

Abstract
The common patatin-like phospholipase domain–containing protein 3 (PNPLA3) variant I148M predisposes to nonalcoholic liver disease but not its metabolic sequelae. We compared the handling of labeled polyunsaturated fatty acids (PUFAs) and saturated fatty acids (SFA) in vivo in humans and in cells harboring different PNPLA3 genotypes. In 148M homozygous individuals, triglycerides (TGs) in very low–density lipoproteins (VLDL) were depleted of PUFAs both under fasting and postprandial conditions compared with 148I homozygotes, and the PUFA/SFA ratio in VLDL-TGs was lower relative to the chylomicron precursor pool. In human PNPLA3-148M and PNPLA3-KO cells, PUFA but not SFA incorporation into TGs was increased at the expense of phosphatidylcholines, and under lipolytic conditions, PUFA-containing diacylglycerols (DAGs) accumulated compared with PNPLA3-148I cells. Polyunsaturated TGs were increased, while phosphatidylcholines (PCs) were decreased in the human liver in 148M homozygous individuals as compared with 148I homozygotes. We conclude that human PNPLA3-I148M is a loss-of-function allele that remodels liver TGs in a polyunsaturated direction by impairing hydrolysis/transacylation of PUFAs from DAGs to feed phosphatidylcholine synthesis.
Keywords: Hepatology, Metabolism
Keywords: Genetic variation, Hepatitis
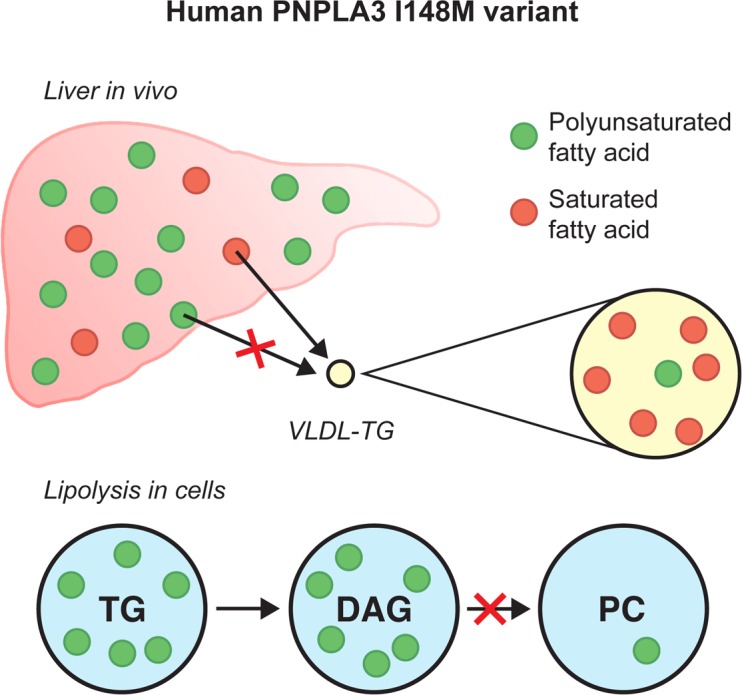
The human PNPLA3-148M variant, the major genetic risk of NAFLD, increases liver polyunsaturated triglycerides and impairs hydrolysis/transacylation of polyunsaturated fatty acids from diacylglycerols.
Introduction
The I148M variant in patatin-like phospholipase domain–containing protein 3 (PNPLA3) is found worldwide in 30%–50% of all subjects (1). It increases intrahepatocellular triglycerides (IHTGs) and risks of nonalcoholic steatohepatitis (NASH), alcoholic and nonalcoholic cirrhosis, and hepatocellular carcinoma (2). Unlike in nonalcoholic fatty liver disease (NAFLD) associated with obesity and metabolic syndrome, carriers with NAFLD due to the I148M gene variant are neither insulin resistant nor predisposed to develop metabolic sequelae, such as cardiovascular disease, compared with noncarriers (1). Indeed, several studies have recently shown the gene variant to be protective against cardiovascular disease (3, 4).
In humans, hepatic TGs are markedly enriched in polyunsaturated fatty acids (PUFA) in carriers of the PNPLA3-I148M variant compared with noncarriers (5). This lipidome differs from the NAFLD lipidome associated with insulin resistance, in which liver is enriched in predominantly saturated or monounsaturated TGs (5). The human lipidomic data of NAFLD in PNPLA3 variant carriers closely resemble the hepatic lipidomic profile of PNPLA3-KO mice (6, 7). In the latter study, PNPLA3 was proposed to act as a transacylase transferring PUFAs from TG to lysophospholipids or as a TG hydrolase hydrolyzing PUFAs from TGs in a remodeling pathway for lipid droplet phospholipids (7).
However, data from mouse models are not easy to reconcile with the human data. PNPLA3-KO mice accumulate polyunsaturated TGs but do not develop hepatic steatosis (6–8). Opposite to humans and knock-in (KI) mice expressing a catalytically inactive PNPLA3 (PNPLA3-S47A–KI), PUFAs are depleted in hepatic TGs in I148M-KI mice — i.e. in mice in which the I148M has been introduced to the endogenous mouse PNPLA3 gene (7). Mouse PNPLA3 is approximately 68% homologous with human PNPLA3 (9). This difference could contribute to the discrepant results in mice as compared with humans. Regarding cell models, hepatic cell lines such as HuH7 and HepG2 are not ideal for studying the function of the PNPLA3-I148M variant, as both cell lines are homozygous for the variant allele (10, 11). There are no studies addressing the function of the PNPLA3-I148M variant in humans in vivo or in vitro in human cells that do not endogenously express the I148M variant and in which the variant has been knocked in rather than overexpressed.
In the present study, we wished to determine why PUFAs are enriched in TGs in the human liver. This is important for understanding the pathogenesis of the most important genetic risk factor of NAFLD. To this end, we compared the hepatic handling of labeled PUFAs (13C-18:2) and saturated fatty acids (SFAs, 13C-16:0) and the composition of very low–density lipoproteins (VLDL) in homozygous carriers and noncarriers of the PNPLA3-I148M variant. Furthermore, by using CRISPR-Cas9, we engineered human cells homozygous for the PNPLA3 148I allele (PNPLA3-148I, WT) to generate cell lines with a homozygous I148M substitution (PNPLA3-148M–KI) or a homozygous PNPLA3 deletion (PNPLA3-KO). In these cells, we employed “click” chemistry of alkyne-labeled C-18:2 and C-16:0 FAs to analyze rapid FA fluxes during lipogenesis and lipolysis. Finally, we compared the lipid composition of human liver biopsies between homozygous carriers and noncarriers of the I148M variant.
Results
Increased IHTGs in homozygous 148M variant allele carriers (PNPLA3148MM) compared with noncarriers (PNPLA3148II).
Characteristics of the PNPLA3148II and PNPLA3148MM groups are shown in Table 1. The PNPLA3148MM group had a significantly, 3.5-fold higher IHTG content than the PNPLA3148II group (6.3% [interquartile range (IQR) 4.5%–14.6%] vs. 1.8% [1.0%–6.7%]) (Table 1). The PNPLA3148II and PNPLA3148MM groups were similar with respect to age, sex, glucose, insulin concentrations, and BMI (Table 1). There were no significant differences between the groups in physical activity or dietary intake, as determined by 1-week accelerometer data and analysis of 3-day dietary records (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.127902DS1).
Table 1. Clinical characteristics of the subjects.

Deficiency of polyunsaturated TGs in VLDL in the PNPLA3148MM as compared with the PNPLA3148II group.
Total concentrations of plasma TG, free fatty acids (FFA), VLDL-TG and chylomicron-TG, and glucose and serum insulin were similar between the PNPLA3148MM and PNPLA3148II groups in the fasting state and postprandially at every time point (Figure 1). Lipidomic analysis of VLDL was performed for detailed characterization of VLDL-TGs in the fasting state and postprandially. The relationship between the number of double bonds in VLDL-TGs and the ratio of the mean absolute concentrations of corresponding VLDL-TGs in the PNPLA3148MM as compared with the PNPLA3148II group are shown in Figure 2. The number of double bonds was inversely related to the ratios of VLDL-TGs in PNPLA3148MM vs. PNPLA3148II in the fasting state and at 120 minutes, 300 minutes, and 420 minutes postprandially (Figure 2). Thus, although total concentrations of VLDL-TGs were similar (Figure 1), the TGs secreted from the liver in VLDL before and during the meal were deficient in polyunsaturated TGs in the PNPLA3148MM as compared with the PNPLA3148II group.
Figure 1. Similar total concentrations of circulating lipids, glucose, and insulin in the PNPLA3148MM vs. PNPLA3148II groups.

Concentrations of (A) plasma TGs, (B) free fatty acids, (C) VLDL-TG, (D) chylomicron-TG, (E) glucose, and (F) serum insulin in the PNPLA3148MM and PNPLA3148II groups in the fasting state (0 minutes) and during the postprandial period. Data are shown as mean ± SEM. The blue lines and circles denote the PNPLA3148II (n = 14) group, and the red lines and squares denote the PNPLA3148MM (n = 12) group. There were no significant differences between the groups as determined using 2-way ANOVA.
Figure 2. Differences between distinct VLDL-TGs in the PNPLA3148MM vs. PNPLA3148II groups according to the number of double bonds.

Panels show linear regression lines between the number of double bonds in VLDL-TGs and the log2 fold-change of absolute concentrations of corresponding VLDL-TGs in the PNPLA3148MM (n = 12) vs. the PNPLA3148II (n = 14) group (A) in the fasting state (0 minutes) and (B) 120 minutes, (C) 300 minutes, and (D) 420 minutes following the meal. Each circle denotes a distinct VLDL-TG species. Significance was determined using linear regression analysis.
Deficiency of PUFAs in VLDL-TG FAs in the PNPLA3148MM as compared with the PNPLA3148II group.
The percentage of distinct FAs of total FAs in VLDL-TGs in the PNPLA3148MM as compared with the PNPLA3148II group in the fasting state are shown in Figure 3A. Saturated palmitate (16:0), monounsaturated oleate (18:1), and polyunsaturated linoleate (18:2) were the most abundant FAs in VLDL-TGs in both groups (Figure 3A). The percentage of 18:2 FA in VLDL-TG FAs was significantly lower in the PNPLA3148MM as compared with the PNPLA3148II group in the fasting state (Figure 3A) and during the entire postprandial period (Figure 3, B and C). In contrast, the percentage of SFA 16:0 in VLDL-TG FAs was significantly higher in the PNPLA3148MM as compared with the PNPLA3148II group in the fasting state (Figure 3A) and during the postprandial period (Figure 3, B and D).
Figure 3. Differences in the composition and handling of VLDL-TG fatty acids in the PNPLA3148MM vs. the PNPLA3148II groups.

(A) Percentage of distinct fatty acids of total unlabelled fatty acids in VLDL-TG in the PNPLA3148MM (red bars, n = 12) and PNPLA3148II (blue bars, n = 14) groups in the fasting state. (B) Fold change in the percentage of distinct fatty acids of total unlabeled fatty acids in VLDL-TGs in the PNPLA3148MM (n = 12) vs. the PNPLA3148II (n = 14) groups in the fasting state (0 minutes) and during the postprandial period. The x-axis denotes distinct fatty acids, and y-axis denotes postprandial time. Each square represents log2 fold-change of the percentage of a distinct fatty acid of total fatty acids in VLDL-TG in the PNPLA3148MM (n = 12) vs. the PNPLA3148II (n = 14) groups at that time point. (C and D) Percentage of unlabeled polyunsaturated fatty acid linoleate (18:2) (C) and saturated fatty acid palmitate (16:0) (D) of total fatty acids in VLDL-TG in the PNPLA3148MM (red squares and lines, n = 12) and PNPLA3148II (blue circles and lines, n = 14) groups in the fasting state and during the 420-minute postprandial period. (E) Linear regression lines between the number of double bonds in VLDL-TG fatty acids and the log2 fold-change of absolute concentrations of corresponding VLDL-TG fatty acid in the PNPLA3148MM (n = 12) vs. the PNPLA3148II (n = 14) groups in the fasting state (0 min; black circles) and at 120 minutes (red squares), 180 minutes (green triangles pointing up), 240 minutes (blue triangles pointing down), 300 minutes (purple diamonds), 360 minutes (orange circles), and 420 minutes (turquoise squares). (F) Ratio of 13C-18:2 to 13C-16:0 fatty acids in plasma VLDL-TG related to the corresponding ratio in the chylomicron precursor pool in the PNPLA3148MM (red squares and lines, n = 12) and the PNPLA3148II (blue circles and lines, n = 14) groups. Data are shown as mean ± SEM. *P < 0.05. Significance was determined using 2-tailed Student’s t test for unpaired data, 2-way ANOVA, and linear regression as appropriate.
The relationship between the number of double bonds in VLDL-TG FAs and the ratio of the mean absolute concentrations of corresponding VLDL-TG FAs in the PNPLA3148MM as compared with the PNPLA3148II group are shown in Figure 3E. The number of double bonds was inversely related to the ratios of VLDL-TG FAs in homozygous carriers vs. noncarriers during the postprandial period (Figure 3E). These data show that there is PUFA deficiency in VLDL-TG in 148M carriers and support the hypothesis that PUFAs are retained in the liver in PNPLA3148MM as compared with the PNPLA3148II group.
In vivo evidence of retention of labeled 13C-18:2 PUFAs in the human liver in PNPLA3148MM compared with PNPLA3148II group.
Dietary FAs are transported from the intestinal lumen to the circulation in chylomicron-TGs, which then undergo hydrolysis and are taken up by the liver (12). VLDL-TGs are produced exclusively by the liver (12). Thus, to compare handling of PUFAs and SFAs in the liver, we calculated their ratio in VLDL-TGs and chylomicron-TGs. The PNPLA3148MM group had a significantly lower 13C-18:2/13C-16:0 ratio in VLDL-TGs compared with the PNPLA3148II group (P < 0.01) when related to the corresponding ratio in the chylomicron precursor pool (Figure 3F). There were no differences in enrichment of either of the FAs in the chylomicron precursor pools between the PNPLA3148MM and the PNPLA3148II groups (Supplemental Figure 1).
Increased storage of neutral lipids in homozygous PNPLA3-148M–KI human cells.
We used human epidermoid carcinoma A431 cells that are readily amenable for genetic manipulation and homozygous for the PNPLA3-148I allele. PNPLA3 is expressed in the skin (13) and in A431 cells at levels roughly comparable with HepG2 cells (Human Protein Atlas; https://www.proteinatlas.org/). We have used these cells in our earlier studies on lipid processing and storage (14, 15). Starting from the WT PNPLA3 cells, we engineered PNPLA3-KO cells, as well as cells expressing the 148M allele as homozygous (PNPLA3-148M–KI), and generated stable cell lines. Under basal culture conditions, the PNPLA3-148M–KI cells exhibited, as expected, elevated neutral lipid levels (TGs and cholesteryl esters, CEs) compared with WT cells (30.67 ± 4.7 [SEM] ng lipid/μg protein vs. 18.27 ± 2.4 [SEM], n = 6). We also engineered cells stably expressing GFP-tagged PNPLA3-148I or PNPLA3-148M on a PNPLA3-KO background. In these cells, both forms of PNPLA3 are expressed at similar levels (Supplemental Figure 2A) and associate with lipid droplets (Supplemental Figure 2B). These results are in keeping with our previous findings in hepatoma cells (16).
Increased storage of PUFAs but not SFAs in PNPLA3-148M–KI and PNPLA3-KO cells.
We then studied how the addition of saturated or increasingly unsaturated exogenous FAs affects lipid storage in A431 cells. Fluorescence microscopy revealed that all 3 cell lines generated lipid droplets when exposed to FAs for 24 hours. Expectedly, oleic and palmitic acid were potent inducers of lipid droplets, and PUFAs were less efficient (Figure 4). However, lipid droplet accumulation induced by PUFAs was strikingly increased in PNPLA3-148M–KI and PNPLA3-KO cells. This effect was not observed with saturated or monounsaturated FA. These results show that lack of PNPLA3 activity results in preferential sequestering of PUFAs to neutral lipids.
Figure 4. Characterization of lipid storage in homozygous PNPLA3-148I, PNPLA3-148M, and PNPLA3-KO A431 cells.

Cells were incubated for 24 hours in the presence of 100 μM palmitate, oleate, linoleate, or mixture of DHA and EPA (50 μM each); they were fixed and stained with lipid droplet dye LD540. Scale bar: 10 μm. The bar graph depicts percentage of cell area occupied by lipid droplets ± SEM. n = 62–76 cells. * P < 0.05 (1-way ANOVA with Dunnett’s correction).
Increased incorporation of PUFAs to TGs and decreased incorporation to PCs in PNPLA3-148M–KI and PNPLA3-KO cells.
Click chemistry is powerful for tracing rapid FA metabolism (15, 17). Using this approach, we compared how alkyne-palmitate and alkyne-linoleate are metabolized into major cellular lipid species during 15 minutes of labeling. The partitioning of alkyne-palmitate to phosphatidylcholines (PCs), diacylglycerols (DAGs) and TGs was similar between PNPLA3 genotypes (Figure 5A). At this time point, the majority of alkyne-linoleate was found in PCs in all cell lines (Figure 5B). However, in PNPLA3-148M–KI and PNPLA3-KO cells, the fraction of alkyne-linoleate in PC was significantly reduced and, in TGs, concomitantly increased as compared with WT (Figure 5B). Moreover, the percentage of FFA was higher in PNPLA3-148M–KI and PNPLA3-KO cells (29.6% ± 2.4% and 29.1% ± 2.5% respectively, n = 9) compared with WT cells (21.3% ± 3.7%, n = 9), implying defects in incorporation of PUFAs into PC.
Figure 5. Partitioning of alkyne-labeled fatty acid in homozygous PNPLA3-148I, PNPLA3-148M, and PNPLA3-KO A431 cells.

(A) Cells were incubated for 15 minutes with 100 μM alkyne-palmitate and then extracted, click-reacted, and analyzed by TLC. Bars represent percentage of incorporated alkyne-palmitate in indicated lipid species ± SEM. n = 9 from 3 individual experiments. (B) Cells were incubated for 15 minutes with 100 μM alkyne-linoleate and analyzed as in A. Bars represent percentage of incorporated alkyne-linoleate in indicated lipid species ± SEM. n = 9–17 from 4–6 individual experiments; * P < 0.05 (1-way ANOVA with Dunnett’s correction). (C) Cells were incubated for a 1-hour minimum with 100 μM alkyne-linoleate in the presence of cholesterol esterification inhibitor. After labeling, cells were either collected (0 minutes chase) or further incubated in lipoprotein-deficient medium supplemented with cholesterol esterification and DGAT inhibitors for 15, 30, or 60 minutes; they were then analyzed as in A. Bars represent percentage of incorporated alkyne-linoleate in indicated lipid species, normalized to PNPLA3-148I cells at 0 minutes chase ± SEM. n = 5–8 from 3–4 individual experiments; *P < 0.05 (1-way ANOVA with Dunnett’s correction).
Accumulation of DAGs in PNPLA3-148M–KI and PNPLA3-KO cells in lipolytic conditions.
Due to the observed differences between PNPLA3 genotypes in PUFA metabolism and the reported PNPLA3 lipase activity (18, 19), we next measured release of linoleic acid from TG stores. To this end, we incubated WT A431 cells with alkyne-linoleate for 1 hour in the presence of a cholesterol esterification inhibitor (PKF-035) to ensure maximal neutral lipid deposition as TGs. The cells were then either immediately harvested (0 minutes chase) or incubated for 15, 30, or 60 minutes (chase) in the presence of diacylglycerol O-acyltransferase 1 (DGAT-1) and -2 inhibitors that prevent TG synthesis as well as a cholesterol esterification inhibitor. As the inhibitors prevent FA reesterification to neutral lipids, under these conditions, cells start to hydrolyze the generated TGs. As expected, alkyne-linoleate containing TGs and DAGs decreased during chase, and this was paralleled by increased partitioning of alkyne-linoleate into phospholipids, with marginal levels of FFAs (Supplemental Figure 3A).
When the effect of PNPLA3 genotype on TG hydrolysis was investigated, we found that both PNPLA3-148M–KI and PNPLA3-KO cells had elevated TG levels at the end of alkyne-linoleate labeling as compared with PNPLA3-148I (Figure 5C). The increase in TGs in PNPLA3-KO and PNPLA3-148M–KI cells is consistent with the idea that the human gene variant increases TGs because it acts as a loss-of-function variant. During the 1-hour chase, TGs started to diminish but stayed elevated in both genotypes relative to PNPLA3-148I cells (Figure 5C). Of note, although TGs stayed elevated in PNPLA3-KO and PNPLA3-148M–KI cells compared with WT PNPLA3-148I cells, we did not observe major differences in the hydrolysis rate of TGs under lipolytic conditions between these cells. This does not favor the idea that PNPLA3 functions mainly in TG hydrolysis.
Strikingly, under lipolytic conditions, we observed a pronounced increase in DAGs in both PNPLA3-148M–KI and PNPLA3-KO cells as compared with WT (Figure 5C). This effect was not observed during the 15-minute labeling (Figure 5B), suggesting that the increase was not due to increased generation of DAGs but rather to their impaired hydrolysis. In PNPLA3-148M–KI and PNPLA3-KO cells, there was also a tendency for decreased incorporation of alkyne-linoleate into PCs, but the difference was not significant (Supplemental Figure 3B). This is not surprising because, during the course of hours, linoleic acid can end up in the PC pool via several pathways.
Polyunsaturated TGs are enriched while PCs are deficient in the human liver in homozygous carriers of the PNPLA3-I148M variant compared with noncarriers.
Since polyunsaturated TGs accumulated in the PNPLA3-148M-KI cells at the expense of PCs, we next asked whether the enrichment of polyunsaturated TGs would be associated with a decrease in PCs in the human liver in homozygous carriers of the PNPLA3-I148M variant as compared with noncarriers. To this end, we reanalyzed previously described data of human liver lipidome (5) in homozygous I148M variant carriers and noncarriers. As previously reported, the livers of the I148M variant carriers were enriched in polyunsaturated TGs, such as TG(56:6) and TG(58:6) (P < 0.05) (Figure 6). Consistent with the in vitro data, the livers of homozygous I148M variant carriers were deficient in multiple PCs such as PC(36:6) and PC(32:2), as compared with noncarriers (P < 0.05) (Figure 6).
Figure 6. Polyunsaturated TGs are enriched, while PCs are deficient in the human liver in homozygous carriers (PNPLA3148MM) as compared with noncarriers (PNPLA3148II) of the PNPLA3-I148M variant.

The x-axis denotes log2 fold-change in hepatic concentration of a given lipid in PNPLA3148MM (n = 7) as compared with PNPLA3148II (n = 64) group. The y-axis denotes negative logarithm of P value of 2-tailed Student’s t test comparing hepatic concentrations of a given lipid in PNPLA3148MM as compared with PNPLA3148II group. Red squares denote TGs, and blue circles denote PCs. Horizontal dashed line represents –log10(0.05). Each symbol represents distinct hepatic lipid species. Lipid species that were significantly decreased in the PNPLA3148MM as compared with the PNPLA3148II group are listed on the left side of the figure, while those that were increased are listed on the right side in the order of significance. Data in this figure are from separate liver biopsy cohort described earlier (5).
Discussion
Liver TGs in human liver biopsies are markedly polyunsaturated in PNPLA3148M gene variant carriers as compared with noncarriers (5). The present data show, using a combination of GC, ultra-high performance liquid chromatography-tandem mass spectrometry (UHPLC-MS), and stable isotope techniques, that this reflects increased retention of PUFAs in the liver and results in PUFA deficiency in VLDL-TG secreted by the liver. The evidence from the in vivo studies can be summarized as follows. In the PNPLA3148MM as compared with the PNPLA3148II group both in the fasting state and postprandially, (a) the number of double bonds in VLDL-TGs measured by UHPLC-MS (Figure 2) and (b) the proportion of 18:2 FA (GC) in VLDL-TGs (Figure 3C) were lower, and (c) PUFAs in VLDL-TGs (GC) were deficient (Figure 3E). When the subjects ingested equal amounts of a saturated (16:0) and a polyunsaturated (18:2) FA in a meal labeled with respective stable isotope tracers, the ratio of labeled 18:2 vs. 16:0 was lower in the PNPLA3148MM than the PNPLA3148II group (Figure 3F). There are no previous studies comparing the composition of VLDL-TG either in the fasting state or postprandially between PNPLA3148MM and PNPLA3148II groups or kinetic studies addressing function of the 148M variant in humans.
To explore the mechanism explaining retention of PUFAs in the human liver, we engineered PNPLA3-KO cells as well as cells expressing the 148M allele as homozygous (PNPLA3-148M–KI) and generated stable cell lines. This KI model of the PNPLA3 variant is the first to our knowledge to examine the impact of endogenous levels of the human PNPLA3 variant on lipid composition and metabolism. In previous studies using human cell lines (16, 20, 21), the gene variant was overexpressed, potentially causing artefacts.
Lipid droplet accumulation induced by PUFAs, but not by saturated or monounsaturated FA, was strikingly increased in PNPLA3-148M–KI and PNPLA3-KO cells. These results suggest that lack of PNPLA3 activity results in preferential sequestering of PUFAs to neutral lipids and that the 148M variant resembles loss of PNPLA3 function in human cells.
Studies using click chemistry allow tracing of rapid changes in FA metabolism (17). In PNPLA3-148M–KI and PNPLA3-KO cells, the fraction of alkyne-linoleate in PC was significantly reduced and, in TGs, increased as compared with WT cells. In human liver samples, concentrations of polyunsaturated PCs were significantly decreased, while TGs were increased in the livers of homozygous carriers of the PNPLA3-I148M variant as compared with noncarriers (Figure 6). These data closely resemble those of KI mice expressing a catalytically inactive PNPLA3 variant (PNPLA3-S47A–KI mice) but oppose those characterizing PNPLA3-148M–KI mice (7). In these mice, very long–chain PUFAs are enriched in TGs and depleted in phospholipids. Therefore, the human PNPLA3-148M variant resembles human and mouse PNPLA3 loss of function.
In PNPLA3-148M and PNPLA3-KO cells, PUFAs containing DAGs accumulated under lipolytic conditions compared with PNPLA3-148I cells. This was paralleled by a tendency for decreased incorporation of PUFAs into PCs in PNPLA3-148M and PNPLA3-KO cells. These data suggest that PNPLA3 promotes transfer of PUFAs from DAGs to generate polyunsaturated PCs, thus refining the model proposed by Mitsche et al. (7). It is conceivable that PNPLA3 acts as a PUFA-specific transacylase, catalyzing the transfer of PUFAs in DAGs generated from TG hydrolysis (such as 2,3-DAGs), to yield DAGs compatible with PC generation (1,2-DAGs). Alternatively, PNPLA3 may act as a PUFA-specific lipase hydrolyzing PUFAs from DAGs, to be used for the synthesis of PUFA-containing PCs. Importantly, the PNPLA3-148M variant had similar effects on lipid metabolism as PNPLA3 deletion. While we cannot exclude the possibility that I148M substitution results in altered substrate specificity of the enzyme, the data strongly suggest that the I148M substitution results in loss of PNPLA3 activity in human cells. Either way, I148M remodels liver TGs in the human liver in a polyunsaturated direction (22). This lipid composition opposes the saturated TG composition characterizing liver in NAFLD associated with insulin resistance (metabolic NAFLD) (5, 23). The retention of polyunsaturated TGs in the liver may explain why carriers of the PNPLA3 gene variant are protected against cardiovascular disease, despite having an increase in liver fat content (3, 4).
Methods
Subjects.
A total of 26 subjects who were homozygous for either the C or the G allele at rs738409 and fulfilled the inclusion and exclusion criteria (vide infra) were recruited among nondiabetic individuals who had previously been genotyped for PNPLA3 at rs738409 in our laboratory (24) or in the population-based National FINRISK 2007 (25).
Inclusion criteria included: (a) age 18–65 years; (b) PNPLA3 genotype CC or GG at rs738409; and (c) alcohol consumption less than 20 g per day for women and less than 30 g per day for men. Exclusion criteria included: (a) known acute or chronic disease other than obesity, NAFLD, or hypertension on the basis of medical history, physical examination, and standard laboratory tests (complete blood count, serum creatinine, or electrolyte concentrations); (b) clinical or biochemical evidence of liver disease other than NAFLD, or clinical signs or symptoms of inborn errors of metabolism; (c) history or current use of toxins or drugs associated with liver steatosis; (d) history or current use of lipid lowering medications; and (e) pregnancy or lactation.
Clinical study design.
The study consisted of (a) a screening visit, (b) a metabolic study visit, and (c) a visit to the imaging center for quantification of IHTG using proton magnetic resonance spectroscopy (1H-MRS).
The screening visit was performed after an overnight fast. A history and physical examination were performed to review the inclusion and exclusion criteria. Blood samples were obtained for measurement of circulating blood count, glucose, Hemoglobin A1c (HbA1c), serum insulin, thyroid-stimulating hormone, hepatitis C virus antibody, plasma glucose, low-density lipoprotein (LDL) cholesterol and high-density lipoprotein (HDL) cholesterol, TG, albumin, thromboplastin time, C-reactive protein, sodium, potassium, aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), and γ glutamyltransferase (GGT) concentrations as described (24).
After the screening visit, the subjects wore portable accelerometers (GT3X, Actigraph) for 7 days to estimate their physical activity. The subjects were asked to collect a 3-day dietary record to determine their baseline dietary composition. The dietary records were analyzed using the AivoDiet software (version 2.0.2.3; Aivo Finland).
Metabolic study visit.
For 3 days prior to the metabolic study day, the subjects were asked to avoid foods naturally enriched in 13C (such as sea food, corn, and sugar), alcohol, and strenuous exercise. In the previous evening before the metabolic study, the subjects consumed a standardized meal (a vegetarian sandwich, 330 kcal, 21 g fat, 25 g carbohydrate, and 9 g protein, produced by Ravioli). The subjects came to the clinical research center after an overnight fast. Body composition (InBody720; BioSpace), weight, height, and waist circumference were measured as described (5). A cannula was inserted into an antecubital vein, and baseline blood samples were taken to measure plasma albumin, AST, ALT, ALP, GGT, bilirubin, C-peptide, glucose, LDL and HDL cholesterol, and TG concentrations, as well as serum insulin concentrations. Participants were then fed a mixed test meal containing 44 g carbohydrate, 10 g protein, and 37 g fat, with 100 mg of [U-13C]palmitic acid and 100 mg of [U-13C]linoleic acid (both from Cambridge Isotopes) added to trace the fate of the dietary FAs (26). The meal consisted of 40 g Kellogg’s Rice Krispies with 200 g skimmed milk and a chocolate milkshake containing 20 g butter, 20 g rapeseed oil, 3 tbs sweetener, and 2.4 g cocoa powder.
Blood samples were taken at 0, 60, 120, 180, 240, 300, 360, and 420 minutes after the consumption of the test meal for measurement of plasma glucose, TG, FFAs, and serum insulin concentrations, and at 0, 120, 180, 240, 300, 360, and 420 minutes for isolation of VLDL and chylomicrons by ultracentrifugation, as described (27). The FA composition and concentrations of 13C-18:2 and 13C-16:0 FAs were determined in total plasma, plasma FFA, and the VLDL and chylomicron fractions (vide infra). Lipidomics analyses (vide infra) were performed using the VLDL fraction using UHPLC-MS at the time points of 0, 120 ,and 420 minutes.
Imaging visit.
Before the metabolic study visit, the subjects underwent 1H-MRS to quantify IHTG content (vide infra).
Isolation of VLDL and chylomicron fractions.
Separations of chylomicron of Svedberg flotation rate (Sf) >400 and VLDL-rich fraction (Sf20-400) were made by sequential flotation using density gradient ultracentrifugation (Beckman L-80, Beckman Coulter) as previously described (23).
FA isotopic enrichment.
To determine the specific FA composition and isotopic enrichment, total lipids were extracted from plasma, and VLDL and chylomicrons, and FA methyl esters (FAMEs) were prepared (28, 29). The FA compositions (μmol/100 μmol total FA) of these fractions were determined by gas chromatography (GC), and the FA concentrations were calculated (27). [U-13C]palmitate and [U-13C]linoleate enrichments were measured in plasma FFA, TG, Sf >400 (chylomicron-TG), Sf20-400–TG, and VLDL-TG FAMEs derivatives using a Delta Plus XP GC-combustion isotope ratio mass spectrometer (Thermo Electron) (30). The tracer-to-tracee ratio (TTR) of a baseline measurement before administration of [U-13C]palmitate was subtracted from the TTR of each sample to account for natural abundance. The TTRs for [U-13C]palmitate and [U-13C]linoleate were multiplied by the corresponding unlabeled concentrations to give plasma and lipoprotein tracer concentrations (26).
Lipidomic analysis of VLDL-TG using UHPLC-MS.
The UHPLC–quadrupole time of flight mass spectrometer (QTOFMS) analyses were done in a similar manner as described earlier with some modifications (31). UHPLC-QTOFMS system was from Agilent Technologies, combining 1290 Infinity system and 6545 QTOFMS, interfaced with a dual jet stream electrospray (dual ESI) ion source. MassHunter B.06.01 software (Agilent Technologies) was used for all data acquisition and MZmine 2 was used for data processing (32). ACQUITY UPLC BEH C18 column (2.1 mm × 100 mm, particle size 1.7 μm) by Waters Corporation was used for the UHPLC separation. The lipidomics methods are described in detail in Supplemental Methods. The analyses covered most of the main molecular lipids, including ceramides, dihydroceramides, TGs, DAGs, sphingomyelins, hexosylceramides, PC, phosphatidylethanolamines (PE), phosphatidylserines (PS), and lysoPC. The lipid identification was based on an internal library, which had been constructed based on accurate mass measurements in combination with tandem mass measurements. For specific lipids, the composition of FA chains had been determined with separate measurements, and for those, the FA composition was specified — e.g., TG(14:0/16:0/18:0).
Measurement of IHTG content by 1H-MRS.
IHTG content was measured by 1H-MRS performed on a clinical 1.5T Siemens Avantofit imager. MRS data was analyzed using jMRUI v5.2 software with AMARES algorithm as described (33).
Lipidomic analysis of the human liver.
We performed reanalysis of TGs and PCs in homozygous carriers (n = 7) and noncarriers (n = 64) of the PNPLA3-I148M variant in previously described data of human liver lipidome of a separate cohort (5). Briefly, human liver biopsies were obtained during laparoscopic surgery and immediately frozen in liquid nitrogen. Subsequently, molecular lipids were measured using a Q-TOF Premier (Waters Corporation) QTOFMS combined with an Acquity Ultra Performance LC (Waters Corporation) as previously described (5).
Other analyses from human samples.
The fasting plasma glucose was measured using the hexokinase method on an autoanalyzer (Roche Diagnostics Hitachi 917, Hitachi Ltd.). Serum insulin concentration was determined by time-resolved fluoroimmunoassay using an Insulin Kit (AUTOdelfia, Wallac). HbA1c was measured with immunoturbidometric method (Abbott Laboratories) and plasma ALT, AST, and GGT concentrations were measured using photometric International Federation of Clinical Chemistry methods (Abbott Laboratories). Plasma ALT, AST, and GGT activities were determined as recommended by the European Committee for Clinical Laboratory Standards, and plasma TG, total, LDL and HDL cholesterol concentrations were measured using enzymatic kits and an autoanalyzer (Roche Diagnostics Hitachi 917, Hitachi Ltd.). Plasma albumin was measured using a photometric method on an autoanalyzer (Modular Analytics EVO; Hitachi High-Technologies Corporation).
Cell culture and generation of CRISPR cell lines.
Human epidermoid carcinoma A431 cells (ATCC) were cultured in DMEM (Lonza) with 10% FBS (Thermo Fisher Scientific) containing L-glutamine (2 mM) (MilliporeSigma) and streptomycin/penicillin (100 U/ml each) (Lonza). PNPLA3-KO and I148M-KI cell lines were constructed using CRISPR/Cas9-mediated genome editing (34). Briefly, a homology-directed repair template was generated by PCR (primer sequences 5′ - ATACACGCGTCCAGTCCAAGGAACCTGTCC - 3′ and 5′ - ATACGTCGACGCAGTAAGTTTTGCTGCCCG - 3′), by using Huh7 genomic DNA as a template, and ligated into pGL3-Basic vector. The construct was transfected into A431 cells, together with a vector encoding Cas9, sgRNA targets flanking the mutation site (sense: 5′ - CACCGTAGAAGGGGATGAAGC - 3′; antisense: 5′ - AAACGCTTCATCCCCTTCTAC - 3′), and a puromycin selection marker. Clones were isolated after transient selection with puromycin by limiting dilution. Homozygous mutations were validated by genomic PCR. With this protocol, we obtained a homozygous PNPLA3-I148M–KI cell line, as well as a homozygous PNPLA3-KO cell line with a 2-bp deletion after 146C, resulting in a frameshift and premature termination of translation (PNPLA3-KO).
Lipid analyses from cells.
For the determination of unlabeled lipids, cells were extracted and lipids analyzed by high-performance thin layer chromatography as in ref. 35. Click labeling and analyses were performed essentially as described (15, 17). Briefly, for alkyne-FA labeling, cells grown in 12-well plates were incubated at +37°C, 5% CO2 for 15 minutes in loading medium containing serum-free DMEM supplemented with 1% FA free BSA, and 100 μM alkyne-linoleate (Cayman Chemical) or alkyne-palmitate (Avanti Polar Lipids). In lipolysis experiments, a cholesterol esterification inhibitor (Sandoz PKF 58-035, 2 μg/ml) was added, and the labeling was performed for 1 hour. For chase, cells were incubated with serum-free DMEM containing 5% lipoprotein-deficient serum (LPDS), prepared as described in ref. 36; PKF 58-035 (2 μg/ml); and DGAT-1 and DGAT-2 inhibitors (5 μM each, MilliporeSigma, PZ0207 and PZ0233) for 15, 30, or 60 minutes. Lipid extraction, click reaction, and analysis of alkyne FA incorporation into selected lipids (TG; 1,2/2,3-DAG; PE; and PC) by thin-layer chromatography was performed as in ref. 17.
Microscopy.
For lipid droplet analyses, cells were incubated for 24 hours with BSA-complexed 100 μM palmitate, oleate, linoleate, or a mixture of docosahexaenoic acid (DHA, 22:6[n-3]) and eicosapentaenoic acid (EPA, 20:5[n-3]) (50 μM each) (MilliporeSigma) for 24 hours. The cells were fixed with 4% PFA, stained with lipid droplet stain LD540 (37) (Princeton BioMolecular Research), and imaged with Nikon Eclipse Ti-E N-STORM epifluorescence microscope. Thresholded lipid droplet area as percentag of total cell area was analyzed from micrographs with ImageJ FIJI (NIH).
Statistics.
Continuous variables were tested for normality using the Kolmogorov-Smirnov test. The independent 2-tailed Student’s t and Mann-Whitney U tests were used to compare normally and nonnormally distributed data, respectively. Normally distributed data were reported in means ± SEM, while nonnormally distributed were reported in medians and interquartile ranges. Pearson χ2 test was used to evaluate if the distributions of categorical variables differ between the groups. The FA composition of VLDL-TG data was analyzed by a 2-way ANOVA. AUC for 13C-18:2/13C-16:0 FAs in chylomicron-TG and VLDL-TG were calculated using the trapezoid method. The former AUC was analyzed with respect to the latter AUC using linear regression. The UHPLC-MS data were analyzed by relating the ratio of a given VLDL-TG in the PNPLA3148MM vs. the PNPLA3148II group to the number of double bonds in that VLDL-TG using linear regression. Statistical analyses were performed by using IBM SPSS Statistics 23.0.0.0 version and GraphPad Prism 7.0d for Mac OS X (GraphPad Software). A P value of less than 0.05 indicated statistical significance. For in vitro experiments, statistical significance was determined by 1-way ANOVA with Dunnett’s correction in GraphPad Prism 7.0.
Study approval.
The study protocols were approved by the ethics committee of the Hospital District of Helsinki and Uusimaa, Helsinki, Finland. The studies were conducted in accordance with the Declaration of Helsinki. Each participant provided written informed consent after being explained the nature and potential risks of the study.
Author contributions
PKL, LH, E. Ikonen, and HYJ were responsible for study concept and design. MHV also participated in study design. MHV, E. Ikonen, and HYJ supervised the study. PKL, E. Isokuortti, and SLB acquired clinical data; AN, MHV, and CT acquired in vitro data; AH and NL acquired and analyzed magnetic resonance data; LH acquired GC and MS data; and TH and MO acquired lipidomics data. MP recruited subjects from the National FINRISK cohort. MOM was responsible for genotyping. PKL, AN, MHV, LH, E. Ikonen, and HYJ analyzed and interpreted data. PKL and YZ carried out statistical analyses. PKL, MHV, E. Ikonen, and HYJ drafted the manuscript. All authors participated in critical revision of the manuscript for important intellectual content.
Supplementary Material
Acknowledgments
We thank Anne Salo, Aila Karioja-Kallio, Päivi Ihamuotila, and Pentti Pölönen for their excellent technical assistance; Siiri Luukkonen for graphical assistance; Shiqian Li for his generous help in designing A431 CRISPR cell lines; and the volunteers for their help. This study was supported by research grants from the Academy of Finland (HY grant 309263 and E. Ikonen grants 282192, 307415, 312491), EU H2020 project Elucidating Pathways of Steatohepatitis (HY EPoS grant 634413), and the H2020-JTI-IMI2 EU project 777377-2 Liver Investigation: Testing Marker Utility in Steatohepatitis (LITMUS) (HY), as well as from EVO (HY), Paulo (PKL), Sigrid Juselius (HY, PKL, E. Ikonen), Finnish Medical (PKL), Alfred Kordelin (PKL), and Liv och Hälsa (MH-V) foundations.
Version 1. 08/22/2019
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: JCI Insight. 2019;4(16):e127902.https://doi.org/10.1172/jci.insight.127902.
Contributor Information
Panu K. Luukkonen, Email: panu.luukkonen@fimnet.fi.
Auli Nick, Email: auli.nick@helsinki.fi.
Maarit Hölttä-Vuori, Email: maarit.holtta-vuori@helsinki.fi.
Christoph Thiele, Email: cthiele@uni-bonn.de.
Elina Isokuortti, Email: elina.isokuortti@helsinki.fi.
Susanna Lallukka-Brück, Email: susanna.lallukka-bruck@helsinki.fi.
You Zhou, Email: you.zhou@helsinki.fi.
Antti Hakkarainen, Email: antti.hakkarainen@gmail.com.
Nina Lundbom, Email: nina.lundbom@hus.fi.
Markku Peltonen, Email: markku.peltonen@thl.fi.
Marju Orho-Melander, Email: Marju.Orho-Melander@med.lu.se.
Tuulia Hyötyläinen, Email: tuulia.hyotylainen@oru.se.
Leanne Hodson, Email: leanne.hodson@ocdem.ox.ac.uk.
Elina Ikonen, Email: elina.ikonen@helsinki.fi.
Hannele Yki-Järvinen, Email: hannele.yki-jarvinen@helsinki.fi.
References
- 1.Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology. 2011;53(6):1883–1894. doi: 10.1002/hep.24283. [DOI] [PubMed] [Google Scholar]
- 2.Trépo E, Romeo S, Zucman-Rossi J, Nahon P. PNPLA3 gene in liver diseases. J Hepatol. 2016;65(2):399–412. doi: 10.1016/j.jhep.2016.03.011. [DOI] [PubMed] [Google Scholar]
- 3.Liu DJ, et al. Exome-wide association study of plasma lipids in >300,000 individuals. Nat Genet. 2017;49(12):1758–1766. doi: 10.1038/ng.3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simons N, Isaacs A, Koek GH, Kuč S, Schaper NC, Brouwers MCGJ. PNPLA3, TM6SF2, and MBOAT7 Genotypes and Coronary Artery Disease. Gastroenterology. 2017;152(4):912–913. doi: 10.1053/j.gastro.2016.12.020. [DOI] [PubMed] [Google Scholar]
- 5.Luukkonen PK, et al. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J Hepatol. 2016;64(5):1167–1175. doi: 10.1016/j.jhep.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 6.Basantani MK, et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J Lipid Res. 2011;52(2):318–329. doi: 10.1194/jlr.M011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitsche MA, Hobbs HH, Cohen JC. Patatin-like phospholipase domain-containing protein 3 promotes transfer of essential fatty acids from triglycerides to phospholipids in hepatic lipid droplets. J Biol Chem. 2018;293(24):9232. doi: 10.1074/jbc.AAC118.004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen W, Chang B, Li L, Chan L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology. 2010;52(3):1134–1142. doi: 10.1002/hep.23812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smagris E, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology. 2015;61(1):108–118. doi: 10.1002/hep.27242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green CJ, et al. Characterization of lipid metabolism in a novel immortalized human hepatocyte cell line. Am J Physiol Endocrinol Metab. 2015;309(6):E511–E522. doi: 10.1152/ajpendo.00594.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Min HK, Sookoian S, Pirola CJ, Cheng J, Mirshahi F, Sanyal AJ. Metabolic profiling reveals that PNPLA3 induces widespread effects on metabolism beyond triacylglycerol remodeling in Huh-7 hepatoma cells. Am J Physiol Gastrointest Liver Physiol. 2014;307(1):G66–G76. doi: 10.1152/ajpgi.00335.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hodson L. Hepatic fatty acid synthesis and partitioning: the effect of metabolic and nutritional state. Proc Nutr Soc. 2019;78(1):126–134. doi: 10.1017/S0029665118002653. [DOI] [PubMed] [Google Scholar]
- 13.Huang Y, et al. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc Natl Acad Sci USA. 2010;107(17):7892–7897. doi: 10.1073/pnas.1003585107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanerva K, et al. LDL cholesterol recycles to the plasma membrane via a Rab8a-Myosin5b-actin-dependent membrane transport route. Dev Cell. 2013;27(3):249–262. doi: 10.1016/j.devcel.2013.09.016. [DOI] [PubMed] [Google Scholar]
- 15.Salo VT, et al. Seipin regulates ER-lipid droplet contacts and cargo delivery. EMBO J. 2016;35(24):2699–2716. doi: 10.15252/embj.201695170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruhanen H, et al. PNPLA3 mediates hepatocyte triacylglycerol remodeling. J Lipid Res. 2014;55(4):739–746. doi: 10.1194/jlr.M046607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thiele C, et al. Tracing fatty acid metabolism by click chemistry. ACS Chem Biol. 2012;7(12):2004–2011. doi: 10.1021/cb300414v. [DOI] [PubMed] [Google Scholar]
- 18.Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J Biol Chem. 2004;279(47):48968–48975. doi: 10.1074/jbc.M407841200. [DOI] [PubMed] [Google Scholar]
- 19.Lake AC, et al. Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. J Lipid Res. 2005;46(11):2477–2487. doi: 10.1194/jlr.M500290-JLR200. [DOI] [PubMed] [Google Scholar]
- 20.He S, et al. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem. 2010;285(9):6706–6715. doi: 10.1074/jbc.M109.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perttilä J, et al. PNPLA3 is regulated by glucose in human hepatocytes, and its I148M mutant slows down triglyceride hydrolysis. Am J Physiol Endocrinol Metab. 2012;302(9):E1063–E1069. doi: 10.1152/ajpendo.00125.2011. [DOI] [PubMed] [Google Scholar]
- 22.Sacks FM, et al. Dietary Fats and Cardiovascular Disease: A Presidential Advisory From the American Heart Association. Circulation. 2017;136(3):e1–e23. doi: 10.1161/CIR.0000000000000510. [DOI] [PubMed] [Google Scholar]
- 23.Luukkonen PK, et al. Saturated Fat Is More Metabolically Harmful for the Human Liver Than Unsaturated Fat or Simple Sugars. Diabetes Care. 2018;41(8):1732–1739. doi: 10.2337/dc18-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sevastianova K, et al. Genetic variation in PNPLA3 (adiponutrin) confers sensitivity to weight loss-induced decrease in liver fat in humans. Am J Clin Nutr. 2011;94(1):104–111. doi: 10.3945/ajcn.111.012369. [DOI] [PubMed] [Google Scholar]
- 25. Borodulin K, et al. Cohort Profile: The National FINRISK Study. Int J Epidemiol [published online ahead of print November 20, 2017]. doi: 10.1093/ije/dyx239. [Google Scholar]
- 26.Hodson L, et al. Greater dietary fat oxidation in obese compared with lean men: an adaptive mechanism to prevent liver fat accumulation? Am J Physiol Endocrinol Metab. 2010;299(4):E584–E592. doi: 10.1152/ajpendo.00272.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hodson L, et al. The contribution of splanchnic fat to VLDL triglyceride is greater in insulin-resistant than insulin-sensitive men and women: studies in the postprandial state. Diabetes. 2007;56(10):2433–2441. doi: 10.2337/db07-0654. [DOI] [PubMed] [Google Scholar]
- 28.Heath RB, Karpe F, Milne RW, Burdge GC, Wootton SA, Frayn KN. Selective partitioning of dietary fatty acids into the VLDL TG pool in the early postprandial period. J Lipid Res. 2003;44(11):2065–2072. doi: 10.1194/jlr.M300167-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Pramfalk C, et al. Sex-Specific Differences in Hepatic Fat Oxidation and Synthesis May Explain the Higher Propensity for NAFLD in Men. J Clin Endocrinol Metab. 2015;100(12):4425–4433. doi: 10.1210/jc.2015-2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chong MF, Fielding BA, Frayn KN. Mechanisms for the acute effect of fructose on postprandial lipemia. Am J Clin Nutr. 2007;85(6):1511–1520. doi: 10.1093/ajcn/85.6.1511. [DOI] [PubMed] [Google Scholar]
- 31.O’Gorman A, et al. Identification of a plasma signature of psychotic disorder in children and adolescents from the Avon Longitudinal Study of Parents and Children (ALSPAC) cohort. Transl Psychiatry. 2017;7(9):e1240. doi: 10.1038/tp.2017.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pluskal T, Castillo S, Villar-Briones A, Oresic M. MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics. 2010;11:395. doi: 10.1186/1471-2105-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sevastianova K, et al. Effect of short-term carbohydrate overfeeding and long-term weight loss on liver fat in overweight humans. Am J Clin Nutr. 2012;96(4):727–734. doi: 10.3945/ajcn.112.038695. [DOI] [PubMed] [Google Scholar]
- 34.Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32(4):347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hölttä-Vuori M, Vainio S, Kauppi M, Van Eck M, Jokitalo E, Ikonen E. Endosomal actin remodeling by coronin-1A controls lipoprotein uptake and degradation in macrophages. Circ Res. 2012;110(3):450–455. doi: 10.1161/CIRCRESAHA.111.256842. [DOI] [PubMed] [Google Scholar]
- 36.Goldstein JL, Basu SK, Brown M. Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 1983;98:241–260. doi: 10.1016/0076-6879(83)98152-1. [DOI] [PubMed] [Google Scholar]
- 37.Spandl J, White DJ, Peychl J, Thiele C. Live cell multicolor imaging of lipid droplets with a new dye, LD540. Traffic. 2009;10(11):1579–1584. doi: 10.1111/j.1600-0854.2009.00980.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.