

Abstract
Background:
Despite robust cholesterol lowering, cardiovascular disease (CVD) risk remains increased in patients with diabetes. Consistent with this, diabetes impairs atherosclerosis regression following cholesterol lowering in humans and mice. In mice, this is attributed in part to hyperglycemia-induced monocytosis, which increases monocyte entry into plaques despite cholesterol lowering. Additionally, diabetes skews plaque macrophages towards an atherogenic inflammatory M1 phenotype instead of towards the atherosclerosis-resolving M2 state typical with cholesterol lowering. Functional high-density lipoprotein (HDL), typically low in patients with diabetes, reduces monocyte precursor proliferation in murine bone marrow and has anti-inflammatory effects on human and murine macrophages. Our study aimed to test if raising functional HDL levels in diabetic mice prevents monocytosis, reduces the quantity and inflammation of plaque macrophages, and enhances atherosclerosis regression following cholesterol lowering.
Methods:
Aortic arches containing plaques developed in Ldlr−/− mice were transplanted into either wild-type (WT), diabetic WT, or diabetic mice transgenic for human apolipoprotein A-I (apoA-I), which have elevated functional HDL. Recipient mice all had low levels of LDL-cholesterol to promote plaque regression. After 2 weeks, plaques in recipient mouse aortic grafts were examined.
Results:
Diabetic WT mice had impaired atherosclerosis regression, which was normalized by raising HDL levels. This benefit was linked to suppressed hyperglycemia-driven myelopoiesis, monocytosis, and neutrophilia. Increased HDL improved cholesterol efflux from bone marrow progenitors, suppressing their proliferation and monocyte and neutrophil production capacity. In addition to reducing circulating monocytes available for recruitment into plaques, in the diabetic milieu, HDL also suppressed the general recruitability of monocytes to inflammatory sites and promoted plaque macrophage polarization to the M2, atherosclerosis-resolving state. There was also a decrease in plaque neutrophil extracellular traps (NETs), which are atherogenic and increased by diabetes.
Conclusions:
Raising apoA-I and functional levels of HDL promotes multiple favorable changes in the production of monocytes and neutrophils and in the inflammatory environment of atherosclerotic plaques of diabetic mice after cholesterol lowering, and may represent a novel approach to reduce CVD risk in people with diabetes.
Keywords: macrophage, diabetes mellitus, arteriosclerosis, high-density lipoprotein, apolipoprotein
Introduction
Patients with diabetes have a two to four-fold greater risk of atherosclerotic cardiovascular disease (CVD) 1. Despite advances in therapies to reduce CVD risk, patients with diabetes remain at increased risk for cardiovascular morbidity and mortality 2. Notably, diabetes not only increases CVD events, but also impairs the resolution, or regression of atherosclerotic disease 3. Consistent with the clinical data, diabetic mice have impaired atherosclerosis regression, as measured by the quantity and inflammatory state of plaque macrophages after aggressive lipid lowering 4–7. Diabetes in mice also increases monocytosis by activation of precursors in bone marrow (BM), which leads to more significant monocyte infiltration and macrophage content in plaques 4, 6. Furthermore, hyperglycemia impairs the polarization of plaque macrophages to the M2, inflammation-resolving state, which generally follows lipid reduction in atherosclerotic mice 8.
The association between diabetes and leukocytosis extends beyond mouse models to clinical populations 6, 9–12. For example, in humans, leukocytosis is an established risk factor for the progression and impaired regression of atherosclerosis 13, 14. Increased white blood cell levels also associate with CVD in patients with Type 1 diabetes 6. Reducing white blood cell count and inflammatory phenotype, particularly of monocytes and macrophages, may therefore represent a novel approach to reduce CVD risk and promote atherosclerosis regression in people with diabetes.
Based on pre-clinical studies, one approach to accomplish these goals may be to increase the levels of functional HDL, normally low in patients with diabetes 15–17. HDL not only suppresses the proliferation of leukocyte bone marrow precursors through its ability to promote the efflux of cholesterol from these cells 18, but it also has several anti-inflammatory properties 19, 20. We therefore hypothesized that increasing the levels of functional HDL in diabetic mice would prevent monocytosis, decrease the entry of monocytes into atherosclerotic plaques after lipid reduction, and consequently promote atherosclerosis regression. In addition to reducing the quantity of plaque macrophages, we also hypothesized that increased levels of functional HDL would favorably affect their quality by dampening their heightened inflammatory state in diabetic mice 4, 6, 8 and promoting polarization to the atherosclerosis-resolving M2 phenotype 21.
To directly test these hypotheses, we have utilized a model in which atherosclerotic plaques are developed in LDL receptor knockout (Ldlr−/−) mice, and then the diseased aortic arches are transplanted to either i) wild-type mice (maximal regression environment); ii) wild-type diabetic mice (impaired regression); or iii) diabetic mice expressing the human apoA-I transgene (Tg), which increases the levels of functional HDL 22, 23. The results showed multiple benefits of raising functional HDL levels in diabetic mice, including reduced monocytosis and monocyte recruitment to plaques, decreased plaque content of atherogenic neutrophil extracellular traps (the formation of which are increased by diabetes 24), enrichment in M2-like macrophages, and improved regression. These data not only convincingly support our hypotheses; they provide the first direct evidence of the positive effects of raising HDL levels on established plaques in a diabetic model. These results suggest that improving HDL function will enhance the currently limited level of cardiovascular risk reduction after robust lipid lowering in patients with diabetes, particularly in the many with dyslipidemia characterized by low plasma HDL.
Methods
The data that support the findings of this study are available from the corresponding author on reasonable request.
Animal Studies
All procedures described were approved by the Animal Use and Care Committee at the NYU School of Medicine. The model of aortic arch transplantation has been previously described 25. Briefly, the aortic arch is excised from a donor mouse (in these studies, Ldlr−/−) and is implanted into the abdominal aorta of a recipient mouse by end-to-side anastomoses, with the intervening abdominal aortic segment tied off to divert all blood flow through the transplanted arch segment. Ldlr−/− mice were weaned at 4-6 weeks onto a high-fat Western Diet (WD, 21% [wt/wt] fat, 0.3% cholesterol, Research Diets). Mice were maintained on WD for 16 weeks, after which a subset was used for baseline measurements, and the remainder were used for transplantation procedures. Transplant recipient mice were C57Bl/6 (WT), diabetic WT or diabetic apoA-I Tg, and the transferred aortic arches were harvested after 2 weeks on chow. Four weeks prior to transplantation, two groups of recipient mice were made diabetic by daily i.p. injections of streptozotocin (STZ; 50 mg/kg, Sigma-Aldrich) or citrate buffer for 5 days to induce diabetes or serve as a control. Mice used in the study were purchased from Jackson Laboratories (C57Bl/6: stock #000664, ApoA-I Tg: stock #001927).
Clinical Data
Informed consent and recruitment were performed under the New York University Langone Medical Center Institutional Review Board. Patient samples from an ongoing study investigating platelet activity were used for analysis 26. For this cohort, subjects ≥21 years old were recruited from New York University Langone Medical Center, Bellevue Hospital, or the Veterans Affairs NY Harbor Healthcare System. Subjects were excluded if they used NSAIDs in the past week, antithrombotic therapy, renal failure (creatinine clearance<30ml/min or on dialysis), presence of co-existing inflammatory disease (e.g. rheumatoid arthritis, lupus, etc.), cancer, active infection, platelet count <100 × 109/L or >450 × 109/L, or any known hemorrhagic diathesis. For human subject samples, peripheral blood samples were drawn using a 19-gauge needle without the use of a tourniquet. After an initial 2 cc discard, blood was collected into tubes containing 3.2% (0.105 moles/L) sodium citrate for monocyte phenotype measurements.
Plasma Lipoprotein Analyses
Total cholesterol, HDL-cholesterol (HDL-C), and triglyceride concentrations were measured using colorimetric assays (Wako Diagnostics, Richmond, VA). Blood glucose and insulin levels were measured after 4 hours of fast with a glucometer (Freestyle lite, Roche), and by ELISA (Linco), respectively. S100A8/A9 levels were measured by ELISA kits from ALPCO.
Size and Concentration of HDL (HDL-PIMA)
HDL-PIMA was quantified by calibrated-ion mobility analysis (IMA), using HDL isolated by ultracentrifugation from EDTA plasma 27. Since electrophoretic mobility depends chiefly on size, IMA data are expressed in terms of particle diameter, which corresponds to the calculated diameter of a singly charged, spherical particle with the same electrophoretic mobility. For each spectrum, three HDL subspecies (small, medium, and large) were deconvoluted from the IMA spectra by unsupervised, iterative curve-fitting. HDL peak areas were directly converted to HDL-PIMA using a calibration curve constructed with a protein standard. For total HDL particle concentration, intra- and interassay coefficients of variation were <10% and <20%, respectively. For the individual subspecies, CVs were <20%.
ApoA-I Infusions
Apolipoprotein A-I (apoA-I) was isolated from fresh plasma of healthy volunteer donors by sequential ultracentrifugation as previously described 27. All subjects gave written informed consent, and the Institutional Review Board of the Cleveland Clinic approved all study protocols. All apoA-I preparations were tested for endotoxin contamination by the Limulus amoebocyte lysate assay (LAL; Charles River Laboratories, Wilmington, USA) after digestion with trypsin for 36 hours at 37°C and heat treatment at 75°C for 20 minutes. All apoA-I preparations used in the study were confirmed to be essentially endotoxin free. Mice were injected subcutaneously (s.c.) between the shoulder blades with either 15 mg/injection of human native apoA-I (apoA-I group) or the carrier (PBS; control group) four times (every other day) prior to termination of the experiment.
Plaque Analysis
At harvest, baseline aortic arches and transplanted arch grafts were perfused with 10% sucrose, frozen in OCT, and serial-sectioned at 6 μm onto glass slides. Following staining, microscopic images were digitized, and morphometric measurements were performed using Image Pro Plus software (Micro Optical Solutions).
Immunostaining
To stain for macrophages, slides were fixed in 100% acetone and immunostained with CD68 (clone 1957, Serotec), followed by biotinylated secondary antibody (Vector Laboratories), with visualization using a Vectastain ABC kit (Vector Laboratories). For mannose receptor (CD206) staining the above protocol was used with an anti-Cd206 primary antibody (MR5D3). Macrophage plaque proliferation was assessed by staining for Ki67 (clone SP6, Abcam), and apoptosis by cleaved caspase 3 (Cell Signaling). Neutrophil extracellular traps (NETS) were identified by staining with histone H3 (citrulline R2 + R8 + R17, Abcam), myeloperoxidase (clone 2D4, Abcam) and Ly6G (Clone 1A8, BD Biosciences). Triple positive areas were determined to be positive for NETs. Image analysis was performed in ImageJ.
Plaque Collagen Quantification
Tissues were stained with picrosirius red and quantified with Image Pro Plus software using polarizing light microscopy.
Monocyte/Macrophage Trafficking
Monocytes were labeled as previously described 4. Briefly, 1μm Fluoresbrite FITC-dyed (YG) plain microspheres (Polysciences Inc.) were diluted in PBS (1:4), and 250μl was injected retro-orbitally into mice to label circulating monocytes. Bead labeling efficiency was assessed by flow cytometry, 24h after bead injection. For the egress study, donor mice were injected with 1 mg (i.p) of EdU 48 h prior to aortic transplantation. Macrophages retained in plaques following transplantation were identified by performing a click reaction with Alexa Fluor 647 nm-azide (Click-iT EdU Imaging Kit, Invitrogen) according to the manufacturer’s instructions. Slides were imaged on a Leica SCN400F slide scanner, and EdU positive cells were quantified.
Flow cytometry
Blood Leukocytes
Blood was collected via tail vein bleeding with EDTA-coated capillary tubes. Red blood cells were lysed with BD Pharm Lyse (BD Biosciences), and blocking achieved with anti-mouse CD16/CD32 (eBioscience). Monocytes were identified by staining with PE anti-mouse CD115 (Biolegend) and APC anti-mouse Ly6G/Ly6C (Biolegend). Neutrophils were identified as CD45hiCD115loLy6-C/Ghi by flow cytometry using a LSRII analyzer. Intracellular ROS was assessed via dihydroethidium (Invitrogen) staining.
Hematopoietic stem and progenitor cells
Hematopoietic stem and progenitor cells were analyzed by flow cytometry as previously described 4. Briefly, bone marrow was harvested from femurs and tibias and red blood cells were lysed with BD Pharm Lyse (BD Biosciences). Cells were incubated with a cocktail of antibodies against lineage-committed (lin) cells (B220, CD19, CD11b, CD3e, TER-119, CD2, CD8, CD4, Ly6-C/Ly6-G, all FITC) and markers to identify the stem and progenitor cells that were identified as LSK (lin−, Sac1+, and ckit+), CMP (lin−, Sca1−, ckit+, CD34int, and FcγRIIint/FcγRIIIint), and GMP (lin−, Sca1−, ckit+, CD34int, and FcγRIIhi/FcγRIIIhi). Cell cycle analysis was performed using DAPI (Sigma Aldrich) to measure cells in the S-G2M phase. Flow cytometry was performed using an LSR II (for analysis) or MoFlo (for sorting). All flow cytometry data were analyzed using FlowJo X software (Tree Star). RNA from sorted cells was extracted with RNeasy Micro kit (Qiagen) and cDNA was obtained with SuperScript VILO (Life Technologies).
Monocyte Type Assessment (Human Samples)
Monocyte characterization was performed by flow cytometry analysis on whole blood collected in sodium citrate. Ten thousand monocytes were collected based on their forward-side-scatter properties. Monocyte subsets were identified by double immunostaining for CD14 and CD16.
Isolation of Monocyte Progenitors & Cholesterol Efflux
Hematopoietic stem and progenitor cells were analyzed by flow cytometry as previously described 4. Briefly, bone marrow was harvested from femurs and tibias of diabetic and control C57B/6 mice, and red blood cells were lysed with BD Pharm Lyse (BD Biosciences). Cells were incubated with a cocktail of antibodies against lineage-committed (lin) cells (B220, CD19, CD11b, CD3e, TER-119, CD2, CD8, CD4, Ly6-C/Ly6-G, all FITC) and markers to identify the progenitor cells (CD34), flow cytometry sorted cells were incubated with chemokines to support the survival of monocyte progenitor populations during the cholesterol efflux, Flt3L, SCF, IL-3 and IL-6 (StemSpan™ CC100, StemCell Technologies). Monocyte progenitors were loaded with [3H]cholesterol (0.5 μCi/ml [3H]cholesterol for 24 h, and equilibrated in 2 mg/ml BSA medium for 2 h). Cholesterol efflux to apoA-I (50 μg/ml) or to the media (2 mg/ml BSA) in the absence of acceptors was carried out for 4 h. Efflux to apoA-I and HDL was established to be linear at this time point (Supplemental Figure 1). Efflux was measured by scintillation counting and is expressed as a percentage of [3H]cholesterol in medium/([3H]cholesterol in medium + [3H]cholesterol in cells) × 100%.
RNA Isolation, cDNA Synthesis and qRT-PCR
For quantitative real-time PCR analysis of specific cell populations in the blood and BM, cells were isolated via FACS sorter directly into RLT lysis buffer, and RNA isolated using RNeasy Micro kit (Qiagen) and cDNA synthesized using SuperScript VILO (Invitrogen). qRT-PCR Samples were normalized to m36b4 or Cyclophilin a. Primer sequences: s100a8 (5′-TGCGATGGTGATAAAAGTGG-3′, 5′-GGCCAGAAGCTCTGCTACTC-3′), s100a9 (5′-CACAGTTGGCAACCTTTATG-3′, 5′-CAGCTGATTGTCCTGGTTTG-3′), Rage (5′-ACTACCGAGTCCGAGTCTACC-3′, 5′-GTAGCTTCCCTCAGACACACA-3′), M36b4 (5′-AGATGCAGCAGATCCGCAT-3′, 5′-GTTCTTGCCCATCAGCACC-3′), Abca1 (5′-GGACATGCACAAGGTCCTGA-3′, 5′-CAGAAAATCCTGGAGCTTCAAA-3′), Abcg1 (5′-CCCTCAAAGCCGTATCTGAC-3′, 5′-TTGACACCATCCCAGCCTAC-3′).
Statistical analysis
Linear regression was performed in R to evaluate the association between HbA1c (%) and the outcome variables (total WBC and HDL-C) adjusting for confounders including sex, age, BMI, total cholesterol, LDL-C, triglycerides, hypertension for diabetic and non-diabetic patients respectively Regression analysis data are reported as β regression coefficient (95% CI) and P value. Clinical non-normal variables were log transformed as appropriate.
Data are presented as mean ± SEM. Statistical tests performed are outlined in the Figure legends, and all analyses were performed in GraphPad Prism 7.05 unless otherwise stated. Prior to analyses, normality of data was assessed with the D’Agostino-Pearson omnibus test. If data were found to not follow a normal distribution, a non-parametric test was performed as indicated.
Results
Hyperglycemia is Correlated with Leukocytosis and Reduced HDL Levels in Humans & Mice with Diabetes
Leukocytosis and low levels of HDL-C and HDL particles are consistently observed in patients with diabetes 10, 11, 16, but studies to determine the association between these two factors and diabetes are limited. Therefore, we determined the association of circulating white blood cell counts with either HbA1c, a long term marker of glucose control, in a human cohort (demographics in Table 1) or with plasma glucose levels in our mouse population.
Table 1:
Patient Demographics
| Demographics | ||
|---|---|---|
| Non Diabetics | Diabetics | |
| n | 148 | 151 |
| Male (%) | 79.7 | 69.5 |
| Age (median (IQR)) | 56 (48, 67) | 64 (57, 75) |
| BMI (median (IQR)) | 26.8 (23.9, 30.7) | 28.5 (24.9, 31.7) |
| Total Cholesterol (mg.dL) (median (IQR)) | 153.0 (125.0, 180.5) | 137.5 (106.0, 174.3) |
| LDL (mg.dL) (median (IQR)) | 82 (57, 112) | 64 (47.3, 94.3) |
| HDL (mg.dL) (median (IQR)) | 44 (36, 51) | 38 (31, 45) |
| Trigylcerides (mg.dL) (median (IQR)) | 97.0 (70.3, 163.8) | 130.0 (93.5, 238.0) |
| WBC count (103/uL) (median (IQR)) | 6.5 (5.7, 8.3) | 6.9 (5.5, 8.8) |
| HbA1c (median (IQR)) | 5.7 (5.4, 6.0) | 7.2 (6.4, 8.5) |
| Insulin (%) | 0 | 50.3 |
| Hypertension (%) | 58.8 | 88.1 |
| Hyperlipidemia (%) | 62.8 | 78.8 |
| Statin (%) | 85.1 | 85.4 |
| Race (%) | ||
| White | 71.6 | 57.6 |
| Black | 9.5 | 18.5 |
| Asian | 8.8 | 7.3 |
| Other | 10.1 | 16.6 |
| Ethnicity (% Hispanic) | 14.2 | 25.8 |
| Smoking (% current) | 14.9 | 11.9 |
BMI, body mass index, HDL, high density lipoprotein, IQR, inter quartile range, WBC, white blood cell
Both groups contained samples from humans and mice with diabetes (Figure 1) and without diabetes (Supplemental Figure 2). The level of HbA1c or blood glucose was positively associated with white blood cell counts (Figure 1A, C, ρ = 0.310,0.384 respectively) and negatively associated with HDL-C in humans and mice with diabetes (Figure 1B, D, ρ = −0.192, −0.427 respectively). A significant association between HbA1c and WBC or HDL-C remained following multivariable adjustment for sex, age, BMI, total cholesterol, LDL-C, triglycerides, and hypertension status in the human diabetic population (Supplementary Table 1 and 2). Overall, these data demonstrate that poor glucose control correlates with leukocytosis and reduced HDL-C, and that our diabetic mouse model resembles the clinical diabetic cohort.
Figure 1. Hyperglycemia correlates with elevated white blood cell count and reduced HDL-C in humans and mice with diabetes.
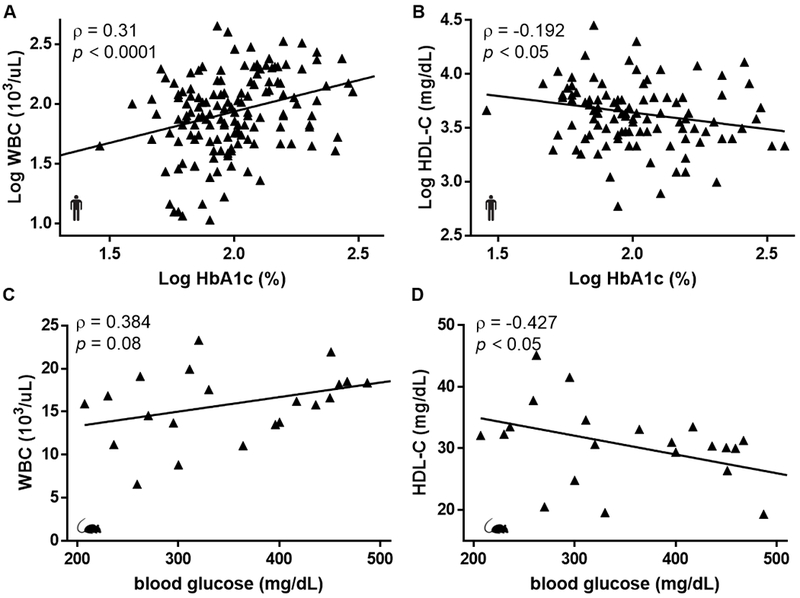
(A) White blood cell count and (B) HDL-C correlates with HbA1c in humans (n=146, n=102). (C) White blood cell count and (D) HDL-C correlates with blood glucose in diabetic mice (n=22). Pearson’s correlation coefficient (ρ) and p values were assessed in R. Black triangles and lines represent observations and the fitted regression lines.
Overexpression of apoA-I/HDL Overcomes Hyperglycemia-Enhanced Leukocytosis
To evaluate if raising HDL particles (HDL-P) levels can overcome hyperglycemia-associated leukocytosis we utilized mice overexpressing apoA-I (“apoA-I Tg”). Mice carrying the human apoA-I transgene under the control of its natural promoter have elevated plasma levels of total apoA-I, which is incorporated into HDL particles and results in increases in both circulating apoA-I and HDL-C. Consistent with the historical data 27, we found increases in circulating HDL-P (Figure 2A) and human apoA-I (Figure 2B). In addition, the mono-distribution of HDL particles in WT mice (Figure 2C) was converted to the subsets of HDL resembling those in human plasma (Figure 2D).
Figure 2. Elevating HDL overcomes hyperglycemia-mediated leukocytosis.
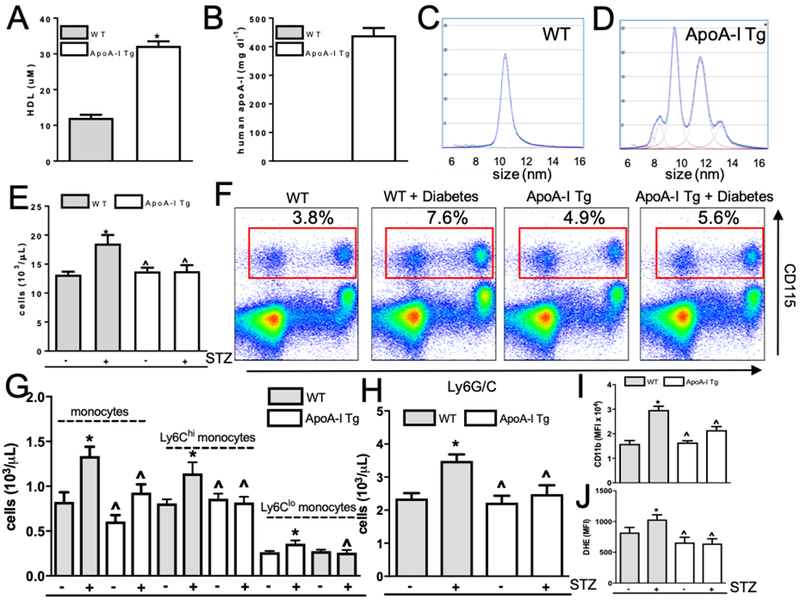
(A) HDL and (B) human apoA-I levels are raised in apoA-I Tg mice compared to wild-type (WT) controls (n=10). (C-D) HDL-P spectra of WT control mice and mice overexpressing apoA-I/HDL (representative images). (E) White blood cell counts in WT, diabetic WT, apoA-I Tg, and diabetic apoA-I Tg mice (n=8–10/grp). (F) Representative flow cytometry plots of blood leukocyte subsets from WT, diabetic WT, apoA-I Tg, and diabetic apoA-I Tg mice. Red box depicts monocyte population. (G) Quantification of monocytes, and monocyte subsets and (H) neutrophils (n=8–10/grp). Neutrophil (I) CD11b and (J) ROS in WT, diabetic WT, apoA-I Tg, and diabetic apoA-I Tg mice (n=8–10/grp). In panel A, data are expressed as mean ± SEM. * p<0.0001 determined by a t-test (two-tailed). Data are expressed as mean ± SEM. * p<0.01 compared to WT control group or ^ p<0.01 compared to WT STZ group as determined via 2-way ANOVA and Tukey’s post-hoc test.
We next investigated the effect of raising apoA-I/HDL on white blood cell counts in STZ-induced diabetic mice. In diabetic WT mice, circulating white blood cell counts were greater than in control nondiabetic WT mice (18.5 ± 1.4 versus 13.2 ± 0.5 103/uL, p<0.001, Figure 2E). In contrast, white blood cell counts were no different between control and diabetic apoA-I Tg mice (apoA-I Tg: 13.7 ± 0.6 versus diabetic apoA-I Tg: 13.7 ± 1.1 × 103/uL). This result could not be attributed to different levels of hyperglycemia in WT and apoA-I Tg mice (Supplemental Figure 3A). Consistent with our previous studies (e.g., 6), we found a significant elevation in circulating monocyte count, particularly in the Ly6Chi inflammatory subset, between diabetic and control WT mice (diabetic WT: 1.2 ± 1.4 versus WT: 0.81 ± 0.04 × 103/uL, Figure 2F–G, Supplemental Figure 3C). In diabetic apoA-I-Tg mice we observed a significant decrease in circulating monocytes (0.92 ± 0.09 × 103/uL) and inflammatory Ly6Chi monocytes (0.81 ± 0.06 × 103/uL) compared to WT diabetic mice (p<0.01 and p<0.05, respectively). Additionally, in diabetic mice overexpressing apoA-I/HDL, we observed a significant reduction in circulating neutrophils compared to WT diabetic mice (diabetic WT: 3.5 ± 0.2 versus diabetic apoA-I Tg: 2.4 ± 0.2 × 103/uL, p<0.01, Figure 2H). Consistently, we observed that monocytes and neutrophils isolated from diabetic apoA-I Tg mice had a reduced inflammatory profile, as assessed by cell surface CD11b expression and reactive oxygen species generation, when compared to WT diabetic mice (Figure 2I–J, Supplemental Figure 3D–E).
Overexpression of apoA-I/HDL Suppresses Diabetes-mediated Myeloproliferation by Promoting Cholesterol Efflux in Common Myeloid and Granulocyte Macrophage Progenitors, and by Suppressing Neutrophil Activation
Previously, hyperglycemia-mediated leukocytosis had been linked to increased content of monocyte and neutrophil progenitors within the bone marrow, namely common myeloid progenitors (CMPs) and granulocyte macrophage progenitors (GMPs) 4, 6. Indeed, in this study we observed significant increases in the CMP (0.32 ± 0.03 versus 0.22 ± 0.01 %, p<0.01) and GMP (1.7 ± 0.04 versus 1.34 ± 0.05 %, p<0.01) populations in WT diabetic mice compared to WT controls (Figure 3A–B, Supplemental Figure 4). ApoA-I/HDL overexpression in diabetic mice significantly reduced the CMP and GMP populations (by 31% and 24%, p<0.01, respectively; Figure 3B), indicating that raising apoA-I/HDL can suppress the production of monocytes in the setting of diabetes by reducing the number of bone marrow progenitors.
Figure 3. Raising apoA-I/HDL in diabetic mice suppresses hyperglycemia-driven myeloproliferation by promoting cholesterol efflux and suppressing neutrophil S100A8/A9 production.
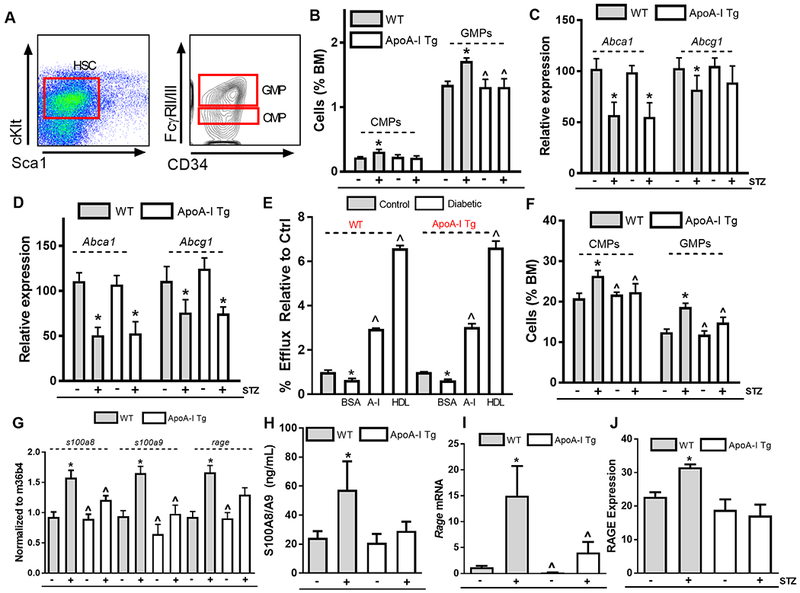
(A) Representative flow cytometry of monocyte and neutrophil precursors, common myeloid progenitors (CMPs), and granulocyte macrophage progenitors (GMPs). (B) Quantification of CMPs and GMPs in the bone marrow in WT, diabetic WT, apoA-I Tg, and diabetic apoA-I Tg mice (n=7–9/grp). Abca1 and Abcg1 expression in (C) CMP and (D) GMP populations within the bone marrow (n=7–9/grp). (E) Cholesterol efflux capacity of monocyte progenitors (CMPs & GMPs) for WT, diabetic WT, apoA-I Tg and diabetic apoA-I Tg mice (n=9 mice/grp). (F) CMP and GMP cell cycle analyses in the bone marrow in WT, diabetic WT, apoA-I Tg, and diabetic apoA-I Tg mice (n=7–9/grp). (G) s1008, s100a9 and rage expression in neutrophils isolated from WT, diabetic WT, apoA-I Tg, and diabetic apoA-I Tg mice (n=6/grp). (H) Circulating S100A8/A9 plasma protein levels, (I) bone marrow rage expression, (J) CMP RAGE surface expression (n=7–9/grp). Data are expressed as mean ± SEM. * p<0.01 compared to WT control group or ^ p<0.01 compared to WT STZ group as determined via 2-way ANOVA and Tukey’s post-hoc test in panel (B)-(D), (F)-(J). * p<0.05 compared to the control group or ^ p<0.05 compared to the diabetic BSA group for WT and ApoA-I Tg mice respectively as determined via 1-way ANOVA and Tukey’s post-hoc test in panel (E).
ABCA1 and ABCG1 play a crucial role in hematopoietic stem cell proliferation 18 by increasing plasma membrane cholesterol content. Consistent with our previous study in diabetic mice 4, we found that Abca1 and Abcg1 were significantly suppressed in the CMP and GMP populations in both diabetic WT and diabetic apoA-I Tg mice (Figure 3C–D). Thus, we hypothesized that impaired cholesterol efflux capacity of the CMP and GMP populations in hyperglycemia is a contributing factor to proliferation of these progenitors. In support of this, we found that the efflux capacity of CMP and GMP isolated from either diabetic WT or diabetic apoA-I Tg mice to be significantly impaired when compared to their respective controls (p<0.05, Figure 3E). Addition of apoA-I or HDL restored cholesterol efflux in CMP and GMP populations of WT or Tg diabetic mice. This is reminiscent of the ability of apoA-I or HDL to restore cholesterol efflux in Abca1−/−Abcg1−/− mice by providing more acceptors in the aqueous diffusion pathway, which predominates in cells not overloaded with cholesterol 28.
These data suggested that supplying additional apoA-I/HDL in the diabetic setting restores plasma membrane cholesterol homeostasis in CMPs and GMPs, which is critical for the regulation of their proliferation 18. Indeed, cell cycle analysis of CMPs and GMPs demonstrated that overexpression of apoA-I/HDL suppresses the percentage of cells in the G2M phase in the diabetic setting (by 20% and 33%, respectively; p<0.01, Figure 3F), indicating that in the setting of diabetes, overexpression of apoA-I/HDL can directly modulate the proliferative status of monocyte progenitor populations, even though the expression of the key efflux transporters is decreased. Further, we found apoA-I infusions also abrogated hyperglycemia-mediated myelopoiesis (Supplemental Figure 5A).
In addition to impaired cholesterol efflux, hyperglycemia-mediated myelopoiesis is, in part, dependent on neutrophil activation and subsequent S100A8/A9 release 6. In our diabetic mouse model, raising apoA-I/HDL significantly suppressed hyperglycemia-driven neutrophil activation (Figure 2I–J), and transcriptional profiling of neutrophils from control and apoA-I/HDL overexpressing diabetic mice revealed that that there is a significant reduction in s100a8 and s100a9 mRNA, as well as their diabetes-relevant receptor, receptor for advanced glycation end products (RAGE) (Figure 3G). Corresponding with these data, we observed a significant reduction in circulating S100A8/A9 levels in diabetic mice overexpressing apoA-I/HDL (p<0.01, Figure 3H). Furthermore, we found that Rage-mRNA and cell surface expression were significantly suppressed on bone marrow CMP populations in diabetic mice overexpressing apoA-I/HDL (Figure 3I–J). We hypothesize that this is a result of suppressed NFκB activation due to a reduction in p65 nuclear localization and target transcript expression (Supplemental Figure 5B–C), a known regulator of RAGE signaling 29.
Consistent with the ability of apoA-I/HDL to suppress leukocytosis in mice, we also found HDL-C to be negatively correlated with total neutrophil and monocyte counts in our patient cohort (Figure 4A,B). In addition, we observed the inflammatory CD14++CD16+ monocyte population to be significantly reduced in patient populations with HDL-C levels greater than 40 mg/dL (p<0.01, Figure 4C, Supplemental Figure 6–7). Plasma S100A9 levels were negatively correlated with HDL-C, providing evidence in to support of the anti-inflammatory capacity of HDL in humans (Figure 4D), as we found in mice (Figure 3).
Figure 4. HDL-C is negatively associated with leukocyte subsets and inflammation.
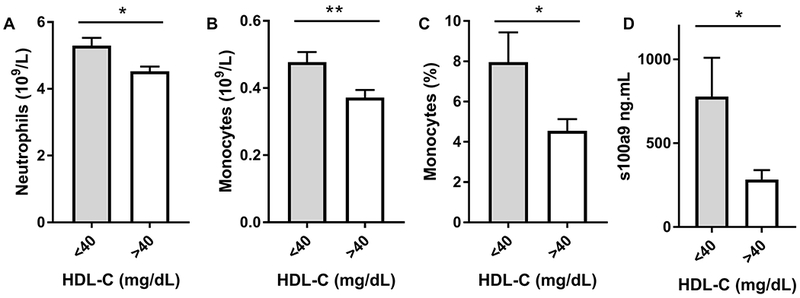
(A) Neutrophil, and (B) monocyte counts stratified by HDL-C (< 40 mg/dL n=127, >40 mg/dL n=157) and (C) CD14+CD16+ monocyte populations as stratified by HDL-C (< 40 mg/dL n=24, >40 mg/dL n=28). (D) Plasma S100A9 concentration as determined via ELISA (< 40 mg/dL n=8, >40 mg/dL n=6). Subjects are derived from the patient cohort present in Table 1. Data are expressed as mean ± SEM. * p<0.01, ** p<0.005 as determined via Mann-Whitney U test.
Raising apoA-I/HDL Promotes Atherosclerosis Regression in the Setting of Diabetes
We next tested whether raising apoA-I/HDL in the setting of diabetes could overcome diabetes-mediated impairments to atherosclerotic plaque inflammation resolution and regression. Thus, diabetic Ldlr−/− mice were fed a high fat-cholesterol diet for 16 weeks to develop advanced plaques. Plaque-laden aortic arches were then transplanted into WT, diabetic WT or diabetic mice over-expressing apoA-I/HDL (Figure 5A). Two weeks post transplantation, aortic arches were excised and plaque characteristics determined. Mice sacrificed at the 16-week time point were used to determine baseline lesion characteristics. Plasma cholesterol levels (Figure 5B) were significantly greater in baseline mice compared to aortic arch transplant recipient groups (baseline: 1224 ± 58 mg/dL; recipients: WT: 57 ± 22, diabetic WT: 75 ± 26; diabetic apoA-I Tg 166±16). HDL-C levels were elevated in the diabetic apoA-I Tg mice compared to the WT recipient groups (Supplemental Figure 8). Glucose levels in diabetic recipient mice were increased over 3-fold compared to controls (Figure 5C). Consistent with our previous observations, circulating leukocyte and monocyte levels in WT and diabetic apoA-I Tg mice were reduced, compared to diabetic WT mice (p<0.01, Figure 5D–E).
Figure 5. Raising apoA-I/HDL promotes atherosclerosis regression in diabetic mice.
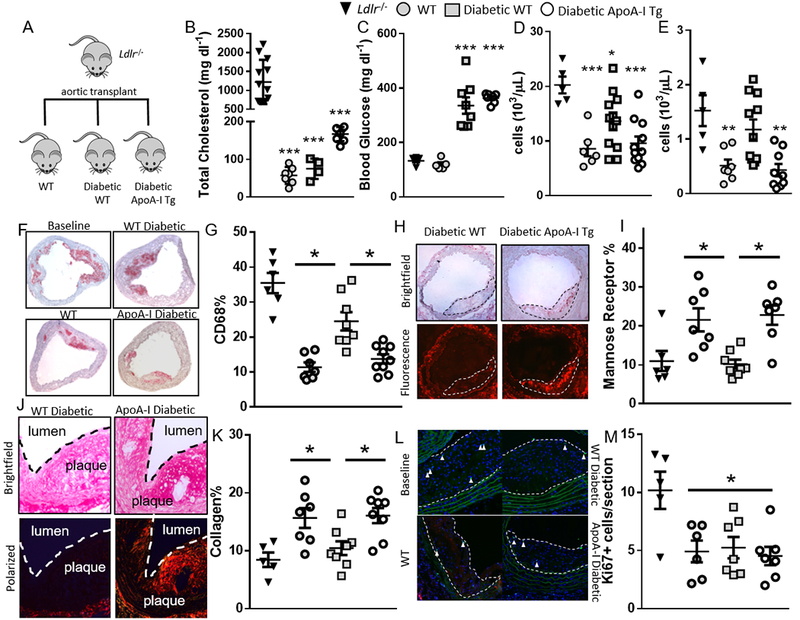
(A) Atherosclerosis regression protocol. (B) Total cholesterol, (C) blood glucose, (D) white blood cell counts (E) monocyte counts in baseline, WT, diabetic WT and diabetic apoA-I Tg mice (n= 7–11 mice/grp). (F) Representative images and (G) quantification of plaque CD68 staining. (H) Representative images and (I) quantification of plaque mannose receptor expression. (J) Representative images and (K) quantification of plaque collagen content. (L) Representative images and (M) quantification of plaque Ki67 staining (n=7–11 mice/grp). Data are expressed as mean ± SEM. *** p<0.001, ** p<0.01, * p<0.05 compared to the baseline group as determined via 1-way ANOVA and Tukey’s post-hoc test in panels (B), (C), (D), (E) and (M). * p<0.01, compared to the WT STZ group as determined via 1-way ANOVA and Tukey’s post-hoc test in panels (G), (I), and (K).
To assess regression, atherosclerotic lesions were immunostained for macrophages using antibodies to the marker CD68. Consistent with our previous findings (e.g., 30), transfer of aortic arches to WT mice resulted in the regression of the atherosclerotic lesions, indicated by a significant reduction in the percentage of plaque area positive for CD68 (p<0.01) when compared to baseline plaques (35.5 ± 2.8% versus 11.2 ± 1.5%; Figure 5F–G). Compared to the WT group, regression was significantly impaired in the diabetic WT recipients (24.5 ± 2.6%, p<0.01). Notably, overexpression of apoA-I/HDL in diabetic mice reduced the CD68+ content to essentially the level achieved in WT mice (13.5 ± 1.6%), indicating that raising apoA-I/HDL in the setting of diabetes promotes plaque regression, despite persistent hyperglycemia.
In regression, changes to plaque macrophage content are typically accompanied by changes in macrophage phenotype (reviewed in 31), but this is attenuated by diabetes 4, 6, 8. Recently, we reported in mice that there is a requirement of macrophages to polarize to the M2 anti-inflammatory state for successful plaque resolution after lipid lowering 21. Furthermore, we and others have found that apoA-I/HDL promotes mouse and human M2 macrophage polarization 32, 33. Thus, we hypothesized that raising apoA-I/HDL in the setting of diabetes would promote macrophage polarization to the M2 state. To determine whether there were phenotypic alterations to plaque macrophages in the regression groups, plaques were immunostained for the M2 marker mannose receptor, which is commonly used in human and mouse studies. Diabetes impaired mannose receptor macrophage expression during the regression phase. However, overexpression of apoA-I/HDL overcame this defect (p<0.005 compared to diabetic WT, Figure 5H–I). Additionally, collagen content, considered to be a plaque stability marker and inversely proportional to the level of matrix metalloprotease activity of macrophages, was greater in regressing plaques (p<0.01, Figure 5J–K). Although transplantation of aortic arches into diabetic WT mice did not result in plaque collagen enrichment, likely because of a sustained content of M1 macrophages and persistent MMP activity, overexpression of apoA-I/HDL significantly increased plaque collagen content despite hyperglycemia (p<0.01).
Overexpression of apoA-I/HDL Suppresses Monocyte Infiltration into Regressing Plaques and Another Site of Inflammation
Differences in the number of macrophages within atherosclerotic lesions can include the following: i) changes in proliferation of macrophages, ii) altered macrophage egress, and, iii) changes in the recruitment of circulating monocytes 31. Plaque macrophage proliferation, quantified by Ki67 staining, was greater in baseline than regressing plaques (approx. 50% reduction, p<0.01). However, neither diabetes nor apoA-I/HDL overexpression affected proliferation of plaque macrophages (Figure 5L–M). Thus, we turned to the other kinetic possibilities.
Utilizing in vivo cell tracking techniques previously established in our lab 4, 21 (Figure 6A–B), we found that transplantation of aortic arches (in which the plaque macrophages had been marked with EdU) into WT recipient mice led to a 75% reduction in EdU positive cells when compared to baseline mice. This demonstrated macrophage egress during the regression phase (p<0.001, Figure 6C), consistent with previous studies 4, 6. Diabetic WT and diabetic apoA-I Tg mice had a similar reduction in EdU positive plaque cells (Figure 6C), meaning that macrophages egress from plaques to similar degrees in the recipient groups.
Figure 6. Increased circulating levels of apoA-I/HDL suppresses monocyte recruitment to diabetic plaques, and to sites of inflammation.
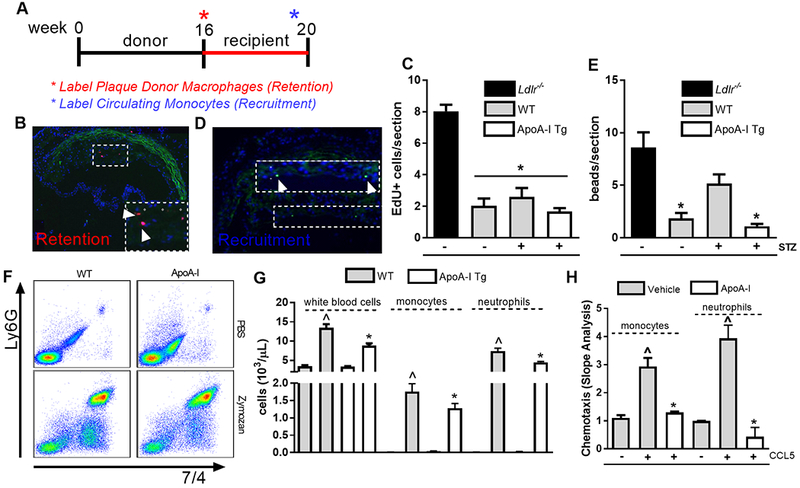
(A) Monocyte and macrophage plaque trafficking protocol. Representative image of (B) macrophage retention and (C) quantification in regressing atherosclerotic plaques (n=6–8 mice/grp). Representative image of (D) monocyte recruitment and (E) quantification in regressing atherosclerotic plaques (n=6–8 mice/grp). Data are expressed as mean ± SEM. * p <0.01, compared to the baseline group as determined via 1-way ANOVA and Tukey’s post-hoc test. (F) Representative flow cytometry plots of monocyte and neutrophil recruitment to the peritoneal cavity following zymozan injection. (G) Quantification of recruited leukocytes, monocytes and neutrophils to the peritoneal cavity following a zymozan challenge in wild-type (WT) and apoA-I Tg mice (n=3–7 mice/grp). Data are expressed as mean ± SEM. * p <0.01, compared to WT group, and ^ p <0.01 compared to zymozan injected WT group as determined via 2-way ANOVA and Tukey’s post-hoc test. (H) Monocyte and neutrophil chemotaxis to CCL5 following pre-treatment with apoA-I (40 ug.mL). Data are expressed as mean ± SEM. * p<0.01, compared to the no chemokine control, and ^ p<0.01, compared to CCL5-stimulated cells as determined via 1-way ANOVA and Tukey’s post-hoc test.
To study monocyte entry into plaques, fluorescent beads that are taken up by circulating monocytes 34 were injected intravenously, as we have done previously 4, into baseline or transplant recipient mice prior to harvest of the aortae. Bead-bearing cells were then counted in plaques to assess the contribution of monocyte recruitment (Figure 6D). As shown in Figure 6E, monocyte recruitment into plaques transplanted into WT mice decreased 78% (p<0.001) when compared to baseline plaques. Hyperglycemia in WT mice was associated with a 3-fold increase in monocyte recruitment to plaques when compared to WT mice; however, overexpression of apoA-I/HDL in the diabetic setting led to a striking 80% reduction in monocyte recruitment when compared to WT diabetic control mice (p<0.002). No differences in monocyte recruitment were observed between WT or diabetic apoA-I Tg mice, indicating that overexpression of apoA-I/HDL in the diabetic setting restores monocyte recruitment into plaques to the level in mice with normoglycemia.
The 80% decrease in monocyte recruitment to plaques transplanted to diabetic apoA-I Tg mice was much greater than we would expect based on the number of circulating monocytes, given that overexpression of apoA-I/HDL in the setting of diabetes reduced diabetes-driven monocytosis by 27% (Figure 2G). This suggested that apoA-I/HDL impeded an intrinsic ability of leukocyte recruitment to sites of inflammation. To test this, WT and apoA-I Tg mice were injected i.p. with zymosan to create a mild peritonitis 35. Post zymosan challenge, the inflammatory cell content of the peritoneal exudates was assessed by flow cytometry (Figure 6F). Total leukocyte recruitment in the apoA-I/HDL overexpressing mice were significantly reduced (by 35%, p<0.002) when compared to WT controls, with a reduction in total monocytes (27%, p<0.01) and neutrophils (40%, p<0.005, Figure 6G). Similarly, we found that ex vivo treatment of monocytes and neutrophils with apoA-I suppressed (56% reduction, p<0.02, and 89% reduction, p<0.01, respectively) in vitro recruitment to a chemokine typically used in chemotaxis studies, CCL5.
ApoA-I/HDL Suppresses Diabetes-driven Neutrophil Extracellular Trap Formation
Neutrophil extracellular traps (NETs) are a key scaffold in pathologic thrombi and fuel cardiovascular, inflammatory and thrombotic diseases in humans and mice (reviewed in 36). Recently, neutrophils isolated from diabetic patients and mice have been shown to be primed to produce NETs (termed NETosis) 24. In progressing plaques, NETs have been shown to promote plaque inflammation, and inhibition of this process suppresses both plaque size and inflammation (reviewed in 37). To date, the role of NETs in plaque regression has not been assessed, nor has the contribution of NETosis to diabetic plaque regression. Given the striking impairments by apoA-I/HDL of neutrophil activation and quantity present under diabetic conditions (Figure 3G), we sought to characterize the contribution of NETs in regressing plaques under diabetic and control conditions, and the contribution of raising apoA-I/HDL.
To identify NETs, plaques were co-stained for citrullinated histone, myeloperoxidase and neutrophils (Ly6G). Triple positive areas were quantified as NETs (Figure 7A). Compared to baseline plaques, we observed a significant decrease in plaque NETs in WT mice (83.6% reduction, p<0.05, Figure 7B). However, consistent with studies demonstrating that the diabetic milieu promotes NETosis, we found plaque NET content to be significantly increased to a similar extent in diabetic WT transplant recipient mice when compared to WT controls (p<0.01). In transplant recipient diabetic mice overexpressing apoA-I/HDL we observed a significant reduction in plaque NET content compared to both baseline and diabetic WT transplant recipient mice (p<0.01, and p<0.02, respectively), demonstrating that raising apoA-I/HDL overcomes diabetes-driven neutrophil activation and neutrophil-associated plaque inflammation. These data provide a new mechanism by which diabetes impedes atherosclerosis regression, by promoting plaque NETosis, and a new facet of apoA-I/HDL’s anti-inflammatory properties, which is a likely contributing factor to improving plaque regression under diabetic conditions.
Figure 7. Plaque neutrophil extracellular traps are elevated in regressing plaques from diabetic mice, and are reduced following raising apoA-I/HDL.
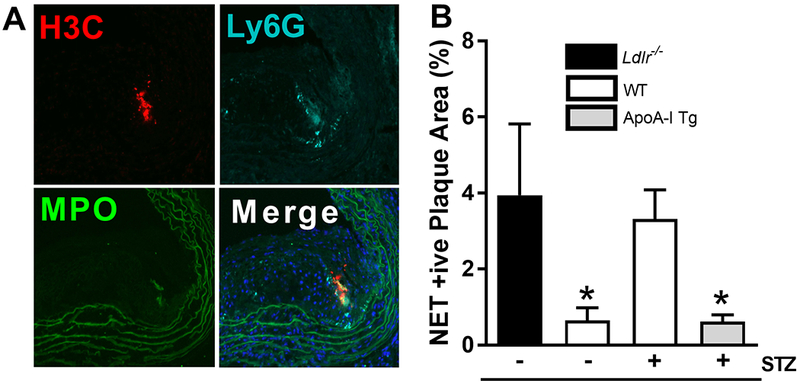
(A) Representative images of plaque neutrophil extracellular traps (NETs), as determined by citrullinated histone h3 (H3C), neutrophil (Ly6G), and myeloperoxidase (MPO) costaining. (B) Quantification of plaque NET content (n=6–10/grp). Data are expressed as mean ± SEM. * p<0.01, compared to the baseline group as determined via 1-way ANOVA and Tukey’s post-hoc test.
Discussion
Despite intensive lipid lowering with statin therapy, patients with diabetes remain at high-risk for cardiovascular events (e.g., 38). One feature of patients with diabetes and metabolic syndrome are reduced levels of HDL-C, HDL-P and apoA-I levels. Additionally, HDL isolated from diabetic patients is consistently demonstrated to be dysfunctional (reviewed in 16). Though the value of HDL-C as a determinant of CVD risk is under dispute, there is still enthusiasm for raising levels of functional HDL to suppress atherosclerosis progression and promote regression 39, 40. Our current study, the first of its kind, aimed to test whether raising apoA-I/HDL and improving the numbers of functional HDL in the diabetic milieu would restore atherosclerosis regression.
The relevance of our pre-clinical models for this investigation are supported by our finding in multiple studies that, as in humans, diabetes in mice impairs atherosclerosis regression even after cholesterol lowering 4, 6, 7. Additionally, our data show similar correlations between glucose, white blood counts, and HDL-C levels in the mice and a clinical patient cohort. The data presented clearly demonstrate that raising apoA-I/HDL in the setting of hyperglycemia improves the regression of pre-existing atherosclerotic plaques. The major consequences of elevating apoA-I/HDL were: i) attenuation of hyperglycemia-driven leukocytosis by suppression of bone marrow progenitor proliferation via promotion of cholesterol efflux and suppression of RAGE-s100a8/a9 signaling; ii) reduced monocyte recruitment to plaques; and, iii) decreased plaque content of NETs, an atherosclerosis and inflammation-promoting structure elevated in human and murine diabetic tissues 24, 36, 37.
We have reported previously that the mechanism in mice by which hyperglycemia induces monocytosis is through BM progenitor proliferation, which in turn results in increased recruitment of monocytes to plaques after reduction in plasma cholesterol levels 4, 6, 8. In our current study we found that raising apoA-I/HDL overcame the diabetes-driven myelopoiesis associated with enhanced cholesterol efflux from leukocyte progenitor populations (Figure 3E). The clinical relevance of these findings is implied by the negative associations in our patient cohort of circulating neutrophil and monocyte (total and CD14++CD16+) levels and HDL-C (Figure 4), with the caveat that HDL function is not always clinically correlated with HDL-C (e.g. 41).
One downstream effect of the suppression of myelopoiesis by apoA-I/HDL is a restored monocyte count, thereby preventing an expansion of the pool of recruitable inflammatory cells to atherosclerotic lesions. Yvan-Charvet et al. 18 linked cholesterol efflux-related genes in the BM progenitors to their proliferation and subsequent production of monocytes by showing that deficiency of Abca1 and Abcg1, even in the nonhyperlipidemic setting, led to monocytosis and leukocyte infiltration into tissues. They hypothesized that greater plasma membrane cholesterol increased responsiveness to proliferative factors, and found that membrane cholesterol and hematopoietic stem cell proliferation could be reduced by apoA-I even in the absence of ABCA1 and ABCG1.
In the present study, hyperglycemia induced a decrease in Abca1 and Abcg1 expression in the BM progenitors (Figure 3C–D), which, as just noted, would be expected to be a proliferative stimulus. Elevating apoA-I/HDL did not influence progenitor Abca1 and Abcg1 expression; it did, however, restore the serum efflux capacity to levels found in non-diabetic mice (Figure 3E), and subsequently suppressed BM proliferation (Figure 3F). We attribute the ability of apoA-I/HDL to promote cholesterol efflux to be independent of changes to transporters, but rather due to enhanced aqueous diffusion of cholesterol 28. Such a conclusion is consistent with the aforementioned studies in Abca1 and Abcg1-deficient mice, in which overexpression of apoA-I/HDL also suppressed leukocytosis 18.
Neutrophil-derived s100a8/a9 is an additional major contributor to monocyte and neutrophil BM progenitor proliferation in diabetic mice 6, 8 by signaling through RAGE to drive leukocytosis. In the current study, we found that increasing apoA-I/HDL reduced s100a8/9 plasma levels (Figure 3H), as well as Rage expression in BM progenitors (Figure 3I–J). The effect of apoA-I/HDL to decrease s100a8/9 expression likely reflects a lower level of neutrophil activation, and given that a major regulator of RAGE expression is NFκB 29, the attenuation of RAGE mRNA and surface expression by apoA-I/HDL is a likely consequence of its ability to inhibit the NFκB pathway (Supplemental Figure 5B–C) 6, 29. This mechanism is likely to work in concert with apoA-I/HDL’s ability to promote cholesterol efflux and, thus, alter signaling processes initiated in lipid rafts or by engagement of lipid raft components to suppress hyperglycemia-mediated inflammation. This is supported by a recent study that RAGE expression suppresses cholesterol efflux and downregulates myeloid Abca1 and Abcg1 42.
In addition to the decrease in monocyte number, we found that another contributing factor to regression in the diabetic setting is reduced recruitment of monocytes when apoA-I/HDL is increased; i.e., raising apoA-I/HDL suppressed exaggerated monocyte recruitment during the regression phase. While this is the first study to demonstrate this, it is also consistent with the effects of apoA-I/HDL on murine and human monocyte/macrophage recruitment to the chemoattractants in studies we recently reported 35. The present results also resemble those in some key aspects from our studies of diabetic mice treated with anti-miR-33 4. Among other effects, this raised the level of HDL particles, reduced monocyte recruitment in plaques after lipid lowering, and promoted the pro-regression/inflammation resolving 4, 6, 8, 21, 32, 40 M2 macrophage phenotype. Thus, apoA-I/HDL increases that are accomplished by multiple strategies may be expected to have similar efficacies.
Downstream of leukocyte progenitor production, neutrophils from diabetic mice overexpressing apoA-I/HDL had a reduced inflammatory profile (Figure 3G), and the plaques in these mice had reduced NET content (Figure 6). This finding is of relevance given recent studies that demonstrate that the diabetic state can induce neutrophil NET formation and impair wound healing. Additionally, plaque NET content has been shown to drive atherogenesis by promoting macrophage and T cell cytokine production. Our findings point to a previously uncharacterized role of apoA-I/HDL, namely its ability to promote diabetic atherosclerosis regression by suppressing neutrophil NETosis. We hypothesize this is driven by apoA-I/HDL’s ability to reduce hyperglycemia-driven neutrophil activation, and heightened efflux of cholesterol from lipid-laden plaque macrophages leading to a reduction in plaque cholesterol crystal content 43. Indeed, very recently Westerterp et al. 44 found increased plaque NETosis in Abca1/Abcg1 deficient mice, and attributed this to endosomal and membrane cholesterol accumulation. This provides support for our findings that raising apoA-I/HDL in a setting of impaired ABCA1/ABCG1-mediated cholesterol efflux can reduce plaque NETosis and thereby contribute in a novel way to resolving inflammation as part of plaque regression.
The changes we observed in murine BM-derived progenitors may mechanistically link diabetes to heightened CVD risk. For example, monocytosis and neutrophilia are also prevalent in people with diabetes and correlates with their risk of CVD. We speculate that therapies that raise functional apoA-I/HDL will be effective in that population to reduce CVD risk through similar effects in human BM. There may also be additional benefits of raising apoA-I/HDL in diabetes through other mechanisms, given that previous preclinical studies have found that HDL improves insulin sensitivity and glucose tolerance, and promotes insulin secretion under high glucose conditions (reviewed in 19). However, by comparing groups of diabetic mice with identical glucose levels, we have shown that increased HDL reduces inflammation exclusive of any changes in glycemic control.
To put our work in a broader context, we note that T1DM is recognized as an autoimmune disease, and recent clinical evidence indicates that T2DM, rather than presenting just as a metabolic disorder, is also characterized by many autoimmune characteristics 45. Many autoimmune diseases, including systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA), are associated with low HDL-C plasma levels, inflammation and premature rates of atherosclerosis 46. It is likely that there is also reduced HDL functionality, so that raising apoA-I/HDL in individuals with these types of conditions may have the potential to achieve cardio-beneficial effects. Previous studies have found that apoA-I injections suppress characteristics of autoimmune diseases including lymph node enlargement, and activation and proliferation of lymphocytes 47.
Throughout this report, we have emphasized the functional aspect of apoA-I and HDL because of recent insights into the limitations of HDL-C as a marker or therapeutic target in CVD risk assessment or reduction, respectively 48, 49. It is important to note that patients with diabetes typically have lower HDL-C levels and fewer HDL particles with reduced anti-inflammatory functions and impaired cholesterol efflux capacity (reviewed in 50). Therefore, based on the mechanisms we have established, we speculate that improving the level of functional HDL will translate to reducing the high level of residual risk of CVD after conventional treatments in patients with diabetes.
Supplementary Material
Clinical Perspective.
What is New?
Poor regression of atherosclerotic plaques after LDL-C lowering in mice with diabetes can be overcome by raising levels of HDL particles by increasing expression of its major protein, apoA-I.
In the setting of diabetes, raising HDL results in reductions in the number of circulating monocytes and neutrophils, known contributors to atherosclerotic plaque growth and inflammation, by suppressing the hyperglycemic-enhanced proliferation of the leukocyte bone marrow precursors.
Raising apoA-I/HDL also dampened the inflammatory activities of leukocytes, including reducing the amount plaque-resident neutrophil extracellular traps (NETS), pro-inflammatory structures known to be at higher levels in lesions of mice and humans with diabetes.
What are the Clinical Implications?
Patients with diabetes often have low HDL particle number and/or function, and have impaired atherosclerosis regression and have high residual risk of CVD after aggressive LDL-C lowering therapy.
Our results suggest that raising functional HDL in patients with diabetes may represent an effective approach to reduce residual CVD risk given its ability to suppress production of leukocytes and their contributions to plaque inflammation.
Acknowledgements
We thank Cyrus Nikain and Stephanie Pena for technical assistance, Dr. Michael Phillips for advice about the cholesterol efflux assays, and Dr. Stan Hazen and his laboratory for providing the apoA-I and help in the experimental design for the infusion studies.
Sources of Funding:
T.J.B., E.A.F., and I.J.G. were supported by NIH Grants R01 DK095684, R01 HL117226, R01 HL084312, PO1 HL092969, R01 HL129433, and P01HL131481. T.J.B is supported by an American Heart Association Career Development Award (18CDA34110203AHA). A.J.M. is supported by the National Health and Medical Research Council Grants (APP1083138 and APP1106154), along with a career development fellowship from the NHMRC (APP1085752) and a future leader fellowship from the National Heart Foundation (100440) and a Centenary Award from CSL. M.S.G is supported by an American Heart Association Career Development Award (18CDA34080540). J.S.B is supported in part by NIH Grants R01 HL114978, R01HL139909, and R35HL144993.
Abbreviations
- ApoA-I
Apolipoprotein A-I
- BM
Bone marrow
- CMP
Common myeloid progenitors
- CVD
Cardiovascular disease
- GMP
Granulocyte macrophage progenitors
- HDL
High density lipoprotein
- HDL-P
High density lipoprotein particle
- NETs
Neutrophil Extracellular Traps
- RAGE
Receptor for advanced glycation end products
- STZ
Streptozotocin
- Tg
Transgenic
Footnotes
Disclosures
None.
References
- 1.Haffner SM, Lehto S, Ronnemaa T, Pyorala K and Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. New Eng J Med. 1998;339:229–234. [DOI] [PubMed] [Google Scholar]
- 2.Cholesterol Treatment Trialists C, Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hiro T, Kimura T, Morimoto T, Miyauchi K, Nakagawa Y, Yamagishi M, Ozaki Y, Kimura K, Saito S, Yamaguchi T, et al. Diabetes mellitus is a major negative determinant of coronary plaque regression during statin therapy in patients with acute coronary syndrome--serial intravascular ultrasound observations from the Japan Assessment of Pitavastatin and Atorvastatin in Acute Coronary Syndrome Trial (the JAPAN-ACS Trial). Circulation journal : official journal of the Japanese Circulation Society. 2010;74:1165–1174. [DOI] [PubMed] [Google Scholar]
- 4.Distel E, Barrett TJ, Chung K, Girgis NM, Parathath S, Essau CC, Murphy AJ, Moore KJ and Fisher EA. miR33 inhibition overcomes deleterious effects of diabetes mellitus on atherosclerosis plaque regression in mice. Circ Res. 2014;115:759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaudreault N, Kumar N, Olivas VR, Eberle D, Stephens K and Raffai RL. Hyperglycemia impairs atherosclerosis regression in mice. Am J Pathol. 2013;183:1981–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, Ramkhelawon B, Distel E, Westerterp M, Huang LS, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell metabolism. 2013;17:695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parathath S, Grauer L, Huang LS, Sanson M, Distel E, Goldberg IJ and Fisher EA. Diabetes adversely affects macrophages during atherosclerotic plaque regression in mice. Diabetes. 2011;60:1759–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feig JE, Parathath S, Rong JX, Mick SL, Vengrenyuk Y, Grauer L, Young SG and Fisher EA. Reversal of hyperlipidemia with a genetic switch favorably affects the content and inflammatory state of macrophages in atherosclerotic plaques. Circulation. 2011;123:989–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohshita K, Yamane K, Hanafusa M, Mori H, Mito K, Okubo M, Hara H and Kohno N. Elevated white blood cell count in subjects with impaired glucose tolerance. Diabetes Care. 2004;27:491–496. [DOI] [PubMed] [Google Scholar]
- 10.Ford ES. Leukocyte count, erythrocyte sedimentation rate, and diabetes incidence in a national sample of US adults. Am J Epidemiol. 2002;155:57–64. [DOI] [PubMed] [Google Scholar]
- 11.Schmidt MI, Duncan BB, Sharrett AR, Lindberg G, Savage PJ, Offenbacher S, Azambuja MI, Tracy RP and Heiss G. Markers of inflammation and prediction of diabetes mellitus in adults (Atherosclerosis Risk in Communities study): a cohort study. Lancet. 1999;353:1649–1652. [DOI] [PubMed] [Google Scholar]
- 12.de Ferranti SD, de Boer IH, Fonseca V, Fox CS, Golden SH, Lavie CJ, Magge SN, Marx N, McGuire DK, Orchard TJ, et al. Type 1 diabetes mellitus and cardiovascular disease: a scientific statement from the American Heart Association and American Diabetes Association. Circulation. 2014;130:1110–1130. [DOI] [PubMed] [Google Scholar]
- 13.Friedman GD, Klatsky AL and Siegelaub AB. The leukocyte count as a predictor of myocardial infarction. New England J Med. 1974;290:1275–1278. [DOI] [PubMed] [Google Scholar]
- 14.Lee CD, Folsom AR, Nieto FJ, Chambless LE, Shahar E and Wolfe DA. White blood cell count and incidence of coronary heart disease and ischemic stroke and mortality from cardiovascular disease in African-American and White men and women: atherosclerosis risk in communities study. Am J Epidemiol. 2001;154:758–764. [DOI] [PubMed] [Google Scholar]
- 15.Garg A Dyslipoproteinemia and diabetes. Endocrinol Metab Clin North Am. 1998;27:613–625, ix–x. [DOI] [PubMed] [Google Scholar]
- 16.Taskinen MR. Diabetic dyslipidaemia: from basic research to clinical practice. Diabetologia. 2003;46:733–749. [DOI] [PubMed] [Google Scholar]
- 17.Wilson PW, Meigs JB, Sullivan L, Fox CS, Nathan DM and D’Agostino RB, Sr. Prediction of incident diabetes mellitus in middle-aged adults: the Framingham Offspring Study. Arch Intern Med. 2007;167:1068–1074. [DOI] [PubMed] [Google Scholar]
- 18.Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, Welch CL, Wang N, Randolph GJ, Snoeck HW, et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010;328:1689–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rye KA and Barter PJ. Cardioprotective functions of HDLs. J Lipid Res. 2014;55:168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saemann MD, Poglitsch M, Kopecky C, Haidinger M, Horl WH and Weichhart T. The versatility of HDL: a crucial anti-inflammatory regulator. Eur J Clin Invest. 2010;40:1131–1143. [DOI] [PubMed] [Google Scholar]
- 21.Rahman K, Vengrenyuk Y, Ramsey SA, Vila NR, Girgis NM, Liu J, Gusarova V, Gromada J, Weinstock A, Moore KJ, et al. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J Clin Invest. 2017;127:2904–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chajek-Shaul T, Hayek T, Walsh A and Breslow JL. Expression of the human apolipoprotein A-I gene in transgenic mice alters high density lipoprotein (HDL) particle size distribution and diminishes selective uptake of HDL cholesteryl esters. Proc Natl Acad Sci U S A. 1991;88:6731–6735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rubin EM, Ishida BY, Clift SM and Krauss RM. Expression of human apolipoprotein A-I in transgenic mice results in reduced plasma levels of murine apolipoprotein A-I and the appearance of two new high density lipoprotein size subclasses. Proc Natl Acad Sci U S A. 1991;88:434–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, Kahn CR and Wagner DD. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015;21:815–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trogan E, Fayad ZA, Itskovich VV, Aguinaldo JG, Mani V, Fallon JT, Chereshnev I and Fisher EA. Serial studies of mouse atherosclerosis by in vivo magnetic resonance imaging detect lesion regression after correction of dyslipidemia. Arterioscler Thromb Vasc Biol. 2004;24:1714–1719. [DOI] [PubMed] [Google Scholar]
- 26.Dann R, Hadi T, Montenont E, Boytard L, Alebrahim D, Feinstein J, Allen N, Simon R, Barone K, Uryu K, et al. Platelet-Derived MRP-14 Induces Monocyte Activation in Patients With Symptomatic Peripheral Artery Disease. J Am Coll Cardiol. 2018;71:53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hutchins PM, Ronsein GE, Monette JS, Pamir N, Wimberger J, He Y, Anantharamaiah GM, Kim DS, Ranchalis JE, Jarvik GP, et al. Quantification of HDL particle concentration by calibrated ion mobility analysis. Clin Chem. 2014;60:1393–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adorni MP, Zimetti F, Billheimer JT, Wang N, Rader DJ, Phillips MC and Rothblat GH. The roles of different pathways in the release of cholesterol from macrophages. J Lipid Res. 2007;48:2453–2462. [DOI] [PubMed] [Google Scholar]
- 29.Yao D and Brownlee M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes. 2010;59:249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, Tacke F, Randolph GJ and Fisher EA. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc Natl Acad Sci U S A. 2006;103:3781–3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moore KJ, Sheedy FJ and Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanson M, Distel E and Fisher EA. HDL induces the expression of the M2 macrophage markers arginase 1 and Fizz-1 in a STAT6-dependent process. PloS one. 2013;8:e74676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Datta G, Kramer PA, Johnson MS, Sawada H, Smythies LE, Crossman DK, Chacko B, Ballinger SW, Westbrook DG, Mayakonda P, et al. Bioenergetic programming of macrophages by the apolipoprotein A-I mimetic peptide 4F. Biochem J. 2015;467:517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iqbal AJ, Barrett TJ, Taylor L, McNeill E, Manmadhan A, Recio C, Carmineri A, Brodermann MH, White GE, Cooper D, et al. Acute exposure to apolipoprotein A1 inhibits macrophage chemotaxis in vitro and monocyte recruitment in vivo. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinod K and Wagner DD. Thrombosis: tangled up in NETs. Blood. 2014;123:2768–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doring Y, Soehnlein O and Weber C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ Res. 2017;120:736–743. [DOI] [PubMed] [Google Scholar]
- 38.Almdal T, Scharling H, Jensen JS and Vestergaard H. The independent effect of type 2 diabetes mellitus on ischemic heart disease, stroke, and death: a population-based study of 13,000 men and women with 20 years of follow-up. Arch Intern Med. 2004;164:1422–1426. [DOI] [PubMed] [Google Scholar]
- 39.Hewing B, Parathath S, Barrett T, Chung WK, Astudillo YM, Hamada T, Ramkhelawon B, Tallant TC, Yusufishaq MS, Didonato JA, et al. Effects of native and myeloperoxidase-modified apolipoprotein a-I on reverse cholesterol transport and atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2014;34:779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feig JE, Rong JX, Shamir R, Sanson M, Vengrenyuk Y, Liu J, Rayner K, Moore K, Garabedian M and Fisher EA. HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte-derived cells. Proc Natl Acad Sci U S A. 2011;108:7166–7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. New Eng J Med. 2011;364:127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Daffu G, Shen X, Senatus L, Thiagarajan D, Abedini A, Hurtado Del Pozo C, Rosario R, Song F, Friedman RA, Ramasamy R, et al. RAGE Suppresses ABCG1-Mediated Macrophage Cholesterol Efflux in Diabetes. Diabetes. 2015;64:4046–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adams CW and Abdulla YH. The action of human high density lipoprotein on cholesterol crystals. Part 1. Light-microscopic observations. Atherosclerosis. 1978;31:465–471. [DOI] [PubMed] [Google Scholar]
- 44.Westerterp M, Fotakis P, Ouimet M, Bochem AE, Zhang H, Molusky MM, Wang W, Abramowicz S, la Bastide-van Gemert S, Wang N, et al. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis and Atherogenesis. Circulation. 2018; 138:898–912. doi: 10.1161/CIRCULATIONAHA.117.032636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Velloso LA, Eizirik DL and Cnop M. Type 2 diabetes mellitus--an autoimmune disease? Nat Rev Endocrinol. 2013;9:750–755. [DOI] [PubMed] [Google Scholar]
- 46.Frostegard J Atherosclerosis in patients with autoimmune disorders. Arterioscler Thromb Vasc Biol. 2005;25:1776–1785. [DOI] [PubMed] [Google Scholar]
- 47.Wilhelm AJ, Zabalawi M, Grayson JM, Weant AE, Major AS, Owen J, Bharadwaj M, Walzem R, Chan L, Oka K, et al. Apolipoprotein A-I and its role in lymphocyte cholesterol homeostasis and autoimmunity. Arterioscler Thromb Vasc Biol. 2009;29:843–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fisher EA, Feig JE, Hewing B, Hazen SL and Smith JD. High-density lipoprotein function, dysfunction, and reverse cholesterol transport. Arterioscler Thromb Vasc Biol. 2012;32:2813–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rader DJ and Hovingh GK. HDL and cardiovascular disease. Lancet. 2014;384:618–625. [DOI] [PubMed] [Google Scholar]
- 50.Lemmers RFH, van Hoek M, Lieverse AG, Verhoeven AJM, Sijbrands EJG and Mulder MT. The anti-inflammatory function of high-density lipoprotein in type II diabetes: A systematic review. J Clin Lipidol. 2017;11:712–724 e715. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.