIMCyp, BCCCyp, co-pellets with CA tubes, but not those containing P90A mutation. (F) BCCCyp co-elutes stably with CA hexamers, but marginally with individual CA. (D) An MxB truncation containing residues 1–83 co-pellets with CA tubes, but the MxB 11RRR13 to 11AAA13 mutation eliminates co-pelleting. (G) MxB1–83 does not co-elute with CA nor hexamers. (E) BCCSPRY or PCNA-SPRY constructs containing 2 or 3 SPRY domains of TRIM5α, respectively, co-pellet with CA tubes. (H) BCCSPRY does not co-elute with hexamers nor CA. See also Figure S1.
All three restriction factors strongly bound in vitro assembled disulfide-crosslinked CA tubes (Pornillos et al., 2009) (Figure 1C–E). In the conditions tested, we observed complete or near complete binding of host factors to CA tubes in a co-pelleting assay. We optimized capsid-binding conditions for each factor and found that TRIM5α and MxB preferred low ionic strength conditions (50–100 mM NaCl). As expected, previously published binding-deficient mutations for TRIMCyp (CAP90A) and MxB (MxB11RRR13>11AAA13) significantly reduced co-pelleting (Figure 1C,D) (Goujon et al., 2015; Schulte et al., 2015; Yoo et al., 1997). CA tube binding by these restriction factors is consistent with previously published results, and confirms that our constructs are capable of recognizing the assembled CA lattice. This also validated the use of intra-hexamer stabilizing disulfide-bonds in both the co-pelleting assay and solution-based biochemical assays (described below).
While CA co-pelleting assays are the standard for demonstrating host factor-CA interactions in many HIV research labs, they reveal little information about host factor-binding mechanisms. Drastic capsid-binding differences between the host factors tested here only became apparent when each was tested against available soluble CA constructs in size-exclusion chromatography (SEC) co-elution assays. The weak CA dimer (Gamble et al., 1997)—which is the natural pre-lattice state of CA—was only recognized by TRIMCyp, and the co-elution was marginal (Figure 1G). We expect the micromolar dissociation constant of the CypA-CA interaction likely made this interaction challenging to observe using this assay (Ylinen et al., 2010). However, the previously described engineered disulfide-linked CA hexamers (Pornillos et al., 2009)—a fundamental repeating unit of the CA lattice—were strongly recognized by TRIMCyp (Figure 1F)(Figure S1B). TRIMCyp coelution appeared much more complete with hexamers than with dimers. Neither MxB nor TRIM5α showed any interaction with either CA dimers or hexamers. (Figure 1G, H)(Figure S1C,D).
From these results, a dichotomy existed within the capsid-binding host factors. One factor (TRIMCyp) targeted CA hexamers and displayed weak affinity towards CA dimers. In an accompanying report, we also found that the viral cofactor FEZ1 specifically targeted CA hexamers (Huang et al., 2019). On the other hand, two factors (TRIM5α and MxB) apparently require either multiple CA hexamers or the interfaces between CA hexamers for capsid recognition. It became apparent that further binding mode analysis of the four factors—and potentially many of the other numerous putative capsid-binding factors—would be challenging due to a lack of soluble CA oligomers that fully represent interfaces observed in the assembled capsid.
Bridging the gap between CA monomers and hexamers: Creation of partial-hexamer assemblies
To more rapidly map the binding modes of host factors that target CA hexamers, we sought to create specific “partial-hexamer” subassemblies that essentially sub-divide a CA-hexamer into smaller pieces. We succeeded in producing two partial-hexamer assemblies best described as 1/3- and 1/2-hexamers, which are composed of two and three CA monomers, respectively (Figure 2A). These CA monomers are arranged with six-fold rotations—just as they are in complete CA hexamers. To stabilize and solubilize these partial-hexamers, we co-opted established CA intra-hexamer cysteine pairs (14C/45C and 42C/54C) and dimer interface mutations (184A/185A, or AA in short) (Table S1) and followed the established hexamer assembly protocol (Pornillos et al., 2009).
Figure 2. Partial-hexamer design, assembly, and validation.

(A) Top: schematics of 1/3-hexamer and 1/2-hexamer design. Two types of disulfide-bonds (S-S) are colored (A14C/E45C in blue, A42C/T54E in red). Bottom: SDS-PAGE demonstrating formation of 1/3- and 1/2-hexamers. On the left, CA samples run in reducing conditions show ~25 kDa monomers. On the right, CA samples run in non-reducing conditions reveal dimer, trimer, and hexamer species as designed. (B) Size-exclusion chromatography (SEC) demonstrates well-behaved partial-hexamer assemblies (marked with schematics). (C) Orthogonal views of the crystal structures (ribbon) of 1/3-hexamerEE (top) and 1/2-hexamerEE-ΔCTD (bottom) in the hexamer envelope (grey surface, PDB ID: 3H47). The structures are highly homologous to the corresponding portions in known hexamer crystal structures. See also Figure S2, Table S1, and Table S2.
To produce 1/3-hexamers, we first separately purified two monomeric CA constructs (CA14C/AA and CA45C/AA) that each contain a single cysteine mutation, only half of what is needed for assembling the cross-linked hexamer (Figure 2A). Since each construct alone does not contain a complementary cysteine to form a complete pair, they do not form disulfide-linked oligomers individually. As designed, however, a disulfide-bonded CA dimer was formed when we assembled CA14C/AA and CA45C/AA together in a 1:1 ratio (Figure 2A). Its 50 kDa molecular weight corresponded to that of a 1/3-hexamer. This design only generates a cross-linked dimer in the 1/3-hexamer configuration. Cross-linked hexamers cannot be formed because only one disulfide bond can be productively formed between 14C and 45C, each of which is on a different CA.
To ensure that these 1/3-hexamers remain discrete and do not proceed to assemble into natural hexamers, we further introduced A42E and T54E mutations at their exposed intrahexamer surfaces (constructs used: CA14C/42E/AA and CA45C/54E/AA). These mutations were designed to cause charge-charge repulsion if two 1/3-hexamer subunits come in close, hexamer-like proximity. Both 1/3-hexamers and 1/3-hexamers-EE (assemblies with -EE suffix contain 42E/54E mutations) demonstrated the appropriate 1/3-hexamer ~50 kDa solution molecular weight as observed by SEC (Figure 2B). It appeared that 42E/54E mutations are not required to keep 1/3-hexamers discrete at the concentrations tested. When any one of the two CA molecules in the 1/3-hexamer contains the wild-type 184W185M dimerization motif, dimeric 1/3-hexamers can potentially be formed to generate CA tetramers that contain the inter-hexamer interface in capsid (Figure S4A,B).
The ability to efficiently “mix and match” CA molecules with various engineered cysteines and solubilizing mutations allowed us to further create discrete 1/2-hexamers (Figure 2A). We incubated a 1:1:1 mixture of three heterologous CA constructs (CA14C/AA, CA45C/54C/AA, and CA42C/AA) built from a combination of 14C/45C and 42C/54C cysteine pairs. Again, we did not expect individual constructs to form disulfide-linked oligomers because they alone lack a complete complementary cysteine pair. When mixed, however, we observed a range of disulfide bonded CA species (Figure S2A). The primary product was a disulfide-bonded CA 3-mer that corresponds to the molecular weight of a 1/2-hexamer. In our design a disulfide-bonded CA 3-mer could only occur if the three disparate CA constructs made intra-hexamer contacts in a precise order--CA45C/54C/AA in the middle, linked on one face to CA14C/AA by the 14C/45C disulfide pair, and on the other to CA42C/AA by the 42C/54C disulfide pair. Numerous unproductive reactions made 1/2-hexamer assembly less efficient than the simpler 1/3-hexamer assembly. Despite this complexity, we readily purified both 1/2-hexamers and 1/2-hexamers-EE (the latter using CA14C/42E/AA, CA45C/54C/AA, and CA42C/54E/AA) to homogeneity, and they both displayed correct 1/2-hexamer solution molecular weight (~75 kDa) when analyzed by SEC (Figure 2B).
To validate their correct CA lattice architecture, we solved crystal structures of representative partial-hexamer assemblies (Table S2). We determined a 1/3-hexamerEE structure at 3.4 Å resolution. Indeed, as we designed, two disulfide-bonded CA monomers arranged as a discrete 1/3-hexamer are observed in the asymmetric unit (Figure 2C, top). This 1/3-hexamer is closely superimposable onto prior disulfide-linked hexamer structures with an overall root-mean-square deviation (RMSD) of 1.06 Å (Pornillos et al., 2009) (Figure 2C). The only deviation is in the positioning of one flexible CTD that does not participate in intra-hexamer contacts. This domain is normally stabilized in a complete hexamer by contacts with a neighboring CA that is not contained in 1/3-hexamers and 1/2-hexamers. 1/3-hexamerEE subunits were stable as discrete units and did not form complete hexamers even with symmetry-related molecules (Figure S2B). The hexamer-preventing 42E/54E mutations did not otherwise alter the CA monomer or partial-hexamer structure.
We also determined the crystal structures of 1/2-hexamer and 1/2-hexamerEE-ΔCTD (-ΔCTD suffix indicates a flexible CA CTD was purposely proteolytically removed for crystallization). As designed, 1/2-hexamerEE-ΔCTD crystallized as a discrete 1/2-hexamer and did not form complete hexamers with symmetry-related molecules. Its structure and 1/2-hexamer architecture overlaid closely with the prior disulfide-bonded hexamer structure (RMSD of 1.48 Å)(Pornillos et al., 2009) (Figure 2C, bottom). The 42E/54E mutations and proteolytic removal of a CA CTD did not affect the overall structure. While the 1/2-hexamer without hexamer-preventing mutations (42E/54E and ΔCTD) behaved as a discrete half-hexamer in solution, it crystallized as a complete hexamer in a 2-dimensional hexamer lattice very similar to that observed in prior CA crystal structures (Figure S2D) (Gres et al., 2015; Pornillos et al., 2009). This structure suggests that 1/2-hexamers have a propensity to reform hexamers in the appropriate conditions in the absence of the 42E/54E mutations. Overall, these 1/3- and 1/2-hexamer crystal structures demonstrate that the small capsid assemblies can reliably complement complete hexamers in studies of host factor capsid-binding analysis.
Partial-hexamer assemblies reveal the flexible and avid capsid binding mode of TRIMCyp
We used our partial-hexamer assemblies to investigate the capsid-binding properties of TRIMCyp. The coiled-coil domains of TRIM5 proteins form an anti-parallel dimer that brings two capsid-binding domains in proximity (SPRY for TRIM5α and cyclophilin A (CypA) for TRIMCyp) (Goldstone et al., 2014). The CypA domains of TRIMCyp can directly bind CA monomers, albeit weakly (Caines et al., 2012; Ylinen et al., 2010). This single-binding event is comparable to the well-studied recruitment of host cell CypA to the capsid surface (Franke et al., 1994; Gamble et al., 1996). It is hypothesized that TRIM5 proteins oligomerize to bring multiple copies of their capsid-binding domain to the capsid surface (Ganser-Pornillos et al., 2011; Li and Sodroski, 2008). This creates a high-affinity interaction through avidity. It is not known how the two CypA domains in TRIMCyp avidly bind two CA molecules on the capsid surface.
We employed SEC and sedimentation velocity analytical ultracentrifugation (AUC) assays to examine the interaction between TRIMCyp and 1/3-hexamerEE. In SEC co-elution assays we observed that TRIMCyp co-eluted efficiently with both 1/3- and 1/2-hexamerEE, comparable to that observed with complete hexamers (Figure 3A). Conversely, we observed very little coelution between monomeric CypA domains and hexamers (Figure S3A). Based on these results, we expect the two CypA domains of TRIMCyp likely simultaneously bind two CA molecules present in 1/3-, 1/2-, and complete hexamers. We validated and quantified this result using AUC. Analysis of the continuous distribution of sedimentation coefficients function c(s) for each component alone showed symmetric peaks with sedimentation coefficients of 3.3 S and 3.9 S for 1/3-hexamerEE and TRIMCyp, respectively, which corresponded to molecular weights of ~49 kD (51 kD expected) and ~84 kD (90 kD expected) (Figure S3B). Analysis of a mixture of 1/3-hexamerEE and TRIMCyp indicates the reaction occurred with fast exchange on the timescale of sedimentation. Four mixtures were analyzed by direct Lamm Equation modelling (Brautigam, 2011) to measure the affinity of the interaction (Figure S3C). The equilibrium dissociation constant KD determined in this manner for the interaction was 6.6 ± 0.9 μM. Using the same method, we determined the affinity for a single crab-eating macaque CypA for 1/3-hexamerEE to be 27.6 ± 9.0 μM, which is similar to the previously reported value (Ylinen et al., 2010). These experiments demonstrate that TRIMCyp indeed exhibits a higher affinity for 1/3-hexamerEE than a single CypA domain.
Figure 3. TRIMCyp has an avid and flexible capsid binding mode.

(A)-(D) BCCCyp efficiently binds partial hexamers containing at least two wild-type (WT) binding sites with residue P90 (green circles). SEC co-elution is indicated by a shift of the elution volume (red relative to the others), with a corresponding shift of the elution fraction bands in SDS-PAGE (bottom). BCCCyp co-elutes with 1/3-hexamerEE (A) and 1/2-hexamerEE containing two WT binding sites (B). (C) Co-elution is abrogated for 1/2-hexamerEE with three or two P90A mutations (red crosses). (D) Restoration of a hexamer from two 1/2-hexamers (cyan and pink cartoons) each containing a single P90 site restores BCCCyp binding. (E) Schematic models of the two CypA domains on TRIMCyp (cartoon) bind to any two CA subunits within a hexamer (surface). See also Figure S3.
An important advantage of our 1/3- and 1/2-hexamer design is that each CA position within an assembly can be individually altered by a desired mutation, which was not possible with the established hexamer constructs. As such, we made single and multiple P90A mutations in the 1/2-hexamerEE assembly to more finely map TRIMCyp binding. Complete TRIMCyp coelution was observed with a 1/2-hexamerEE that contained two native CypA loops (Figure 3B). A single P90A mutation at either the center (WT-90A-WT) or lateral (90A-WT-WT) CA position did not reduce TRIMCyp coelution as compared to complete hexamers or native 1/2-hexamer-EE. When there is a single or no wild-type binding site, a 1/2-hexamerEE with either two or three P90A mutations showed no coelution with TRIMCyp (Figure 3C).
We further used a 1/2-hexamer construct (lacking hexamer-ablating -EE mutations) that had two P90A mutations. 1/2-hexamers without EE mutations are able to reform a complete hexamer (as we observed in the crystal structure), which would have precisely two native CypA-binding loops across the hexamer center. We indeed observed significant coelution between this 1/2-hexamer and TRIMCyp (Figure 3D), suggesting that TRIMCyp was able to bind across and stabilize two 1/2-hexamers in a hexamer-like orientation. These data demonstrate that TRIMCyp can avidly bind any two CA molecules within a hexamer with different relative orientations and separations, likely due to flexibility of the two CypA modules in the TRIMCyp dimer (Figure 3E).
Trapping the 2- and 3-fold inter-hexamer interfaces in one soluble platform—the “hexamer-2”
The success of our ‘mix and match’ approach to stabilize partial-hexamer assemblies enabled us to build more complex capsid assemblies containing native inter-hexamer surfaces. These are likely required for the binding of numerous host factors that have little affinity towards CA hexamers, such as MxB and TRIM5α. To develop a soluble construct containing the native 2- and 3-fold inter-hexamer surfaces, we added two native dimerization motifs to our previously described 1/3-hexamerEE to attempt to form a “trimer of dimers” (Figure 4A). This structure has been computationally modeled to be the most stable lattice interface and the key nucleating structure during capsid maturation (Chen and Tycko, 2011; Grime and Voth, 2012; Tsiang et al., 2012). Importantly, the trimer of dimers structure contains the six CA monomers that surround the CA three-fold interface, with all native inter-hexamer lattice interfaces. We envisioned this assembly as a powerful counterpart to traditional hexamers for analyzing capsid-host interactions. As such, we will refer to it as “hexamer-2.”
Figure 4. Assembly of hexamer-2foldon containing the native 3-fold and 2-fold capsid interfaces.

(A) Left: Schematic of the six CA monomers (purple) centered at the interface between three CA hexamers. Middle: Design of hexamer-2 (purple cartoon) as a trimer of 14C/45C (yellow spheres) disulfide-crosslinked 1/3-hexamers. The 42E/54E solubilizing mutations are shown as cyan spheres. Right: Side view of hexamer-2foldon design highlighting the fused trimeric foldon domain at the C-terminus of CA. (B) SDS-PAGE (left) and SEC (right) analysis of purified 1/3-hexamer and hexamer-2foldon assemblies. In non-reducing SDS-PAGE analysis 1/3-hexamers and hexamer-2foldon assemblies run as ~50 kDa dimers. In reducing conditions, they run as ~25 kDa monomers. (C) Negative-stain EM micrograph (top), 2D class averages (middle), and 3D reconstruction (bottom) of hexamer-2foldon. (D) Crystal structure of hexamer-2foldon with the 2Fo-Fc electron density as grey surface (1□ level). (E) Superposition of the hexamer-2foldon crystal structure with the corresponding regions in native (PDB ID: 4XFX) and cross-linked (PDB ID: 3H47) CA crystal structures. Most differences between the structures are due to flexibility at the NTD-CTD hinge that connects the two CA domains. See also Figure S4 and Table S3.
During the course of our studies, it became apparent that the three native dimerization motifs in 1/3-hexamerEE-WM would need further stabilization. 1/3-hexamerEE-WM was not a stable trimer in solution (Figure 4B) and eluted at a position similar to 1/3-hexamerEE lacking native dimerization motifs. To stabilize its weak native interfaces, we directly fused it to the bacteriophage T4 foldon domain. Foldon is a small (30 residue), naturally trimeric protein with dimensions matching the central three CA units within hexamer-2 (Berthelmann et al., 2014). In our design, the foldon domain is fused to the inner capsid surface and, thus, will not alter host factor binding to the outer surface. The precise constructs used to assemble hexamer-2foldon are described in Table S3. For most assays, we used a hexamer-2foldon construct lacking several flexible C-terminal CA residues and containing a reported CA lattice mutant (CAA204D) (Zhao et al., 2013). These modifications were added to prevent further hexamer-2foldon oligomerization and to improve stability. These modifications do not alter the native inter-hexamer surfaces within hexamer-2foldon and did not appear to alter hexamer-2foldon solution behavior (Figure 4B).
As designed, hexamer-2foldon appeared on non-reducing SDS PAGE as disulfide-linked 1/3-hexamers at ~50 kDa molecular weight (Figure 4B). Strikingly, a monodispersed ~150 kDa species was observed by SEC and SEC-MALS (Figure 4B, Figure S4C). This demonstrates that the ~50-kDa 1/3-hexamer subunits within hexamer-2foldon form stable trimers. We analyzed the architecture of hexamer-2foldon by negative-stain electron microscopy and observed triangular, ~90 Å-wide assemblies (Figure 4C) that matched our design predictions.
To further validate the designed hexamer-2foldon architecture, we determined the crystal structure of hexamer-2foldon at 4.2Å resolution (Figure 4D)(Table S2). The asymmetric unit contained one copy of the designed 1/3-hexamer-foldon. Upon application of crystallographic three-fold symmetry, the six CA monomers centered around the 3-fold inter-hexamer interface were observed. Crystal packing created a two-dimensional CA lattice very similar to that of native hexameric CA (Gres et al., 2015). Consistent with its designed flexible tethering role, the foldon domain was mostly disordered with little density observed in the predicted location (Figure S4D,E). This confirms that the linkage between CA and foldon is flexible, and that CA architecture within hexamer-2-foldon is likely not unnaturally constrained by the presence of the foldon domain.
We compared our hexamer-2foldon structure to the crystal structures of native CA (PDB 4XFX) and disulfide-linked hexamers (PDB 3H47) (Figure 4E)—all of which crystallize in a flat CA lattice comparable to that of native capsid (Gres et al., 2015; Pornillos et al., 2009). In all structures, individual 1/3-hexamer subunits align closely with RMSD of ~2.1 Å. The corresponding hexamer-2 regions align with RMSD of 3.8 Å between hexamer-2foldon and native hexamer, comparable to the ~3.6 Å value for the same regions between native and cross-linked hexamers. We believe the differences between structures presented here are likely an example of the malleability required in the native CA lattice to provide curvature, especially since the observed differences are most prominent at CA regions known to be flexible (NTD-CTD hinge, CTD dimer interface) (Mattei et al., 2016; Zhao et al., 2013). The solution biophysical analysis and structural validation described here supports the correct architecture of hexamer-2foldon assemblies and their use in the analysis of capsid-host factor interactions (examples below).
Stabilizing large, multi-hexamer/pentamer assemblies by the SpyCatcher/SpyTag isopeptide bond system
The least understood capsid-binding host factors are those that span multiple hexamers for capsid recognition. These factors form large oligomers over the capsid surface to avidly bind repeating motifs of the CA lattice. Specifically, the detailed mechanism of TRIM5α-CA recognition as a chief block to interspecies retroviral transmission remains elusive. Other factors, like TRIMCyp and MxB, likely also span multiple hexamers (Alvarez et al., 2017; Fribourgh et al., 2014; Li et al., 2016). To study these host factors, we co-opted additional protein engineering strategies to stabilize large, multi-hexamer/pentamer assemblies.
We used a combination of native CA dimerization motifs and the SpyCatcher/SpyTag isopeptide bond system to stably link multiple hexamers in a capsid lattice arrangement (Figure 5A). SpyCatcher is a beta-barrel protein, derived from the 13-kDa Streptococcus pyogenese protein FbaB, with one strand missing. The missing strand is termed SpyTag, a 13-amino acid polypeptide. After mixing, SpyCatcher and SpyTag are rapidly and stably linked by a natural isopeptide bond between two amino acid side chains (Zakeri et al., 2012). SpyTag and SpyCatcher fusions have been extensively used to stabilize otherwise weak protein complexes. A CA construct with a C-terminally fused SpyCatcher (CA14C/45C-SpyCat) rapidly reacted with a CA construct with a C-terminally fused SpyTag (CA14C/45C-SpyTag) to form a covalent dimer (Figure 5A).
Figure 5. Producing soluble multi-hexamer/pentamer assemblies.

(A) A schematic of di-hexamer assembly (left) and SDS PAGE analysis of the CASpyCat-CASpyTag reaction (right). The SpyTag/SpyCatcher reaction proceeded essentially to completion. (B) Hexamer-SpyCatcher/Tag assembly and purification. CA and CA-Spy fusions are mixed at an appropriate ratio to assemble hexamers (top), purified by anion-exchange chromatography (middle), and analyzed by non-reducing SDS-PAGE (bottom). Hexameric species containing specific amounts of CA-SpyTag/Catcher fusion molecules were efficiently separated. (C) Schematics of producing Tri-hexamer (top) and Hepta-hexamer (bottom right) assemblies. (D) Schematics of pentamer-hexamer assemblies. (E-H) SEC (SEC-MALS in (E)), non-reducing SDS-PAGE, and negative-stain EM analysis of assembled Di-hexamers (E), Tri-hexamers (F), Hepta-hexamers (G), and Pentamer-hexamers (H). Sample micrographs are shown, as indicated, with associated 2-D class averages. In (E) and (F), the 2-D class averages of constructs containing 184A/185A mutant interfaces show non-contacting hexamer subunits. See also Figure S5.
We purified disulfide-linked hexamers that contained either single or multiple SpyCatcher or SpyTag fusions. This was achieved by mixing CA14C/45C/AA and CA14C/45C-SpyCat in an appropriate ratio to assemble hexamers. The resultant hexamers were purified using anion exchange chromatography (Figure 5B). We observed distinct elution peaks that correlated with disulfide-bonded hexamers containing zero, one, or two+ CASpyCat molecules as observed by SDS-PAGE (Figure 5B). Since each CASpyCat molecule was designed to contain a native dimerization interface, each elution peak also correlated with hexamers containing specific numbers of native dimerization interfaces. Notably, the amount of zero, one, and two+ CASpyCat incorporation events closely followed theoretical predictions using a simple binomial distribution model (Figure S5A). Thus, the ratio of incorporation events could be fine-tuned to experimental needs by adjusting the mixture ratio. The SpyTag-containing hexamers were produced in the same manner, with an MBP tag added to facilitate purification.
These purified hexamer-SpyCatcher/Tag constructs served as building blocks to assemble multi-hexamers (Figure 5C, Figure S5B–D). Covalent two-hexamer assemblies (referred to further as di-hexamers) were readily formed when 1-SpyTag-containing hexamers and 1-SpyCatcher-containing hexamers were reacted at a 1:1 ratio. Covalent three-hexamer assemblies (referred to further as tri-hexamers) were formed by reacting 2-SpyCatcher-containing hexamers and 1-SpyTag-containing hexamers at a 1:2 ratio. For both assemblies, the SpyCatcher/SpyTag isopeptide reaction occurred efficiently at all tested concentrations. Covalent ~300 kDa (di-hexamer MW) and ~450 kDa (tri-hexamer MW) assemblies were purified to homogeneity as observed by non-reducing SDS PAGE and SEC-MALS (Figure 5E, F; Figure S5E).
The lattice-like architecture of di- and tri-hexamers was apparent when we analyzed their structure using negative-stain EM (Figure 5E, F). Their hexamer building blocks were packed closely with a spacing consistent with that found in known capsid structures. Tri-hexamer assemblies displayed three orientations of hexamer building blocks—termed triangular, bent, or linear. These orientations are a result of the random incorporation of two CASpyTag molecules into two of the six positions of a hexamer. Each of the three tri-hexamer architectures represent patterns found in native HIV-1 capsids. The close packing between hexamers in di- and trihexamers was significantly affected when we mutated the native dimerization motifs in their CASpyCat/Tag molecules (Figure 5E, F). In these mutated assemblies, the hexamer building blocks were widely spaced with apparent flexibility between hexamers. This data strongly supports the notion that di- and tri-hexamers are bridged by native inter-hexamer surfaces. The SpyCatcher/SpyTag domains stabilize the assembly only by increasing the local concentration of the native dimerization motifs.
We further assembled megadalton, 7-hexamer assemblies (referred to as hepta-hexamers) by reacting 6-SpyTag-containing hexamers with an excess (approximately 8–12 fold) of 1-SpyCatcher-containing hexamers (Figure 5C, lower panel). However, the assembly tended to aggregate and precipitate making it challenging to purify. To address this problem, we incorporated a described anti-capsid nanobody in our assembly procedure (Gray et al., 2017). This nanobody binds the CA CTD with a reported 300 nM affinity. Based on the published nanobody-CA CTD structure, the nanobody should not interfere with CA hexamer formation but would block inter-hexamer interactions (Figure S5F). When incorporated at the appropriate concentration, the nanobody may prevent non-specific propagation/aggregation of multi-hexamer assemblies. We pre-bound a ratio of six nanobodies to one 6-SpyTag-containing hexamer and three nanobodies to one 1-SpyCatcher-containing hexamer. This is enough nanobody to theoretically coat the exposed outer surface of hepta-hexamer CTDs.
Putative reacted hepta-hexamers with bound nanobody remained soluble and eluted as a single, albeit broad, peak on SEC (Figure 5G). Negative-stain EM analysis confirmed that most assemblies contained seven hexamers (Figure 5G). The conformations of the hepta-hexamers were heterogenous, however, as only a fraction appeared 6-fold symmetric and many contained hexamers that were loosely packed. This is likely due to a fraction of nanobodies binding at inter-hexamer surfaces within hepta-hexamers instead of on the hepta-hexamer outer surface. Substantial optimization of the nanobody-CA ratio may be required to find a balance between hepta-hexamer aggregation prevention and correct architecture. Nonetheless, this demonstrates that the nanobody is an effective tool to prevent CA oligomerization for creating otherwise aggregation-prone CA lattice assemblies. The current hepta-hexamer assemblies may be valuable for the study of host factors that target large regions of capsid.
We also used this approach to stably link the previously reported disulfide-bonded CA pentamers (Pornillos et al., 2011) with hexamers. Similar to the assembly of multi-hexamers, defined number of SpyCatcher motifs were incorporated into assembled pentamers and reacted at the appropriate ratio with hexamers with appropriate SpyTags (Figure 5D). The correct pentamer-hexamer architectures were apparent when directly observed using negative-stain EM (Figure 5H). We did not use the pentamer-containing assemblies in host factor binding analysis since it is unclear whether the disulfide-linked pentamer architecture faithfully represents to that found in native cores. Pentamers observed in the cryo-EM tomography analysis of native HIV-1 cores displayed a different architecture than disulfide-linked pentamers (Mattei et al., 2016). Nonetheless, the results demonstrate the effectiveness of our assembly procedure, and the use of the current pentamer assemblies may still provide valuable insights into understanding pentamer incorporation into capsids and to potentially help identify host factors that prefer pentamer-hexamer interfaces.
MxB specifically recognizes the 3-fold inter-hexamer interfaces on HIV-1 capsid
The creation of a variety of capsid assemblies enabled us to better define the lattice sensing abilities of the capsid-targeting restriction factor MxB. Several capsid mutations (P207S, G208R, T210K) near the 3-fold inter-hexamer interface in hexamer-2foldon have been shown to reduce MxB restriction activity and capsid binding (Busnadiego et al., 2014; Opp et al., 2016). On the MxB side, its N-terminus has been shown to drive interaction with the HIV-1 capsid (Schulte et al., 2015). An MxB triple arginine motif (MxB-11RRR13) in this region has been shown to be critical to CA recognition and viral restriction (Goujon et al., 2015; Schulte et al., 2015). The N-terminal region of MxB is not observed in the cryo-electron microscopy structure of full-length MxB (Alvarez et al., 2017). These data suggest that the MxB binding motif is likely an unstructured peptide instead of a folded domain.
It has been proposed that MxB-CA binding requires inter-hexamer surfaces on capsid (Fribourgh et al., 2014). Consistent with previous studies, we demonstrated no co-elution of an N-terminal construct of MxB (MxB(1–83)-MBP) with either disulfide-linked CA hexamers or wild-type dimeric CA in SEC-binding assays (Figure 1G). In contrast, we observed significant co-elution of MxB(1–83)-MBP with hexamer-2foldon (Figure 6A, left). This co-elution was abolished with the MxB(1–83)-11AAA13-MBP mutant (Figure 6A, right). Co-elution was also abolished when the foldon domain was not present to stabilize hexamer-2, which alone is not stable at the concentrations used in this assay (Figure S6A). Using isothermal titration calorimetry, we found that MxB(1–83)-MBP bound hexamer-2foldon with a dissociation constant of ~10 μM under our experimental conditions (Figure 6C; Figure S6B,C). These data directly link MxB antiviral and capsid-binding properties to a specific high-order capsid motif. Moreover, while both the traditional hexamer and our hexamer-2foldon contain six CA molecules, only hexamer-2foldon confers the ability of MxB-binding, suggesting that MxB recognizes the 3-fold inter-hexamer interface contained in hexamer-2foldon.
Figure 6. MxB and TRIM5 proteins recognize inter-hexamer interfaces.

(A) MxB1–83 (left), but not the 11AAA13 mutant (right), co-elutes with hexamer-2foldon (red) in SEC (top) and SDS-PAGE (bottom) analyses. (B) MxB1–83 does not co-elute with di-hexamers in SEC. (C) MxB1–83 binds to hexamer-2foldon with a 9.6 ± 1.1 μM Kd by ITC. (D) Model of full-length MxB (cartoon) wedging its unstructured N-termini into two disparate three-fold inter-hexamer interfaces (surface). (E) A PCNA trimer fused to the SPRY domain of rhesus TRIM5α co-elutes with hepta-hexamers (red) in SEC. (F) BCCCyp shows marginal binding to a di-hexamer containing only one wild-type P90 site (left), but significant binding to a di-hexamer with two wild-type P90 sites on adjacent hexamers (right). P90 is indicated with green circles and P90A as red crosses. (G) A model of flexible TRIMCyp (cartoon) binding between hexamers (surface), and within hexamers (Figure 2). See also Figure S6.
We further tested MxB(1–83)-MBP binding to our multi-hexamer assemblies. In agreement with our hexamer-2foldon binding data, we observed preferred MxB coelution with the tri-hexamer assemblies, some of which contain the 3-fold inter-hexamer interface (Figure S6D,E). The interaction appeared weaker than that with hexamer-2foldon, which is likely due to the fact that only ~1/3 of tri-hexamers are of the triangular variety that contain a three-fold interface. Additionally, not all CA dimer interfaces at the three-fold surface are native due to design restraints, and this could reduce the observed affinity. In contrast, the di-hexamer assembly containing the native 2-fold interface showed no detectable interaction with MxB(1–83)-MBP, similar to single hexamers (Figure 6B). These data firmly establish that MxB specifically targets the 3-fold interface on HIV-1 capsid (Figure 6D).
TRIM5α requires a large surface area for capsid interaction
We hoped that our multi-hexamer assemblies would enable a binding mode analysis of TRIM5α. TRIM5α molecules dimerize and further oligomerize to form a hexagonal network on HIV-1 capsid, which significantly strengthens the interaction through avidity (Ganser-Pornillos et al., 2011; Li et al., 2016). It has been proposed that the individual SPRY domains of TRIM5α recognize multiple CA NTDs across inter-hexamer surfaces (Biris et al., 2012; Ganser-Pornillos et al., 2011; Goldstone et al., 2014; Morger et al., 2018; Yang et al., 2012). This has proven challenging to test, however, due to the very low CA binding affinity of individual SPRY domains and the lack of inter-hexamer surfaces in the prior soluble CA oligomers.
We tested our dimeric MBP-BCCSPRY construct in SEC coelution assays with hexamers, di-hexamers, tri-hexamers, and hepta-hexamers (Figure S6F). However, we were unable to observe significant MBP-BCCSPRY coelution with any multi-hexamer assembly. We expect that the binding affinity of this construct is too low to observe strong coelution in the conditions of this assay. We did, however, observe significant coelution between our artificial SPRY domain trimer (PCNA-SPRY) and hepta-hexamers (Figure 6E). Because hepta-hexamers contain multiple, redundant inter-hexamer surfaces, we expect that the three SPRY domains of PCNA-SPRY may avidly bind three independent capsid sites. Because an interaction of this nature is inherently complicated due to redundancies, it is difficult to surmise which capsid surface is targeted by a single SPRY domain. However, we have shown a recombinant TRIM5α construct clearly binding a capsid oligomer in the low micromolar concentration range. This is a necessary early step for future biochemical and structural studies.
TRIMCyp can bridge two hexamers in HIV-1 capsid
Although we demonstrated that TRIMCyp constructs can flexibly, yet avidly bind two CA molecules within a hexamer, we took advantage of the mixed protein composition in our di-hexamer assemblies to determine if TRIMCyp can also bridge two hexamers. The edge subunits of di-hexamers are composed of CA14C/45C/AA molecules, whereas the dimer interface between hexamers is composed of reacted CA-SpyTag and CA-SpyCatcher fusion molecules. Thus, each di-hexamer is composed of three different CA molecules, and each can be independently mutated to P90A to significantly reduce CypA binding.
As a negative control, we first mutated all di-hexamer CA positions to P90A and observed no coelution between this di-hexamer and TRIMCyp (Figure S6G). Next, we reverted the CA-SpyTag fusion molecule from P90A back to its native form (P90), so that the di-hexamers would have only a single WT CypA binding loop. Only very weak TRIMCyp co-elution was observed with this di-hexamer construct (Figure 6F, left), similar to the interaction between individual CA and CypA molecules. Finally, we reverted both CA-Spy fusions to WT CypA loops. These newly assembled di-hexamers have precisely two WT CypA loops, one on each hexamer centered at the native dimerization interface. TRIMCyp substantially co-eluted with these di-hexamers (Figure 6F, right). The results show that TRIMCyp is capable of avidly binding two CA molecules on neighboring hexamers. This further reinforces the idea that the two CypA domains of the TRIMCyp dimer are not rigidly oriented relative to the coil-coiled domains and can avidly target many pairs of CA molecules with a variety of distances and orientations (Figure 2H, 6G).
Discussion
Previously described disulfide-bonded CA hexamers and assembled CA tubes have been the most powerful in vitro tools for the analysis of the interactions between HIV-1 capsid and capsid-binding host factors. However, both of these CA assemblies have considerable limitations. Individual hexamers are only effectively recognized by a small subset of capsid-binding host factors. Targeting inter-hexamer surfaces may be evolutionarily beneficial for host factors, perhaps as a means to associate only with assembled capsids that contain the viral genome (as opposed to free CA). CA tubes contain the hexamer and inter-hexamer surfaces that largely represent those in infectious viral capsids. However, they are heterogeneous and many host factors do not bind them with enough uniformity to make quantitative or high-resolution structural analysis feasible. Furthermore, because CA tubes are collective structures with many interface patterns, they cannot be used to determine mechanistically the precise interface targeted by a host factor.
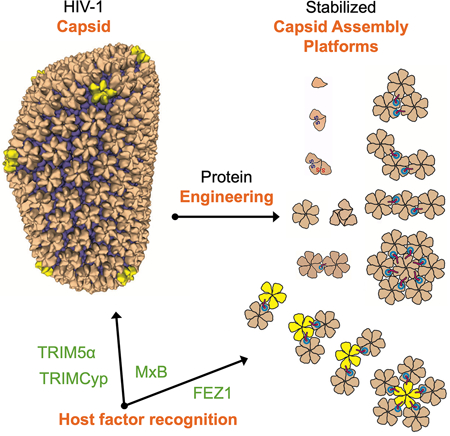
The study reported herein provides a comprehensive guide to the design and production of stable, discrete HIV-1 capsid assemblies, ranging in size from 50 kDa-1MDa (2–42 CA molecules), that fully encompass the unique interfaces present in assembled mature HIV-1 capsids (Figure 7). The simple design and preparation of these capsid assemblies make them amenable to investigations using a variety of established solution-based techniques, which cannot be achieved using existing CA hexamers or tubes. They allow for more in-depth mechanistic studies of known capsid-interacting host factors, enable rapid understanding of emerging capsid factors after discovery, and can be used to facilitate the discovery of new factors that sense capsid patterns. Furthermore, these assemblies may lead to increased efficiency of developing new capsid-labeling/monitoring reagents and capsid-targeting antiviral drugs. Beyond HIV, the design principles and the assembling techniques displayed here can help in the capture of diverse protein oligomers for biochemical analysis of other challenging biological systems.
Figure 7. A complete toolkit of soluble, discrete capsid complexes that mimic every intra-and inter-hexamer/pentamer interface found on the assembled HIV-1 capsid.

On the left, small capsid assemblies presented in this study and prior publications are represented. On the right, multi-hexamer/pentamer assemblies established in this study are represented by negative-stain electron microscopy class averages
We were able to use these capsid assemblies to gain deeper mechanistic insights into the unique binding modes of four host factors that play critical roles in HIV infection. MxB uses a triple-arginine motif in its N-terminal region to target the three-fold interface between capsid hexamers. The TRIMCyp dimer uses both of its CypA domains to avidly and flexibly bind numerous pairs of CA molecules within the assembled lattice. It tolerates a diverse range of different distances and orientations between the two CA molecules. TRIM5α remains the most challenging to study, as it appears to require more than three complete hexamers for avid binding. In an accompanying report, we show that the viral cofactor FEZ1 binds at the central pore within a hexamer. This highlights that both restriction factors and cofactors can be exquisitely sensitive to high-order capsid lattice architecture.
The data provided here, along with numerous previous studies, paint a picture of nearly every exposed capsid surface as a potential target of a host factor. HIV-1 recruits a variety of host factors to its capsid surface and each binding site must be fine-tuned for the requirements of the virus and the abundance of the proteins in the cell. Conversely, host immune factors like TRIMCyp, TRIM5α and MxB are also evolving and must compete with the recruited cofactors and counter-evolution of the viral capsid surface. The capsid surface therefore represents a hotbed of activity and evolution. The importance of our engineered capsid assemblies becomes even more apparent as new capsid-binding factors are discovered at a rapid pace.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yong Xiong (yong.xiong@yale.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All molecular cloning was carried out in E. coli XL10-Gold Ultracompetent cells. All recombinant proteins were expressed and purified from the E. coli strain BL21(DE3). Both cell lines were routinely cultured at 37 °C while shaking at > 220 RPM. XL10-Gold cells were grown in Luria Broth and BL21(DE3) cells were grown in either Luria Broth (starter culture) or Terrific Broth (for protein expression).
METHOD DETAILS
Cloning and expression
All CA constructs were cloned into pET-11a (EMD Millipore). CA-SpyCatcher fusions contained a C-terminal 6xHis tag. CA-SpyTag fusions contained a C-terminal MBP tag followed by a 6xHis tag. A SARS-n main protease (Mpro) protease cleavage site between SpyTag and MBP allowed for removal of the MBP and 6xHis (Yang et al., 2003). A five amino acid gly-ser linker separate CA from the Spy fusion domains. CA-Foldon was generated by direct fusion of foldon to the C-terminus of CA. The B-box, coiled-coil, and cyclophilin domains from Macaca fascicularis TRIMCyp (residues 89–468) were cloned into pRSFDuet-1 (EMD Millipore) with an N-terminal 6xHis tag. The B-box, coiled-coil, and SPRY domains of Macaca mulatta TRIM5α (residues (88–497) were cloned into a pMAL-derived vector (NEB) with an N-terminal 6xHis, MBP, and Mpro protease site. The TRIM5α SPRY domain (residues 280–497) was cloned into pRSFDuet-1 with an N-terminal 6xHis tag and PCNA to generate PCNA-SPRY. Human MxB residues 1–83 were cloned into pETDUET-1 (EMD Millipore) with an N-terminal 6xHis and C-terminal MBP tag. The nanobody 37E7 was cloned into pET28-a (EMD Millipore) with a C-terminal mpro cleavage site followed by a 6xHis tag. The cyclophilin domain of TRIMCyp was cloned into pET28-a with an N-terminal 6xHis and thrombin cleavage site. Mutations were made by site-directed mutagenesis using KOD Hot Start DNA polymerase (Novagen). Oligos were purchased from Integrated DNA Technologies. The SpyCatcher DNA was a gift from Lynn Regan at Yale University.
CA proteins were overexpressed in E. coli BL21(DE3) cells at 25° C for 12 h by induction with 0.5 mM IPTG at OD600 0.6–0.8. TRIMCyp, MxB, and nanobody constructs were similarly overexpressed at 18° for 16h. TRIM5 α constructs were co-transformed with the pGro7 plasmid (Takara) and overexpressed at 18° for 16h with 0.5 mM IPTG and 2 mg/mL arabinose.
Protein purification
Bacterial cells were harvested by centrifugation at 5000 rpm. Cells were resuspended in lysis buffer (50 mM TRIS pH 8, 300 mM NaCl, 0.1 mM TCEP) and lysed using a microfluidizer. Roche cOmplete protease inhibitor tablets were added to TRIMCyp, TRIM5α, and MxB purifications. Cell debris was clarified by centrifugation at 15,000 RPM for 35 minutes. TRIMCyp and PCNA-SPRY constructs were purified by nickel affinity, anion exchange and size exclusion chromatography. MxB constructs were purified by nickel affinity, cation exchange, and size exclusion chromatography. BCCSPRY domain containing TRIM5α constructs were purified by nickel affinity, MBP affinity, anion exchange, and size exclusion chromatography. Nanobody constructs were purified by nickel affinity, anion exchange, and size exclusion chromatography. All size exclusion chromatography was performed in 50 mM TRIS pH 8, 300 mM NaCl. Untagged CA proteins were purified by 25% w/v ammonium sulfate precipitation, dialysis into low-salt buffer (25 mM HEPES pH 7, 0.1 mM TCEP), and cation exchange chromatography. CA-foldon fusions were purified by 35% w/v ammonium sulfate precipitation and anion exchange chromatography. CA-Spy fusions were purified by nickel affinity and anion exchange chromatography. All CA constructs were dialyzed into CA storage buffer prior to freezing or further experiments (50 mM TRIS pH 8, 75 mM NaCl, 40 mM BME). All purification steps were monitored by SDS-PAGE gel electrophoresis (gels purchased from ConnSTEM and Invitrogen).
Capsid tube co-pelleting assays
Disulfide cross-linked CA tubes were assembled by dialyzing (in Slide-A-Lyzer dialysis cassettes) CA14C/45C into 50 mM TRIS pH 8, 1M NaCl at 15 mg/mL for one to two nights, followed by dialysis into 50 mM TRIS for another one to two nights. For co-pelleting assays, host factors were incubated in 21 uL reactions with CA tubes for 30 minutes at room temperature, then spun at 20000 × g for 10 minutes at 4°C. Total, soluble, and pellet fractions were taken at appropriate times and analyzed via SDS-PAGE. TRIMCyp constructs were incubated at 1.5 uM monomeric concentration with 37.5 uM tube-producing CA. MxB constructs were incubated at 3 uM with 75 uM tube-producing CA. TRIM5α constructs were incubated at 3 uM monomeric concentration with 150 uM tube-producing CA. TRIMCyp pelleting assays were performed in 25 mM TRIS pH 8, 150 mM NaCl. MxB pelleting assays were performed in 25 mM TRIS pH 8, 100 mM NaCl. TRIM5α pelleting assays were performed in 25 mM TRIS pH 8, 75 mM NaCl.
Assembly and purification of 1/3-hexamers, 1/2-hexamers, and hexamer-2-foldon
1/3-hexamers and hexamer-2-foldon were assembled using a 1:1 molar ratio of the appropriate CA proteins typically between 10–40 mg/mL of total protein. Mixtures were dialyzed overnight (using Thermo Slide-a-lyzer dialysis cassettes) in 50 mM TRIS pH 8, 1M NaCl. Mixtures were dialyzed for a second night in 50 mM TRIS pH 8. For 1/2-hexamer assembly, purified CA constructs were mixed at 1:1:1 molar ratios. Assemblies were purified using HiTrap Q HP anion exchange columns (GE Healthcare). 1/3- and 1/2-hexamer assemblies usually eluted between 100–200 mM NaCl. Hexamer-2-foldon assemblies eluted from an anion exchange column at approximately 250 mM NaCl. Each assembly was polished using a Superdex 200PG or Superdex 200GL size- exclusion chromatography column (GE Healthcare) ran with buffer containing 50 mM TRIS pH 8 and, 300 mM NaCl. All assemblies could be concentrated to at least 50 mg/mL and frozen at −80°C for long-term storage.
1/2-hexamer-EE-ΔCTD was produced by adding a Mpro protease cleavage sequence between the CA NTD and CTD (CA144-RMYS—KLQAGF—IRQG-CA157). The Mpro cleavage sequence drastically reduced disulfide-bonded hexamer formation in the 14C/45C/AA background, but did little to alter the assembly of 1/3- and 1/2-hexamers. The CTD cleavage is efficient in monomeric CA but is reduced in 1/3- and 1/2-hexamers. Cleavage reactions proceeded at least overnight at 4 degrees and were usually incomplete. Anion exchange using a HiTrap Q (GE Healthcare) was required to separate uncut and cut species.
Assembly and purification of multi-hexamer assemblies
Hexamers with incorporated SpyTag-MBP or SpyCatcher were assembled by dialysis for one night in 50 mM TRIS pH 8, 1M NaCl and for one night in 50 mM TRIS pH 8. To obtain the highest amount of one and two SpyTag or SpyCatcher incorporations, a ratio of one CASpyTag (or CASpyCat) to four CA14C/45C/AA was used. The binomial distribution function in Microsoft Excel was used to predict likelihood of hexamer incorporation of CA-Spy fusion molecules. After dialysis, the assembly mixtures were applied directly to an anion exchange column. Hexamers with various amounts of SpyTag/Catcher incorporation were eluted by a linear NaCl gradient. Assemblies generally eluted between 100–200 mM NaCl.
To assemble hepta-hexamers, Hexamer-6-SpyTag-MBP was assembled like above except for the presence of stoichiometric amount of nanobodies bound to its CTDs. After dialysis, it was directly applied to a Superdex 200PG column (GE Healthcare) for purification. Nanobody was added to purified hexamer-1SpyCatcher in a ratio of 3 nanobodies per hexamer. Hexamer-1-SpyCat+nanobody assemblies were reacted overnight with one hexamer-6-SpyTag-MBP+nanobody at 8–12 :1 ratio and approximately 10 mg/mL total protein.
SpyCatcher/Tag reactions generally proceeded overnight as isopeptide formation was occasionally not complete after a few hours. The reaction was performed in 50 mM TRIS pH 8 and a range of NaCl concentrations without apparent reduction in efficiency (from 50 mM to 300 mM NaCl). Only after the reaction was complete was the MBP tag on CA-SpyTag-MBP removed by Mpro digestion (digestion occurred for 2 hrs or overnight). Di-hexamers, tri-hexamers, and hepta-hexamers were purified using a Superdex 200PG (GE Healthcare) followed by a Superose 6 column (GE Healthcare) in 50 mM TRIS pH 8, 300 mM NaCl. Large molecular weight assemblies were analyzed by performing SDS-PAGE with NuPAGE 3–8% Tris-Acetate gels (ThermoFisher) following the manufacturer’s provided protocol.
Crystal structure determination and refinement
All small assemblies were screened for crystallization using the microbatch under-oil method using a 2:1 ratio of paraffin to silicon oil (Chayen et al., 1992). Protein concentrations ranged from 0.5–3 mg/mL. Most crystallization screening were performed at room temperature. 1 uL of protein solution was mixed with 1 uL of precipitant solution.
All small assemblies crystallized in numerous conditions. 1/3-hex-EE crystallized at room temperature between 1–2 mg/mL in 0.2 M Calcium Acetate Hydrate, 0.1 M MES: NaOH, pH 6, and 25 % (w/v) PEG 8000. The crystals were frozen with paratone oil as cryo-protectant. Diffraction data were collected at the NE-CAT beamline 24ID-E at the Advanced Photon Source. 1/2-hexEE-ΔCTD crystallized at room temperature at 1.5 mg/mL in 0.1M Sodium Citrate pH 5 and 8% PEG 8000. The crystals were frozen in paratone oil. Diffraction data were collected at NE-CAT beamline 24ID-C at the Advanced Photon Source. 1/2-hexamers crystallized at room temperature at 1 mg/mL in 0.1 M PCB buffer pH 8 and 25% (w/v) PEG 1500 and were cryo-protected in 25% ethylene glycol. Diffraction data were collected at NE-CAT beamline 24-IDC at the Advanced Photon Source. Hexamer-2foldon/204D/(1–221) crystallized at room temperature at 0.75 mg/mL in 0.2M NaCl, 0.1M HEPES pH 7.5, and 12% PEG 8000. Crystals were cryo-protected in 25% glycerol. Diffraction data were collected at Brookhaven National Laboratory beamline 17-ID1 (AMX).
Both HKL2000 and XDS were used for data processing (Kabsch, 2010a, b). Molecular replacement CA search models were made from either a disulfide-hexamer structure (PDB 3H47) or a native CA structure (PDB 4XFX) (Gres et al., 2015; Pornillos et al., 2009). Molecular replacement was performed using the CCP4 program Phaser (Collaborative Computational Project, 1994; McCoy et al., 2007; Vagin and Teplyakov, 2000; Winn et al., 2011). Iterative rounds of refinement in REFMAC and PHENIX were carried out, along with model building in COOT (Adams et al., 2010; Collaborative Computational Project, 1994; Murshudov et al., 1997). Individual disulfide bonds could not be resolved in the 1/2-hexamer crystal structure due to the intrinsic rotational averaging within the crystal lattice. Alignment RMSD values were generated using the SSM align module in COOT and the CCP4 program LSQKAB (Kabsch W. Acta. Cryst. A32 922–923 (1976).). Figure images were generated in Pymol, Chimera, and Coot, and Meshlab.
Size-exclusion chromatography co-elution assays
Host factors were mixed with CA assemblies for 30 minutes to 1 hour on ice in the same buffer used for CA-tube co-pelleting assays (unless specified). For TRIMCyp assays, mixtures were in a 500 uL volume with 36.6 uM monomeric concentration of TRIMCyp and 27.5 uM monomeric concentration of CA (from the appropriate assembly). All TRIMCyp binding tests were performed on GE S200 10/300 GL columns. MxB binding tests with small capsid assemblies were performed in 500 uL reaction volumes with 56 uM MxB and 78 uM monomeric concentration of CA (from the appropriate assembly). Samples were run on a Superose 6 GL (GE). For MxB coelution assays with multi-hexamer assemblies a Yarra SEC3000 column was used. The column was run in 25 mM phosphate buffer at pH 7, 100 mM NaCl. Binding reactions were in a volumn of 60 uL and contained 56 uM MxB-MBP protein and 78 uM monomeric concentration of CA (from appropriate multi-hexamer assembly). For TRIM5α binding assays, mixtures were in a 50 uL volume with 65 uM monomeric concentration of TRIM5α and 157 uM monomeric concentration of CA (from appropriate assembly). Binding mixtures were run a Superdex 200 5/150 GL column (GE).
Negative-stain electron microscopy
To observe capsid assemblies using negative-stain EM, we glow discharged a 400 mesh Cu grid, carbon coated, for 30 seconds at 25 mA. Sample was applied for 30 seconds and excess was removed by blotting with filter paper. We performed negative staining by applying 2% uranyl acetate to the grid, blotting immediately, applying again, incubating 30 seconds, and finally blotting residual stain with a filter paper. 50–100 images were collected at 73k magnification on a Tecnai T12 or Talos L120C microscope. We analyzed images and performed 2-D classification, and 3-D reconstruction for hexamer-2, using Relion (Scheres, 2012a, b).
Isothermal titration calorimetry
All ITC experiments were performed using a TA Instruments NanoITC machine. Binding reactions were performed at 6°C in 25 mM phosphate pH 7, 75 mM NaCl, and 5% glycerol. MxB was stable in these conditions after overnight dialysis and during the course of experiments. Capsid assemblies were in the cell and MxB was injected. Data were analyzed using the NanoAnalyze (TA Instruments) software. All curves were fit with an independent one-site binding model.
Analytical Ultracentrifugation
Binding reactions were performed at various BCCCyp, CypA, and 1/3-hexamer concentrations in 50 mM Tris pH 8.0, 150 mM NaCl buffer: BCCCyp concentrations ranged from 2–20 μM, CypA concentrations ranged from 1–50 μM, and 1/3-hexamerEE concentrations ranged from 0.5–10 μM. Samples were centrifuged in a Beckman An-60TI rotor in a Beckman Optima XL-I analytical ultracentrifuge for at least 12 hours at 42,000 rpm at 20°C. Sedimentation of different species was followed by absorption at 280 nm. Individual proteins were analyzed using SEDFIT to obtain continuous sedimentation coefficient distributions (Schuck, 2000). The sedimentation coefficients and molecular weights of the individual proteins obtained were noted and used in the following analysis. Reaction data was processed using SEDPHAT to obtain solutions to the Lamm Equation coupled to reaction fluxes (Brautigam, 2011) using the following parameters: vbar = 0.73 cm3/g, buffer density = 1.0058 g/mL, buffer viscosity = .010312 Poise, sBCCCyp = 3.93 S, MWBCCCyp = 84,300 Da, s1/3-hexEE= 3.32 S, MW1/3-hexEE = 48,500 Da, sM.fasc cypA = 1.92, MWM.fasc cypA = 21,500, εBCCCyp = 44,920 M−1cm−1, ε1/3-hexEE = 56,004 M−1cm−1, and εM.fasc cypA = 8,480 M−1cm−1. Log(Ka), log(koff), and scomplex were allowed to vary during alternating rounds of Simplex and Marquardt-Levenburg fitting. The confidence levels reported were calculated using the error surface projection method with a 68.3% (1σ) confidence interval (Brautigam, 2011). Figures were plotted using GUSSI (Brautigam, 2015).
Size exclusion chromatography linked to multi-angle light scattering
Each tested capsid assembly was loaded onto either a Superdex 200 GL or Superose 6 column (GE Healthcare). Multiangle laser light-scattering experiments were performed in buffer (50 mM Tris-HCl pH 8, 150 mM NaCl) at room temperature. Light-scattering data were collected on a Dawn Eos spectrometer (Wyatt Technology) coupled to an Opti-lab Dsp (Wyatt Technologies) interferometric refractometer. Samples (100 uL) at 1 mg/mL were injected and run over gel filtration columns at a flow rate of 0.5 ml/min. Multiangle laser light scattering (690 nm), absorbance (280 nm), and the refractive index were monitored after elution. Before samples were run, the system was calibrated and normalized to monomeric bovine serum albumin. The dn/dc value (change in solution refractive index with respect to protein concentration) is relatively constant for proteins (Wen, 1996), and the value for all experiments and analysis reported was set to 0.19. Data were processed using the software ASTRA as previously described (Wyatt, 1993).
QUANTIFICATION AND STATISTICAL ANALYSIS
Pelleting assays were repeated three times with different protein preparations. Band density was quantified using Image Studio Lite. The analysis is present in Figure legend 1.
ITC experiments were repeated three times with different protein preparations. The mean and standard deviation values were calculated using Microsoft Excel.
DATA AND SOFTWARE AVAILABILITY
The crystal structures and diffraction data presented here have been deposited in the Protein Data Bank. The accession codes of 1/3-hexamerEE, 1/2-hexamerEE-ΔCTD, hexamer-2foldon, and 1/2-hexamer are 6EC2, 6ECN, 6ECO, and 6OBH, respectively.
Supplementary Material
Highlights:
The self-polymerizing HIV-1 capsid protein ‘trapped’ in discrete, soluble oligomers
Engineered capsid protein assemblies faithfully mimic the infectious capsid surface
Host factors MxB, TRIMCyp, TRIM5α, and FEZ1 recognize unique capsid patterns
Capsid-binding by proteins and small molecules can be rapidly analyzed
ACKNOWLEDGEMENTS
We thank by W. Eliason for assistance in performing size-exclusion chromatography-multi-angle light scattering experiments and J. Wang for assistance in X-ray data processing. We thank W. Mothes for discussion. We thank I. Wang and E. Perets for preliminary experiments. We thank Dr. Chad Brautigam for assistance in AUC data analysis. We also thank the staff at the Advanced Photon Source beamlines 24-ID and E, and the National Synchrotron Light Source II beamline AMX. This work was by NIH, NSF, and DOE grants P50GM082251 (Y.X.), P30 GM124165 (NE-CAT beamlines), S10RR029205 (Pilatus detector), S10OD021527 (Eiger detector) and DE-AC02–06CH11357 (APS Synchrotron source). B.J.S. was supported by the Predoctoral Program in Biophysics NIH T32 GM008283 and the Gruber Science Fellowship. K.M.D. was supported by the Interdisciplinary Immunology Training Program NIH T32 AI007019. S.S.S. was supported by the Predoctoral Training Program in Virology NIH T32 AI055403. I.H. was supported by the Taiwan Ministry of Education Scholarship to Study Abroad. There are no conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
ACCESSION NUMBERS
The structures and diffraction data presented here have been deposited in the Protein Data Bank. The accession codes of 1/3-hexamerEE, 1/2-hexamerEE-ΔCTD, hexamer-2foldon, and 1/2-hexamer are 6EC2, 6ECN, 6ECO, and 6OBH, respectively.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez FJD, He S, Perilla JR, Jang S, Schulten K, Engelman AN, Scheres SHW, and Zhang P (2017). CryoEM structure of MxB reveals a novel oligomerization interface critical for HIV restriction. Sci Adv 3, e1701264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthelmann A, Lach J, Grawert MA, Groll M, and Eichler J (2014). Versatile C(3)-symmetric scaffolds and their use for covalent stabilization of the foldon trimer. Org Biomol Chem 12, 2606–2614. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A, Alam SL, Fricke T, Zadrozny K, Sedzicki J, Taylor AB, Demeler B, Pornillos O, Ganser-Pornillos BK, Diaz-Griffero F, et al. (2014). Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc Natl Acad Sci U S A 111, 18625–18630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biris N, Yang Y, Taylor AB, Tomashevski A, Guo M, Hart PJ, Diaz-Griffero F, and Ivanov DN (2012). Structure of the rhesus monkey TRIM5alpha PRYSPRY domain, the HIV capsid recognition module. Proc Natl Acad Sci U S A 109, 13278–13283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautigam CA (2011). Using Lamm-Equation modeling of sedimentation velocity data to determine the kinetic and thermodynamic properties of macromolecular interactions. Methods 54, 4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautigam CA (2015). Calculations and Publication-Quality Illustrations for Analytical Ultracentrifugation Data. Methods Enzymol 562, 109–133. [DOI] [PubMed] [Google Scholar]
- Brennan G, Kozyrev Y, and Hu SL (2008). TRIMCyp expression in Old World primates Macaca nemestrina and Macaca fascicularis. Proc Natl Acad Sci U S A 105, 3569–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs JA, Simon MN, Gross I, Krausslich HG, Fuller SD, Vogt VM, and Johnson MC (2004). The stoichiometry of Gag protein in HIV-1. Nat Struct Mol Biol 11, 672–675. [DOI] [PubMed] [Google Scholar]
- Busnadiego I, Kane M, Rihn SJ, Preugschas HF, Hughes J, Blanco-Melo D, Strouvelle VP, Zang TM, Willett BJ, Boutell C, et al. (2014). Host and viral determinants of Mx2 antiretroviral activity. J Virol 88, 7738–7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byeon IJ, Meng X, Jung J, Zhao G, Yang R, Ahn J, Shi J, Concel J, Aiken C, Zhang P, et al. (2009). Structural convergence between Cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell 139, 780–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caines ME, Bichel K, Price AJ, McEwan WA, Towers GJ, Willett BJ, Freund SM, and James LC (2012). Diverse HIV viruses are targeted by a conformationally dynamic antiviral. Nat Struct Mol Biol 19, 411–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EM, and Hope TJ (2015). HIV-1 capsid: the multifaceted key player in HIV-1 infection. Nat Rev Microbiol 13, 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chayen NE, Shaw Stewart PD, and Blow DM (1992). Microbatch crystallization under oil — a new technique allowing many small-volume crystallization trials. Journal of Crystal Growth 122, 176–180. [Google Scholar]
- Chen B, and Tycko R (2011). Simulated self-assembly of the HIV-1 capsid: protein shape and native contacts are sufficient for two-dimensional lattice formation. Biophysical journal 100, 3035–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project N (1994). The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Diaz-Griffero F, Qin XR, Hayashi F, Kigawa T, Finzi A, Sarnak Z, Lienlaf M, Yokoyama S, and Sodroski J (2009). A B-box 2 surface patch important for TRIM5alpha self-association, capsid binding avidity, and retrovirus restriction. J Virol 83, 10737–10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich EA, Brennan G, Ferguson B, Wiseman RW, O’Connor D, and Hu SL (2011). Variable prevalence and functional diversity of the antiretroviral restriction factor TRIMCyp in Macaca fascicularis. J Virol 85, 9956–9963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis AC, and Melikyan GB (2018). Single HIV-1 Imaging Reveals Progression of Infection through CA-Dependent Steps of Docking at the Nuclear Pore, Uncoating, and Nuclear Transport. Cell Host Microbe 23, 536–548 e536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank GA, Narayan K, Bess JW Jr., Del Prete GQ, Wu X, Moran A, Hartnell LM, Earl LA, Lifson JD, and Subramaniam S (2015). Maturation of the HIV-1 core by a non-diffusional phase transition. Nat Commun 6, 5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke EK, Yuan HE, and Luban J (1994). Specific incorporation of cyclophilin A into HIV-1 virions. Nature 372, 359–362. [DOI] [PubMed] [Google Scholar]
- Fribourgh JL, Nguyen HC, Matreyek KA, Alvarez FJD, Summers BJ, Dewdney TG, Aiken C, Zhang P, Engelman A, and Xiong Y (2014). Structural Insight into HIV-1 Restriction by MxB. Cell host & microbe 16, 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke T, White TE, Schulte B, de Souza Aranha Vieira DA, Dharan A, Campbell EM, Brandariz-Nuñez A, and Diaz-Griffero F (2014). MxB binds to the HIV-1 core and prevents the uncoating process of HIV-1. Retrovirology 11, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble TR, Vajdos FF, Yoo S, Worthylake DK, Houseweart M, Sundquist WI, and Hill CP (1996). Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87, 1285–1294. [DOI] [PubMed] [Google Scholar]
- Gamble TR, Yoo S, Vajdos FF, von Schwedler UK, Worthylake DK, Wang H, McCutcheon JP, Sundquist WI, and Hill CP (1997). Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science 278, 849–853. [DOI] [PubMed] [Google Scholar]
- Ganser-Pornillos BK, Chandrasekaran V, Pornillos O, Sodroski JG, Sundquist WI, and Yeager M (2011). Hexagonal assembly of a restricting TRIM5alpha protein. Proc Natl Acad Sci U S A 108, 534–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser-Pornillos BK, Cheng A, and Yeager M (2007). Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell 131, 70–79. [DOI] [PubMed] [Google Scholar]
- Ganser BK, Li S, Klishko VY, Finch JT, and Sundquist WI (1999). Assembly and analysis of conical models for the HIV-1 core. Science 283, 80–83. [DOI] [PubMed] [Google Scholar]
- Goldstone DC, Walker PA, Calder LJ, Coombs PJ, Kirkpatrick J, Ball NJ, Hilditch L, Yap MW, Rosenthal PB, Stoye JP, et al. (2014). Structural studies of postentry restriction factors reveal antiparallel dimers that enable avid binding to the HIV-1 capsid lattice. Proc Natl Acad Sci U S A 111, 9609–9614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goujon C, Greenbury RA, Papaioannou S, Doyle T, and Malim MH (2015). A triple-arginine motif in the amino-terminal domain and oligomerization are required for HIV-1 inhibition by human MX2. J Virol 89, 4676–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goujon C, Moncorge O, Bauby H, Doyle T, Barclay WS, and Malim MH (2014). Transfer of the amino-terminal nuclear envelope targeting domain of human MX2 converts MX1 into an HIV-1 resistance factor. J Virol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goujon C, Moncorge O, Bauby H, Doyle T, Ward CC, Schaller T, Hue S, Barclay WS, Schulz R, and Malim MH (2013). Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 502, 559–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray ER, Brookes JC, Caillat C, Turbe V, Webb BLJ, Granger LA, Miller BS, McCoy LE, El Khattabi M, Verrips CT, et al. (2017). Unravelling the Molecular Basis of High Affinity Nanobodies against HIV p24: In Vitro Functional, Structural, and in Silico Insights. ACS Infect Dis 3, 479–491. [DOI] [PubMed] [Google Scholar]
- Gres AT, Kirby KA, KewalRamani VN, Tanner JJ, Pornillos O, and Sarafianos SG (2015). STRUCTURAL VIROLOGY. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science 349, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grime JM, and Voth GA (2012). Early stages of the HIV-1 capsid protein lattice formation. Biophysical journal 103, 1774–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P-T, Summers BJ, Xu C, Perilla JR, Malikov V, Nagahavi MH, and Xiong Y (2019). FEZ1 is recruited to a conserved cofactor site on capsid to promote HIV-1 trafficking. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010a). Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr 66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010b). Xds. Acta Crystallogr D Biol Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane M, Yadav SS, Bitzegeio J, Kutluay SB, Zang T, Wilson SJ, Schoggins JW, Rice CM, Yamashita M, Hatziioannou T, et al. (2013). MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 502, 563–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Hill CP, Sundquist WI, and Finch JT (2000). Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 407, 409–413. [DOI] [PubMed] [Google Scholar]
- Li X, and Sodroski J (2008). The TRIM5alpha B-box 2 domain promotes cooperative binding to the retroviral capsid by mediating higher-order self-association. J Virol 82, 11495–11502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YL, Chandrasekaran V, Carter SD, Woodward CL, Christensen DE, Dryden KA, Pornillos O, Yeager M, Ganser-Pornillos BK, Jensen GJ, et al. (2016). Primate TRIM5 proteins form hexagonal nets on HIV-1 capsids. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Perilla JR, Ning J, Lu M, Hou G, Ramalho R, Himes BA, Zhao G, Bedwell GJ, Byeon IJ, et al. (2016). Cyclophilin A stabilizes the HIV-1 capsid through a novel non-canonical binding site. Nat Commun 7, 10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Pan Q, Ding S, Qian J, Xu F, Zhou J, Cen S, Guo F, and Liang C (2013). The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe 14, 398–410. [DOI] [PubMed] [Google Scholar]
- Mamede JI, Cianci GC, Anderson MR, and Hope TJ (2017). Early cytoplasmic uncoating is associated with infectivity of HIV-1. Proc Natl Acad Sci U S A 114, E7169–E7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattei S, Glass B, Hagen WJ, Krausslich HG, and Briggs JA (2016). The structure and flexibility of conical HIV-1 capsids determined within intact virions. Science 354, 1434–1437. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morger D, Zosel F, Buhlmann M, Zuger S, Mittelviefhaus M, Schuler B, Luban J, and Grutter MG (2018). The Three-Fold Axis of the HIV-1 Capsid Lattice Is the Species-Specific Binding Interface for TRIM5alpha. J Virol 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, and Dodson EJ (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Opp S, Vieira DA, Schulte B, Chanda SK, and Diaz-Griffero F (2016). MxB Is Not Responsible for the Blocking of HIV-1 Infection Observed in Alpha Interferon-Treated Cells. J Virol 90, 3056–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perilla JR, and Gronenborn AM (2016). Molecular Architecture of the Retroviral Capsid. Trends Biochem Sci 41, 410–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, and Yeager M (2009). X-ray structures of the hexameric building block of the HIV capsid. Cell 137, 1282–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pornillos O, Ganser-Pornillos BK, and Yeager M (2011). Atomic-level modelling of the HIV capsid. Nature 469, 424–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AJ, Jacques DA, McEwan WA, Fletcher AJ, Essig S, Chin JW, Halambage UD, Aiken C, and James LC (2014). Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog 10, e1004459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayah DM, Sokolskaja E, Berthoux L, and Luban J (2004). Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 430, 569–573. [DOI] [PubMed] [Google Scholar]
- Scheres SH (2012a). A Bayesian view on cryo-EM structure determination. Journal of molecular biology 415, 406–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH (2012b). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P (2000). Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophysical journal 78, 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte B, Buffone C, Opp S, Di Nunzio F, De Souza Aranha Vieira DA, Brandariz-Nunez A, and Diaz-Griffero F (2015). Restriction of HIV-1 Requires the N-Terminal Region of MxB as a Capsid-Binding Motif but Not as a Nuclear Localization Signal. J Virol 89, 8599–8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, and Sodroski J (2004). The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427, 848–853. [DOI] [PubMed] [Google Scholar]
- Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, Diaz-Griffero F, Anderson DJ, Sundquist WI, and Sodroski J (2006). Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc Natl Acad Sci U S A 103, 5514–5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stremlau M, Perron M, Welikala S, and Sodroski J (2005). Species-specific variation in the B30.2(SPRY) domain of TRIM5alpha determines the potency of human immunodeficiency virus restriction. J Virol 79, 3139–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiang M, Niedziela-Majka A, Hung M, Jin D, Hu E, Yant S, Samuel D, Liu X, and Sakowicz R (2012). A trimer of dimers is the basic building block for human immunodeficiency virus-1 capsid assembly. Biochemistry 51, 4416–4428. [DOI] [PubMed] [Google Scholar]
- Vagin A, and Teplyakov A (2000). An approach to multi-copy search in molecular replacement. Acta Crystallogr D Biol Crystallogr 56, 1622–1624. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, and Engelman AN (2017). Capsid-Dependent Host Factors in HIV-1 Infection. Trends Microbiol 25, 741–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Ji X, Zhao G, Ning J, Zhao Q, Aiken C, Gronenborn AM, Zhang P, and Xiong Y (2012). Structural insight into HIV-1 capsid recognition by rhesus TRIM5alpha. Proc Natl Acad Sci U S A 109, 18372–18377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, Sun L, Mo L, Ye S, Pang H, et al. (2003). The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci U S A 100, 13190–13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylinen LM, Price AJ, Rasaiyaah J, Hue S, Rose NJ, Marzetta F, James LC, and Towers GJ (2010). Conformational adaptation of Asian macaque TRIMCyp directs lineage specific antiviral activity. PLoS Pathog 6, e1001062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S, Myszka DG, Yeh C, McMurray M, Hill CP, and Sundquist WI (1997). Molecular recognition in the HIV-1 capsid/cyclophilin A complex. Journal of molecular biology 269, 780–795. [DOI] [PubMed] [Google Scholar]
- Yu Z, Dobro MJ, Woodward CL, Levandovsky A, Danielson CM, Sandrin V, Shi J, Aiken C, Zandi R, Hope TJ, et al. (2013). Unclosed HIV-1 capsids suggest a curled sheet model of assembly. Journal of molecular biology 425, 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakeri B, Fierer JO, Celik E, Chittock EC, Schwarz-Linek U, Moy VT, and Howarth M (2012). Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc Natl Acad Sci U S A 109, E690–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning J, Ahn J, Gronenborn AM, Schulten K, Aiken C, et al. (2013). Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 497, 643–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The crystal structures and diffraction data presented here have been deposited in the Protein Data Bank. The accession codes of 1/3-hexamerEE, 1/2-hexamerEE-ΔCTD, hexamer-2foldon, and 1/2-hexamer are 6EC2, 6ECN, 6ECO, and 6OBH, respectively.