

Abstract
Alternative splicing, the process of removing introns and joining exons of pre-mRNA, is critical for growth, development, tissue homeostasis, and species diversity. Dysregulation of alternative splicing can initiate and drive disease. Aberrant alternative splicing has been shown to promote the “hallmarks of cancer” in both hematological and solid cancers. Of interest, recent work has focused on the role of alternative splicing in prostate cancer and prostate cancer health disparities. We will provide a review of prostate cancer health disparities involving the African American population, alternative RNA splicing, and alternative splicing in prostate cancer. Lastly, we will summarize our work on differential alternative splicing in prostate cancer disparities and its implications for disparate health outcomes and therapeutic targets.
Keywords: alternative splicing, prostate cancer, cancer disparities, drug resistance
Introduction
Alternative splicing of pre-mRNA plays a major role in both normal development and cancer progression. By hijacking and leveraging the complex and tightly regulated process of alternative splicing, cancer cells are able to acquire many of the “hallmarks of cancer” (David and Manley 2010). Prostate cancer (PCa), the most diagnosed cancer in men in the United States, is no exception. There have been several comprehensive review articles detailing the important role of alternative splicing in PCa progression and aggressiveness (Antonopoulou and Ladomery 2018; David and Manley 2010; Hagen and Ladomery 2012; Lapuk et al. 2014; Munkley et al. 2017; Rajan et al. 2009; Sette 2013). These reviews, however, do not address the critical topic of alternative splicing in PCa health disparities. PCa exhibits dramatic race/ethnic disparities as African American (AA) men have significantly higher risk, morbidity, and mortality compared to European American (EA) men. In this review, we will summarize some of the major molecular mechanisms and alternative splice events in PCa, as well as introduce our recent study elucidating the important role of differential alternative splicing in mediating PCa disparities.
Prostate cancer health disparities
PCa is the most diagnosed cancer in men in the United States and accounts for over one-fifth of all newly diagnosed cancers in men (Siegel, Miller, and Jemal 2018). More than 164,000 new cases are diagnosed each year and PCa is the second leading cause of male-cancer related deaths annually. PCa also has the highest heritability of any cancer at 10% (Lynch et al. 2016). In addition to family history, well established risk factors of PCa include Lynch syndrome, age, and race/ethnicity (Brawley 2012; Powell 2007). Despite increased screening and overall decreasing mortality rates of PCa, AA men have significantly higher rates of PCa incidence, high-risk cancer, and mortality (Cooperberg 2013). AA men are 1.7 times more likely to be diagnosed with PCa and have a 2.4 times greater mortality rate compared to EA men (DeSantis et al. 2016). This mortality ratio is the largest of any other malignancy in the United States (Rebbeck 2017). Additionally, PCa appears to develop at an earlier age in AA men who present with significantly higher prostate specific antigen (PSA) plasma levels, more clinically advanced disease, and develop higher grade metastatic disease at a three-four fold greater rate (Chornokur, Dalton, Borysova, and Kumar, 2011; Martin, Starks, and Ambs, 2013; Oltean et al. 2006; Powell and Bollig-Fischer 2013). This health disparity has been attributed to epidemiological differences in socioeconomic status, health seeking behavior, access to healthcare, and treatment plans (Chornokur et al. 2011; Martin, Starks, and Ambs 2013). Even after adjusting for clinical and epidemiological factors, however, AA men still have significantly higher occurrence and mortality rates (Evans et al. 2008; Robbins, Whittemore, and Thom 2000; Tyson and Castle 2014). This disease disparity suggests that genetic ancestry plays an important role in PCa incidence, progression, and aggressiveness.
Molecular differences in African American prostate cancer
Multiple studies have shown genetic and biological differences in prostate tumors in AA and EA patient populations. TMPRSS2-ERG gene fusions and PTEN deletions were once thought to be characteristic of all prostate tumors. However, recent reports have shown these genetic alterations occur at a much lower frequency in AA PCa. Only 20–30% of AA PCa tumors contain TMPRSS2-ERG gene fusions compared to 40–50% in EA patients (Magi-Galluzzi et al. 2011) and loss of PTEN was observed in 34% of EA men and only 18% of AAs (Tosoian et al. 2017).
Genome-wide association studies (GWAS) have identified multiple loci that confer a greater risk for PCa in AA men compared to EA men. The rs1447295 variant at the 8q24 locus has been associated with earlier diagnosis and increased risk in AA patients (Schumacher et al. 2007). Six other variants (rs16901979, rs7000448, rs6983267, rs111906932, rs114798100, and rs111906923) have also been linked to increased PCa risk in AA men (Haiman et al. 2007; Han et al. 2016). African ancestry-specific PCa risk alleles have been identified at chromosomes 13q34 and 22q12 (Conti et al. 2017). Additionally, a risk variant at the 17q21 locus has been found more frequently in men of African descent compared to other populations (Haiman et al. 2011). Many of these alleles reside within long coding RNA sequences.
Single nucleotide polymorphisms (SNPs) in genes that regulate androgen and testosterone metabolism have also been linked to PCa disparity in AAs. Polymorphisms in the cytochrome p450 enzyme CYP17 increases the risk of PCa in AA men by 60% (Taioli et al. 2013). A homozygous ‘CC’ genotype in the 5’ promoter region (rs743572) in AA men is clinically associated with advanced PCa disease (Kittles et al. 2001).
In terms of the cancer transcriptome, AA PCa has been shown to exhibit increased expression of genes that promote growth (e.g. EGFR and AKT1) and metastasis (e.g. CXCR4 and BMP2) compared to EA PCa (Powell et al. 2013; Shuch et al. 2004; Wallace et al. 2008). For the IL-6 gene, a race-specific and anti-correlated expression pattern is observed during PCa progression. Namely, EA PCa has increased expression of IL-6 compared to EA normal prostate, while IL-6 is downregulated in AA PCa compared to AA normal prostate (Teslow et al. 2018). Exogenous treatment with IL-6 downregulated TP53 in AA PCa cell lines and upregulated expression of a splice variant of MBD2, promoting a cancer stem-like cell phenotype (Teslow et al. 2018). Additionally, AA PCa exhibits an increased inflammatory signature, including increased expression of inflammatory genes (e.g. CCR7) and more frequent copy number variations of genes related to the immune response (e.g. IL-27, ITGAL, ITGAM) (Hardiman et al. 2016; Powell et al. 2013; Rose et al. 2010; Wallace et al. 2008).
AA PCa cell lines and patient specimens have distinct miRNA profiles compared to EA PCa. AA PCa cell lines have increased expression of hsa-miR-26a compared to EA cell lines derived from tumors of similar stage and grade (Theodore et al. 2010). Theodore et al. (2014) showed decreased expression of five miRNAs due to hypermethylation of CpG islands within promoter regions in AA PCa. Of particular interest, miR-152 had significantly lower expression in AA patients versus EA patients (in both non- and malignant tissue). Ectopic over-expression of miR-152 in PCa cell lines downregulated expression of DNMT1 by binding to the 3’UTR of the mRNA, leading to decreased proliferation, migration, and invasion.
Ten miRNAs have been identified that exhibit enriched or depleted expression in AA versus EA PCa (Wang et al. 2015). These miRNAs, including miR-133a (AA depleted), miR-513c (AA depleted), and miR-96 (AA enriched), were computationally predicted and experimentally shown to target key genes known to promote cancer, such as MCL1, STAT1, and FOXO3A. Ectopic treatment of PCa cell lines with AA-depleted miRNA mimics (for miR-133a and −513c) or AA-enriched miRNA antagomirs (for miR-96) resulted in decreased proliferation, invasion, and caspase activity. In agreement with these in vitro findings, AA PCa specimens showed significantly increased expression of MCL-1 and STAT1 and decreased expression of FOXO3A compared to EA PCa samples.
The role of epigenetics in PCa disparities is also being explored. Using quantitative pyrosequencing, Kwabi-Addo et al. (2010) and Devaney et al. (2015) revealed increased gene promoter methylation in AA PCa specimens compared to EA PCa. RARβ2, SPARC, TIMP3, NKXX2–5, ABCG5, and SNRPN genes were all found to be highly methylated in AA PCa samples and cell lines. Tang et al. (2013) identified an association between increased RARB and APC methylation and increased PCa risk in AA men.
Alternative splicing
An area of research that has recently garnered considerable attention with the advent of genome-wide approaches (e.g. exon arrays, RNA-Seq) is the role of alternative splicing (AS) in cancer and cancer disparities. AS is the major mechanism for post-transcriptional regulation of gene expression, mRNA diversity, and protein modification. During AS, introns are typically excised from the precursor mRNA (pre-mRNA) and the remaining exons can be joined together in different combinations to produce multiple unique mature mRNA transcripts from a single gene. It is estimated that over 90% of human genes transcribe pre-mRNAs that undergo AS with an average of five unique mRNA variants per coding gene. This generates a proteomic complexity of ~100,000 distinct protein isoforms from ~20,000 protein-coding genes. Types of splicing events include exon skipping (removal of specific exons), cryptic exon expression, selection between two mutually exclusive exons, exon scrambling, intron retention, alternative 5’ or 3’ splice sites (altering boundaries between introns and exons), alternative promoters (which can alter reading frames), and alternative polyadenylation sites (Fig. 1). This is a highly complex and flexible system that responds to cell type, tissue type, developmental stage, physiological system, and disease state.
Fig. 1. Schematic representations of different types of splicing events.
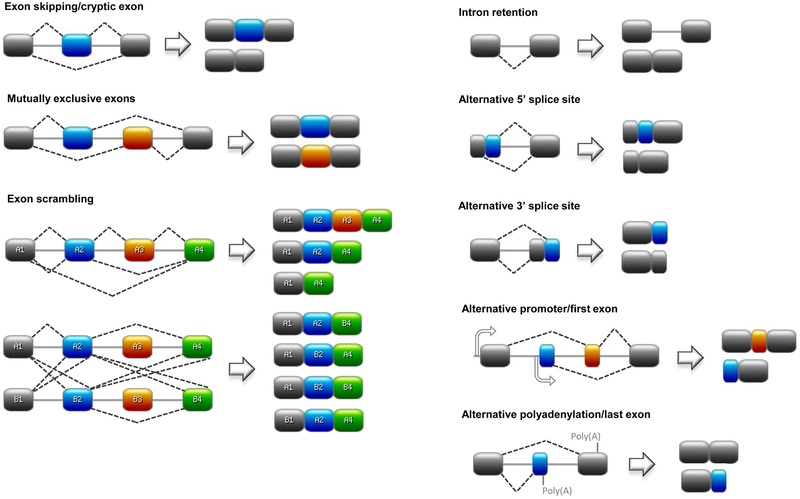
Exons are depicted as rectangles and introns as solid lines. Broken lines represent splicing events. Abbreviations: Poly(A), polyadenylation site. Designed on https://prosite.expasy.org.
AS generates a variety of protein isoforms with different sequences and altered functions from the same gene, promoting diversification of the transcriptome and proteome at both the species and interspecies levels. Although not all AS variants are functional, many can have similar or different functions, different stability kinetics, alternative subcellular localizations, or encode isoforms that are susceptible to different post-translational modifications (e.g. phosphorylation, ubiquitination). By altering the repertoire of splice variants within a cell in a time- and/or spatial-dependent manner, AS can lead to protein isoforms with different interactome networks by promoting or inhibiting different DNA-protein, protein-protein, protein-ligand, and protein-drug interactions.
Splicing events are regulated by cis-acting sequences (splice sites, splicing enhancers or silencers, and branch points) located within the pre-mRNA and 30–500 trans-acting factors of the spliceosome, including small nuclear RNAs (snRNAs) and RNA-binding proteins (RBPs). AS is also strongly influenced by RNA polymerase kinetics, chromatin modifications, chromatin structure, epigenetic modifications (e.g. DNA and/or RNA methylation), nucleosome occupancy, location of cis-elements, secondary structure of pre-mRNA, and sequence editing (Baralle and Giudice 2017; Gallego-Paez et al. 2017).
Cis-regulatory sequences are divided into two groups: splice sites that are required for spliceosome binding and binding sites for other RBPs. Sequences within exons (5’ and 3’ splice sites) and within introns (branch point and polypyrimidine sequences) designate exon-intron boundaries for the spliceosome. These splice sites can be constitutive (always recognized as splice sites) or alternative. The strength of a splice site is important for splicing accuracy and frequency. Strong splice sites contain consensus sequences that are well recognized by the spliceosome, thereby undergo splicing at a high rate. Weak splice sites rely on cis-acting sequences and cell context for splicing to occur. Splicing regulatory elements (SREs) include intronic or exonic splicing enhancers (ISE, ESE) or silencers (ISS, ESS). These provide binding sites for trans-acting factors, such as splicing factors (SF).
The spliceosome is composed of five snRNPs and over 200 SFs and auxiliary proteins. SnRPs (U1, U2, U4, U5, and U6) are the core components of the spliceosome ribozyme and are responsible for recognizing splice sites. The spliceosome also contains DEAD/H-box RNA-dependent ATPases that allows for changes in RNA-RNA base pairing (Will and Lührmann 2011). A splicing event begins with U1 binding the 5’ splice site, the SF3b complex within U2 binding the branch point site, and U2AF1 and U2AF2 auxiliary proteins binding the 3’ splice site. U1 and U2 interact to form the pre-spliceosome. Next, U4, 5, and 6 are recruited, the spliceosome rearranges, U1 and U4 are released, and the spliceosome becomes activated. In the first splicing reaction, the phosphodiester bond at the 5’ splice site is cleaved via nucleophilic attack from the adenosine in the branch point site. The intron then forms an intermediate lariat structure and the phosphodiester bond at the 3’ splice site is cleaved via nucleophilic attack by the free 3’ hydroxyl group on the phosphate of the 3’ splice site. Finally, the two exons are ligated together and the intron lariat is released (Wongpalee and Sharma 2014).
Trans-acting RBPs, such as SFs and auxiliary proteins (e.g. SF1, U2AF), complex with the spliceosome to add additional flexibility and complexity to the splicing process. RBPs bind cis-regulatory sites to promote or inhibit splice site recognition which is dependent on location of binding (e.g. within intronic or exonic sequences, upstream of an alternative exon, within a downstream intron), cellular context, regulation by other RBPs, and expression level of the RBP (Fu and Ares 2014; Rossbach et al. 2014). The most well studied trans-acting factors are the serine/arginine-rich SF (SRSF) and heterogeneous nuclear ribonucleoprotein particle (hnRNP) families. SRSFs are composed of two RNA recognition motifs at the N-terminus and a serine-rich domain at the C-terminus that is involved in protein-protein interactions. SRSFs are generally considered positive splicing regulators. They promote exon inclusion by preferentially binding to purine-rich ESE or ISE sequences and recruiting U1 to 5’ splice sites and U2AF to 3’ splice sites (Zhou and Fu 2013). SRSF protein kinases (SRPKs) and CDC-like kinases (CLKs) activate SRSFs by phosphorylation in the cytoplasm or nucleus, respectively. The hnRNP family is largely classified as negative splicing regulators. Like SRSFs, they have two RNA recognition motifs, however their protein-protein interaction domains are unstructured. HnRNPs promote exon skipping by binding ESS and ISS sequences and inhibiting recognition of splice sites. They may also prevent spliceosome assembly after 3’ splice site recognition via steric hindrance of snRNPs.
SRSFs and hnRNPs have more nuanced roles than exclusively positive or negative splicing regulators (Huelga et al. 2012; Pandit et al. 2013). Their effect on splicing can depend on serval factors, such as the location of the binding site. For example, SRSFs enhance splicing when binding to sequences within exons and repress splicing when bound to introns (Zhou and Fu 2013). The functional consequences of SF binding can also be influenced by cell differentiation, cell fate, tissue-identity, organ development, and disease state (Wang et al. 2008).
Alternative splicing and cancer
All components of the splicing process are tightly regulated and any alteration can lead to disease causation and progression. The involvement of splicing dysregulation in oncogenic processes is known to activate oncogenes and inactivate tumor suppressors. Gene expression program changes via aberrant splicing in cancer cells select for functional changes that promote the malignant progression of the tumor (Gonçalves, Pereira, and Jordan 2017). Modifications in the splice sites or splicing machinery can lead to DNA damage, genomic instability, changes in epigenetics, alterations in transcriptional elongation, and changes in gene expression thus helping to promote any of the “hallmarks of cancer” (Dvinge et al. 2016; Ryan et al. 2016; Sebestyén et al. 2016). Splice variants are being used to characterize tumor subtypes and are targets of interest for cancer biomarkers and therapeutics (Oltean and Bates 2014). Due to the potential for functional differences, individual AS variants need to be studied separately to better understand each variant’s role in disease progression. In addition, an understanding of the overall splicing changes, as a change in one trans-acting factor can affect the splicing of hundreds of transcripts, will be instrumental in identifying the role of AS in cancer.
The Cancer Genome Atlas (TCGA) data has been used to identify genome-wide AS events in cancer versus normal tissues and between different tumor subtypes and stages. Globally, AS events occur more frequently than somatic mutations in driver genes. AS also occurs more often in cancer related pathways and in genes that are frequently mutated in cancers (Climente-González et al. 2017; Wang et al. 2017). Analysis of TCGA data has also identified key somatic mutations in splice sites that affect exon-intron boundaries, resulting in changes in expression of oncogenes and tumor suppressors in cancer (Supek et al. 2014). In general, splicing of proto-oncogenes generates constitutively active or gain of function variants that confer an increased oncogenic advantage. Synonymous mutations, which can alter splice sites, are also more highly enriched in oncogenes. Conversely, AS of tumor suppressors can introduce premature stop codons and altered reading frames, resulting in decreased protein levels via nonsense mediated decay or decreased function. Cancer cells have increased levels of intron retention in tumor suppressor transcripts which promote premature termination, nonsense-mediated decay, and tumor suppressor inactivation (Dvinge and Bradley 2015; Hu, Yau, and Ahmed 2017; Jung et al. 2015). Mutations in splice sites or splice site choice can result in isoform switching or generation of novel splice variants (Alsafadi et al. 2016). Thus, somatic mutations in key genes or splice sites involved in AS may be a major driver in many cancers.
Differential splicing can generate variants with opposing functions or shift the balance between two isoforms. For example, while the full length isoform of caspase-9 is pro-apoptotic, a shorter isoform missing exons 3–6 is anti-apoptotic and has been identified in cancers, including non-small cell lung carcinoma (Seol and Billiar 1999). SRSF1, which is overexpressed in many cancers, binds within intron 6 to promote inclusion of exons 3–6 to generate the long variant (Shultz et al. 2011). Conversely, hnRNPL binds an ESS in exon 3 and induces splicing exclusion of exons 3–6 to generate the short variant (Goehe et al. 2010). Kinases such as AKT are predicted to phosphorylate and activate both SRSF1 and hnRNPL (Vu et al. 2013). The known tumor suppressor gene TP53 has over seven different splice variants that have been detected in a variety of cancers (Chen and Weiss 2015). Splicing events are concentrated in the 5’ and 3’ ends and result in alternative promoter selection, exon skipping, intron retention, alternative 5’ and 3’ splice sites, or alternative reading frames. These P53 isoforms can inhibit full length P53, impair growth or senescence suppression, and are associated with decreased patient survival.
Frequently, tumors display alterations in trans-acting factors. Perturbations in the expression level, localization, activity, or degradation of RBPs, SFs, or their upstream regulators can vary dramatically between different cancers. While hnRNPA2/B1 is an oncogenic driver in glioblastoma via splicing of tumor suppressors IG20 and MST1R (RON) (Golan-Gerstl et al. 2011), RBM4 controls apoptosis, proliferation, and migration as a tumor suppressor in a variety of other solid tumors (Wang et al. 2014). The most common SF mutations in hematological and solid tumors are heterozygous missense gain or alteration of function mutations in SF3B1, U2AF1, and SRSF2 and homozygous loss of function mutations in ZRSR2 (Agrawal et al. 2018; Dvinge et al. 2016). SF3B1 is a member of the SF3b complex within the U2 snRNP of the spliceosome. Mutations have been observed in the 3’ splice site of the SF3B1 pre-mRNA resulting in nonsense mediated decay, which commonly occurs in breast cancer (Maguire et al. 2015). Mutations in the zinc finger domains of U2AF1, a U2 small nuclear RNA auxiliary factor, are frequently identified in non-small cell lung cancer (Imielinski et al. 2012). Missense mutations are often observed in SRSF2 that change its binding affinity to ESE sequences and are common in chronic myelomonocytic leukemia (Yoshida et al. 2011). ZRSR2 is a zinc finger RBP in the U12 minor spliceosome complex. Mutations that introduce in-frame stop codons or disrupt the reading frame are common in myelodysplastic syndrome (Madan et al. 2015).
Due to the pivotal role of AS in cancer, many researchers are focusing their efforts on identifying or developing molecules that target aberrant AS in cancers (Bates et al. 2017; Dvinge et al. 2016; Gallego-Paez et al. 2017). Potential targets for these therapies include mutations in splice sites, cis-regulatory elements, and promoter or coding regions of trans-acting factors. Due to the high mutation rates observed in cancers, SF3B1 is a major target for splicing-modulating drugs. In addition, upstream factors such as SF kinases and specific splice isoforms of oncogenes or tumor suppressors are also attractive targets. Cancers that rely on splicing activity are ideal candidates for AS-targeted therapy. For example, MYC-driven cancers rely on the spliceosome, through BUD31, for promoting oncogenesis (Hsu et al. 2015). BUD31 associates with SF3B1, U2AF1, and other core spliceosome factors. Inhibition of the spliceosome via spliceosome inhibitors or BUD31 depletion downregulates survival, tumor growth, and metastatic potential of breast cancers driven by MYC.
There are a variety of types of therapeutic compounds used to target AS. The most well-known are antisense oligonucleotides (ASOs) which are composed of nucleotides or analogs that hybridize with a complimentary nucleic acid sequence. By coding for the complimentary sequence of the target, ASOs can potentially block splice sites via steric hindrance, target mRNAs for degradation, redirect splicing, or prevent trans-factors from binding. ASOs have gained traction in treating Duchenne Muscular Dystrophy (DMD), spinal muscular atrophy (SMA), and amyotrophic lateral sclerosis (ALS) (Gallego-Paez et al. 2017). In oncology, two ASOs, AZD9150 targeting STAT3 and AZD4785 targeting KRAS, are in clinical trials for solid advanced and metastatic diseases (Hong et al. 2015; Ross et al. 2017).
Small molecule inhibitors (SMIs) have been designed to target SF kinases and spliceosome components. SRPIN340 which targets SRPKs and TG-003 which targets CLKs cause decreased activity of SFs and subsequent decreased expression of “splice-correct” signaling proteins, such as VEGF and p70-S6K (Araki et al. 2015; Siqueira et al. 2015). ML315, a chemically modified quinazoline probe, selectively inhibits the CLK family as well (Coombs et al. 2010). Cp028 has been shown to inhibit intermediate stage spliceosome assembly by causing the early release of U4/U6 (Sidarovich et al. 2017).
Natural products and their derivatives have also shown promise in targeting different stages of AS. Leucettine L41, derived from the marine sponge product leucettamine B, is an ATP-competitive inhibitor against CLK1 and CLK3 and has been shown to inhibit phosphorylation of SRSF4 and SRSF6 (Debdab et al. 2011). Another natural product, N-palmitoyl-l-leucine targets late stage spliceosome assembly (Effenberger et al. 2015). Indole derivatives, such as benzopyridoindoles and pyridocarbazoles, alter SF-ESE-dependent splicing in key oncogenic genes such as MST1R (Ghigna et al. 2010; Soret et al. 2005). The RBP RBM39 is a target of sulfonamides derived from para-aminobenzoic. Mutations in RBM39 and resistance to indisulam are common in leukemia and lymphomas. These mutations block the complex formation of RBM39 with the CUL4-DCAF15 ubiquitin ligase complex, halting the normal proteasomal degradation of RBM39 and resulting in aberrant pre-mRNA splicing (Han et al. 2017). SRPIN340 is an isonicotinamide compound shown to inhibit expression of SRPK1 and a pro-angiogenic VEGF variant (Gammons et al. 2014). Derivatives of the natural compound FR901464 have shown promising ability to inhibit SF3B. These analogs include spliceostatin A, meayamycin, and sudemycin (Albert et al. 2009; Convertini et al. 2014; Kaida et al. 2007).
Pladienolide-scaffold derivatives have had the most success in clinical trials. E7107, derived from pladienolide B, inhibits SAP130 of the SF3B complex (Kotake et al. 2007). This weakens the binding interaction between U2 and the pre-mRNA by locking SF3B1 in an inactive conformation and sterically preventing binding to the branch point adenosine (Finci et al. 2018). E7107 was one of the first splicing modulator drugs to enter clinical trials in solid tumors in 2007 (Eskens et al. 2013; Hong et al. 2014), however further studies in humans were suspended due to unexpected toxicity. H3B-8800, another pladienolide-derivative, selectively inhibits wildtype and mutated SF3B1 isoforms and enriches for intron retention in SF-coding mRNAs (Seiler et al. 2018). Trials using H3B-8800 in hematological cancers have been ongoing since 2016.
Our current limited understanding of overall “splicing sickness”, restoration of normal splicing, and downstream effects of spliceosomal mutations need to be addressed in order to develop new AS drugs. Overcoming issues of systemic delivery, toxicity, off-target effects, efficacy, and targeting the desired cell type will be key in splice-modulating therapies becoming a safe and efficacious therapeutic option for cancer patients.
Alternative splicing in prostate cancer
A number of genes undergoing AS have been associated with PCa development and progression. The androgen receptor (AR), a steroid nuclear hormone that plays a major role in normal prostate homeostasis and PCa development, is the primary target for early PCa treatment. PCa tumors, however, develop AR-targeted treatment resistance (i.e. castrate-resistant) as the disease progresses. One mechanism in which PCa tumors develop drug resistance is through AS of the AR. Among the 20 different AR splice variants identified, ARv7 is the most clinically frequent and relevant variant. The ARv7 variant is generated by inclusion of a cryptic exon within exon 3 that encodes a protein isoform with a truncation of the entire C-terminal ligand binding domain (LBD). The LBD is important for AR activation by androgens and subsequent translocation of the AR into the nucleus for transcriptional regulation of androgen-dependent genes. The ARv7 isoform acts independently of androgen binding and is constitutively present in the nucleus of prostate cells, regardless of androgen stimulation (Cao et al. 2014). Levels of ARv7 mRNA in PCa patients can help predict responsiveness to anti-androgen therapies, such as abiraterone and enzalutamide (Antonarakis et al. 2014). The SF hnRNPA1 and RBP SAM68 are believed to contribute to regulation of the ARv7 variant. Relocalization of hnRNPA1 from the nucleus to the cytoplasm decreases expression of ARv7 in PCa cells and resensitizes them to enzalutamide (Antonarakis et al. 2014; Ko et al. 2014; Nadiminty et al. 2015; Tummala et al. 2017). SAM68 preferentially increases expression of ARv7 in PCa cells, via SAM68 stabilization of the ARv7 mRNA via direct RNA-protein binding and indirect mediation by SRSF1 (Stockley et al. 2015).
A second clinically relevant AR splice variant, ARv567es, has been identified in PCa cells where exons 5–7 (of 8 total) are skipped, truncating the majority of the LBD. Similar to the ARv7 isoform, ARv567es is constitutively active and androgen independent (Sun et al. 2010). This variant is highly expressed in metastatic and malignant prostate tissue (Hörnberg et al. 2011). ARv567es regulates oncogenes involved in cell cycle progression including UBE2C, which codes for a ubiquitin-conjugating protein involved in the machinery that inactivates the mitotic checkpoint and promotes proliferation (Liu et al. 2015).
The fibroblast growth factor receptor (FGFR) 2 undergoes AS of the third Ig-like extracellular domain, generating two isoforms: FGFR2IIIb and FGFR2IIIc. FGFR2IIIb is expressed highly in normal prostate epithelial cells and is a known tumor suppressor. FGFR2IIIc is involved in autocrine signaling and expressed more highly in mesenchymal cells. While no change in overall FGFR2 protein expression is observed as PCa progresses (Sahadevan et al. 2007), a switch in FGFR2 isoforms occurs due to AS. Decreased expression of the IIIb isoform and exclusive expression of the IIIc isoform is associated with epithelial to mesenchyme transition (EMT) and loss of AR sensitivity (Carstens et al. 1997). This increase in FGFR2IIIc expression correlates with an increase in fibroblast growth factor (FGF) 8b, a ligand associated with PCa (Gnanapragasam et al. 2003).
The vascular endothelial growth factor (VEGF) is largely responsible for cellular growth and survival via angiogenesis in both normal and cancerous conditions. “Canonical” splicing of VEGF produces a VEGF isoform that is pro-angiogenic, while an alternative 3’ splice site event generates an anti-angiogenic isoform VEGF165b, which is the main isoform. VEGF165b differs from pro-angiogenic VEGF in the last six amino acids and acts as an antagonist of the VEGF receptor (Woolard et al. 2004). Expression of pro-angiogenic VEGF is an early driver of PCa and increased expression corresponds with later stage PCa and increased expression of SRSF1 (Rennel et al. 2008). Inhibition of the SF kinase SRPK1, a known activator of SRSF1, causes splice switching of VEGF165b in PCa cells and decreased tumor formation in PCa mouse models (Mavrou et al. 2015).
Bcl-x plays a pivotal role in regulating apoptosis. Alternative 5’ splice site usage within exon 2 of the BCL2L1 pre-mRNA generates two variants that have opposing functions. The long anti-apoptotic isoform, Bcl-x(L), is associated with cell survival, while the shorter isoform (Bcl-x(S) promotes apoptosis, cell death, and sensitivity to chemotherapeutics in PCa (Mercatante, Mohler, and Kole 2002). High Bcl-x(L) to Bcl-x(S) ratios have been observed in PCa and overexpression of the short isoform induces apoptosis-mediated cell death in cancer cells (David and Manley 2010; Rajan et al. 2009). SAM68 selectively favors the upstream 5’ splice site, thus favoring production of the BCL2L1 long variant and preventing apoptosis. Increased expression of Bcl-x(L) has been identified in PCa patients and cell lines, resulting in decreased apoptotic-induced cell death and decreased sensitivity to cytotoxic therapeutics (Busà et al. 2007).
Splice variants of another apoptotic-related gene SH3GLB1, which codes for the BAX binding protein Bif-1, has recently been implicated in the transition of adenocarcinoma to aggressive treatment-induced neuroendocrine (t-NE) PCa. Bif-1a, the pro-apoptotic protein isoform encoded by a variant lacking exon 6, is the predominant isoform expressed in adenocarcinoma specimens (Gan et al. 2018). Bif-1b (encoded by a variant containing a short version of exon 6) and Bif-1c (encoded by a variant containing a long version of exon 6), however, become highly expressed in t-NE PCa. This switch in dominant variant expression is regulated by the SF SRRM4.
Cyclin D1 (CCDN1) associates with cyclin-dependent kinase 4 (CDK4) to promote cell cycle progression through the G1 phase. Two alternative splice variants of CCDN1 have been identified: the cyclin D1a mRNA, which is the full length and more common variant, and the cyclin D1b variant, in which intron 4 is retained leading to early termination. The cyclin D1b protein isoform plays a distinct role as an AR co-regulator to promote expression of AR-dependent genes associated with tumor growth and metastasis in PCa, specifically SNAI1 (Augello et al. 2013). Additionally, increased expression of SRSF1 in PCa cells correlates with enhanced expression of cyclin D1b, but not D1a (Olshavsky et al. 2010; Paronetto et al. 2010).
ST6GalNac1 is an enzyme that synthesizes the sialyl-T (sTn) antigen and modifies the glycosylation pattern of cell surface glycoproteins that play a role in cell adhesion and metastasis. ST6GalNac1 is androgen-sensitive, thus indicating a role for this enzyme in PCa. Recently, RNA-seq data have identified a shorter splice variant of ST6GalNac1 that has only been reported in PCa (Munkley et al. 2015). The short isoform results from the inclusion of an additional exon (exon 2) within the 5’ UTR that generates a new start codon and encodes a longer mRNA variant but a shorter, fully functional protein isoform that has increased expression compared to the full length protein isoform missing exon 2. In vitro studies suggest a role for the short isoform in promoting EMT through decreased cell adhesion and increased cell motility.
A new splice variant of PCSK6 has been identified in PCa. PCSK6 codes for the proprotein convertase PACE4 that modifies proprotein substrates in secretory and known oncogenic pathways. Couture et al. (2017) identified a variant of PCSK6 with a shorter 3’UTR via AS of exon 25 (variant known as PACE4-altCT). While both the full length PACE4 and the shorter PACE4-altCT are expressed in PCa specimens, PACE4-altCT showed increased expression in higher grade tumors. Additionally, PACE4-altCT appears less susceptible to degradation and secretion, is more stable, more rapidly activated, and increases growth and proliferation when compared to the full length protein. PACE4-altCT directly increases the processing of pro-GDF15 (i.e. prostate differentiation factor), a TGFβ ligand with a known role in immunosuppression, protection against radiation-induced cell death, and neovascularization.
Three splice variants of CLK1 have been identified: full length CLK1, CLK1T1 (skipping of exon 4), and CLK1T2 (retention of intron 4). CLK1 is responsible for phosphorylating and activating SRSFs and other SFs. Both T1 and T2 isoforms lack the catalytic domain and are inactive. The CLK1T2 has been found to be the more prominent isoform in PCa. Treating PCa cells with a CLK1 inhibitor shifts the CLK1 variant expression ratio to favor both the expression of full-length active CLK1, as well as expression of the pro-apoptotic variants of CASP9, MCL-1, BCL2L1, and survivin (Uzor et al. 2018).
The HSD17B4 gene encodes 17β-hydroxysteroid dehydrogenase type 4 (17βHSD4), an enzyme involved in testosterone and dihydrotestosterone metabolism. Recently, five splice variants of HSD17B4 were identified, four of which encode enzyme isoforms that do not inactivate testosterone and dihydrotestosterone via conversion to inert steroid products (Ko et al. 2018). The remaining isoform, isoform 2, is the major enzyme expressed in prostate tissue and is able to inactivate androgens. The splice variant encoding isoform 2 of 17βHSD4 is missing part of exon 2 and all of exon 3, which code for sections of the short-chain alcohol dehydrogenase domain. This isoform was found to be functionally suppressed in metastatic castration-resistant PCa.
As outlined above, AS plays an important role in PCa development, progression and drug resistance. While it is fairly well accepted that AA PCa is genetically different from EA PCa, the role of AS in PCa disparities is less clear. Over 60% of the studies cited above used only cell lines derived from EA patients (Table 1). Of the remaining studies, two cell lines (22RV1 and M12) of mixed or self-reported AA ancestry were utilized. The 22RV1 cell line was derived from the CWR22 line, a primary prostatic carcinoma serially transplanted in nude mice (Sramkoski et al. 1999; Wainstein et al. 1994.). A recent study determined 22RV1 was only 41% AA ancestry (Woods-Burnham et al. 2017). The M12 line was immortalized from the P69SV40T cell line via transfection with SV40 T antigen and passaged in nude mice (Bae et al. 1994, 1998). While the ancestry of the M12 line has not been confirmed by genotyping, the parental cell line was reported to be derived from prostate epithelial cells from 63 year old AA man. None of these studies use an AA PCa cell line with over 50% AA genetic ancestry, such as MDA PCa 2b or RC77 T/E (74% and 73%, respectively) (Woods-Burnham et al. 2017). Forty-one percent of the studies cited analyzed primary prostatic samples, but none specified the ancestry (genotyped or self-reported) of the patients.
Table 1. Summary of splicing events in prostate cancer.
Cell lines and patient samples with known African American ancestry are in bold and underlined. Abbreviations: ARV7, androgen receptor splice variant 7; hnRNPA1, heterogeneous nuclear ribonucleoprotein A1; SRSF1, serine/arginine splicing factor 1; ARv567es, androgen receptor variant (exons) 5,6,7 exon skipping; MED1, mediator complex subunit 1; FGFR2, fibroblast growth factor receptor 2; VEGF, vascular endothelial growth factor; BCL2L1, B-cell lymphoma 2-like 1; Sam68, Src-associated substrate in mitosis of 68 kDa; SH3GLB1, SH3 domain-containing GRB2-like protein B1; SRRM4, serine/arginine repetitive matrix 4; CCND1, cyclin D1; SRSF1, serine/arginine splicing factor 1; ST6GalNAc1, ST6 N-acetylgalactosaminide alpha-2,6-sialyltransferase 1; PCSK6, proprotein convertase sutilisin/kexin type 6; CLK1, CDC like kinase 1; 17βHSD4, 17β-Hydroxysteroid dehydrogenase type 4; PIK3CD; phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit delta; AA, African American; EA, European American.
| Reference | Spliced gene (Splicing factor) |
Alternative splicing event |
Cell lines and/or patient samples | |
|---|---|---|---|---|
| Cao et al. 2014 | ARV7 | Cryptic exon | LNCaP, 22Rv1, PC-3 | |
| Antonarakis et al. 2014 | ARV7 (hnRNPA1) | Cryptic exon | Patients (ethnicity N/A) | |
| Ko et al. 2014 | ARV7 (hnRNPA1) | Cryptic exon | PC-3 | |
| Nadiminty et al. 2015 | ARV7 (hnRNPA1) | Cryptic exon | LNCaP, 22Rv1, VCaP, C4-2B | |
| Tummala et al. 2017 | ARV7 (hnRNPA1) | Cryptic exon | 22Rv1, C4-2B | |
| Stockley et al. 2015 | ARV7 (Sam68, SRSF1) | Cryptic exon | 22Rv1, VCaP, CWR22, PC3-M | |
| Sun et al. 2010 | ARv567es | Exon skipping | M12, LNCaP, LuCaP | |
| Hörnberg et al. 2011 | ARv567es | Exon skipping | Patients (ethnicity N/A) | |
| Liu et al. 2015 | ARv567es (MED1) | Exon skipping | LNCaP, M12 | |
| Sahadevan et al. 2007 | FGFR2 | Mutually exclusive exons | PC-3, DU145, patients (ethnicity N/A) | |
| Carstens et al. 1997 | FGFR2 | Mutually exclusive exons | LNCaP, PC-3, DU145, DUP9479, DUKAP-1, DUPKAP-2 | |
| Gnanapragasam et al. 2003 | FGFR2 | Mutually exclusive exons | Patients (ethnicity N/A) | |
| Woolard et al. 2004 | VEGF | Alternative 3’ splice site | Patients (ethnicity N/A) | |
| Rennel et al. 2008 | VEGF | Alternative 3’ splice site | PC-3, patients (ethnicity N/A) | |
| Mavrou et al. 2015 | VEGF | Alternative 3’ splice site | PC3, LNCaP, DU145 | |
| Mercatante, Mohler, & Kole 2002 | BCL2L1 | Alternative 5’ splice site | PC-3 | |
| Busà et al. 2007 | BCL2L1 (SAM68) | Alternative 5’ splice site | LNCaP, patients (ethnicity N/A) | |
| Gan et al. 2018 | SH3GLB1 (SRRM4) | Alternative 5’ splice site, exon skipping | LNCaP, 22RV1, PC-3, DU145, NCI-H660, LNCaP95, patients (ethnicity N/A) | |
| Augello et al. 2013 | CCND1 | Intron retention | LNCaP, VCaP, LAPC4, PC-3, C4-2 | |
| Olshavsky et al. 2010 | CCND1 (SRSF1) | Intron retention | LNCaP, LAPC4 | |
| Paronetto et al. 2010 | CCND1 | Intron retention | PC3, LNCaP | |
| Munkley et al. 2015 | ST6GalNAc1 | Exon skipping | LNCaP, VCaP, PC-3, 22Rv1, DU145, LNCaP-AI, and LNCaP-cdxR, BPH-1 | |
| Couture et al. 2017 | PCSK6 | Exon skipping | DU145, LNCaP, patients (ethnicity N/A) | |
| Uzor et al. 2018 | CLK1 | Exon skipping, intron retention | PC-3, DU145, VCaP | |
| Ko et al. 2018 | 17βHSD4 | Alternative 5’ splice site, exon skipping | LNCaP, LAPC4, VCaP, PC-3, DU145, RWPE-1, 22Rv1, patients (ethnicity N/A) | |
| Wang et al. 2017 | PIK3CD | Exon skipping | MDA PCa 2b, VCaP, LNCaP, PC-3, Patients (AA, EA) |
The lack of AA cell lines and patient samples used in AS PCa studies reflects the lack of minority subjects across all cancer, and specifically PCa, research (Spratt et al. 2016). In order to better understand PCa disparities, eliminate the disproportionate disease burden, provide novel biomarkers, and improve survival and quality of life in AA PCa patients, we must increase the use of AA PCa cell lines and specimens in our research.
Differential alternative splicing of PIK3CD in prostate cancer disparities
In order to further our understanding of AS in PCa health disparities, we applied a functional genomics approach to investigate differential AS (dAS) events in AA and EA PCa patients (Wang et al. 2017). Twenty AA and 15 EA PCa tumor and matched normal specimens (treatment naïve, Gleason score 6–8, age 49–81) were collected and samples were analyzed using the Affymetrix Human GeneChip exon array to identify genes undergoing AS (Table 2). We identified 158 unique genes that underwent AS in both AA and EA PCa. It can be concluded that these genes, which included TMPRSS2 and AR, are important for PCa development regardless of race. In comparing AA versus EA PCa, 1,876 unique genes undergoing dAS were identified, including RASGRP2, NF1, and BAK1. Over 2,200 unique genes underwent dAS in AA versus EA normal prostate tissue, suggesting a differential role for these genes in normal prostate homeostasis. We identified splicing events involving 644 genes, including PIK3CD, ITGA4, and MET, that were present in both AA PCa and AA normal tissue, but were absent in EA specimens. We also identified 1,575 unique genes (e.g. FGFR3 and TSC2) undergoing dAS in AA PCa versus AA normal, but not EA PCa versus EA normal. These last two comparisons identify two important groups of splicing events: splicing events involving 644 genes that are inherited based on African ancestry, and splicing events involving 1,575 genes that occur de novo during PCa progression solely in AA men. Over 70% of dAS events identified in AA PCa versus EA PCa occur in pathways known to contribute to oncogenesis (e.g. cell growth, proliferation, cell survival, cell adhesion, DNA repair) and the majority were in-frame exon skipping events. Further validation of a subset of genes identified as potential targets for dAS was performed in an additional cohort of 22–25 AA and 21–24 EA specimens. Ninety one percent of genes chosen for validation via RT-PCR were confirmed. The exon array results also identified 886 differentially expressed genes in AA versus EA PCa (compared to 1,876 dAS genes). These data suggest dAS is playing a much greater role in AA PCa disparities than differential gene expression.
Table 2. Examples of differential alternative splicing events in AA PCa, EA PCa, AA normal, and EA normal specimens.
Abbreviations: AA, African American; EA, European American; PCa, prostate cancer; TMPRSS2, transmembrane protease serine 2; AR, androgen receptor; RASGRP2, RAS guanyl releasing protein 2; NF1, neurofibromin 1; BAK1, BCL2 antagonist/killer 1; MTOR, mechanistic target of rapamycin kinase; EGFR, endothelial growth factor receptor; BCL2L1, B-cell lymphoma 2-like 1; PIK3CD, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit delta; ITGA4, integrin subunit alpha 4; MET, MET proto-oncogene, receptor tyrosine kinase; FGFR3, fibroblast growth factor receptor 3; TSC2, tuberous sclerosis complex subunit 2.
| Patient groups | # of AS genes | Examples |
|---|---|---|
| AA PCa and EA PCa | 158 | TMPRSS2, AR |
| AA PCa vs. EA PCa | 1,876 | RASGRP2, NF1, BAK1 |
| AA normal vs. EA normal | 2,205 | MTOR, EGFR, BCL2L1 |
| AA PCa and AA normal | 644 | PIK3CD, ITGA4, MET |
| AA PCa vs. AA normal | 1,575 | FGFR3, TSC2 |
Of the dAS genes identified, we focused on PIK3CD. PIK3CD codes for the p110δ (or PIK3Cδ) catalytic domain of the class I PI3Ks that bind the p85 inhibitory subunit. Upon activation by a receptor tyrosine kinase, p110δ phosphorylates phosphatidylinositol 4,5-bisphosphate (PftdIns(4,5)P2), generating phosphatidylinositol 3,4,5-trisphosphate (PIP3) which recruits AKT1 to the cell membrane thus activating downstream signaling cascades involved in cell growth, survival, and proliferation. The delta subunit of p110 is highly expressed in leukocytes (Clayton et al. 2002; Jou et al. 2002). Four PIK3CD splice variants were identified: PIK3CD-L includes all 24 exons; PIK3CD-Si is missing exon 8 (encoding a domain between the Ras-binding and C2 domains); PIK3CD-Sii is missing exon 20 (encoding part of catalytic domain) (Fig. 2); PIK3CD-Siii is missing exon 8 and 20; and PIK3CD-Siv has a large deletion that encodes the helical domain and part of catalytic domain.
Fig. 2. Alternative splicing of PIK3CD.
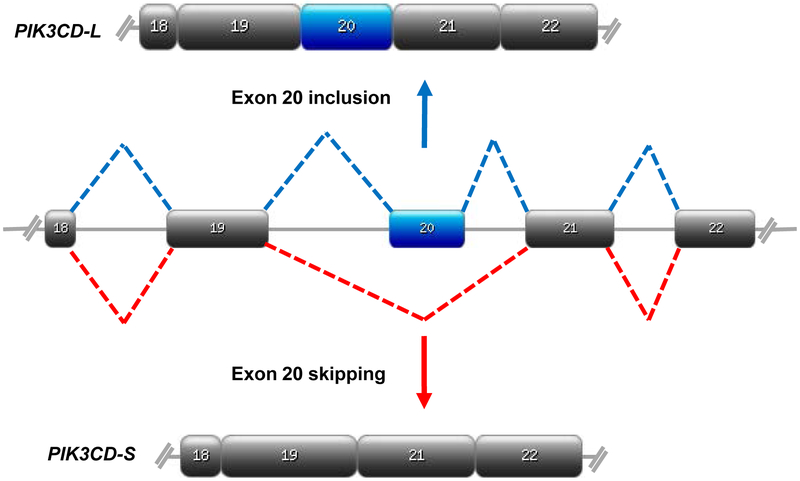
Schematic representations of alternative splicing of PIK3CD pre-mRNA. EA patients predominantly express PIK3CD-L which includes exon 20 (blue), while exon 20 is skipped (red) in AA patients to generate PIK3CD-S. Designed on https://prosite.expasy.org.
We selected the PIK3CD-Sii variant (identified as PIK3CD-S from here on) for further characterization for two reasons. First, the PIK3CD-Sii variant encodes a protein isoform missing 56 amino acids (Fig. 3) of the catalytic domain which has an important role in p110δ activity. Second, analysis of 494 PCa patients from The Cancer Genome Atlas (TCGA) revealed significantly decreased disease-free survival in patients with high PIK3CD-S/PIK3CD-L expression ratios (p=0.0052). Although this analysis was performed irrespective of race or tumor grade, these data provide evidence for the clinical relevance of PIK3Cδ-S in PCa.
Fig. 3. Protein isoforms of p110δ due to alternative splicing.
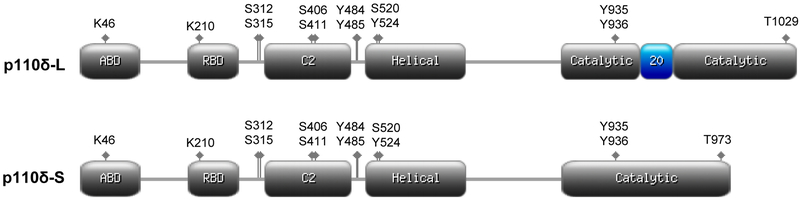
Schematic representations of long isoform due to exon 20 inclusion (top) and short isoform due to exon 20 skipping (bottom) of p110δ. Adaptor (p85) binding domain (ABD), RAS binding domain (RBD), C2, helical, and catalytic domains are shown in grey. Key phosphorylation and ubiquitin lysine (K), serine (S), tyrosine (Y), and threonine (T) sites are indicated as diamonds. The catalytic domain region encoded by exon 20 is shown in blue. Designed on https://prosite.expasy.org.
We performed siRNA-mediated knockdown of the PIK3CD-L variant in an EA PCa cell line VCaP (which has little to no expression of PIK3CD-S) and knockdown of either the PIK3CD-L or PIK3CD-S variant in an AA PCa cell line MDA PCa 2b (which expresses both variants). Knockdown of PIK3CD-L in VCaP cells results in a significant decrease in invasion, proliferation and phosphorylation of key downstream signaling proteins (i.e. AKT, mTOR, and S6). In the AA cell line, knockdown of PIK3CD-L enriches expression of PIK3CD-S, leading to increased proliferation, invasion, and phosphorylation of AKT, mTOR, and S6. Not surprisingly, we observe no significant effects on invasion, proliferation, nor phosphorylation of signaling proteins after “knockdown” of PIK3CD-S in the EA cell line. However, a significant decrease in invasion, proliferation, and phosphorylation was observed after knockdown of PIK3CD-S (thereby enriching for PIK3CD-L) in the AA cell line.
In order to determine the effect of both variants on drug resistance, we ectopically overexpressed either PIK3Cδ-L or PIK3Cδ-S in two EA cell lines, VCaP and PC-3, and treated cells with the SMI CAL-101. CAL-101 (idelalisib (Zydelig®)) targets p110δ and is approved for treatment of hematological malignancies such as chronic lymphocytic leukemia, follicular B-cell non-Hodgkin lymphoma, and small lymphocytic lymphoma. Treatment of PCa cells overexpressing PIK3Cδ-L with CAL-101 results in a significant decrease in proliferation and AKT and S6 phosphorylation. CAL-101 treatment of PIK3Cδ-S overexpressing cells results in no significant suppression of proliferation or AKT and S6 phosphorylation compared to vehicle treated cells. In addition, PIK3Cδ-S expressing cells also have greater baseline proliferation compared to PIK3Cδ-L expressing cells.
Next, we investigated the effect of both PIK3Cδ isoforms on tumor formation, metastasis, and responsiveness to CAL-101 treatment in non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice. We observe significantly decreased tumor formation in mice injected subcutaneously with PIK3Cδ-L-expressing PCa cells and treated with 50mg/kg CAL-101 compared to mice injected with PIK3Cδ-S-expressing cells and treated with CAL-101. Additionally, mice injected with PIK3Cδ-L- expressing cells via tail vein and treated with CAL-101 develop significantly less lung metastases compared to mice injected with PIK3Cδ-S-expressing cells and treated with CAL-101. These data suggest that CAL-101 is not effective against the PIK3Cδ-S isoform in vivo.
In order to test the functional differences between the two PIK3Cδ isoforms, we performed co-immunoprecipitation (co-IP) and cell-free kinase assays. Co-IP experiments demonstrate that the PIK3Cδ-L isoform binds with a significantly higher affinity to the p85α regulatory subunit compared to PIK3Cδ-S. We also observe higher activity of PIK3Cδ-S in a cell free kinase assay with and without the p85α subunit present and with or without wortmannin or CAL-101 treatment compared to PIK3Cδ-L. This suggests that PIK3Cδ-S activity is not as tightly suppressed by p85α and retains kinase activity even in the presence of SMIs such as CAL-101.
What has not been investigated up to this point is the mechanism responsible for the preferential expression of the PIK3CD-S variant in AA PCa. Therefore, we returned to gene expression data generated from previous studies (Wang et al. 2015; Wang et al. 2013) to investigate which upstream SFs may be playing a role in the generation of the PIK3CD-S variant in AA PCa. Interestingly, we identified several SFs, including SRSF2, SRSF7, and HNRNPF, with increased expression in AA PCa compared to EA PCa at both the mRNA and protein levels (data not shown). Moreover, the intronic regions surrounding exon 20 of the PIK3CD pre-mRNA have computationally predicted binding sites for these three SFs. We hypothesized that binding of SRSF2, SRSF7 and/or hnRNPF to flanking regions of exon 20 in the PIK3CD pre-mRNA may facilitate exon 20 skipping, leading to the generation of PIK3CD-S. In order to test this hypothesis, we treated MDA PCa 2b cells with siRNAs targeting SRSF2, SRSF7, or HNRNPF, and observe a decrease in expression of PIK3CD-S and an enrichment of PIK3CD-L (Fig. 4a). We refer to this phenomenon as splice switching. SiRNA-mediated SF knockdowns are confirmed by western blot analysis (Fig. 4b). Our findings suggest that aberrant expression of SFs may be playing a role in the dAS observed in AA PCa.
Fig. 4. Knockdown of overexpressed splicing factors causes splice switching of PIK3CD variants.
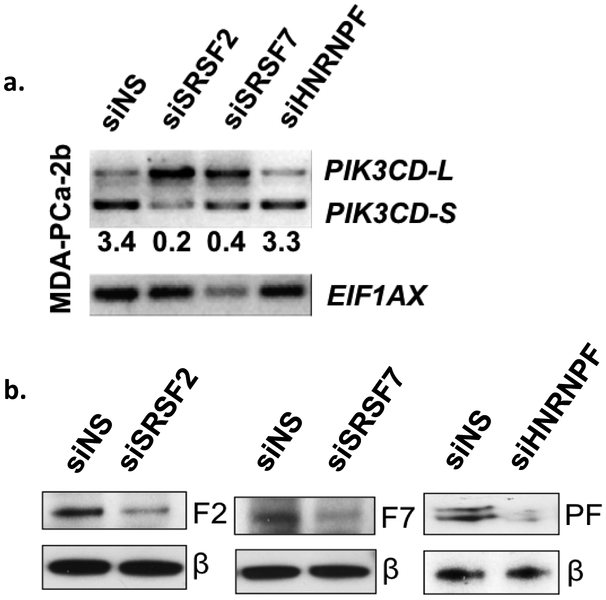
a) siRNA-mediated knockdown of three splicing factors in an AA cell line switches predominant expression of PIK3CD from the –S to the –L variant. Blots were quantified by densitometry and numbers underneath blots represent the -S/-L expression ratio. Shown are representative blots from 3-4 independent experiments. b) Western blot confirms knockdown of splicing factors at the protein level. Abbreviations: F2, SRSF2; F7, SRSF7; PF, hnRNPF; β, β-actin. Shown are representative blots from 3–4 independent experiments.
Thus, we propose a mechanism in which “normal” expression of specific SFs (SRSF2, SRSF7, and/or hnRNPF) in EA PCa promotes inclusion of exon 20 in the final transcript of PIK3CD-L and generation of the PIK3Cδ-L protein (Fig. 5). This protein isoform is sensitive to CAL-101 and has high binding affinity to the p85α regulatory subunit. In AA PCa, however, increased expression of SFs SRSF2, SRSF7, and/or hnRNPF results in dAS of the PIK3CD pre-mRNA, leading to skipping of exon 20, and subsequent generation of the PIK3Cδ-S protein isoform, which exhibits increased oncogenic signaling and decreased sensitivity to SMIs such as CAL-101 (idelalisib). Of interest, ~50% of patients treated second line with idelalisib for certain B-cell malignancies (e.g. chronic lymphocytic leukemia) will exhibit primary resistance to this SMI (Brown et al. 2014; Gopal et al. 2014; Shah and Mangaonkar 2015). The mechanism of resistance is currently unknown. We propose that expression of the PIK3Cδ-S protein isoform may be responsible, in part, for this resistance. We are currently performing a high throughput chemical library screen to identify a SMI that will effectively suppress PIK3Cδ-S activity.
Fig. 5. Proposed mechanism for role of aberrant splicing of PIK3CD in PCa disparities.
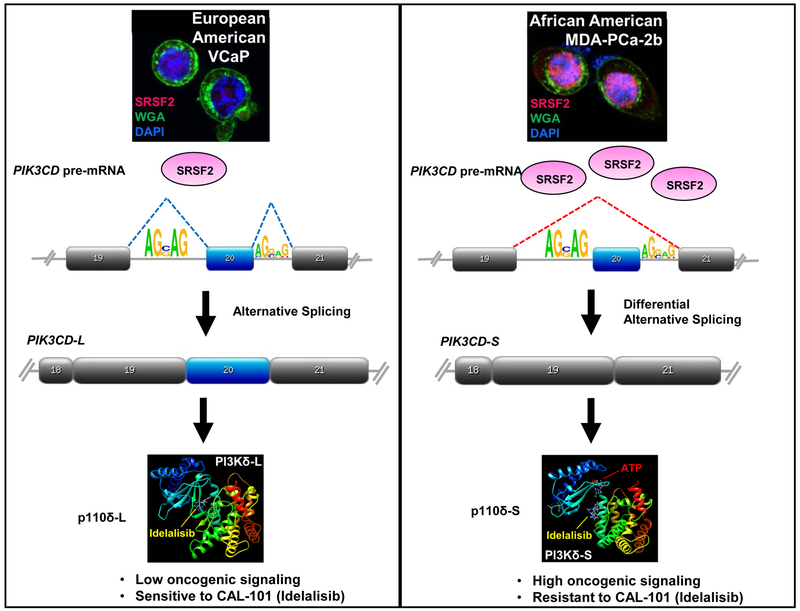
EA cell lines (e.g. VCaP) and patient specimens show “normal”/low expression of SFs, such as SRSF2 (left panel). Normal splicing of PIK3CD pre-mRNA generates the long variant containing exon 20, which encodes the p110δ-L protein that has low oncogenic properties. Aberrant over-expression of SRSF2 in AA cell lines (MDA PCa 2b) and patient samples results in differential alternative splicing of PIK3CD (right panel). This generates p110δ-S protein that has higher oncogenic signaling and is resistant to CAL-101 (idelalisib).
Conclusion
While studies focusing on PCa disparities have increased over the past 10 years, the RNA splicing landscape has not been fully characterized as a potential mechanism for race-related PCa aggressiveness. Our recent study has highlighted genome-wide dAS events occurring specifically in AA PCa. The dAS events in AA PCa are overrepresented in known oncogenic signaling pathways, possibly providing a mechanistic explanation for PCa disparities. While further studies are needed to fully understand the oncogenic capacity of other variants identified in our study (e.g. FGFR3, MET, TSC2), these AS variants could serve as useful biomarkers for prognostic predictions and in identifying non-responsive patients to SMIs. Further characterization of dAS variants in AA PCa patients will provide greater, and much needed, insight into the mechanisms responsible for PCa disparities and possible new leads for therapeutic intervention.
Acknowledgements:
This work is supported by NIH R01 CA204806 (N.H.L.)
References
- Agrawal Anant A, Yu Lihau, Smith Peter G, and Buonamici Silvia. 2018. “Targeting Splicing Abnormalities in Cancer.” Current Opinion in Genetics & Development 48: 67–74. [DOI] [PubMed] [Google Scholar]
- Albert Brian J et al. 2009. “Meayamycin Inhibits Pre-Messenger RNA Splicing and Exhibits Picomolar Activity against Multidrug-Resistant Cells.” Molecular cancer therapeutics 8(8): 2308–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsafadi Samar et al. 2016. “Cancer-Associated SF3B1 Mutations Affect Alternative Splicing by Promoting Alternative Branchpoint Usage.” Nature Communications 7: 10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis Emmanuel S. et al. 2014. “AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer.” New England Journal of Medicine 371(11): 1028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonopoulou Effrosyni and Ladomery Michael. 2018. “Targeting Splicing in Prostate Cancer.” International Journal of Molecular Sciences 19(5): 1287–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki Shinsuke et al. 2015. “Inhibitors of CLK Protein Kinases Suppress Cell Growth and Induce Apoptosis by Modulating Pre-mRNA Splicing” ed. Yan Chunhong. PLOS ONE 10(1): e0116929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augello Michael A et al. 2013. “Convergence of Oncogenic and Hormone Receptor Pathways Promotes Metastatic Phenotypes.” The Journal of Clinical Investigation 123(1): 493–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae Victoria L et al. 1994. “Tumorigenicity of SV40 T Antigen Immortalized Human Prostate Epithelial Cells: Association with Decreased Epidermal Growth Factor Receptor (EGFR) Expression.” International Journal of Cancer 58(5): 721–29. [DOI] [PubMed] [Google Scholar]
- Bae Victoria L 1998. “Metastatic Sublines of an SV40 Large T Antigen Immortalized Human Prostate Epithelial Cell Line.” The Prostate 34(4): 275–82. [DOI] [PubMed] [Google Scholar]
- Baralle Francisco E and Giudice Jimena. 2017. “Alternative Splicing as a Regulator of Development and Tissue Identity.” Nature Reviews Molecular Cell Biology 18(7): 437–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates David O, Morris Jonathan C, Oltean Sebastian, and Donaldson Lucy F. 2017. “Pharmacology of Modulators of Alternative Splicing.” Pharmacological reviews 69(1): 63–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brawley Otis W 2012. “Prostate Cancer Epidemiology in the United States.” World Journal of Urology 30(2): 195–200. [DOI] [PubMed] [Google Scholar]
- Brown Jennifer R et al. 2014. “Idelalisib, an Inhibitor of Phosphatidylinositol 3-Kinase p110δ, for Relapsed/refractory Chronic Lymphocytic Leukemia.” Blood 123(22): 3390–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busà R et al. 2007. “The RNA-Binding Protein Sam68 Contributes to Proliferation and Survival of Human Prostate Cancer Cells.” Oncogene 26(30): 4372–82. [DOI] [PubMed] [Google Scholar]
- Cao Bo et al. 2014. “Androgen Receptor Splice Variants Activating the Full-Length Receptor in Mediating Resistance to Androgen-Directed Therapy.” Oncotarget 5(6): 1646–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstens RP et al. 1997. “Alternative Splicing of Fibroblast Growth Factor Receptor 2 (FGF-R2) in Human Prostate Cancer.” Oncogene 15(25): 3059–65. [DOI] [PubMed] [Google Scholar]
- Chen J and Weiss WA. 2015. “Alternative Splicing in Cancer: Implications for Biology and Therapy.” Oncogene 34(1): 1–14. [DOI] [PubMed] [Google Scholar]
- Chornokur Ganna, Dalton Kyle, Borysova Meghan E, and Kumar Nagi B 2011. “Disparities at Presentation, Diagnosis, Treatment, and Survival in African American Men, Affected by Prostate Cancer.” The Prostate 71(9): 985–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton Elizabeth et al. 2002. “A Crucial Role for the p110delta Subunit of Phosphatidylinositol 3-Kinase in B Cell Development and Activation.” The Journal of Experimental Medicine 196(6): 753–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Climente-González Héctor, Porta-Pardo Eduard, Godzik Adam, and Eyras Eduardo 2017. “The Functional Impact of Alternative Splicing in Cancer.” Cell Reports 20(9): 2215–26. [DOI] [PubMed] [Google Scholar]
- Conti David V et al. 2017. “Two Novel Susceptibility Loci for Prostate Cancer in Men of African Ancestry.” Journal of the National Cancer Institute 109(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Convertini Paolo et al. 2014. “Sudemycin E Influences Alternative Splicing and Changes Chromatin Modifications.” Nucleic acids research 42(8): 4947–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombs Thomas C et al. 2010. Probe Reports from the NIH Molecular Libraries Program Identification of Selective Inhibitors of cdc2-like Kinases 1 and 4 (Clk1, Clk4). [PubMed] [Google Scholar]
- Cooperberg Matthew R 2013. “Re-Examining Racial Disparities in Prostate Cancer Outcomes.” Journal of Clinical Oncology 31(24): 2979–80. [DOI] [PubMed] [Google Scholar]
- Couture Frédéric et al. 2017. “PACE4 Undergoes an Oncogenic Alternative Splicing Switch in Cancer.” Cancer Research 77(24): 6863–79. [DOI] [PubMed] [Google Scholar]
- David Charles J, and Manley James L. 2010. “Alternative Pre-mRNA Splicing Regulation in Cancer: Pathways and Programs Unhinged.” Genes & Development 24(21): 2343–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debdab Mansour et al. 2011. “Leucettines, a Class of Potent Inhibitors of cdc2-like Kinases and Dual Specificity, Tyrosine Phosphorylation Regulated Kinases Derived from the Marine Sponge Leucettamine B: Modulation of Alternative Pre-RNA Splicing.” Journal of medicinal chemistry 54(12): 4172–86. [DOI] [PubMed] [Google Scholar]
- DeSantis Carol E et al. 2016. “Cancer Statistics for African Americans, 2016: Progress and Opportunities in Reducing Racial Disparities.” CA: A Cancer Journal for Clinicians 66(4): 290–308. [DOI] [PubMed] [Google Scholar]
- Devaney JM et al. 2015. “Genome-Wide Differentially Methylated Genes in Prostate Cancer Tissues from African-American and Caucasian Men.” Epigenetics 10(4): 319–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge Heidi and Bradley Robert K. 2015. “Widespread Intron Retention Diversifies Most Cancer Transcriptomes.” Genome Medicine 7(1): 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge Heidi, Kim Eunhee, Abdel-Wahab Omar, and Bradley Robert K. 2016. “RNA Splicing Factors as Oncoproteins and Tumour Suppressors.” Nature Reviews Cancer 16(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effenberger Kerstin A et al. 2015. “The Natural Product N-Palmitoyl-L-Leucine Selectively Inhibits Late Assembly of Human Spliceosomes.” The Journal of biological chemistry 290(46): 27524–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskens Ferry A L M et al. 2013. “Phase I Pharmacokinetic and Pharmacodynamic Study of the First-in-Class Spliceosome Inhibitor E7107 in Patients with Advanced Solid Tumors.” Clinical Cancer Research 19(22): 6296–6304. [DOI] [PubMed] [Google Scholar]
- Evans Simon et al. 2008. “Investigating Black-White Differences in Prostate Cancer Prognosis: A Systematic Review and Meta-Analysis.” International Journal of Cancer 123(2): 430–35. [DOI] [PubMed] [Google Scholar]
- Finci Lorenzo I et al. 2018. “The Cryo-EM Structure of the SF3b Spliceosome Complex Bound to a Splicing Modulator Reveals a Pre-mRNA Substrate Competitive Mechanism of Action.” Genes & development 32(3–4): 309–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Xiang Dong, and Ares Manuel. 2014. “Context-Dependent Control of Alternative Splicing by RNA-Binding Proteins.” Nature Reviews Genetics 15(10): 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego-Paez LM et al. 2017. “Alternative Splicing: The Pledge, the Turn, and the Prestige: The Key Role of Alternative Splicing in Human Biological Systems.” Human Genetics 136(9): 1015–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammons MV et al. 2014. “Targeting SRPK1 to Control VEGF-Mediated Tumour Angiogenesis in Metastatic Melanoma.” British Journal of Cancer 111(3): 477–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Yu et al. 2018. “Roles of Alternative RNA Splicing of the Bif-1 Gene by SRRM4 During the Development of Treatment-Induced Neuroendocrine Prostate Cancer.” EBioMedicine 31: 267–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghigna Claudia et al. 2010. “Pro-Metastatic Splicing of Ron Proto-Oncogene mRNA Can Be Reversed: Therapeutic Potential of Bifunctional Oligonucleotides and Indole Derivatives.” RNA biology 7(4): 495–503. [DOI] [PubMed] [Google Scholar]
- Gnanapragasam VJ et al. 2003. “FGF8 Isoform B Expression in Human Prostate Cancer.” British Journal of Cancer 88(9): 1432–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehe Rachel Wilson et al. 2010. “hnRNP L Regulates the Tumorigenic Capacity of Lung Cancer Xenografts in Mice via Caspase-9 Pre-mRNA Processing.” Journal of Clinical Investigation 120(11): 3923–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan-Gerstl R et al. 2011. “Splicing Factor hnRNP A2/B1 Regulates Tumor Suppressor Gene Splicing and Is an Oncogenic Driver in Glioblastoma.” Cancer Research 71(13): 4464–72. [DOI] [PubMed] [Google Scholar]
- Gonçalves Vânia, Pereira Joana F. S., and Jordan Peter 2017. “Signaling Pathways Driving Aberrant Splicing in Cancer Cells.” Genes 9(1): 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal Ajay K. et al. 2014. “PI3Kδ Inhibition by Idelalisib in Patients with Relapsed Indolent Lymphoma.” New England Journal of Medicine 370(11): 1008–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen Rachel M and Ladomery Michael R. 2012. “Role of Splice Variants in the Metastatic Progression of Prostate Cancer.” Biochemical Society Transactions 40(4): 870–74. [DOI] [PubMed] [Google Scholar]
- Haiman Christopher A et al. 2007. “Multiple Regions within 8q24 Independently Affect Risk for Prostate Cancer.” Nature Genetics 39(5): 638–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haiman Christoper A 2011. “Genome-Wide Association Study of Prostate Cancer in Men of African Ancestry Identifies a Susceptibility Locus at 17q21.” Nature Genetics 43(6): 570–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Ting et al. 2017. “Anticancer Sulfonamides Target Splicing by Inducing RBM39 Degradation via Recruitment to DCAF15.” Science 356(6336): eaal3755. [DOI] [PubMed] [Google Scholar]
- Han Ying et al. 2016. “Prostate Cancer Susceptibility in Men of African Ancestry at 8q24.” Journal of the National Cancer Institute 108(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardiman Gary et al. 2016. “Systems Analysis of the Prostate Transcriptome in African–American Men Compared with European–American Men.” Pharmacogenomics 17(10): 1129–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong DS et al. 2014. “A Phase I, Open-Label, Single-Arm, Dose-Escalation Study of E7107, a Precursor Messenger Ribonucleic Acid (Pre-mRNA) Splicesome Inhibitor Administered Intravenously on Days 1 and 8 Every 21 Days to Patients with Solid Tumors.” Investigational New Drugs 32(3): 436–44. [DOI] [PubMed] [Google Scholar]
- Hong David et al. 2015. “AZD9150, a next-Generation Antisense Oligonucleotide Inhibitor of STAT3 with Early Evidence of Clinical Activity in Lymphoma and Lung Cancer.” Science Translational Medicine 7(314): 314ra185–314ra185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hörnberg Emma et al. 2011. “Expression of Androgen Receptor Splice Variants in Prostate Cancer Bone Metastases Is Associated with Castration-Resistance and Short Survival” ed. Dent Paul. PLoS ONE 6(4): e19059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu Tiffany YT et al. 2015. “The Spliceosome Is a Therapeutic Vulnerability in MYC-Driven Cancer.” Nature 525(7569): 384–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Zhiyuan, Yau Christopher, and Ahmed Ahmed Ashour 2017. “A Pan-Cancer Genome-Wide Analysis Reveals Tumour Dependencies by Induction of Nonsense-Mediated Decay.” Nature Communications 8: 15943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelga Stephanie C et al. 2012. “Integrative Genome-Wide Analysis Reveals Cooperative Regulation of Alternative Splicing by hnRNP Proteins.” Cell Reports 1(2): 167–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski Marcin et al. 2012. “Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing.” Cell 150(6): 1107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jou Shiann-Tarng et al. 2002. “Essential, Nonredundant Role for the Phosphoinositide 3-Kinase p110delta in Signaling by the B-Cell Receptor Complex.” Molecular and Cellular Biology 22(24): 8580–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Hyunchul et al. 2015. “Intron Retention Is a Widespread Mechanism of Tumor-Suppressor Inactivation.” Nature Genetics 47(11): 1242–48. [DOI] [PubMed] [Google Scholar]
- Kaida Daisuke et al. 2007. “Spliceostatin A Targets SF3b and Inhibits Both Splicing and Nuclear Retention of Pre-mRNA.” Nature chemical biology 3(9): 576–83. [DOI] [PubMed] [Google Scholar]
- Kittles Rick A et al. 2001. “CYP17 Promoter Variant Associated with Prostate Cancer Aggressiveness in African Americans.” Cancer Epidemiology, Biomarkers & Prevention 10: 943–47. [PubMed] [Google Scholar]
- Ko Chia-Chen et al. 2014. “Chemical Proteomics Identifies Heterogeneous Nuclear Ribonucleoprotein (hnRNP) A1 as the Molecular Target of Quercetin in Its Anti-Cancer Effects in PC-3 Cells.” The Journal of Biological Chemistry 289(32): 22078–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko Hyun-Kyung et al. 2018. “Loss of an Androgen-Inactivating and Isoform-Specific HSD17B4 Splice Form Enables Emergence of Castration-Resistant Prostate Cancer.” Cell Reports 22(3): 809–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotake Yoshihiko et al. 2007. “Splicing Factor SF3b as a Target of the Antitumor Natural Product Pladienolide.” Nature chemical biology 3(9): 570–75. [DOI] [PubMed] [Google Scholar]
- Kwabi-Addo Bernard et al. 2010. “Identification of Differentially Methylated Genes in Normal Prostate Tissues from African American and Caucasian Men.” Clinical Cancer Research 16(14): 3539–47. [DOI] [PubMed] [Google Scholar]
- Lapuk Anna V, Volik Stanislav V, Wang Yuzhuo, and Collins Colin C 2014. “The Role of mRNA Splicing in Prostate Cancer.” Asian Journal of Andrology 16(4): 515–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Gang et al. 2015. “MED1 Mediates Androgen Receptor Splice Variant Induced Gene Expression in the Absence of Ligand.” Oncotarget 6(1): 288–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch Henry T et al. 2016. “Screening for Familial and Hereditary Prostate Cancer.” International Journal of Cancer 138(11): 2579–91. [DOI] [PubMed] [Google Scholar]
- Madan Vikas et al. 2015. “Aberrant Splicing of U12-Type Introns Is the Hallmark of ZRSR2 Mutant Myelodysplastic Syndrome.” Nature Communications 6(1): 6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magi-Galluzzi Cristina et al. 2011. “TMPRSS2-ERG Gene Fusion Prevalence and Class Are Significantly Different in Prostate Cancer of Caucasian, African-American and Japanese Patients.” The Prostate 71(5): 489–97. [DOI] [PubMed] [Google Scholar]
- Maguire Sarah L et al. 2015. “SF3B1 Mutations Constitute a Novel Therapeutic Target in Breast Cancer.” The Journal of Pathology 235(4): 571–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin Damali N, Starks Adrienne M, and Ambs Stefan 2013. “Biological Determinants of Health Disparities in Prostate Cancer.” Current Opinion in Oncology 25(3): 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrou A et al. 2015. “Serine-Arginine Protein Kinase 1 (SRPK1) Inhibition as a Potential Novel Targeted Therapeutic Strategy in Prostate Cancer.” Oncogene 34(33): 4311–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercatante Danielle R, Mohler James L, and Kole Ryszard 2002. “Cellular Response to an Antisense-Mediated Shift of Bcl-X Pre-mRNA Splicing and Antineoplastic Agents.” The Journal of Biological Chemistry 277(51): 49374–82. [DOI] [PubMed] [Google Scholar]
- Munkley Jennifer et al. 2015. “The Androgen Receptor Controls Expression of the Cancer-Associated sTn Antigen and Cell Adhesion through Induction of ST6GalNAc1 in Prostate Cancer.” Oncotarget 6(33): 34358–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munkley Jennifer, Livermore Karen, Rajan Prabhakar, and Elliott David J 2017. “RNA Splicing and Splicing Regulator Changes in Prostate Cancer Pathology.” Human Genetics 136: 1143–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadiminty Nagalakshmi et al. 2015. “NF-κB2/p52:c-Myc:hnRNPA1 Pathway Regulates Expression of Androgen Receptor Splice Variants and Enzalutamide Sensitivity in Prostate Cancer.” Molecular Cancer Therapeutics 14(8): 1884–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshavsky Nicholas A et al. 2010. “Identification of ASF/SF2 as a Critical, Allele-Specific Effector of the Cyclin D1b Oncogene.” Cancer Research 70(10): 3975–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltean Sebastian et al. 2006. “Alternative Inclusion of Fibroblast Growth Factor Receptor 2 Exon IIIc in Dunning Prostate Tumors Reveals Unexpected Epithelial Mesenchymal Plasticity.” PNAS 103(38): 14116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltean Sebastian, and Bates David O.. 2014. “Hallmarks of Alternative Splicing in Cancer.” Oncogene 33(46): 5311–18. [DOI] [PubMed] [Google Scholar]
- Pandit Shatakshi et al. 2013. “Genome-Wide Analysis Reveals SR Protein Cooperation and Competition in Regulated Splicing.” Molecular Cell 50(2): 223–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paronetto Maria Paola et al. 2010. “Alternative Splicing of the Cyclin D1 Proto-Oncogene Is Regulated by the RNA-Binding Protein Sam68.” Cancer Research 70(1): 229–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell Isaac J. 2007. “Epidemiology and Pathophysiology of Prostate Cancer in African-American Men.” Journal of Urology 177(2): 444–49. [DOI] [PubMed] [Google Scholar]
- Powell Isaac J 2013. “Genes Associated with Prostate Cancer Are Differentially Expressed in African American and European American Men.” Cancer Epidemiology, Biomarkers & Prevention 22(5): 891–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell Isaac J and Bollig-Fischer Aliccia 2013. “Minireview: The Molecular and Genomic Basis for Prostate Cancer Health Disparities.” Molecular Endocrinology 27(6): 879–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan Prabhakar, Elliott David J, Robson Craig N, and Leung Hing Y 2009. “Alternative Splicing and Biological Heterogeneity in Prostate Cancer.” Nature Reviews Urology 6(8): 454–60. [DOI] [PubMed] [Google Scholar]
- Rebbeck Timothy R 2017. “Prostate Cancer Disparities by Race and Ethnicity: From Nucleotide to Neighborhood.” Cold Spring Harbor Perspectives in Medicine: a030387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennel Es et al. 2008. “The Endogenous Anti-Angiogenic VEGF Isoform, VEGF165b Inhibits Human Tumour Growth in Mice.” British Journal of Cancer 98(7): 1250–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins AS, Whittemore AS, and Thom DH 2000. “Differences in Socioeconomic Status and Survival among White and Black Men with Prostate Cancer.” American Journal of Epidemiology 151(4): 409–16. [DOI] [PubMed] [Google Scholar]
- Rose Amy E et al. 2010. “Copy Number and Gene Expression Differences between African American and Caucasian American Prostate Cancer.” Journal of Translational Medicine 8(1): 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross Sarah J et al. 2017. “Targeting KRAS-Dependent Tumors with AZD4785, a High-Affinity Therapeutic Antisense Oligonucleotide Inhibitor of KRAS.” Science translational medicine 9(394): eaal5253. [DOI] [PubMed] [Google Scholar]
- Rossbach Oliver et al. 2014. “Crosslinking-Immunoprecipitation (iCLIP) Analysis Reveals Global Regulatory Roles of hnRNP L.” RNA biology 11(2): 146–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan Michael et al. 2016. “TCGASpliceSeq a Compendium of Alternative mRNA Splicing in Cancer.” Nucleic acids research 44(D1): D1018–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahadevan K et al. 2007. “Selective over-Expression of Fibroblast Growth Factor Receptors 1 and 4 in Clinical Prostate Cancer.” The Journal of Pathology 213(1): 82–90. [DOI] [PubMed] [Google Scholar]
- Schumacher Fredrick R et al. 2007. “A Common 8q24 Variant in Prostate and Breast Cancer from a Large Nested Case-Control Study.” Cancer Research 67(7): 2951–56. [DOI] [PubMed] [Google Scholar]
- Sebestyén Endre et al. 2016. “Large-Scale Analysis of Genome and Transcriptome Alterations in Multiple Tumors Unveils Novel Cancer-Relevant Splicing Networks.” Genome Research 26(6): 732–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler Michael et al. 2018. “H3B-8800, an Orally Available Small-Molecule Splicing Modulator, Induces Lethality in Spliceosome-Mutant Cancers.” Nature Medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seol Dai-Wu and Billiar Timothy R 1999. “A Caspase-9 Variant Missing the Catalytic Site Is an Endogenous Inhibitor of Apoptosis.” The Journal of biological chemistry 274(4): 2072–76. [DOI] [PubMed] [Google Scholar]
- Sette Claudio 2013. “Alternative Splicing Programs in Prostate Cancer.” International Journal of Cell Biology 2013: 458727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah Arpita, and Mangaonkar Abhishek. 2015. “Idelalisib: A Novel PI3Kδ Inhibitor for Chronic Lymphocytic Leukemia.” Annals of Pharmacotherapy 49(10): 1162–70. [DOI] [PubMed] [Google Scholar]
- Shuch Brian et al. 2004. “Racial Disparity of Epidermal Growth Factor Receptor Expression in Prostate Cancer.” Journal of Clinical Oncology 22(23): 4725–29. [DOI] [PubMed] [Google Scholar]
- Shultz JC et al. 2011. “SRSF1 Regulates the Alternative Splicing of Caspase 9 Via A Novel Intronic Splicing Enhancer Affecting the Chemotherapeutic Sensitivity of Non-Small Cell Lung Cancer Cells.” Molecular Cancer Research 9(7): 889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidarovich Anzhalika et al. 2017. “Identification of a Small Molecule Inhibitor That Stalls Splicing at an Early Step of Spliceosome Activation.” eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel Rebecca L, Miller Kimberly D, and Jemal Ahmedin 2018. “Cancer Statistics, 2018.” CA: A Cancer Journal for Clinicians 68(1): 7–30. [DOI] [PubMed] [Google Scholar]
- Siqueira Raoni Pais et al. 2015. “Potential Antileukemia Effect and Structural Analyses of SRPK Inhibition by N-(2-(Piperidin-1-Yl)-5-(Trifluoromethyl)Phenyl)Isonicotinamide (SRPIN340)” ed. Buratti Emanuele. PLOS ONE 10(8): e0134882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soret J et al. 2005. “Selective Modification of Alternative Splicing by Indole Derivatives That Target Serine-Arginine-Rich Protein Splicing Factors.” Proceedings of the National Academy of Sciences 102(24): 8764–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratt Daniel E et al. 2016. “Racial/Ethnic Disparities in Genomic Sequencing.” JAMA Oncology 2(8): 1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sramkoski R Michael et al. 1999. “A New Human Prostate Carcinoma Cell Line, 22Rv1.” In Vitro Cellular & Developmental Biology - Animal 35(7): 403–9. [DOI] [PubMed] [Google Scholar]
- Stockley Jacqueline et al. 2015. “The RNA-Binding Protein Sam68 Regulates Expression and Transcription Function of the Androgen Receptor Splice Variant AR-V7.” Scientific Reports 5(1): 13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Shihua et al. 2010. “Castration Resistance in Human Prostate Cancer Is Conferred by a Frequently Occurring Androgen Receptor Splice Variant.” The Journal of Clinical Investigation 120(8): 2715–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek Fran et al. 2014. “Synonymous Mutations Frequently Act as Driver Mutations in Human Cancers.” Cell 156(6): 1324–35. [DOI] [PubMed] [Google Scholar]
- Taioli Emanuela et al. 2013. “Polymorphisms in CYP17 and CYP3A4 and Prostate Cancer in Men of African Descent.” The Prostate 73(6): 668–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Deliang et al. 2013. “Methylation of the RARB Gene Increases Prostate Cancer Risk in Black Americans.” The Journal of Urology 190(1): 317–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teslow Emily A et al. 2018. “Exogenous IL-6 Induces mRNA Splice Variant MBD2_v2 to Promote Stemness in TP53 Wild-Type, African American PCa Cells.” Molecular Oncology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodore Shaniece C et al. 2014. “MicroRNA Profiling of Novel African American and Caucasian Prostate Cancer Cell Lines Reveals a Reciprocal Regulatory Relationship of miR-152 and DNA Methyltransferase 1.” Oncotarget 5(11): 3512–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodore Shaniece C, Rhim Johng S, Turner Timothy, and Yates Clayton 2010. “MiRNA 26a Expression in a Novel Panel of African American Prostate Cancer Cell Lines.” Ethnicity and Disease 20(1 Suppl 1): S1-96–100. [PMC free article] [PubMed] [Google Scholar]
- Tosoian Jeffrey J et al. 2017. “Prevalence and Prognostic Significance of PTEN Loss in African-American and European-American Men Undergoing Radical Prostatectomy.” European Urology 71(5): 697–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tummala Ramakumar, Lou Wei, Gao Allen C, and Nadiminty Nagalakshmi 2017. “Quercetin Targets hnRNPA1 to Overcome Enzalutamide Resistance in Prostate Cancer Cells.” Molecular Cancer Therapeutics 16(12): 2770–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson Mark D and Castle Erik P 2014. “Racial Disparities in Survival for Patients With Clinically Localized Prostate Cancer Adjusted for Treatment Effects.” Mayo Clinic Proceedings 89(3): 300–307. [DOI] [PubMed] [Google Scholar]
- Uehara Taisuke et al. 2017. “Selective Degradation of Splicing Factor CAPERα by Anticancer Sulfonamides.” Nature Chemical Biology 13(6): 675–80. [DOI] [PubMed] [Google Scholar]
- Uzor Simon et al. 2018. “Autoregulation of the Human Splice Factor Kinase CLK1 through Exon Skipping and Intron Retention.” Gene 670: 46–54. [DOI] [PubMed] [Google Scholar]
- Vu Ngoc T et al. 2013. “hnRNP U Enhances Caspase-9 Splicing and Is Modulated by AKT-Dependent Phosphorylation of hnRNP L.” Journal of Biological Chemistry 288(12): 8575–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainstein Mark A et al. 1994. “CWR22: Androgen-Dependent Xenograft Model Derived from a Primary Human Prostatic Carcinoma1.” Cancer Research 54(23): 6049–52. [PubMed] [Google Scholar]
- Wallace TA et al. 2008. “Tumor Immunobiological Differences in Prostate Cancer between African-American and European-American Men.” Cancer Research 68(3): 927–36. [DOI] [PubMed] [Google Scholar]
- Wang Bi-Dar et al. 2013. “Androgen Receptor-Target Genes in African American Prostate Cancer Disparities.” Prostate Cancer 2013: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Bi-Dar et al. 2015. “Identification and Functional Validation of Reciprocal microRNA-mRNA Pairings in African American Prostate Cancer Disparities.” Clinical Cancer Research 21(21): 4970–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Bi-Dar et al. 2017. “Alternative Splicing Promotes Tumour Aggressiveness and Drug Resistance in African American Prostate Cancer.” Nature Communications 8(15921). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Bi-Dar Dar et al. 2015. “Identification and Functional Validation of Reciprocal microRNA-mRNA Pairings in African American Prostate Cancer Disparities.” Clinical Cancer Research 21(21): 4970–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Eric T et al. 2008. “Alternative Isoform Regulation in Human Tissue Transcriptomes.” Nature 456(7221): 470–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Yang et al. 2014. “The Splicing Factor RBM4 Controls Apoptosis, Proliferation, and Migration to Suppress Tumor Progression.” Cancer Cell 26(3): 374–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will Cindy L and Lührmann Reinhard 2011. “Spliceosome Structure and Function.” Cold Spring Harbor perspectives in biology 3(7): a003707–a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wongpalee Somsakul Pop and Sharma Shalini. 2014. “The Pre-mRNA Splicing Reaction.” In Methods in Molecular Biology (Clifton, N.J.), 3–12. [DOI] [PubMed] [Google Scholar]
- Woods-Burnham Leanne et al. 2017. “The 22Rv1 Prostate Cancer Cell Line Carries Mixed Genetic Ancestry: Implications for Prostate Cancer Health Disparities Research Using Pre-Clinical Models.” The Prostate 77(16): 1601–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolard J et al. 2004. “VEGF165b, an Inhibitory Vascular Endothelial Growth Factor Splice Variant: Mechanism of Action, in Vivo Effect on Angiogenesis and Endogenous Protein Expression.” Cancer Research 64: 7822–35. [DOI] [PubMed] [Google Scholar]
- Yoshida Kenichi et al. 2011. “Frequent Pathway Mutations of Splicing Machinery in Myelodysplasia.” Nature 478(7367): 64–69. [DOI] [PubMed] [Google Scholar]
- Zhou Zhihong, and Fu Xiang-Dong 2013. “Regulation of Splicing by SR Proteins and SR Protein-Specific Kinases.” Chromosoma 122(3): 191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]