

Abstract
Cancer cells from a primary tumor can disseminate to other tissues, remaining dormant and clinically undetectable for many years. Little is known about the cues that cause these dormant cells to “awaken,” resume proliferating and develop into metastases. Studying mouse models, we found that sustained lung inflammation caused by tobacco smoke exposure or nasal instillation of lipopolysaccharide converted disseminated, dormant cancer cells to aggressively growing metastases. Sustained inflammation induced the formation of neutrophil extracellular traps (NETs), and these were required for awakening dormant cancer. Mechanistic analysis revealed that two NET-associated proteases, neutrophil elastase and matrix metalloproteinase 9, sequentially cleaved laminin. The proteolytically remodeled laminin induced proliferation of dormant cancer cells by activating integrin alpha-3beta-1 signaling. Antibodies against NET-remodeled laminin prevented awakening of dormant cells. Therapies aimed at preventing dormant cell awakening could potentially prolong the survival of cancer patients.
Most cancer patients die not from their original primary tumor but from metastases that arise in distant tissues. Often, metastatic disease occurs after a prolonged period of dormancy, when disseminated cancer cells are present but clinically undetectable (1). Disseminated cancer cells can remain dormant for years, even decades, before recurring, or “awakening”, as metastatic cancer. T cells and natural killer cells can eliminate the disseminated cancer cells as they start proliferating, preventing them from reaching clinically detectable levels (2–5). Increased extracellular matrix (ECM) deposition and sprouting angiogenesis have been shown to trigger awakening and metastasis in experimental models (6, 7).
It is still unclear what triggers a change in the balance between signals that keep disseminated tumor cells from growing and those that cause awakening and frank metastases. In breast cancer survivors, elevated plasma levels of C-reactive protein, a non-specific marker of chronic inflammation, are associated with reduced disease-free survival (8), suggesting that inflammation may play a role in the switch between dormancy and metastasis. Inflammation has many causes: for example, smoking induces chronic inflammation in the lung, but the association between smoking and breast cancer risk has been controversial. Nevertheless, two recent, large, pooled analysis studies showed that current smoking or prior heavy smoking was significantly associated with an elevated risk of breast cancer recurrence and death from breast cancer (9, 10). In mice, tobacco smoke exposure increased lung metastasis two-fold (11).
Inflammation is commonly mediated by neutrophils (also called polymorphonuclear leukocytes or PMNs), and these cells are critical for awakening in experimental models (12). Still, it remains unclear how neutrophils cause awakening.
Neutrophils are well known for their ability to kill harmful microorganisms. They do so via: 1) phagocytosis, whereby bacteria or fungi are engulfed and digested; 2) degranulation of cytotoxic enzymes and proteases into the extracellular space; or 3) the formation of neutrophil extracellular traps (NETs)—scaffolds of chromatin with associated cytotoxic enzymes and proteases that are released into the extracellular space where they can trap microorganisms (13). NETs are generated through a signaling process that involves citrullination of histones by the protein arginine deiminase (PAD) 4 enzyme, chromatin decondensation, and disintegration of the nuclear membrane. Contents from the neutrophil’s secretory granules—including neutrophil elastase (NE), cathepsin G (CG), and matrix metalloproteinase 9 (MMP9)—associate with the decondensed chromatin. Finally, the plasma membrane ruptures, and the protease-associated chromatin fibers are released into the extracellular space (14, 15). A growing body of evidence indicates a role for NETs not just in infections, but also in noninfectious inflammatory diseases (13), thrombosis (16, 17), and impaired wound healing in diabetes (18). NETs formed in response to systemic bacterial infection or after surgical stress promote cancer dissemination (19, 20). Using mouse models, we set out to identify how NET structures facilitate metastasis after a period of dormancy.
Inflammation-activated neutrophils drive cancer cell awakening
To determine if local inflammation in the lung could directly drive awakening of disseminated, dormant cancer cells, we studied two models of dormancy. We injected luciferase-and mCherry-expressing breast cancer cells (murine D2.0R and human MCF-7 cell lines) intravenously into syngeneic BALB/c or nude mice, respectively. Even 240 days after injection, tumors did not form. Instead, single, non-proliferative cancer cells were found in the lungs (Fig. 1A and fig. S1, A and B (7, 21)). To explore the effect of lung inflammation on dormancy, we nasally instilled lipopolysaccharide (LPS, also called endotoxin, a potent inducer of inflammation) into mice bearing dormant cancer cells. One LPS instillation, which models a short infection, did not awaken the dormant D2.0R and MCF-7 cancer cells; however, three injections, which model sustained, bacterially induced lung inflammation, led to aggressive lung metastasis (Fig. 1B and fig. S1, C to H). Small clusters of cancer cells appeared between the second and third instillations of LPS, indicating their escape from dormancy, while cancer cells remained as single, non-proliferative cells in control mice (Fig. 1, A to C and fig. S1, A and B). The same results were observed when starting LPS instillation one month, instead of 7 days, after D2.0R and MCF-7 cells’ intravenous injection (fig. S1, I to N), showing that dormant cells remained sensitive to external stimuli even at later time points.
Fig. 1. NETs promote dormant cancer cell awakening after sustained lung inflammation.
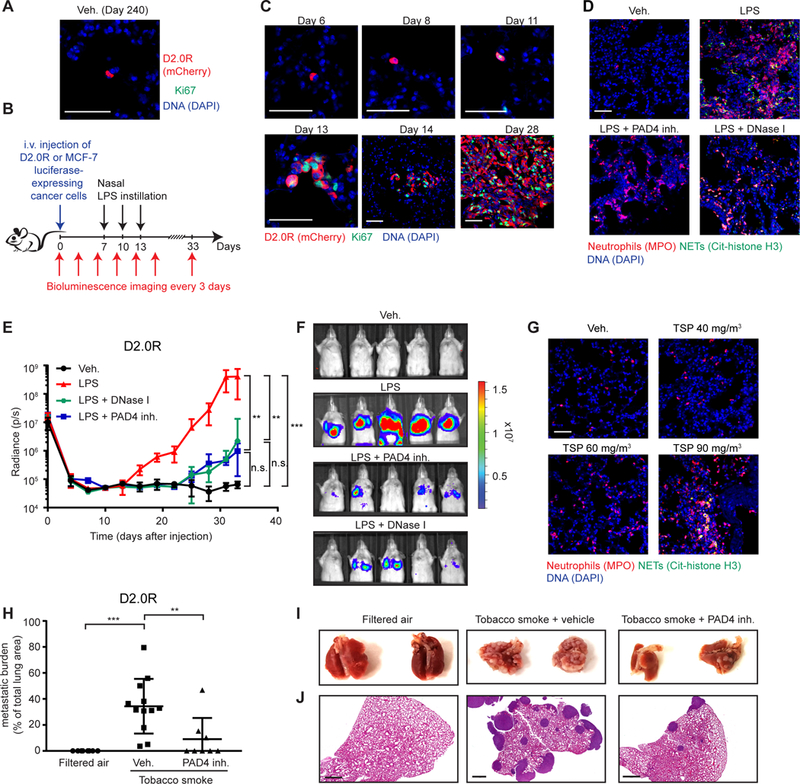
(A, B) D2.0R cells stayed dormant in the lungs of mice for months. (A) Representative micrographs of D2.0R cells (red, mCherry) in Ki67 (green) and DAPI (blue) stained lung sections from untreated mice at day 240. Scale bar: 50 μm. (B) Schematic showing experimental design. (C) Sustained lung inflammation promoted awakening. Representative immunostaining of lungs for D2.0R cells (red, mCherry) and Ki67 (green) with DAPI (blue), at indicated time after injection. Scale bars: 50 μm. (D) Nasal LPS instillation induced NETs in lungs. Representative immunostaining for myeloperoxidase (MPO, red), citrullinated histone H3 (Cit-histone H3, green) and DAPI (blue) in the lungs of mice treated as indicated. Scale bar: 50 μm. (E, F) Targeting NETs reduced inflammation-induced awakening. (E) Mice with D2.0R cells, treated as indicated, were monitored by bioluminescence imaging (BLI) (n=10 mice for vehicle and LPS groups; n=5 mice for DNase I and PAD4 inhibitor groups; mean±SD). (F) Representative BLI images at day 33. (G) Exposure to tobacco smoke (TSP for total suspended particles) induced NETs in the lungs. Representative immunostaining for myeloperoxidase (red), citrullinated histone H3 (green) and DAPI (blue) in the lungs of mice treated as indicated. Scale bar: 50 μm. (H-J) Targeting NETs reduced smoking-induced awakening. (H) Mice with D2.0R cells were treated as indicated, and lung metastatic burden quantified from hematoxylin and eosin (H&E) staining (n=8 mice for filtered air and tobacco smoke + PAD4 groups; n=12 mice for tobacco smoke + vehicle group; mean±SD). (I) Lungs from mice treated as indicated at day 30. (J) Representative images of H&E staining of lungs from panel (H) at day 30. Scale bar: 700 μm.
LPS instillation caused dramatic neutrophil recruitment (movies S1 and S2), both after one and three instillations (fig. S2, A to C). Still, the relationship between the awakening of D2.0R proliferation and neutrophil recruitment and activation remained unclear. To assess these dynamics, we used confocal intravital lung imaging (22) with a fluorescence ubiquitination-based cell cycle indicator (FUCCI) system, which fluorescently labels cells red in G0 and G1 phases and green in S/G2/M phases (fig. S2D, (23)). Neutrophil infiltration and activation were tracked using the LysM-EGFP transgene and the NE 680 FAST probe, which fluoresces after NE cleavage.
In control mice instilled with PBS, NE activity was low and all D2.0R cells were red in the G0/G1 cell cycle phases at days 8, 11, and 21 (movies S3 to S5). In contrast, beginning 5 days after the first LPS instillation (day 11), D2.0R cells became yellow, indicating that they had entered the G1/S transition of the cell cycle, and this transition correlated with high neutrophil recruitment and NE activity (movie S6 and fig. S2, E and F). We detected small clusters of proliferating cells at day 14 and established proliferative lung metastasis at day 21 (movies S7 and S8, and fig. S2F).
We next investigated whether neutrophils contributed to awakening of dormant cancer cells. Indeed, neutrophil depletion completely prevented inflammation-induced awakening of dormant D2.0R and MCF-7 cancer cells (fig. S3, A to I). Thus, sustained inflammation induced by LPS causes dormant cancer cells to re-enter the cell cycle, and this effect requires neutrophils.
NETs awaken dormant cancer cells in mice
After LPS instillation, we detected numerous NETs in the lungs within 4 hours, persisting 24 hours later; in contrast, NETs were absent in normal lung tissue (Fig. 1D and fig. S3, J and K). NETs were also present in the plasma after LPS treatment, as assessed by double stranded DNA (dsDNA) assays or an ELISA against DNA-bound NE (fig. S3, L and M).
To determine whether NETs contributed to cancer cell awakening, we blocked NET formation with a PAD4 inhibitor or digested the NET-DNA scaffold with DNase I (free or coated on nanoparticles) (Fig. 1D and fig. S4, A to F). Both treatments prevented or decreased LPS-induced awakening of dormant D2.0R and MCF-7 cancer cells (Fig. 1, E and F, and fig. S4, G to M). Results were similar when LPS instillation was started one month after intravenous injection of the cancer cells (fig. S1, I to N), showing that NETs remained a powerful external stimulus of awakening even at later time points. DNase I was most effective as a preventative treatment, immediately before LPS instillation, but also reduced end-point metastatic burden when administered at day 14, after the appearance of small clusters of cancer cells in the lungs (fig. S4N). In all cases, the NET-targeting treatments reduced neutrophil recruitment (fig. S4, O and P), suggesting that NETs promote further inflammation.
The cancer cell dormancy models described above were generated by intravenous injection of cancer cells, so we used two additional approaches to explore whether NETs could also induce proliferation after natural dissemination of cancer cells. First, we used the RapidCaP prostate cancer model (24), where prostate cancer develops from normal epithelial tissues within an intact organ. The model has a low incidence of lung metastasis (24), and we observed no lung metastasis in control mice during the observation period, in spite of the presence of single, disseminated cancer cells in the lungs. In contrast, LPS instillations resulted in lung metastasis in 3 of 5 mice. However, 0 of 5 mice developed macroscopic metastasis when NET formation was blocked with the PAD4 inhibitor, even after three additional LPS instillations (fig. S5, A to C, p=0.02). In the second approach, we allowed MCF-7 cells to form primary mammary tumors and spontaneously disseminate. We then resected those primary tumors. Under these conditions also, LPS-induced NETs awakened the cancer cells (fig. S5, D to H).
Repeated LPS instillation models a sustained bacterially-induced inflammation. Smoking similarly induces chronic lung inflammation and has been associated with increased risk of breast cancer recurrence (9, 10). To examine NETs and cancer cell dormancy under these conditions, we exposed mice to three different concentrations of tobacco smoke for three weeks. This resulted in a dose-dependent increase in neutrophil infiltration in the lungs. NETs formed in the lungs at the highest tobacco smoke exposure level (a level almost equivalent to moderate, active smoking and well above secondhand exposure to tobacco smoke; Fig. 1G and fig. S5, I and J). NETs, as well as LPS, were also found in the plasma of the tobacco smoke-exposed mice (fig. S5, K to M), suggesting that tobacco exposure causes systemic exposure to both agents.
We next tested whether the tobacco smoke could awaken dormant D2.0R cells. Indeed, aggressive metastasis developed when mice were exposed to tobacco smoke at the level that induced NET formation, and this was prevented by PAD4 inhibitor treatment. Cells remained dormant in mice exposed to filtered air (Fig. 1, H to J and fig. S5N). Thus, NETs formed during inflammation induced the awakening of dormant cancer cells in multiple mouse models.
NETs awaken slow-cycling cells in vitro in ECM models
To determine how NETs induced awakening of dormant cancer cells, we turned to 3-dimensional (3D) culture systems, where the isolated effect of NETs could be tested in the absence of the many cell types present in lungs. Both D2.0R and MCF-7 cells become slow-cycling when cultured on basement membrane matrix (matrigel) (25), whereas metastatic D2.A1 cells, isolated from the same mammary lesion as the D2.0R cells, proliferated (fig. S6, A and B). To generate NETs, we cultured freshly isolated neutrophils and stimulated them with LPS, phorbol 12-myristate 13-acetate (PMA), or N-formyl-methionyl-leucyl-phenylalanine (fMLP) (Fig. 2A, and fig. S6, C and D). We found that NETs were also induced by co-culturing neutrophils with metastatic D2.A1 cells but not with the dormant D2.0R cells NETs (fig. S6C). This is similar to our previous findings using another pair of cell lines (22), the metastatic/NET-inducing 4T1 and the non-metastatic/non-NET-inducing 4T07 cell lines. Finally, to compare the effects of NETs with that of neutrophil degranulation, we induced degranulation by culturing with complement 5a (c5a). We next tested the effect of the NET-containing conditioned media (CM) from the different neutrophil culture conditions on luciferase-expressing D2.0R and MCF-7 cancer cells cultured on matrigel (Fig. 2A). NET-containing CM led to the awakening and proliferation of dormant cancer cells, whereas CM from degranulated or inactivating conditions had no effect (Fig. 2, B to D, fig. S6E, and fig. S7, A to C). LPS, PMA, fMLP, and c5a added directly to the cancer cells had no effect on awakening in our 3D culture model systems and neither did CM from D2.A1 or D2.0R cancer cells (fig. S7, D and E). Treatment of D2.0R cells with LPS before intravenous injection also did not lead to metastasis (fig. S7, F and G), further suggesting that it was the NETs in the CM and not LPS that activated the slow-cycling cells. When the neutrophil cultures were treated with a PAD4 inhibitor or DNase I during NET-activating conditions, NETs did not form or were digested (fig. S6, C and D), and the CM no longer induced awakening of the dormant cancer cells (Fig. 2D and fig. S7B).
Fig. 2. NETs induce awakening in vitro in the absence of other host cells.
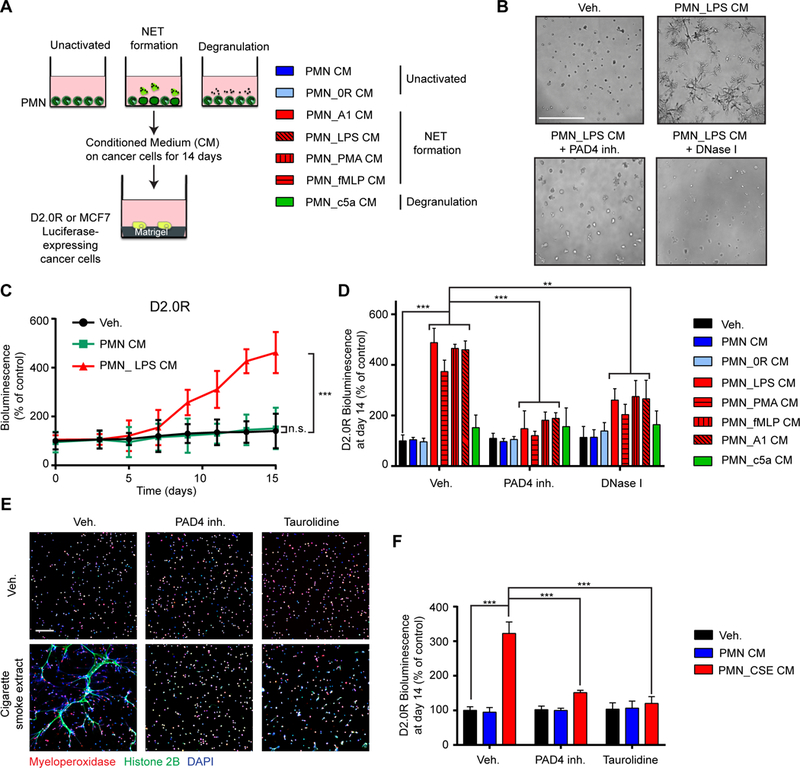
(A) Experimental design for inducing NETs or degranulation neutrophils (PMNs). Conditioned medium (CM) was collected 20 hours after activation and added to cultures of luciferase-expressing cancer cells plated on top of matrigel. (B-D) NET-containing CM induced awakening of D2.0R cells on matrigel. (B) Representative images of 3D cultures at day 15, treated as indicated. Scale bar: 250 μm. (C) BLI quantification over 15 days with indicated treatments (n=3; mean±SD). (D) BLI signal 14 days after indicated treatments. PAD4 inhibitor and DNase I were used during neutrophil culture to block and digest NET formation, respectively (n=3; mean±SD). (E, F) Cigarette smoke extract (CSE) induced the formation of NETs and subsequent awakening in vitro. (E) Immunostaining of mouse neutrophils cultured as indicated. DAPI (blue), anti-MPO (red), and anti-histone H2B (green) staining were used to assess NET formation. Scale bar: 100 μm. (F) BLI signal 14 days after indicated treatments. Taurolidine and PAD4 inhibitor were used during neutrophil culture to block NET formation (n=3; mean±SD).
Given that nicotine can induce NETs in vitro (26), we next tested the effect of cigarette smoke extract (CSE) on neutrophils. We found that CSE induced NETs in vitro in a dose-dependent manner and that this NET-containing CM could also awaken the slow-cycling cells (Fig. 2, E and F, and fig. S7, H and I). Because we had detected LPS in the plasma of tobacco smoke-exposed mice, we assessed the effect of taurolidine, a compound which neutralizes the effect of LPS (27). Taurolidine and PAD4 inhibition both reduced CSE-induced formation of NETs, and the CM was no longer able to awaken the cancer cells (Fig. 2, E and F, and fig. S7, H and I), suggesting that LPS present in CSE drives smoking-induced NETs. CSE had no effect on awakening when it was added directly to the cancer cells in vitro, and caused cancer cell death at the highest concentrations (fig. S7J). Altogether, these results show that NETs induced by multiple means promote cancer cell awakening in vitro in the presence of an artificial ECM and in the absence of lung cells, other immune cells, and vasculature.
NET-associated proteases awaken cancer cells
Degradation of the ECM, activation of cell surface receptors, and the release and activation of cytokines and growth factors by proteases are necessary steps in metastasis (28, 29). We hypothesized that NET-associated protease activity was responsible for the effects of NETs on awakening. Using our 3D culture systems, we analyzed the effects of inhibiting three major NET-associated proteases: cathepsin G (CG), neutrophil elastase (NE), and matrix metalloproteinase 9 (MMP9). Inhibiting NE or MMP9, but not CG activity, in the NET-containing CM prevented awakening of the slow-cycling cancer cells (Fig. 3A and fig. S8, A to D). We also isolated neutrophils from NE and MMP9 knock-out mice and stimulated them to form NETs. CM from these cultures did not induce awakening (fig. S8, E and F). We found that neutrophils, but not cancer cells, were the main source of these proteases. NET-containing CM did not induce expression of the proteases in cancer cells (fig. S9, A to C), and shRNA-mediated knock-down of NE and MMP9 in D2.0R cells did not affect the capacity of these cells to be awakened by NET-containing CM in vitro (Fig. S9, D to F).
Fig. 3. NET-associated NE and MMP9 induce awakening from dormancy through extracellular matrix remodeling.
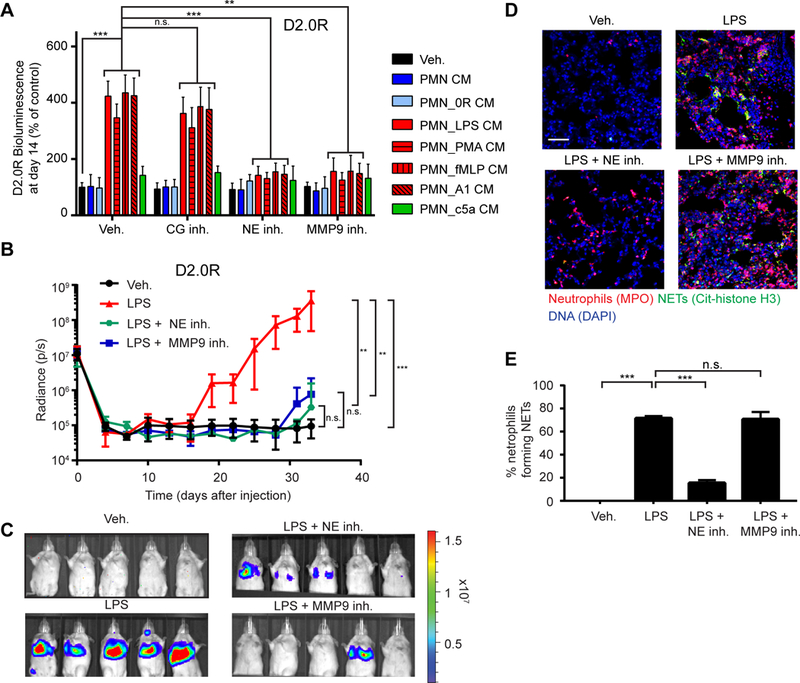
(A) NE and MMP9 were required for NET-induced awakening of D2.0R cells in vitro. BLI signal of luciferase-expressing cells 14 days after indicated treatments. CG, NE, and MMP9 inhibitors were used during cancer cell culture (n=3; mean±SD). (B, C) NE and MMP9 activity were required for LPS-induced awakening in vivo. (B) Mice with D2.0R cells were treated as indicated and monitored by BLI (n=10 mice for vehicle and LPS groups; n=5 mice for NE inhibitor and MMP9 inhibitor groups; mean±SD). (C) Representative BLI images at day 33. (D, E) NE, but not MMP9 inhibition, prevented formation of NETs after LPS-induced lung inflammation. (D) DAPI (blue), anti-MPO (red), and anti-citrullinated histone H3 (green) staining were used to assess NET formation in the lungs of mice treated as indicated. Scale bar: 50 μm. (E) Quantification of NET-forming neutrophils (n=3 mice; mean±SD).
In vivo, inhibiting NE and MMP9 activity also prevented LPS-induced cancer recurrence (Fig. 3, B and C, and fig. S9, G to J). NE inhibition prevented NET formation in vivo, consistent with the known requirement for NE activity during NET formation (30, 31). In contrast, NETs still formed in vivo after MMP9 inhibition, but cells remained dormant, suggesting that the activity of this NET-protease is critical for awakening (Fig. 3, D and E). Similar to the prevention of NET formation by PAD4 inhibition, inhibition of NE, but not MMP9, also reduced neutrophil recruitment to the lungs after LPS-induced inflammation (fig. S9K).
To determine whether the NET-proteases induced awakening by acting directly on the cancer cells or by altering their microenvironment, we pre-incubated matrigel with NET-containing CM before cancer cell plating (fig. S9L). This was sufficient to induce awakening, but required both NE and MMP9 activity (fig. S9M, red bars). In contrast, if the proteases were inhibited during cancer cell culture, after NET-mediated matrigel remodeling had already occurred, then awakening was not inhibited (fig. S9M yellow bars). These data suggest that NET-associated NE and MMP9 induce awakening of dormant cancer cells through ECM remodeling.
NETs awaken cancer cells by laminin remodeling
We next sought to identify the ECM substrate of NE and MMP9 during NET-mediated awakening of dormant cancer cells. According to the manufacturer, matrigel is composed mainly of laminin-111 (60%), collagen type IV (30%), and entactin (8%). Proteolysis of laminin-111, which consists of the α−1, β−1, and γ−1 laminin chains, has previously been linked with regulation of proliferation and cellular architecture in 3D cultures (32). Both D2.0R and MCF-7 cells remained slow-cycling when cultured on purified laminin-111 in vitro (fig. S10, A and B), but were awakened by NET-containing CM (Fig. 4A, and fig. S10, C to E).
Fig. 4. NETs facilitate laminin-111 remodeling.
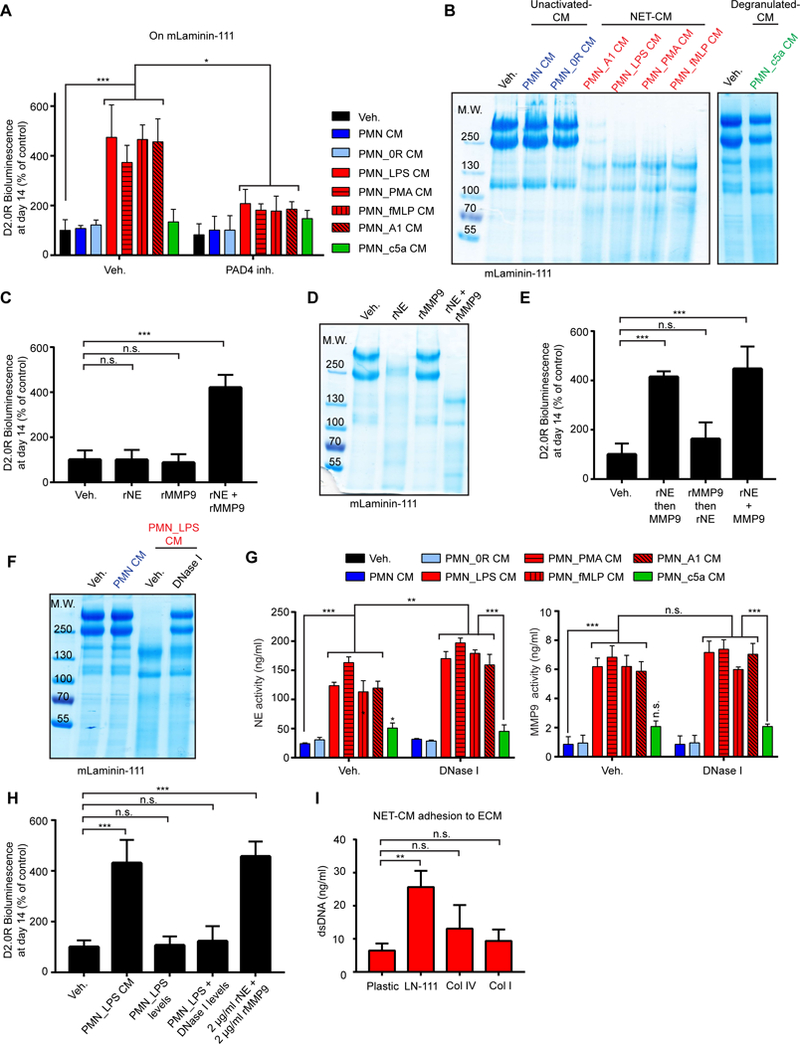
(A) Culturing cancer cells on laminin-111 allowed NET-dependent control of dormancy and awakening. BLI signal of luciferase-expressing D2.0R cells after 14 days under indicated conditions (PAD4 inhibitor was used during PMN culture, n=3; mean±SD). (B) NET-containing CM cleaved laminin-111. Cleavage of laminin-111 after incubation with indicated CM was detected by SDS-PAGE, under reducing and denaturing conditions, and coomassie blue staining. (C-E) Both NE and MMP9 were required for awakening and laminin-111 cleavage. Laminin-111 was incubated with recombinant proteases alone (C) or added sequentially (E), and D2.0R cells were cultured on the remodeled matrix (C, E) or laminin-111 was analyzed by SDS-PAGE (D). BLI was used to quantify the cancer cells after 14 days (for experimental design, see Fig. S22B) (n=3; mean±SD). (F) NET-associated proteases had higher laminin-111 cleavage activity than DNase-released NE and MMP9. Laminin-111 was incubated as indicated before SDS-PAGE. DNase I was added after PMN culture. (G) NE and MMP activity toward soluble fluorescent substrates was not reduced by DNase I treatment of NET-containing CM. NE and MMP activity in the indicated PMN CM. DNase I was used after PMN activation (n=3; mean±SD). (H) NET-associated proteases had higher awakening activity than free NE and MMP9. Laminin-111 was incubated as indicated before D2.0R cell culture. BLI quantification of cancer cells at day 14 (n=3; mean±SD). (I) NETs bound preferentially to laminin-111. CM from neutrophils stimulated with LPS to form NETs was incubated on plastic or ECM-coated plates as indicated. dsDNA bound to ECM proteins were quantified (n=3; mean±SD).
To test whether NET-associated proteases could cleave laminin, we used commercially available purified laminin-111. While NET-containing CM led to laminin-111 cleavage, CM from degranulating or non-activating conditions did not (Fig. 4B and fig. S10F). Using recombinant (r) NE and MMP9, we found that both proteases in combination were necessary to induce cancer cell awakening (Fig. 4C and fig. S10, G to I). Laminin-111 was cleaved by rNE alone, but not by rMMP9; however, when both proteases were present, the cleavage fragments were different (Fig. 4D and fig. S10J). Similarly, when laminin-111 was treated with NET-containing CM, cleavage was completely blocked after NE inhibition, but only partially after MMP9 inhibition (fig. S10K). Therefore, we hypothesized that laminin-111 is cleaved sequentially – first by NE and subsequently by MMP9 – to trigger awakening. Indeed, proteolysis of laminin-111 first by rNE and then by rMMP9, but not in the reverse order, led to cancer cell awakening (Fig. 4E and fig. S10G).
Our in vitro experiments suggest that laminin-111 is the primary substrate of NET proteases in 3D culture. However, laminin-111 has not consistently been detected in adult lungs. Therefore, we assessed the expression of laminin in lung tissue. We confirmed the presence of mRNA coding for the three laminin-111 chains by qPCR and in situ hybridization, and we detected laminin-111 protein using multiple antibodies by immunofluorescence and Western blot (Fig. S11, A to D). Laminin-111 mRNA expression was not affected by nasal LPS instillation, but cleaved laminin-111 was readily detectable in lung tissue lysate after inducing inflammation with LPS (fig. S11, A and D). A polyclonal anti-laminin-111 antibody was used for western blot and immunofluorescence analysis; thus, we could not exclude the possibility that this reagent recognized not only laminin-111 but also other α−1, β−1, or γ−1 containing laminin isoforms. Hence, we tested whether NET-containing CM also could awaken cancer cells cultured on other laminin isoforms. Indeed, we found that laminin-211, −411, and −511 were all sufficient to support NET-induced awakening (fig. S11E), suggesting that awakening can occur in any tissue containing one of these laminins, e.g., at the perivascular niche.
NET-DNA acts as a proteolysis scaffold
Our data showed that proteolytic remodeling is required for cancer cell awakening; yet, digesting NET-DNA with DNase I also prevented awakening in vivo and in vitro (Fig. 1, E and F, Fig. 2B and D, fig. S4, G to I, fig. S4, L and M, and fig. S7B). Consistently, laminin-111 was readily cleaved by incubation with NET-containing CM, and this cleavage was prevented by DNase I (Fig. 4F). Nevertheless, DNase I digestion did not reduce the protease activity against soluble fluorescent substrates (Fig. 4G). However, adding recombinant proteases at the activities measured in the NET-containing CM (120 ng/ml rNE, 6 ng/ml MMP9) or in NET-containing CM after DNase I treatment (160 ng/ml rNE, 6 ng/ml MMP9) had no effect on awakening. Instead, much higher concentrations of the recombinant proteases (2 μg/ml each, the concentrations used in Fig. 4, C to E, fig. S10, H to K, in accordance with the literature) were necessary to induce cancer cell awakening (Fig. 4H). We found areas of NE co-localizing with MMP9 on the DNA fibers of NETs (fig. S11F). Furthermore, NET-DNA preferentially bound to laminin-111 over other tested ECM proteins (Fig. 4I and fig. S11G). These findings suggested that the NET-DNA scaffold allowed the NET-proteases to cleave their substrate more efficiently than when the proteases freely diffused because the DNA binds to laminin and because the two proteases co-localize to the same DNA-scaffold.
Thrombospondin-1 (TSP-1), a large glycoprotein present in the basement membrane surrounding mature blood vessels, regulates cancer cell dormancy (6). TSP-1 secretion by bone marrow-derived Gr1+ cells (which include neutrophils) generates a metastasis-resistant microenvironment, which can be overcome through neutrophil-mediated proteolysis of TSP-1 (33, 34). We found that TSP-1 was also a substrate for NET-associated NE and MMP9 in vitro (fig. S11H) and that it was degraded in vivo after LPS-induced lung inflammation (fig. S11I). TSP-1 degradation alone did not convert slow-cycling cells to proliferating cells in vitro (fig. S11J). However, intact TSP-1 decreased proliferation caused by cleaved laminin-111 (fig. S11, K and L). Our data suggest that TSP-1 modulates cancer cell awakening caused by proteolytic remodeling of laminin-111.
NET-remodeled laminin activates integrin a3β1 signaling
We next focused on how NET-remodeling of laminin-111 led to cancer cell awakening. Integrins are cell surface ECM receptors that recognize conformational changes in the ECM, and signaling through integrin β1 has previously been shown to induce awakening (7, 35–37). Consistently, a marker for cell proliferation, Ki67, was associated with integrin β1 activity and reorganization of the actin cytoskeleton after incubating D2.0R or MCF-7 cells with NET-containing CM or rNE and rMMP9 (Fig. 5A, and fig. S12, A and B). The integrin β1 outside-in signaling pathway can activate the downstream mediators focal adhesion kinase (FAK), MAP kinase ERK kinase (MEK), extracellular signal regulated kinase (ERK), myosin light chain kinase (MLCK), myosin light chain 2 (MLC2), and yes-associated protein (YAP). Accordingly, NET-induced awakening led to activation of FAK, ERK, and MLC2, which required NE and MMP9 activity (Fig. 5B). Moreover, targeting the integrin β1 outside-in signaling pathway at different signaling points using chemical inhibitors or RNA interference inhibited NET-induced awakening in vitro (Fig. 5, C and D, and fig. S12, C and D). Inhibition of integrin β1, FAK, MEK, MLCK, myosin, and YAP activities all led to the loss of FAK, ERK, and MLC2 phosphorylation upon stimulation with NET-containing CM (Fig. S12, E to K), in accordance with the positive feedback loop within this signaling pathway (38). To identify which β1-containing integrin pair was involved in cancer cell awakening, we tested D2.0R cells for expression of α-integrins known to associate with integrin β1 (fig. S13A) (39). Through shRNA mediated knock-down (fig. S13B), we found awakening of dormant cancer cells by NETs required integrin α3, integrin β1, and YAP in the cancer cells (Fig. 5, E to G, and fig. S13, C and D).
Fig. 5. Integrin β1 signaling pathway regulates NET-induced awakening.
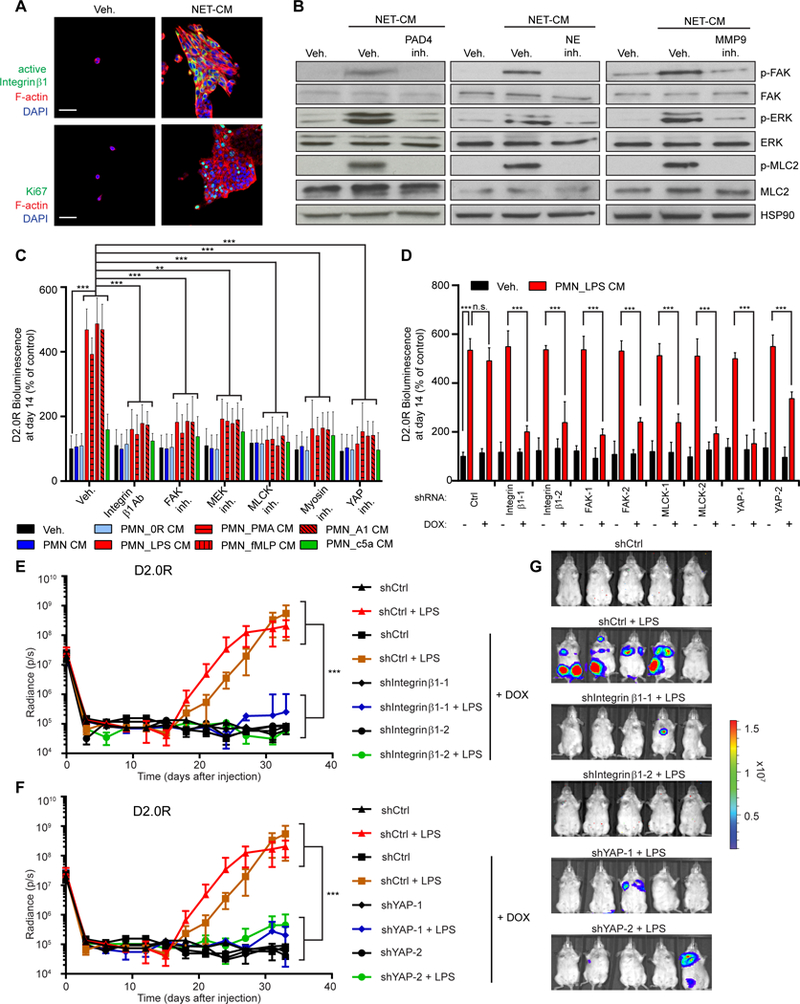
(A) NETs activated integrin β1 and proliferation in dormant cancer cells. D2.0R cells cultured on matrigel-coated 0.2 kPa hydrogel stained for active integrin β1 (green, top) or Ki67 (green, bottom), phalloidin (red), and DAPI (blue). Scale bar: 50 μm. (B) NET-containing CM activated the integrin β1 signaling pathway. D2.0R cells cultured on matrigel for 10 days under indicated conditions, analyzed for phospho-FAK, phospho-ERK½, and phospho-MLC2 by Western blot. Controls: total FAK, ERK½, MLC2, and Heat Shock Protein 90 (HSP90). (C, D) Activation of the integrin β1 pathway is required for NET-induced awakening. (C) BLI of luciferase-expressing D2.0R cells cultured on matrigel for 14 days under indicated conditions (n=3; mean±SD), and (D) cultured on matrigel using doxycycline to induce expression of indicated shRNAs (n=3; mean±SD). (E-G) Inhibition of integrin-mediated YAP activation prevents LPS-induced awakening. (E, F) Mice with inducible shRNA-expressing D2.0R cells were monitored by BLI. Doxycycline treatment was started on day 5 (n=5 mice per group; mean±SD). (G) Representative BLI images at day 33.
LPS-induced lung inflammation resulted in laminin cleavage detectable in lung lysate, and this cleavage depended on neutrophil recruitment, NET formation, and NE and MMP9 activities (fig. S14A). Laminin cleavage was blocked after NE inhibition, but only partially after MMP9 inhibition, consistent with sequential laminin cleavage by NE and MMP9 in vivo. However, no discernable changes in laminin immunofluorescent staining patterns were observed in lungs after LPS-induced inflammation (fig. S10H), and under non-reducing and non-denaturing conditions, we did not observe many laminin-111 fragments after rNE and rMMP9 cleavage, but rather a reduction in its apparent molecular weight (Fig. 6A). These results suggest that NET-mediated proteolysis induced a change in the 3D structure of laminin. We hypothesized that a new laminin-111 epitope was revealed after NET-mediated proteolysis and that the dormant cancer cells sensed this new epitope through integrin α3β1, leading to cancer cell proliferation. To test this idea, we generated monoclonal antibodies (mAbs) from rats immunized against purified rNE and rMMP9 cleaved laminin-111 (fig. S14B). Three laminin-111-recognizing antibody clones (Ab19, Ab25, and Ab28) highly blocked NET-induced integrin β1 activation and subsequent cancer cell awakening in vitro (Fig. 6B and fig. S14C). Of these antibodies, Ab28 specifically recognized NET-remodeled laminin-111 but not intact laminin, while Ab19 and Ab25 recognized both laminin-111 forms (Fig. S14, D and E). We cannot exclude the possibility that other laminin isoforms share the antibody epitopes after NET-mediated remodeling and also are recognized by the antibodies.
Fig. 6. A NET-generated laminin epitope promotes awakening.
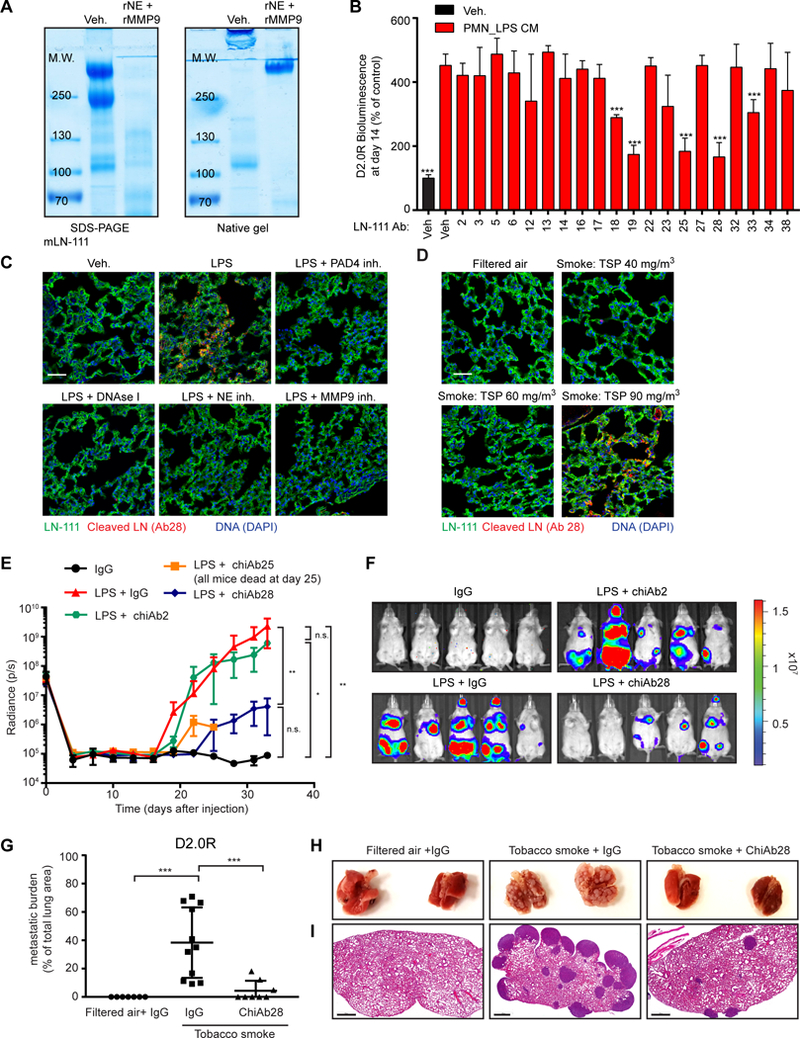
(A) Laminin-111 was not fully degraded by NET-associated proteases. Laminin-111 was incubated with rNE and rMMP9, and cleavage was assessed by SDS-PAGE and coomassie blue staining under reducing/denaturing or non-reducing/non-denaturing conditions. (B) Antibodies against NE- and MMP9-cleaved laminin-111 prevented NET-mediated awakening. BLI of luciferase-expressing D2.0R cells cultured on laminin-111, 14 days after treatment with indicated antibody clone (n=3; mean±SD). (C, D) Lung inflammation and NETs induce production of cleaved laminin. Immunostaining of lungs for DAPI (blue), full-length laminin-111 (ab11575 from Abcam, green), and cleaved laminin antibody 28 (red), treated as indicated. Total suspended particles (TSP) in cigarette smoke exposure conditions indicated. Scale bars: 50 μm. (E-I) Antibodies against NET-remodeled laminin prevented inflammation-induced awakening in vivo. (E) Mice with D2.0R cells were treated as indicated and monitored by BLI (n=5 mice per group; mean±SD). (F) Representative BLI images at day 33. (G) Metastatic burden in lungs from mice exposed to tobacco smoke quantified with H&E staining. (H) Representative photos of lung. (I) Representative H&E images of lungs from panel (H) at day 30 (n=7 mice for filtered air + IgG, n=8 mice for tobacco smoke + ChiAb28 inhibitor, n=11 mice for tobacco smoke + IgG; mean±SD). Scale bar: 700 μm.
Using Ab28, we detected the NET-remodeled laminin epitope in inflamed lungs. It was not present after neutrophil depletion, or after inhibiting NETs formation or NE or MMP9 activities (Fig. 6C and fig. S14F). The epitope was also present in lung tissue from tobacco smoke-exposed mice, but only at an exposure level that induced NETs (Fig. 6D). Moreover, the NET-remodeled laminin epitope was only detectable in lungs after three LPS instillations, the number of doses required to awaken cancer cells (fig. S15A).
The NET-remodeled laminin epitope was detectable in the same lung regions as the NETs (fig. S15A). Furthermore, using D2.0R cells expressing the FUCCI cell cycle reporter (fig. S2D), we observed that all green cells (indicating S, G2, or M cell cycle phase) or cells that were part of a cluster, were located next to remodeled laminin, whereas cells close to intact laminin all remained red (indicating G0/G1 cell cycle phase) (fig. S15, B and C). This co-localization pattern suggested that the NET-remodeled laminin epitope was driving awakening of cancer cells in vivo. To test this, we engineered rat monoclonal antibodies as chimeric mouse IgG2a antibodies (chiAbs), which can be used in mice without eliciting an immune response. ChiAb28 inhibited cancer cell awakening in vitro (fig. S15D) and in both in vivo systems (LPS- and tobacco smoke-induced inflammation, Fig. 6, E to I, and fig. S15, E and F). Together, our data identify a mechanism by which sustained lung inflammation, induced by either tobacco smoke or LPS exposure, can lead to NET-induced proteolytic laminin remodeling that can drive cancer recurrence in the lungs of mice.
Discussion
Neutrophils recruited during lung inflammation can initiate awakening of dormant cancer cells, as shown here and in previous work (12). Now, we report that NETs formed by neutrophils during LPS- or tobacco smoke-induced lung inflammation are required to awaken dormant cancer cells and cause metastasis in mice. We propose that NETs concentrate neutrophil proteases, NE and MMP9, at their substrate, laminin, allowing sequential cleavage and generating an epitope that triggers cancer cell awakening.
Previous studies pointed to a central role for integrin β1 signaling in the awakening of dormant cancer cells (7, 21, 35, 40), but it was unclear what types of stimuli could activate integrin signaling in dormant cancer cells. We have found that NET-remodeled laminin formed during lung inflammation serves as an integrin α3β1 activating epitope, leading to FAK/ERK/MLCK/YAP signaling in cancer cells and to awakening. Based on these results, we developed an inhibitory antibody against NET-remodeled laminin that prevented dormant cancer cells from awakening in response to LPS- or tobacco smoke-induced lung inflammation. Given that our antibody preserves normal integrin β1 signaling, it may be a potent strategy to prevent cancer recurrence and more broadly serve as a treatment for other NET-associated pathologies, such as transfusion-related acute lung injury (41).
Our in vitro experiments identified laminin-111, −211, −411, and −511 as key ECM proteins that cause NET-induced cancer cell awakening. It is particularly intriguing that NETs target laminin-411 and −511, present in the perivascular niche, because this niche has been shown to regulate breast cancer dormancy (6). In previous studies. TSP-1, which is also present in this niche, was identified as a regulator of tumor dormancy and metastasis (6, 33, 34). In vitro, NET-mediated degradation of TSP-1 did not induce awakening. Rather, intact TSP-1 inhibited NET-induced awakening, and NET-mediated degradation of TSP-1 overcame the inhibition. Thus, TSP-1 degradation is likely required in parallel with laminin remodeling in the intact tissue to create a permissive niche for the awakening of dormant cancer cells.
Stiffness of the environment can lead to cell tension-mediated YAP activity, and we have demonstrated that YAP activity is critical for inflammation-induced cancer cell awakening. Our results therefore suggest that changes in cell tension occur after binding to NET-remodeled laminin and play a role in awakening of disseminated dormant cancer cells. Consistently, experimental lung fibrosis, which is associated with increased ECM stiffness, induces cancer cell awakening from dormancy (7). Furthermore, D2.0R and MCF-7 cells are dormant in soft environments in vitro—such as on matrigel, laminin-111 gel, and 0.2 kPa hydrogel (which mimics the stiffness of the lungs)—but these cells proliferate on stiff 2D cell culture dishes. In vivo, dormant, disseminated tumor cells are preferentially located within the soft niches of stiff tissues such as bones, i.e., the bone marrow (42).
NETs drive dormant cancer cells to initiate proliferation, but T cells and natural killer cells may recognize these disseminated cells as they start to proliferate (2, 3, 43). The role of the adaptive immune system in our mouse models is unclear, but LPS increases glucocorticoid levels (44), inhibiting adaptive immune cells. Therefore, LPS and smoking may spur the growth of dormant cells by providing signals to both initiate proliferation, via NETs, and to overcome immune control, through glucocorticoids.
Chronic inflammation and smoking are well known risk factors in metastatic recurrence (8, 9). Obesity is also associated with chronic, low-grade inflammation and with elevated NET release (45), and neutrophils contribute to obesity-associated metastasis in mice (46). Our findings set the stage for epidemiological studies examining whether there are any correlations between inflammation/smoking/obesity, NETs, and cancer recurrence after a long period of dormancy in patients. If such correlations exist, we envision that NETs and their downstream effectors could be targeted to reduce the risk of cancer recurrence.
Methods summary
Proliferation assay
96-well culture-plates were coated with 50 μL of growth factor reduced matrigel (#356231, Corning) or mLN-111 (#3446–005-01, R & D Systems) and incubated at 37 °C for 30 min. The murine D2.0R cell line was used on matrigel or mLN-111 as indicated in the figures, and the human MCF-7 cell line was used on matrigel. mCherry-luciferase cells (2×103) were resuspended in 100 μL of DMEM supplemented with 1% FCS and 2% matrigel or 2% mLN-111 and grown on the coated wells. The next day, media were replaced with 100 μL of conditioned medium (CM) from neutrophils cultured at indicated conditions. The CM was changed every four days. After 14 days, 100 μl of medium containing 5 μg/ml of luciferin (#Luck-1G, Goldbio) were added to the wells and proliferation was measured by bioluminescence imaging (BLI) using a plate reader (SpectraMax i3, Molecular Devices). To measure the proliferation of D2.0R cells expressing integrin, NE, and MMP9 shRNA, CellTiter 96® AQueous One Solution Cell Proliferation Assay (#G3580, Promega) was used, following the manufacturer’s instructions.
Animals
Female BALB/c, nude, and C57BL/6J mice were purchased from Charles River Laboratories. Female NE Knock Out (KO) (#6112) and MMP9 KO (#7084) mice were purchased from The Jackson Laboratory. RapidCaP mice consisted of PtenloxP/loxP; Trp53loxP/loxP; TdTomatoloxP/+ transgenic mice into which a Luc-Cre lentiviral plasmid (Tyler Jacks, #2090, Addgene, purchased from The University of Iowa Viral Vector Core) was injected into the prostate, as previously described (24). All procedures were approved by the Cold Spring Harbor Laboratory or University of California, Davis Institutional Animal Care and Use Committee (IACUC) and were conducted in accordance with the NIH’s “Guide for the Care and Use of Laboratory Animals.”
Activation of neutrophils and NET formation assays
Isolated neutrophils (250,000) were cultured in 24-well plates containing 500 μl of serum-free DMEM and activated overnight with LPS, PMA, fMLP, or CSE to induce NETs. Cancer cells were also co-cultured with neutrophils to induce NETs, as previously described (22), using Transwell with the two chambers separated by a 0.4 μm porous membrane (#353495, Corning). Neutrophils were placed on the bottom of the lower chamber, while the cancer cells (100,000 D2.0R or D2.A1 cells) were placed on the membrane of the upper chamber. To induce degranulation, recombinant complement 5a was used. The next day, the neutrophil CM were collected. GSK484 and DNase I were added 30 min before neutrophil activation to inhibit PAD4 or to digest NET-DNA, respectively. To assess NET formation, neutrophils grown on poly-L-lysine-coated coverslips (#354085, Corning) were fixed with 4% paraformaldehyde (PFA) for 20 min at room temperature, rinsed twice in PBS, incubated in 50 mM of NH4Cl for 10 min and permeabilized with 0.5% Triton X-100 (#BB151–500, Thermo Fisher Scientific) for 5 min. Cells were next blocked in PBS containing 1% bovine serum albumin (BSA, #A3294, Sigma) for 30 min and incubated with anti-H2B (1:200) and anti-myeloperoxidase (1:400) antibodies in blocking buffer overnight at 4 °C. After two washes in PBS, cells were incubated in the presence of fluorochrome-conjugated secondary antibodies (1:250, Invitrogen) for 40 min, rinsed twice in PBS, stained with 4’,6-diamidino-2-phenylindole (DAPI, #D1306, Thermo Fisher Scientific) for 5 min, rinsed in water, and the coverslips were mounted onto glass slides using mounting media (#17985–16, Electron Microscopy Sciences).
Analysis of recombinant laminin-111 degradation
mLN-111 (90 μg) or hLN-111 (1 μg) were incubated with neutrophil conditioned media or recombinant proteases at 37 °C for six hours before adding lysis buffer (25 mM Tris [pH 6.8], 2% SDS, 5% glycerol, 1% β-mercaptoethanol, 0.01% bromophenol blue). For mLN-111, samples were then loaded on SDS-polyacrylamide gel electrophoresis, and the gel was stained with coomassie blue in a small plastic box. For hLN-111, samples were processed as described in the section “Western blot”. For analysis on native gel, SDS and β-mercaptoethanol were omitted from the lysis and running buffers.
Effects of experimental inflammation on dormant cancer cells in vivo
To generate dormant disease, we intravenously injected mCherry-luciferase-D2.0R or -MCF-7 cells (500,000 per mouse) in 100 μl of PBS into eight-week-old female BALB/c or nude mice, respectively. Nude mice were implanted with 17β-estradiol 0.36 mg/pellet (#SE-121, Innovative Research of America) 48 h before MCF-7 cell injection. To determine how lung inflammation reactivates dormant tumor cells in the lung, we induced inflammation through intranasal instillation of LPS derived from Escherichia coli strain 0111:B4 (#L4391, Sigma). LPS (50 μL at a concentration of 0.25 mg/ml, or PBS as control) was administered intranasally on days 7, 10, and 13 or on days 30, 33, and 36 with a P200 pipette under anesthesia (2.5% isoflurane) into mice that had been injected with cancer cells at day 0. Neutrophils were depleted by administering anti-Ly6G antibodies intraperitoneally (clone 1A8, #BP0075–1, Bio X Cell, 200 μg/mouse). Anti-Ly6G antibodies were first given one day before the first LPS administration, and the treatment continued three times weekly until the end of the experiment on day 33. Rat IgG2A isotype was used as a control (clone 2A3, #BP0089, Bio X Cell, 200 μg/mouse). Inhibitors were intraperitoneally injected, starting on the first day of LPS treatment. On days when the mice received LPS administration, they were treated three times with inhibitors (30 min before LPS administration, three hours after administration, and six hours after administration). On all other days, one daily treatment was given, continuing until the end of the experiment on day 33. The doses of the inhibitors were: 50 mg/kg sivelestat (NE inhibitor, #3535, Tocris); 15,000 units/kg DNase I (#04536282001, Roche); 20 mg/kg GSK484 (PAD4 inhibitor, #17488, Cayman Chemical), 50 mg/kg SB3-CT (MMP9 inhibitor, #HY-12354, Medchem Express). Vehicle consisted of 10% dimethyl sulfoxide (DMSO) in PBS for the mice treated with sivelestat, GSK484, and SB3-CT; and PBS for mice treated with DNase I. Immobilization of an enzyme on the surface of nanoparticles can increase enzyme stability, therefore, we also generated and used DNase I-coated nanoparticles, as previously described (22). The nanoparticles were suspended such that each μL contained 1 unit of DNase I; 75 units of DNase I-coated nanoparticles were administered intraperitoneally per mouse. Uncoated nanoparticles were used as control. To follow metastatic disease, mice were injected intraperitoneally with luciferin (5 mg per mouse, #Luck-1G, Goldbio) and imaged using the IVIS Spectrum in vivo imaging system (#128201, PerkinElmer) every three days.
For experiments analyzing the effect of inhibitors on lung inflammation, cancer cells were not injected, and mice were treated with LPS at days 0, 3, and 6. On days when the mice received LPS administration, they were treated three times with inhibitors (30 min before LPS administration, three hours after administration, and six hours after administration). On all other days, one daily treatment was given. Mice were sacrificed at day 7 for tissue processing or at the times indicated in the figure legends.
Effects of tobacco smoke exposure on dormant cancer cells in vivo
To generate dormant disease, mCherry-luciferase-D2.0R (500,000 per mouse) in 100 μl of PBS was intravenously injected into eight-week-old female BALB/c mice. To determine how tobacco smoke (TS) exposure reactivates dormant tumor cells in the lung, the mice were exposed to tobacco smoke or filtered air for three weeks starting one week after injection using a smoke exposure system (Dr. Kent Pinkerton Laboratory, University of California, Davis). For the TS exposed group, mice were exposed five days/week for three weeks to an average concentration of 75 +11 mg/m3 of tobacco smoke particulate for 6 h/day using 3R4F research cigarettes (Tobacco Research Institute, University of Kentucky) that were burned at a rate of four cigarettes every 10 min with a puff volume of 35 mL over a duration of 2 s, once per minute. Both side-stream and mainstream cigarette smoke were collected via a chimney and passed to a dilution and aging chamber to allow the animals to acclimate to TS exposure. The TS exposure started at an initial exposure concentration of 60 mg/m3 and was increased, ultimately achieving a final target concentration of 90 mg/m3. After each six-hour exposure to TS, the mice were then kept in the same chamber but in filtered air. For the control group, mice were handled in the same way, but were instead exposed to filtered air for 24 hours/day, 7 days/week for the duration of the study. The concentration of carbon monoxide in the exposure chambers was monitored and averaged 215 ± 36 ppm over the three weeks of exposure. Mice were treated with vehicle (10% DMSO in PBS) or PAD4 inhibitor (GSK484, #17488, Cayman Chemical) intraperitoneally at a dose of 20 mg/kg/day to inhibit NET formation, starting on the first day of TS exposure and every day until the end of the experiment. ChiAb28 was administered intravenously at a dose of 200 μg/mouse to target NET-remodeled laminin in the lungs, starting on the first day of environmental TS exposure. ChiAb28 was given every three days until the end of the experiment. Mouse IgG2A isotype was used as a control (Clone C1.18.4, #BE0085, Bio X Cell, 200 μg/mouse).
Confocal intravital lung imaging
We used a modified version of our previously described method for lung imaging (22). Mice were anesthetized by intraperitoneal injection of ketamine/dexdomitor (100 mg/kg; 0.5 mg/kg). Saline (500 μl) was injected peritoneally prior to surgery, then tracheotomies were performed, and mechanical ventilation was initiated (MiniVent ventilator for mice, Model 845, Harvard Apparatus) with the mice on a heated stage. From this point forward, the mice were kept anesthetized at 0.8–1.0% isoflurane delivered through the ventilator at 80–100 breaths per minute, with 25 ml of water maintaining positive end expiratory pressure. A thoracotomy was then performed by making an incision between the 4th and 5th rib, and a 3D printed thoracic suction window with an 8 mm coverslip was inserted (47). The window was secured to the stage with single-axis translator micromanipulators (#MSH1.5, Thor Labs), and the left lung was held in place by vacuum pressure, which was adjusted to 15–20 mmHg. Mice were imaged with our custom-built spinning disk confocal microscope modified to upright configuration, as described previously (22). The microscope was controlled by MicroManager software, and data analysis and time-lapse movies were generated using Imaris software.
To visualize neutrophil recruitment and neutrophil elastase activity, LysM-EGFP mice (on BALB/c background, (48)) were administered intranasally with LPS (50 μL, at a concentration of 0.25 mg/ml) or PBS as control as indicated and intravenously with neutrophil elastase 680 FAST probe (4 nmol in 100 ul, #NEV11169, PerkinElmer, 3–6 hours before imaging). At the time of imaging, DAPI was administered (5 ug/ml) intravenously, and images were captured for 2–8 hours. To monitor cancer cells exiting dormancy and undergoing cell cycle progression, D2.0R cells labeled with FUCCI reporter (500,000) were injected intravenously into BALB/c mice at day 0, and the mice were treated with LPS on days 7, 10, and 13, as described above. On days of imaging, the neutrophil elastase 680 FAST probe was administered intravenously 3–6 hours before imaging.
Enzyme-linked immunosorbent assay (ELISA) for NETs
96-well Enzyme ImmunoAssay/Radio ImmunoAssay (EIA/RIA) plates (#3590, Costar) were coated overnight at 4 °C with an anti-elastase antibody (1:250, #sc-9521, Santa Cruz Biotechnology) in 15 mM of Na2CO3, 35mM of NaHCO3, at pH 9.6. The next day, the wells were washed three times with PBS, blocked in 5% BSA for two hours at room temperature, and washed three times with PBS. Then, 50 μl of plasma samples were added to the wells, incubated for two hours at room temperature on a shaker, and plates were washed three times with wash buffer (1% BSA, 0.05% Tween 20 in PBS). Next, anti-DNA-Peroxidase conjugated antibody (1:50, #11774425001, Roche) in 1% BSA in PBS was added to the wells for two hours at room temperature, and the wells were washed five times with wash buffer before the addition of 2,2’-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid (ABTS, #37615, Thermo Fisher Scientific). Optical density was read 40 min later at 405 nm using a plate reader (SpectraMax i3, Molecular Devices).
Generation of anti-NET-remodeled laminin-111 antibodies
mLN-111 (6 mg) was incubated with recombinant NE and MMP9 (2 μg/ml each) at 37 °C for six hours. To remove the recombinant proteases, the cleaved laminin-111 solution was purified using a 100 kDa molecular weight cut-off (MWCO) column (#VS0641, Vivaspin 6, Sartorius), following the manufacturer’s instructions. The presence of laminin-111 and the absence of NE and MMP9 in the upper fraction (>100 kDa) was then assessed using Western blot analysis. The upper fraction containing the cleaved laminin-111 was next dialyzed in PBS using 20,000 MWCO dialysis cassettes (#87735, Thermo Fisher Scientific). Three six-week-old Sprague Dawley rats (Taconics) were immunized with 1.5 mg of cleaved laminin-111 (100 μg per animal per boost for five weekly boosts). Immune response was monitored by ELISA to measure the serum anti-NET remodeled laminin-111 IgG titer from blood samples. After a 60-day immunization course, the rat with the strongest anti-cleaved laminin-111 immune response was terminated, and 108 splenocytes were collected for making hybridomas by fusing with the rat myeloma cell line YB2/0, following the standard method (49). All procedures were approved by the Cold Spring Harbor Laboratory Institutional Animal Care and Use Committee (IACUC). The antibodies generated were tested and endotoxin free.
Generation of chimeric antibodies
To make antibodies for in vivo experiments, we engineered the rat mAbs into mouse chimera antibodies (chiAbs), as this reduces the likelihood of the mice developing neutralization antibodies. In brief, the rat mAb V-domains from the heavy and light chains were cloned into the mice IgG2a framework. The resulting chimeric IgG2a antibody therefore comprised rat V-domains in frame with the constant regions of mice IgG2a. The recombinant chimeric antibodies were then produced from HEK293 cells transfected with the chimeric IgG2a expression constructs, followed by standard purification procedures from the culture supernatants.
Statistical analysis
Data from in vivo BLI quantification were analyzed using one-way ANOVA followed by Tukey’s procedure, except for Fig. S7F, where a two-sided t-test was used. Data from in vitro BLI were analyzed using two-way ANOVA followed by Tukey’s procedure, except for Figs. 4C, 4E, 6B, S7J and S15D. The data represented in these figures were analyzed using one-way ANOVA followed by Dunnett’s procedure, with the reference group indicated on the figure.
Data from NE and MMP9 activity in vitro were analyzed using two-way ANOVA followed by Tukey’s procedure, while data from NE and MMP9 activity in vivo were analyzed using one-way ANOVA followed by Dunnett’s procedure, where the reference group was indicated on the figure. Assays on dsDNA, plasma NET and LPS, number of DTCs/mm2, metastatic burden, neutrophils’ number per field, percentage of neutrophils forming NETs, and percentage of KI67-positive cancer cells in vitro—except for Fig. S7G—were analyzed using one-way ANOVA followed by Dunnett’s procedure, with the reference group indicated on the figure. Fig. S7G was analyzed using a two-sided t-test.
Data from qPCR quantification were analyzed using one-way ANOVA followed by Dunnett’s procedure, where the reference group was indicated on the figure, except for Fig. S10F, where a two-sided t-test was used. To analyze lung metastasis-free survival (Fig. S5C), Kaplan-Meier curves of RapidCaP mice were analyzed using a log-rank (Mantel-Cox) test. For Fig. S5C, an exact p-value was computed using Fisher’s exact test to evaluate the likelihood that the metastatic types (macrometastasis, micrometastasis, single DTC) differed among groups. The analyses on figs. S2E and S15B had the cancer cell as the experimental unit, with each cell characterized on the two variables of interest for the figure. For fig. S2E, cells were characterized based on experimental group (Vehicle, day 11, day 12, day 13) and by cell feature (G0/G1 single cells, S/G2/M single cells, clusters of cells). A total of 182 cells were characterized and Fisher’s exact test was used to assess whether the cell feature distribution differed among the different experimental groups. The exact test was significant (p< 0.0001): Over time, G0/G1 single cells and S/G2/M single cells decreased and clusters of cells increased. For fig. S15B, cells were characterized based on cell type (PBS & G0/G1 single cells, LPS & G0/G1 single cells, LPS &S/G2/M single cells, LPS & clusters of cells) and next to Ab28 (yes, no). A total of 182 cells were characterized. Fisher’s exact test was used to assess whether the percentage of cells that were “next to Ab28” in each cell type group were different among the groups. The exact test was significant (p< 0.0001). All PBS cells were G0/G1 single cells and were not “next to Ab28”. LPS & G0/G1 single cells were equally likely to be “next to Ab28” or “not next to Ab28”. All LPS & S/G2/M single cells and all LPS & clusters of cells were “next to Ab28”. Analysis for figs. S5C, S2E and s16B were generated using Proc Freq in the SAS/STAT software, Version 9.4 of the SAS system for Windows.
A p-value less than 0.05 was considered significant, and p-values are indicated in figures as ***P<0.001, **P<0.01, and *P<0.05. All statistical analyses were performed using GraphPad Prism software version 7 unless otherwise stated.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. Green (NIH) for the D2.0R and D2.A1 cells and Z. Werb (University California, San Francisco) for the MMP9 antibody.
Funding: This work was supported by the CSHL Cancer Center Support P30-CA045508 (J.T.H.Y, C.B., K.C., S.K.L., and P.A.G.); to M.E.: the Department of Defense (W81XWH-14–1-0078) and the Pershing Square Sohn Cancer Research Alliance Prize); to J.A.: the Association pour la Recherche sur le Cancer (AE20141202131), the Terri Brodeur Breast Cancer Foundation, the European Molecular Biology Organization (ALTF 1425–2015), and the Susan G. Komen Foundation (PDF16376754); to D.T.F: the Lustgarten Foundation, the National Cancer Institute and the Cedar Hill Foundation; R01 NIH Research Grant (5R01CA137050, to L.C.T. and A.A.); CSHL and Northwell Health (M.A.S., E.M.C., C.G.P, M.S.G., and M.E.); a Starr Centennial Scholarship and a George A. & Marjorie H. Anderson Scholarship from the Watson School of Biological Sciences (to E.B. and L.M.); a Boehringer Ingelheim Fonds Ph.D. fellowship (to L.M.); a Deutsche ForschungsGemeinschaft research fellowship (KU 3264/1–1, to V.K.); UC Davis NIEHS Environmental Health Science Center (P30 ES023513, to K.E.P., M.E.P., P.U. and D.L.U.); and an AACR-Bayer Healthcare Basic Cancer Research Fellowship (to M.A.S.).
Footnotes
Competing interests:D.T.F. is a Co-Founder of Myosotis LLC (a company developing cancer immunotherapies) and is on the Scientific Advisory Boards of iTEOS Therapeutics (a company developing immuno-oncology drugs), IFM Therapeutics, LLC (a company developing therapies targeting the innate immune system), and Kymab (a company developing therapeutic antibodies). The authors declare no other competing interests.
Data and materials availability: All data are available in the main text or the supplementary materials. The Ab28 monoclonal antibody against cleaved laminin is available upon signing a Material Transfer Agreement.
References and Notes
- 1.Sosa MS, Bragado P,Aguirre-Ghiso JA, Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 14, 611–622 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malladi S. et al. , Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell 165, 45–60 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Romero I,Garrido F,Garcia-Lora AM, Metastases in immune-mediated dormancy: a new opportunity for targeting cancer. Cancer Res 74, 6750–6757 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Farrar JD et al. , Cancer dormancy. VII. A regulatory role for CD8+ T cells and IFN-gamma in establishing and maintaining the tumor-dormant state. J Immunol 162, 2842–2849 (1999). [PubMed] [Google Scholar]
- 5.Muller M. et al. , EblacZ tumor dormancy in bone marrow and lymph nodes: active control of proliferating tumor cells by CD8+ immune T cells. Cancer Res 58, 5439–5446 (1998). [PubMed] [Google Scholar]
- 6.Ghajar CM et al. , The perivascular niche regulates breast tumour dormancy. Nat Cell Biol 15, 807–817 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barkan D. et al. , Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res 70, 5706–5716 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pierce BL et al. , Elevated biomarkers of inflammation are associated with reduced survival among breast cancer patients. J Clin Oncol 27, 3437–3444 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pierce JP et al. , Lifetime cigarette smoking and breast cancer prognosis in the After Breast Cancer Pooling Project. J Natl Cancer Inst 106, djt359 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu AH et al. , The California Breast Cancer Survivorship Consortium (CBCSC): prognostic factors associated with racial/ethnic differences in breast cancer survival. Cancer Causes Control 24, 1821–1836 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murin S,Pinkerton KE, Hubbard NE, Erickson K,The effect of cigarette smoke exposure on pulmonary metastatic disease in a murine model of metastatic breast cancer. Chest 125, 1467–1471 (2004). [DOI] [PubMed] [Google Scholar]
- 12.De Cock JM et al. , Inflammation Triggers Zeb1-Dependent Escape from Tumor Latency. Cancer Res 76, 6778–6784 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jorch SK, Kubes P,An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med 23, 279–287 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Branzk N,Papayannopoulos V,Molecular mechanisms regulating NETosis in infection and disease. Semin Immunopathol 35, 513–530 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carmona-Rivera C,Zhao W,Yalavarthi S,Kaplan MJ, Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann Rheum Dis 74, 1417–1424 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fuchs TA et al. , Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A 107, 15880–15885 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brill A. et al. , Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost 10, 136–144 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong SL et al. , Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med 21, 815–819 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cools-Lartigue J. et al. , Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest 123, 3446–3458, doi: 10.1172/JCI67484(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tohme S. et al. , Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res 76, 1367–1380 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barkan D. et al. , Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res 68, 6241–6250 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park J. et al. , Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med 8, 361ra138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koh SB et al. , A quantitative FastFUCCI assay defines cell cycle dynamics at a single-cell level. J Cell Sci 130, 512–520 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Cho H. et al. , RapidCaP, a novel GEM model for metastatic prostate cancer analysis and therapy, reveals myc as a driver of Pten-mutant metastasis. Cancer Discov 4, 318–333 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barkan D,Green JE, An in vitro system to study tumor dormancy and the switch to metastatic growth. J Vis Exp, pii: 2914 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hosseinzadeh A,Thompson PR, Segal BH, Urban CF, Nicotine induces neutrophil extracellular traps. J Leukoc Biol 100, 1105–1112 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watson RW, Redmond HP, Mc Carthy J,Bouchier-Hayes D,Taurolidine, an antilipopolysaccharide agent, has immunoregulatory properties that are mediated by the amino acid taurine. J Leukoc Biol 58, 299–306 (1995). [DOI] [PubMed] [Google Scholar]
- 28.Egeblad M,Werb Z,New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2, 161–174 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Kessenbrock K,Plaks V,Werb Z,Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papayannopoulos V,Metzler KD, Hakkim A,Zychlinsky A,Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol 191, 677–691 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Metzler KD, Goosmann C,Lubojemska A,Zychlinsky A,Papayannopoulos V,A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep 8, 883–896 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beliveau A. et al. , Raf-induced MMP9 disrupts tissue architecture of human breast cells in three-dimensional culture and is necessary for tumor growth in vivo. Genes Dev 24, 2800–2811 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Catena R. et al. , Bone marrow-derived Gr1+ cells can generate a metastasis-resistant microenvironment via induced secretion of thrombospondin-1. Cancer Discov 3, 578–589 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.El Rayes T. et al. , Lung inflammation promotes metastasis through neutrophil protease-mediated degradation of Tsp-1. Proc Natl Acad Sci U S A 112, 16000–16005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aguirre Ghiso JA, Inhibition of FAK signaling activated by urokinase receptor induces dormancy in human carcinoma cells in vivo. Oncogene 21, 2513–2524 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Aguirre Ghiso JA, Kovalski K,Ossowski L,Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J Cell Biol 147, 89–104 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu D,Aguirre Ghiso J,Estrada Y,Ossowski L,EGFR is a transducer of the urokinase receptor initiated signal that is required for in vivo growth of a human carcinoma. Cancer Cell 1, 445–457 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Calvo F. et al. , Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol 15, 637–646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hynes RO, Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Shibue T,Brooks MW, Weinberg RA, An integrin-linked machinery of cytoskeletal regulation that enables experimental tumor initiation and metastatic colonization. Cancer Cell 24, 481–498 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caudrillier A. et al. , Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest 122, 2661–2671 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pantel K,Alix-Panabieres C,Bone marrow as a reservoir for disseminated tumor cells: a special source for liquid biopsy in cancer patients. Bonekey Rep 3, 584 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pommier A. et al. , Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science 360, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakano K,Suzuki S,Oh C,Significance of increased secretion of glucocorticoids in mice and rats injected with bacterial endotoxin. Brain Behav Immun 1, 159–172 (1987). [DOI] [PubMed] [Google Scholar]
- 45.Delgado-Rizo V. et al. , Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol 8, 81 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quail DF et al. , Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nat Cell Biol 19, 974–987 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Headley MB et al. , Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature 531, 513–517 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Faust N,Varas F,Kelly LM, Heck S,Graf T,Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood 96, 719–726 (2000). [PubMed] [Google Scholar]
- 49.Greenfield EA, Antibodies : a laboratory manual. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, ed. Second edition, 2014), pp. xxi, 847 pages. [Google Scholar]
- 50.Knott SRV et al. , A computational algorithm to predict shRNA potency. Mol Cell 56, 796–807 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.