

Abstract
GLI-Similar 1–3 (GLIS1–3), a subfamily of Krüppel-like zinc finger transcription factors, function as key regulators of several biological processes important to oncogenesis, including control of cell proliferation, differentiation, self-renewal, and epithelial-mesenchymal transition. This review provides a short overview of the critical roles genetic changes in GLIS1–3 play in the development of several malignancies. This includes intrachromosomal translocations involving GLIS2 and ETO2/CBFA2T3 in the development of pediatric non-Down’s syndrome, acute megakaryoblastic leukemia, a malignancy with very poor prognosis, and an association of interchromosomal translocations between GLIS3, GLIS1, and PAX8, and between GLIS3 and CLPTM1L with hyalinizing trabecular tumors and fibrolamellar hepatocellular carcinoma, respectively. Targeting upstream signaling pathways that regulate GLIS signaling may offer new therapeutic strategies in the management of cancer.
Keywords: GLIS Krüppel-like zinc finger proteins, transcription, leukemogenesis, oncogenesis, chromosomal translocation
GLIS1–3 Krüppel-like zinc finger proteins
Krüppel-like zinc finger proteins (see Glossary), one of the largest families of transcription factors, are important in the regulation of many cellular processes and pathologies, including oncogenesis [1, 2]. GLIS1–3 constitute a subfamily of Krüppel-like zinc finger transcription factors, which physiological roles in cancer are just beginning to be recognized [3–7]. These proteins function as activators and repressors of gene transcription by recognizing a G-rich DNA binding sequence, referred to as GLIS binding site (GLISBS), in regulatory regions of target genes. This binding is mediated by a DNA binding domain (DBD) consisting of five zinc finger motifs that are highly conserved among GLIS members [8–15]. GLIS DBDs exhibit high homology with members of the closely-related glioma-associated GLI subfamily. GLI transcription factors are part of the hedgehog signaling pathway and have been implicated in the initiation and maintenance of many cancers [1, 16, 17]. Since GLIS and GLI proteins can recognize similar DNA binding sequences, these transcription factors can compete for GLISBS binding and interfere with each other’s regulation of gene expression [18, 19] and biological functions, as has been reported for the regulation of Wnt family 4 (WNT4) [20]. The transcriptional activation of target genes by GLIS1–3 is mediated by their C-terminal transactivation domain (TAD) through the recruitment of co-activator complexes [3, 19, 21]. In addition to its C-terminal TAD, GLIS2 also contains a putative TAD and repressor domain at its N-terminus [20, 22, 23].
GLIS1–3 are key regulators of a number of physiological processes and are implicated in a variety of pathologies [3, 24]. GLIS3 plays a critical role in the control of cell proliferation, maturation, and/or differentiation of pancreatic beta cells, thyroid follicular cells, renal tubule cells, neural stem cells, and spermatogonial stem cells [9, 24–30]. GLIS3-deficiency in humans and mice leads to a multi-organ phenotype that includes the development of neonatal diabetes, congenital hypothyroidism, infertility, and polycystic kidney disease. GLIS2 is required for the maintenance of normal renal functions and architecture [9, 31]. Loss of GLIS2 function causes nephronophthisis, a kidney disorder characterized by renal atrophy, inflammation, and fibrosis involving apoptosis and epithelial-mesenchymal transition (EMT). Both GLIS1 and GLIS3 have been shown to greatly enhance the reprogramming of somatic cells into pluripotent stem cells (iPSCs), whereas expression of GLIS2 reduces reprogramming efficiency [32–34]. Aberrant control of cellular differentiation, cell proliferation, migration, apoptosis, EMT, and stem cell renewal plays a critical role in cancer development and progression. The regulation of these biological processes by GLIS family members [3], together with the well-established role of the closely-related GLI proteins in oncogenesis [35], suggested that GLIS proteins might be involved in oncogenesis as well. This hypothesis was supported by several recent studies implicating genetic changes in GLIS1–3 in a number of malignancies, including a subtype of acute megakaryoblastic leukemia and hyalinizing trabecular tumors [4–7]. This review provides a short overview of the emerging roles of these genes in cancer.
CBFA2T3/ETO2 and GLIS2 Translocation in Non-DS-AMKL
Acute megakaryoblastic leukemia (AMKL) is a heterogeneous subgroup of acute myeloid leukemia (AML) constituting about 10% of pediatric and 1% of all adult AML patients. Recently, a novel rearrangement involving the CBFA2/RUNX1 Translocation Partner 3 gene (CBFA2T3, also referred to as ETO2 or MTG16) and GLIS2 was found to be associated with 20–30% of pediatric non-Down’s syndrome AMKL (non-DS-AMKL), which constitutes an aggressive subtype of AMKL with a very poor prognosis [4, 5, 7, 36–43]. This translocation was not found in pediatric DS-AMKL or adult patients. About 40% of the CBFA2T3-GLIS2 AMKL patients were reported to carry a rearrangement between the Desert Hedgehog (DHH) and Ras Homologue Enriched in Brain Like 1 (RHEBL1) genes, which leads to the generation of a DHH-RHEBL1 fusion transcript and protein [36, 44]. The combination of these two rearrangements was found to be associated with an even poorer prognosis than CBFA2T3-GLIS2 AMKL patients lacking the DHH-RHEBL1 translocation. However, additional studies are needed to further support these conclusions.
CBFA2T3 and GLIS2 are localized in inverted orientation at opposite ends of chromosome 16 (Fig. 1A). The CBFA2T3/GLIS2 translocation (inv(16)(p13.3q24.3)) comprises an inversion of the telomeric regions of chromosome 16 that fuses the 5’ portion of CBFA2T3 in frame with the 3’ region of GLIS2. Multiple chimeric transcripts have been reported in which exon 10 or 11 of CBFA2T3 was found in frame with either exon 1, 2 or 3 of GLIS2 [4, 5, 7, 37, 43, 45]. The most common transcript is a fusion between exon 10/11 of CBFA2T3 and exon 3 of GLIS2 (Fig. 1A). The expression of the CBFA2T3-GLIS2 chimeric gene remains under the control of the CBFA2T3 promoter, which results in high expression of the fusion gene.
Figure 1.
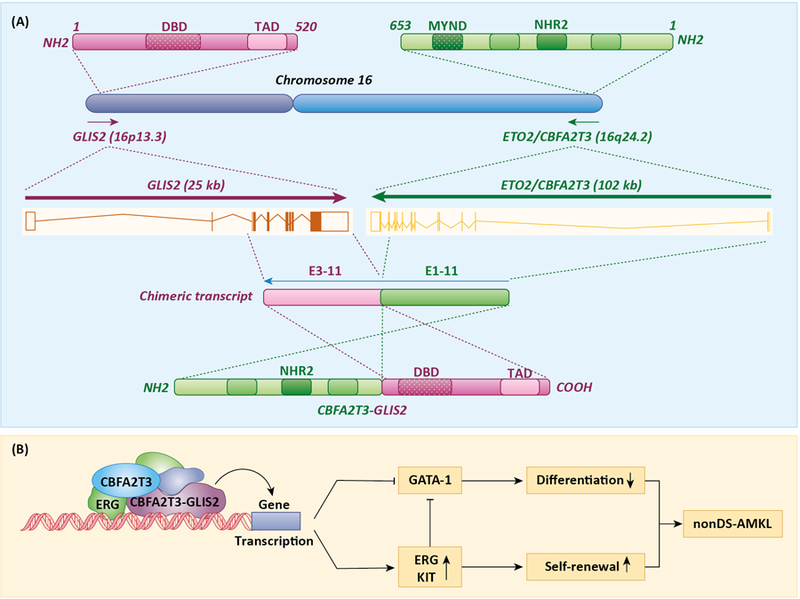
Association of CBFA2T3/GLIS2 translocations with non-DS-AMKL. A. Inversion of the telomeric regions of chromosome 16 fuses the 5’ portion of CBFA2T3 (ETO2) in frame with the 3’ region of GLIS2. Fusion between exon 11 of CBFA2T3 and exon 3 of GLIS2 is one of the most common translocations generating a CBFA2T3-GLIS2 fusion protein containing the NHR2 domain of CBFA2T3 and the DBD and TAD of GLIS2. The DBD, TAD, MYND and NHR2 domains are indicated. The bars within the DBD indicate the five zinc finger motifs. B. CBFA2T3-GLIS2 interacts with ERG and CBFA2T3 to form a transactivation complex that enhances ERG and represses GATA-1 expression, which subsequently promote self-renewal and inhibit megakaryocyte differentiation, two key characteristics of nonDS-AMKL.
Regulation of Gene Expression by CBFA2T3-GLIS2 Fusion Protein
CBFA2T3, a member of the Eight Twenty One (ETO) family, functions as a nuclear co-repressor as part of mSin3-HDAC1/2 co-repressor complexes. It contains four nervy homology regions (NHRs) that mediate oligomerization and interaction with other transcriptional mediators, and promote stabilization of co-repressor complexes [46, 47]. The NHR4 contains a MYND (myeloid, nervy, and Deaf-1)-type zinc finger domain that directly interacts with the N-CoR and SMRT co-repressors, while the other NHRs mediate homodimerization and interaction with other transcriptional regulators, such as GATA binding protein 1 (GATA-1), ETS transcription factor (ERG), and Runt-related transcription factor 1 (RUNX1). CBFA2T3 is expressed in haematopoietic stem and progenitor cells (HSPCs) as well as in megakaryoblastic cells. It plays a critical role in the regulation of the cell cycle, self-renewal capacity, and differentiation of hematopoietic progenitor cells [46, 48]. GLIS2 was reported normally not to be expressed in the hematopoietic system [4, 43]. However, a recent study demonstrated that shRNA knockdown of GLIS2 reduced HSPC (Lineage−Sca-1+c-Kit+ cells) repopulation after their transplantation in mice, in which hematopoietic cells were ablated by ionizing radiation [49]. This study suggested that GLIS2 functions as a regulator of HSPC engraftment and maintenance of hematopoiesis. Clearly additional studies are needed to determine whether GLIS2 might have a role in a distinct HPSC subpopulation during normal hematopoiesis.
The chimeric transcripts generated from the CBFA2T3-exon 10/11 to the GLIS2-exon 1/2/3 translocation result in the generation of a CBFA2T3-GLIS2 fusion protein that contains 3 of the 4 NHRs of CBFA2T3, but lacks the MYND-containing NHR4 domain thereby losing the ability to recruit co-repressors (Fig. 1A). The presence of the remaining NHRs still allows the fusion protein to homodimerize and interact with other transcriptional regulators, such as ERG [50, 51]. In addition, the fusion protein contains the DBD as well as the C-terminal TAD of GLIS2 [19]. Therefore, one might expect that the fusion protein is able to regulate gene transcription and megakaryocyte differentiation through both CBFA2T3(NHR2)- and GLIS2-dependent mechanisms.
Gene expression profiling of CBFA2T3-GLIS2 AMKL samples revealed that they display a pattern of differentially expressed genes that is distinct from that of other non-DS-AMKL samples [4, 5, 7, 51]. Gene expression analysis in transfected human erythroleukemic HEL cells showed that only 37% of the genes differentially regulated by CBFA2T3-GLIS2 overlap with those regulated by CBFA2T3, GLIS2, or both, while the vast majority of the genes regulated by GLIS2 (88%) and by CBFA2T3 (75%) overlap with those of CBFA2T3-GLIS2. This supports the concept that CBFA2T3-GLIS2 regulates gene expression in part through a CBFA2T3- and GLIS3-mediated mechanism, while a majority of the genes that are specifically controlled by CBFA2T3-GLIS2 are aberrantly regulated involving different mechanisms. Among the CBFA2T3-GLIS2-specific genes were genes with established roles in leukemia and HSPC self-renewal, including genes linked to the Notch, SHH, WNT, and TGFβ/BMP signaling pathways [4, 7, 51]. Expression of ERG, which is critical for hematopoietic stem cell renewal and associated with poor AML prognosis, was among the genes induced by CBFA2T3-GLIS2 [46, 51], whereas expression of GATA-1, which promotes megakaryocyte differentiation and reduces proliferation, was decreased. Consequently, these changes in gene expression result in increased self-renewal of progenitor cells and reduced differentiation, key elements in the development of AMLK (Fig. 1B).
Genome-wide CBFA2T3-GLIS2 binding studies showed that about 20% of CBFA2T3-GLIS2-bound genes were differentially expressed, half of which were regulated specifically by the fusion protein, while the other half were also regulated by CBFA2T3, GLIS2 or both [51]. More than half of the CBFA2T3-GLIS2 binding regions were not found in normal cells. These observations are consistent with aberrant regulation of gene expression by the fusion protein in leukemic cells that might in part be due to the assembly of different transcriptional complexes by CBFA2T3-GLIS2 and changes in affinity for CBFA2T3 and GLIS2 binding sites. The vast majority (about 70%) of the CBFA2T3-GLIS2 peaks correlated with transcriptionally active chromatin (H3K4me3 peaks) and super-enhancer regions (H3K27ac peaks), and included the regulatory regions of CBFA2T3, ERG, and KIT. Motif analysis showed that the majority of CBFA2T3-GLIS2 peaks was associated with motifs for known CBFA2T3 partners, including ERG and GATA-1, and overlapped significantly with G/C-rich GLIS2 binding motifs (GLISBS) [51, 52]. These findings indicate that CBFA2T3-GLIS2 regulates gene transcription in coordination with these transcription factors and in part through its interaction with GLIS binding sites. In CBFA2T3-GLIS2 HEL cells, EGR is required for the regulation of many differentially expressed genes, such as transcriptional activation of KIT, as well as for the proliferation and survival of CBFA2T3-GLIS2 leukemic cells in vivo, consistent with the conclusion that ERG is critical for the development of CBFA2T3-GLIS2 AMKL.
CBFA2T3-GLIS2, Self-renewal and Leukemogenesis
Xenotransplantation of CBFA2T3-GLIS2 expressing AMKL cells in mouse recipients showed consistent infiltration of liver, spleen and spinal cord [7, 51]. As CBFA2T3, ectopic expression of CBFA2T3-GLIS2 in murine hematopoietic progenitors resulted in a marked increase in self-renewal capacity; however, transplantation of these cells in syngeneic mice did not result in the development of leukemia [4, 53]. It was concluded that expression of CBFA2T3-GLIS2 in normal murine cells may not be sufficient for leukemogenesis in mice and require the cooperation of other genetic mutations.
Although ectopic expression of CBFA2T3-GLIS2 enhances self-renewal of hematopoietic progenitors, ectopic expression of CBFA2T3 in bone marrow progenitors promotes self-renewal of KIT+ progenitors, but not megakaryocyte differentiation, while expression of CBFA2T3-GLIS2 or GLIS2 induces both self-renewal and differentiation with GLIS2 being significantly more effective in promoting maturation than the fusion protein [51]. Consistent with this, the differentiation promoting activity of CBFA2T3-GLIS2 was found to be dependent on the DBD of GLIS2, while the increase in self-renewal required the NHR2 domain of CBFA2T3. Moreover, CBFA2T3-GLIS2 transcriptional activation was found to required interaction with wild type CBFA2T3. Co-expression with a NHR2 peptide inhibited the dimerization of CBFA2T3-GLIS2 with CBFA2T3, and possibly itself, and resulted in a greatly reduced transcriptional activation, ERG expression and cell proliferation, and increased GATA1 expression and differentiation [51]. The NHR2 peptide also inhibited the development of leukemia in immunodeficient mice transplanted with CBFA2T3-GLIS2 AMKL cells. These observations suggest that competition with NHR2 domain peptides might offer future possibilities for a new therapeutic strategy.
Treatment with inhibitors of the serine/threonine kinase Aurora kinase A (AURKA), which functions as a potent oncogene by regulating cell proliferation and self-renewal [54], have been reported to inhibit proliferation and induce differentiation of megakaryoblastic leukemic cells [55]. AURKA inhibitors were also able to enhance apoptosis and promote megakaryocyte maturation in isolated CBFA2T3-GLIS2 leukemic cells and reduced tumor burden and increased survival of recipient mice transplanted with CBFA2T3-GLIS2 AMLK leukemic cells suggesting potential for an alternative chemotherapeutic strategy [37, 56]. Recent studies showed that treatment with the GLI antagonist, GANT61, induced G1 cell cycle arrest and apoptosis in CBFA2T3-GLIS2 AMLK leukemic cells and enhanced their chemosensitivity [56, 57]. Although there is no evidence that GANT61 directly affects GLIS2 activity, these anti-leukemic effects are likely mediated via the inhibition of the SHH/GLI signaling pathway, which has been reported to be induced in CBFA2T3-GLIS2 AMKL cells [4, 7, 51, 58].
Link Between Ring1A/1B, GLIS2 and AML
Recently, in a study of MOZ-TIF2 AML cells, a different relationship was identified between GLIS2 expression and the regulation of leukemic stem cell self-renewal and differentiation [59]. An inv(8)(p11q13) inversion involving the histone acetyltransferase MOZ (KAT6A) and the nuclear receptor co-activator TIF2 (NCOA2) has been reported to be associated with the development of AML, while ectopic expression of the MOZ-TIF2 fusion protein in myeloid progenitors promotes self-renewal and induces leukemia after cells were transplanted into irradiated mice [59–61]. Deletion of the ubiquitin ligases, Ring1A and Ring1B, which mediate histone H2A ubiquitination (H2AK119Ub) as components of the polycomb transcriptional repressor complex 1 (PRC1) [62], reduced self-renewal capacity and enhanced apoptosis and macrophage differentiation in MOZ-TIF2 AML cells. Moreover, in contrast to MOZ-TIF2 progenitor cells, mice transplanted with MOZ-TIF2 cells lacking Ring1A/1B did not develop AML. Interestingly, in addition to the induction of macrophage-specific markers, deletion of Ring1A/1B greatly increased the expression of GLIS2. In contrast to other reports [4, 51], this study suggests that GLIS2 expression correlates inversely with stem cell renewal and positively with differentiation, and identifies GLIS2 as an important target of Ring1A/1B suppression [59, 63]. The latter was supported by data showing that Ring1B was associated with the GLIS2 promoter region and that deletion of both Ring1A/1B greatly diminished the association of H2AK119Ub and H3K27me3, markers of inactive chromatin, with the GLIS2 promoter resulting in de-repression and activation of GLIS2 transcription. Exogenous expression of GLIS2 in MOZ-TIF2 progenitors induced differentiation into mature myeloid cells, including F4/80+ macrophage-like cells; however, overexpression of GLIS2 did not totally abolish self-renewal capacity in all progenitors and delayed, but did not prevent AML development in mice after transplantation. It is possible that the progenitors that escaped GLIS2-induced differentiation gained additional oncogenic mutations or expressed lower levels of GLIS2. This differentiation-inducing ability is consistent with the differentiation-promoting effects in bone marrow progenitors by ectopic expression of GLIS2 [51]. The importance of GLIS2 in controlling the balance between self-renewal and differentiation in MOZ-TIF2 progenitors was further supported by data showing that deletion of GLIS2 in MOZ-TIF2 leukemic cells lacking Ring1A/1B restores the self-renewal capacity in these cells [59, 63]. Future studies need to determine whether GLIS2 also has function in normal differentiation in vivo.
Role of GLIS2 in Other Malignancies
Two additional studies have provided evidence for a link between GLIS2 and oncogenesis. In one study, an association between high levels of GLIS2 expression and chemoresistance and worse prognosis in gastric cancer was observed [64]. Ectopic expression of GLIS2 increased cell survival of several gastric cancer cell lines when treated with chemotherapeutic agents, such as cisplatin and doxorubicin. The mechanism by which GLIS2 expression enhances survival is not yet understood.
A second study identified a correlation between GLIS2 and the regulation of Tumor-Associated, Calcium Signal Transducer 2 (TACSTD2 or TROP2), a transmembrane glycoprotein with a role in cell-cell adhesion [65]. TACSTD2 is overexpressed in many cancers and associated with enhanced cell proliferation, EMT, and metastasis that involves increased expression and activation of several kinase pathways, including JAK2/STAT3, PI3K/AKT, PKA, NF-κB, and ERK MAP kinase. GLIS2 deficiency increases TACSTD2 expression in kidneys of GLIS2-deficient mice suggesting that TACSTD2 expression is normally repressed by GLIS2. The reported increased EMT in GLIS2-deficient kidneys is consistent with TACSTD2 promoting EMT and might contribute to development of nephronophthisis in these mice [9, 31]. Whether this relationship plays a role in metastasis in renal cancer needs further study.
GLIS1 and GLIS3 Translocations and Cancer
Recently, several studies have linked genetic changes in GLIS3 and GLIS1 to several cancers. Interchromosomal translocations between paired box 8 (PAX8) on chromosome 2q14.1 and GLIS1 on chromosome 1p32.3, and between PAX8 and GLIS3 on chromosome 9p24.2 were reported to be frequently associated with hyalinizing trabecular tumors (HTTs), a rare thyroid neoplasm derived from thyroid follicular cells [6]. PAX8-GLIS1 and PAX8-GLIS3 rearrangements were found in, respectively, 7% and 93% of HTTs, whereas no PAX8-GLIS3 rearrangements and one PAX8-GLIS1 rearrangement were detected among 220 papillary thyroid carcinomas examined. The PAX8-GLIS3 rearrangement places exon 1 and 2 of PAX8 in-frame with exon 3 of GLIS3 generating a fusion protein consisting of an alternative N-terminus of 65 aa and the 801 aa C-terminal region of GLIS3 (Fig. 2A). The PAX8-GLIS1 rearrangement places exons 1 and 2 of PAX8 in-frame with exon 2 of GLIS1 creating a fusion protein consisting of the first 8 aa of PAX8, an alternative 89 aa sequence, and the entire GLIS1 protein (Fig. 2A). In both rearrangements the expression of the fusion transcript remains under the control of the PAX8 promoter, which results in a 20- and 7-fold increase in GLIS1 and GLIS3 mRNA expression, respectively. GLIS3 is normally expressed in thyroid follicular cells, where it activates the transcription of several thyroid hormone biosynthetic and extracellular matrix genes, and is required for the proliferation of these cells [27]. The DBD and TAD of GLIS3 or GLIS1 are preserved in both fusion proteins suggesting that they still can effectively function as transcriptional activators. This is supported by data showing that the overexpression of the GLIS3 fusion protein in HTTs results in enhanced cell proliferation and increased expression of the iodide transporter gene, SLC5A5, and extracellular matrix genes [6]. The latter is consistent with the regulation of extracellular matrix and cell cycle genes by GLIS3 in the thyroid gland and the identification of SLC5A5 as a direct transcriptional target of GLIS3 [27].
Figure 2.
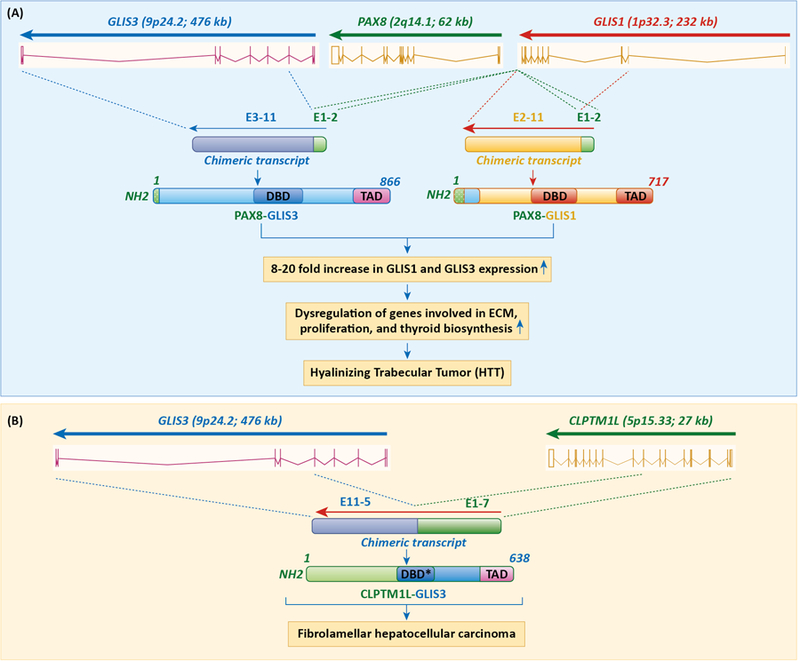
Translocations involving GLIS1 and GLIS3 are associated with several cancers. A. Interchromosomal rearrangements between PAX8 on chromosome 2q14.1 and GLIS3 on chromosome 9p24.2 and between PAX8 and GLIS1 on chromosome 1p32.3 are frequently associated with hyalinizing trabecular tumors (HTTs). These translocations cause overexpression of GLIS1 or GLIS3 fusion proteins, which then interact with GLISBS in GLIS target genes and recruitment of co-activators, such as CBP. This subsequently leads to activation of proliferation-regulatory, ECM, and thyroid hormone biosynthetic genes, target genes of GLIS3, and increased tumorigenicity of thyroid follicular cells. B. Association of a translocation between exons 1–7 of CLPTM1L on chromosome 5 and exons 5–11 of GLIS3 with fibrolamellar hepatocellular carcinoma (FHCC). This translocation results in the generation of a CLPTM1L-GLIS3 fusion protein, in which the extracellular N-terminal region (1–297) of the CLPTM1L protein is in frame with the 416–775 region of GLIS3. DBD* indicates GLIS3 DBD lacking the first three zinc finger motifs.
A different study identified a translocation between chromosome 5 and chromosome 9 involving exons 1–7 of cleft lip and palate transmembrane protein 1-like (CLPTM1L) and exons 5–11 of GLIS3 in a patient with fibrolamellar hepatocellular carcinoma (FHCC), a rare form of hepatocellular carcinoma that typically affects young adults (Fig. 2B)[66]. This translocation leads to the generation of a CLPTM1L-GLIS3 fusion protein, in which the extracellular N-terminal region (1–297) of the CLPTM1L protein is in frame with the 416–775 region of GLIS3 (ENST00000381971.8; GLIS3–202) that includes part of its third zinc finger motif, the 4th and 5th zinc finger motifs and its C-terminal TAD. The absence of the first three zinc finger motifs abolishes the normal DNA-binding function of GLIS3. The CLPTM1L-GLIS3 fusion protein was largely localized in the cytoplasm when expressed in HeLa cells in contrast to CLPTM1L, which is transmembrane protein, and GLIS3, which is largely nuclear. This indicates that, in contrast to the PAX8-GLIS3 fusion protein, the normal transcriptional activity of GLIS3 is lost in CLPTM1L-GLIS3. Expression of the fusion protein in Hep3B and C3A hepatocellular carcinoma cells caused an increase in colony-forming ability and cell proliferation consistent with the concept that the fusion protein functions as an oncogene and participates in the development of FHCC in this patient [66].
Additional Links Between GLIS1/3 and Cancer
Overexpression of GLIS3 has been observed in a variety of tumor cell types, including glioblastoma, ependymoma, ductal breast carcinomas, chromophobe renal cell carcinoma, colorectal cancer, and melanoma [67–73]. High GLIS3 mRNA expression in ependymomas, which constitute 3–9% of all neuroepithelial malignancies, was associated with high proliferation indices. Elevated levels of GLIS3 expression were also found to correlate with invasive mesenchymal-like melanoma and reduced levels of E-cadherin expression, while down-regulation of GLIS3 expression by siRNA inhibited migration of cultured melanoma cells and melanoma invasion in a chicken transplantation model [72]. These studies appear to suggest that increased expression of GLIS3 promotes proliferation, migration and metastasis. The precise mechanisms by which GLIS3 enhanced migration and invasiveness has yet to be determined, but may at least in part be due to a stimulation of epithelial-mesenchymal transition [9, 31].
In contrast to these studies, two other reports revealed a negative correlation between GLIS3 expression and cancer progression. Study of the tumor suppressor, N-myc downstream regulated gene 1 (NDRG1) [74], which expression is down-regulated in multiple cancers, including pancreatic cancer, identified an inverse correlation between GLIS3 expression and oncogenesis. Ectopic expression of NDRG1 in pancreatic cancer cell lines caused a significant increase in GLIS3 and the HECT domain E3 ubiquitin ligase, neural precursor cell-expressed, developmentally-downregulated 4-like (NEDD4L), as well as an up-regulation of the tumor suppressors, PTEN and SMAD4. Down-regulation of GLIS3 expression was also observed in juvenile granulosa cell tumors containing mutations in the oncogene AKT1 [75].
In addition to the role of GLIS1 rearrangements in HTTs, GLIS1 has been implicated in the regulation of several other features associated with malignancy, including risk of relapse and EMT. Acquisition of additional mutations is believed to be a major factor in the development of resistance to chemo- and radiotherapy, the continued presence of stem cells, and relapse in leukemia. High hyperdiploid (51–68 chromosomes) acute lymphoblastic leukemia (ALL) encompasses the largest subgroup (about 30%) of pediatric B-cell precursor ALL, which constitutes one of the most common malignancies in children [76–79]. It is characterized by the non-random gain of chromosomes X, 4, 6, 10, 14, 17, 18, and 21. About 20% of children suffering from high hyperdiploid ALL develop recurrent disease. This relapse is frequently associated with oncogenic mutations in genes with roles in the receptor tyrosine kinase (RTK)/RAS pathway and epigenetic regulation, and with variations in cell fate determining genes. Recently, nucleotide variations downstream of GLIS1, which has been shown to promote reprogramming of somatic cells into induced pluripotent stem cells [32, 33], was reported to be associated with increased risk of relapse in patients with high hyperdiploid ALL [79]. Deleterious mutations in GLIS1 have also been identified in several recurrent ETV6-RUNX1-positive ALL cases.
Several studies have reported an association between GLIS1 expression and tumorigenic phenotype. In MMTV-Cut-like Homeobox 1 (CUX1) transgenic mice, which develop various histologically different types of mammary tumors, GLIS1 was found to be highly expressed in a subset of adenosquamous mammary carcinomas that exhibited high-level of WNT gene expression [80]. Activation of the WNT/β-catenin pathway has been associated with a variety of cancers, including breast cancer. Elevated WNT expression in human breast cancer was found associated with increased levels of GLIS1 and CUX1 mRNA expression. This correlated with increased expression of several EMT-related genes, including Snail1 (SNAI1), vimentin (VIM), and Twist family BHLH transcription factor 1 (TWIST1), and lower expression of the epithelial marker E-cadherin (CDH1). Exogenous expression of GLIS1 in an MMTV-CUX1 cancer cell line and in MCF10A cells caused a significant increase in the expression of several WNT genes, including WNT2, WNT10a, and WNT7b, and in β-catenin/TCF transcriptional activity. These observations suggest that in cells that express high levels of GLIS1 and CUX1, these two proteins cooperate to optimally induce WNT gene expression that subsequently leads to increased EMT-related gene expression, cell migratory behavior, and invasiveness. A different study showed that hypoxia significantly enhanced GLIS1 expression in a number of carcinoma cell lines, including mammary carcinoma MCF7 and lung carcinoma A549 [81]. Hypoxia-inducing factors, HIF1a and HIF2a, together with JUN were shown to enhance GLIS1 transcription, whereas expression of the von Hippel–Lindau (VHL) protein in renal carcinoma cells, which enhances the proteasome-mediated degradation of HIF1a, reduced the level of GLIS1 expression. The activation domain, but not the DNA binding domain of HIF2a, was found to be required for GLIS1 activation suggesting a noncanonical mechanism of transcriptional regulation of GLIS1. Hypoxia has been reported to induce WNT signaling, and promote EMT and cancer progression [82], this up-regulation of WNT might be partially mediated by increased GLIS1 expression.
Concluding Remarks
In summary, GLIS1–3 transcription factors regulate the expression of many genes involved in the control of cell proliferation, stem cell renewal, differentiation, inflammatory responses, and epithelial-mesenchymal transition, all processes that are of critical importance in oncogenesis. Genetic alterations in GLIS1–3, as well as changes in the expression of these genes, are linked to several malignancies, including non-DS-AMKL, hyalinizing trabecular tumors, and fibrolamellar hepatocellular carcinoma. In the case of the role of CBFA2T3-GLIS2 rearrangements in leukemia, a lot of progress has been made in understanding the molecular mechanisms that underlie the changes in gene expression and signaling pathways induced by the CBFA2T3-GLIS2 fusion protein that lead to the development of non-DS-AMKL. However, these studies raised also additional questions (see outstanding questions). These studies may also open possibilities for several new therapeutic strategies, including treatment with NHR2 peptides and AURKA inhibitors. For many of the other malignancies, in which GLIS1–3 have been implicated, the challenge will be to obtain greater insights into the molecular mechanisms by which translocations and overexpression promote a tumorigenic phenotype and into the upstream pathways that regulate GLIS1–3 activity. By targeting GLIS1–3 pathways, such molecular insights might lead to the development of new approaches in the management of cancer.
Outstanding Questions.
Does GLIS2 have a regulatory role during normal hematopoiesis and what is its molecular mechanism?
Is regulation of the hedgehog-GLI pathways involved in any aspect of the (GLIS1–3)-associated malignancies?
What are the regulatory mechanisms by which GLIS proteins regulate self-renewal, differentiation, and cellular proliferation and how do they relate to the development of various cancers?
Which upstream signaling pathways regulate GLIS activity and can targeting GLIS1–3 signaling pathways offer new therapeutic strategies for leukemia and other malignancies?
It there any association between GLIS1–3 single nucleotide polymorphisms and an increased risk of developing cancer?
What roles might regulation of immune responses and inflammation by GLIS1–3 play in (GLIS1–3)-dependent oncogenesis?
Highlights.
Genetic alterations in GLIS1–3 are associated with the development of several malignancies.
CBFA2T3 (ETO2) and GLIS2 rearrangements are associated with 20–30% of pediatric non-Down’s syndrome AMKL.
The CBFA2T3/ETO2-GLIS2 fusion protein regulates gene transcription in part through CBFA2T3- and GLIS3-mediated mechanisms, while a majority of the genes are aberrantly regulated involving different mechanisms.
Increased self-renewal of progenitor cells and reduced differentiation are key elements in the development of CBFA2T3/ETO2-GLIS2 AMLK.
Translocations between PAX8 and GLIS3 are frequently associated with hyalinizing trabecular tumors (HTTs), a thyroid neoplasm derived from thyroid follicular cells.
Targeting GLIS1–3 signaling pathways may present novel opportunities in the treatment of various cancers.
Acknowledgments:
I would like to thank David Scoville and Hong Soon Kang (NIEHS) for their comments on the manuscript. This research was supported by the Intramural Research Program of the NIEHS, NIH Z01-ES-101585.
Glossary
- Krüppel-like zinc finger proteins
DNA binding proteins containing three or more zinc finger motifs, that regulate gene transcription
- Krüppel-like zinc finger motifs
A protein structural motif consisting of a tetrahedral configuration of 2 cysteines and 2 histidines stabilized in the center by a zinc ion. These motifs interact with G/C-rich DNA sequences and mediate protein-protein interactions
- Hedgehog signaling pathway
A signal transduction mechanism in which Hedgehog signaling proteins bind to membrane receptors and regulate gene expression via activation of GLI transcription factors. This pathway is involved in the regulation of embryonic development, cell proliferation and differentiation, and plays a critical role in many cancers
- Transactivation domain
A protein domain that mediates the activation of gene transcription through its recruitment of co-activator proteins
- Translocation
Exchange of genomic regions within the same chromosome or between different chromosomes causing loss of (a) gene(s) or the generation of a fusion protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The author declares no conflict of interest.
References
- 1.Aberger F and Ruiz IAA (2014) Context-dependent signal integration by the GLI code: the oncogenic load, pathways, modifiers and implications for cancer therapy. Semin. Cell. Dev. Biol 33, 93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tetreault MP et al. (2013) Kruppel-like factors in cancer. Nat. Rev. Cancer 13, 701–713. [DOI] [PubMed] [Google Scholar]
- 3.Jetten AM (2018) GLIS1–3 transcription factors: critical roles in the regulation of multiple physiological processes and diseases. Cell. Mol. Life Sci 75, 3473–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gruber TA et al. (2012) An Inv(16)(p13.3q24.3)-encoded CBFA2T3-GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell 22, 683–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masetti R et al. (2013) CBFA2T3-GLIS2 fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not restricted to FAB M7 subtype. Blood 121, 3469–3472. [DOI] [PubMed] [Google Scholar]
- 6.Nikiforova MN et al. (2019) GLIS Rearrangement is a Genomic Hallmark of Hyalinizing Trabecular Tumor of the Thyroid Gland. Thyroid 29, 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thiollier C et al. (2012) Characterization of novel genomic alterations and therapeutic approaches using acute megakaryoblastic leukemia xenograft models. J. Exp. Med 209, 2017–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kang HS et al. (2010) Gli-similar (Glis) Krüppel-like zinc finger proteins: insights into their physiological functions and critical roles in neonatal diabetes and cystic renal disease. Histol. Histopath 25, 1481–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim YS et al. (2008) Kruppel-like zinc finger protein Glis2 is essential for the maintenance of normal renal functions. Mol. Cell. Biol 28, 2358–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamar E et al. (2001) Identification of NKL, a novel Gli-Kruppel zinc-finger protein that promotes neuronal differentiation. Development 128, 1335–1346. [DOI] [PubMed] [Google Scholar]
- 11.Lichti-Kaiser K et al. (2014) Transcription Factor Gli-Similar 3 (Glis3): Implications for the Development of Congenital Hypothyroidism. J. Endocrinol. Diabetes Obes 2, 1024. [PMC free article] [PubMed] [Google Scholar]
- 12.Lichti-Kaiser K et al. (2012) Gli-similar proteins: their mechanisms of action, physiological functions, and roles in disease. Vitam. Horm 88, 141–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakashima M et al. (2002) A novel gene, GliH1, with homology to the Gli zinc finger domain not required for mouse development. Mech. Dev 119, 21–34. [DOI] [PubMed] [Google Scholar]
- 14.Zhang F and Jetten AM (2001) Genomic structure of the gene encoding the human GLI-related, Kruppel-like zinc finger protein GLIS2. Gene 280, 49–57. [DOI] [PubMed] [Google Scholar]
- 15.Zhang F et al. (2002) Characterization of Glis2, a novel gene encoding a Gli-related, Kruppel-like transcription factor with transactivation and repressor functions. Roles in kidney development and neurogenesis. J. Biol. Chem 277, 10139–10149. [DOI] [PubMed] [Google Scholar]
- 16.Pak E and Segal RA (2016) Hedgehog Signal Transduction: Key Players, Oncogenic Drivers, and Cancer Therapy. Dev. Cell 38, 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toftgard R (2000) Hedgehog signalling in cancer. Cell. Mol. Life Sci 57, 1720–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim YS et al. (2003) GLIS3, a novel member of the GLIS subfamily of Kruppel-like zinc finger proteins with repressor and activation functions. Nucleic Acids Res 31, 5513–5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vasanth S et al. (2011) Identification of nuclear localization, DNA binding, and transactivating mechanisms of Kruppel-like zinc finger protein Gli-similar 2 (Glis2). J. Biol. Chem 286, 4749–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li B et al. (2011) Increased hedgehog signaling in postnatal kidney results in aberrant activation of nephron developmental programs. Hum. Mol. Genet 20, 4155–4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.ZeRuth GT et al. (2013) The Kruppel-like protein Gli-similar 3 (Glis3) functions as a key regulator of insulin transcription. Mol. Endocrinol 27,1692–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim SC et al. (2005) Kruppel-like zinc finger protein Gli-similar 2 (Glis2) represses transcription through interaction with C-terminal binding protein 1 (CtBP1). Nucleic Acids Res 33, 6805–6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim YS et al. (2007) The Kruppel-like zinc finger protein Glis2 functions as a negative modulator of the Wnt/beta-catenin signaling pathway. FEBS Lett 581, 858–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dimitri P et al. (2015) Expanding the Clinical Spectrum Associated With GLIS3 Mutations. J. Clin. Endocrinol. Metab 100, E1362–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kang HS et al. (2016) Transcription Factor GLIS3: A New and Critical Regulator of Postnatal Stages of Mouse Spermatogenesis. Stem Cells (Dayton, Ohio) 34, 2772–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang HS et al. (2009) Transcription factor Glis3: a novel critical player in the regulation of pancreatic β-cell development. Mol. Cell. Biol 29, 6366–6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang HS et al. (2017) GLIS3 is indispensable for TSH/TSHR-dependent thyroid hormone biosynthesis and follicular cell proliferation. J. Clin. Invest 127, 4326–4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Senee V et al. (2006) Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat. Genet 38, 682–687. [DOI] [PubMed] [Google Scholar]
- 29.Jeon K et al. (2019) GLIS3 Transcriptionally Activates WNT Genes to Promote Differentiation of Human Embryonic Stem Cells into Posterior Neural Progenitors. Stem Cells 37, 202–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Y et al. (2009) The Kruppel-like zinc finger protein Glis3 directly and indirectly activates insulin gene transcription. Nucleic Acids Res 37, 2529–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Attanasio M et al. (2007) Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat. Genet 39, 1018–1024. [DOI] [PubMed] [Google Scholar]
- 32.Maekawa M et al. (2011) Direct reprogramming of somatic cells is promoted by maternal transcription factor Glis1. Nature 474, 225–479. [DOI] [PubMed] [Google Scholar]
- 33.Scoville DW et al. (2017) GLIS1–3: emerging roles in reprogramming, stem and progenitor cell differentiation and maintenance. Stem Cell Investig DOI: 10.21037/sci.2017.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee SY et al. (2017) Glis family proteins are differentially implicated in the cellular reprogramming of human somatic cells. Oncotarget 8, 77041–77049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raleigh DR and Reiter JF (2019) Misactivation of Hedgehog signaling causes inherited and sporadic cancers. J. Clin. Invest 129, 465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Masetti R et al. (2013) DHH-RHEBL1 fusion transcript: a novel recurrent feature in the new landscape of pediatric CBFA2T3-GLIS2-positive acute myeloid leukemia. Oncotarget 4, 1712–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thiollier C et al. (2012) [Novel ETO2-GLIS2 fusion and therapeutic strategy in acute megakaryoblastic leukemia]. Med. Sci. (Paris) 28, 1013–1016. [DOI] [PubMed] [Google Scholar]
- 38.de Rooij JD et al. (2017) Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat. Genet 49, 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gruber TA and Downing JR (2015) The biology of pediatric acute megakaryoblastic leukemia. Blood 126, 943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Rooij JDE et al. (2016) Recurrent abnormalities can be used for risk group stratification in pediatric AMKL: a retrospective intergroup study. Blood 127, 3424–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hara Y et al. (2017) Prognostic Impact of Specific Molecular Profiles in Pediatric Acute Megakaryoblastic Leukemia in Non-Down Syndrome. Genes Chromosomes Cancer 56, 394–404. [DOI] [PubMed] [Google Scholar]
- 42.Ishibashi M et al. (2016) Clinical Courses of Two Pediatric Patients with Acute Megakaryoblastic Leukemia Harboring the CBFA2T3-GLIS2 Fusion Gene. Turk. J. Haematol 33, 331–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thirant C et al. (2017) Molecular pathways driven by ETO2-GLIS2 in aggressive pediatric leukemia. Mol. Cell. Oncol DOI: 10.1080/23723556.2017.1345351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mitsui-Sekinaka K et al. (2018) A pediatric case of acute megakaryocytic leukemia with double chimeric transcripts of CBFA2T3-GLIS2 and DHH-RHEBL1. Leuk. Lymphoma 59, 1511–1513. [DOI] [PubMed] [Google Scholar]
- 45.Masetti R et al. (2019) CBFA2T3-GLIS2-positive acute myeloid leukaemia. A peculiar paediatric entity. Br. J. Haematol 184, 337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fujiwara T et al. (2013) Role of transcriptional corepressor ETO2 in erythroid cells. Exp. Hematol DOI: 10.1016/j.exphem.2012.10.015. [DOI] [PubMed] [Google Scholar]
- 47.Lutterbach B et al. (1998) ETO, a target of t(8;21) in acute leukemia, interacts with the N-CoR and mSin3 corepressors. Mol. Cell. Biol 18, 7176–7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fischer MA et al. (2012) Myeloid translocation gene 16 is required for maintenance of haematopoietic stem cell quiescence. Embo J 31, 1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holmfeldt P et al. (2016) Functional screen identifies regulators of murine hematopoietic stem cell repopulation. J. Exp. Med 213, 433–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hamlett I et al. (2008) Characterization of megakaryocyte GATA1-interacting proteins: the corepressor ETO2 and GATA1 interact to regulate terminal megakaryocyte maturation. Blood 112, 2738–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thirant C et al. (2017) ETO2-GLIS2 Hijacks Transcriptional Complexes to Drive Cellular Identity and Self-Renewal in Pediatric Acute Megakaryoblastic Leukemia. Cancer Cell 31, 452–465. [DOI] [PubMed] [Google Scholar]
- 52.Beak JY et al. (2008) Functional analysis of the zinc finger and activation domains of Glis3 and mutant Glis3(NDH1). Nucleic Acids Res 36, 1690–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dang J et al. (2017) AMKL chimeric transcription factors are potent inducers of leukemia. Leukemia 31, 2228–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goldenson B and Crispino JD (2015) The aurora kinases in cell cycle and leukemia. Oncogene 34, 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wen Q et al. (2012) Identification of regulators of polyploidization presents therapeutic targets for treatment of AMKL. Cell 150, 575–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Masetti R et al. (2017) Hh/Gli antagonist in acute myeloid leukemia with CBFA2T3-GLIS2 fusion gene. J. Hematol. Oncol DOI: 10.1186/s13045-017-0396-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Long B et al. (2016) Targeting GLI1 Suppresses Cell Growth and Enhances Chemosensitivity in CD34+ Enriched Acute Myeloid Leukemia Progenitor Cells. Cell. Physiol. Biochem 38, 1288–1302. [DOI] [PubMed] [Google Scholar]
- 58.Wellbrock J et al. (2015) Expression of Hedgehog Pathway Mediator GLI Represents a Negative Prognostic Marker in Human Acute Myeloid Leukemia and Its Inhibition Exerts Antileukemic Effects. Clin. Cancer Res 21, 2388–2398. [DOI] [PubMed] [Google Scholar]
- 59.Shima H et al. (2018) Ring1A and Ring1B inhibit expression of Glis2 to maintain murine MOZ-TIF2 AML stem cells. Blood 131, 1833–1845. [DOI] [PubMed] [Google Scholar]
- 60.Largeot A et al. (2016) Expression of the MOZ-TIF2 oncoprotein in mice represses senescence. Exp. Hematol DOI: 10.1016/j.exphem.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carapeti M et al. (1998) A novel fusion between MOZ and the nuclear receptor coactivator TIF2 in acute myeloid leukemia. Blood 91, 3127–3133. [PubMed] [Google Scholar]
- 62.Iwama A (2017) Polycomb repressive complexes in hematological malignancies. Blood 130, 23–29. [DOI] [PubMed] [Google Scholar]
- 63.Heery DM (2018) A fellowship of Ring1 maintains AML stem cells. Blood 131, 1771–1773. [DOI] [PubMed] [Google Scholar]
- 64.Yuan J et al. (2019) GLIS2 redundancy causes chemoresistance and poor prognosis of gastric cancer based on coexpression network analysis. Oncol Rep 41, 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guerra E et al. (2013) The Trop-2 signalling network in cancer growth. Oncogene 32, 1594–1600. [DOI] [PubMed] [Google Scholar]
- 66.Xu L et al. (2015) Genomic analysis of fibrolamellar hepatocellular carcinoma. Hum. Mol. Genet 24, 50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cooper LA et al. (2010) The proneural molecular signature is enriched in oligodendrogliomas and predicts improved survival among diffuse gliomas. PLoS One DOI: 10.1371/journal.pone.0012548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lukashova-v Zangen I et al. (2007) Ependymoma gene expression profiles associated with histological subtype, proliferation, and patient survival. Acta Neuropathol. (Berl.) 113, 325–337. [DOI] [PubMed] [Google Scholar]
- 69.Charafe-Jauffret E et al. (2006) Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene 25, 2273–2284. [DOI] [PubMed] [Google Scholar]
- 70.Rami F et al. (2016) Alteration of GLIS3 gene expression pattern in patients with breast cancer. Adv. Biomed. Res DOI: 10.4103/2277-9175.178803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Al-Temaimi RA et al. (2016) Identification of 42 Genes Linked to Stage II Colorectal Cancer Metastatic Relapse. Int. J. Mol. Sci DOI: 10.3390/ijms17050598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jayachandran A et al. (2016) Identifying and targeting determinants of melanoma cellular invasion. Oncotarget 7, 41186–41202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yusenko MV and Kovacs G (2009) Identifying CD82 (KAI1) as a marker for human chromophobe renal cell carcinoma. Histopathology 55, 687–695. [DOI] [PubMed] [Google Scholar]
- 74.Kovacevic Z et al. (2013) The iron-regulated metastasis suppressor NDRG1 targets NEDD4L, PTEN, and SMAD4 and inhibits the PI3K and Ras signaling pathways. Antioxid. Redox Signal 18, 874–887. [DOI] [PubMed] [Google Scholar]
- 75.Auguste A et al. (2015) Molecular analyses of juvenile granulosa cell tumors bearing AKT1 mutations provide insights into tumor biology and therapeutic leads. Hum. Mol. Genet 24, 6687–6698. [DOI] [PubMed] [Google Scholar]
- 76.Malinowska-Ozdowy K et al. (2015) KRAS and CREBBP mutations: a relapse-linked malicious liaison in childhood high hyperdiploid acute lymphoblastic leukemia. Leukemia 29, 1656–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Paulsson K et al. (2015) The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat. Genet 47, 672–676. [DOI] [PubMed] [Google Scholar]
- 78.Paulsson K and Johansson B (2009) High hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer 48, 637–660. [DOI] [PubMed] [Google Scholar]
- 79.Chen C et al. (2015) Next-generation-sequencing of recurrent childhood high hyperdiploid acute lymphoblastic leukemia reveals mutations typically associated with high risk patients. Leuk. Res 39, 990–1001. [DOI] [PubMed] [Google Scholar]
- 80.Vadnais C et al. (2014) Autocrine Activation of the Wnt/beta-Catenin Pathway by CUX1 and GLIS1 in Breast Cancers. Biol. Open 3, 937–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Khalesi E et al. (2013) The Kruppel-like zinc finger transcription factor, GLI-similar 1, is regulated by hypoxia-inducible factors via non-canonical mechanisms. Biochem. Biophys. Res. Commun 441, 499–506. [PubMed] [Google Scholar]
- 82.Gonzalez DM and Medici D (2014) Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal DOI: 10.1126/scisignal.2005189. [DOI] [PMC free article] [PubMed] [Google Scholar]