

Abstract
Alzheimer disease (AD) is a heterogeneous disease with a complex pathobiology. The presence of extracellular amyloid-β deposition as neuritic plaques and intracellular accumulation of hyperphosphorylated tau as neurofibrillary tangles remain the primary neuropathologic criteria for AD diagnosis. However, a number of recent fundamental discoveries highlight important pathological roles for other critical cellular and molecular processes. Despite this, no disease modifying treatment currently exists and numerous phase 3 clinical trials have failed to demonstrate benefit. We review here recent advances in our understanding of AD pathobiology and discuss current treatment strategies, highlighting recent clinical trials and opportunities for developing future disease modifying therapies.
INTRODUCTION
Dementia describes an intra-individual pattern of decline in memory and thinking impairing at least two domains of cognition (McKhann et al., 2011). Alzheimer disease (AD) is the most common cause of dementia. The majority of cases occur after age 65, constituting late-onset AD (LOAD), while cases occurring earlier than age 65 are considerably more rare, constituting less than 5% of all cases and are termed early-onset AD (EOAD) (Alzheimer’s Association, 2019). Approximately 1-2% of AD is inherited in an autosomal dominant fashion (ADAD) and can present with very early age of onset (EOAD) and more rapid rate of progression, and sometimes is associated with other neurologic symptoms seen less frequently in sporadic AD (Bateman et al., 2012). Clinical syndromes consistent with AD are defined by classical symptoms and cognitive profiles. However, AD as a distinct entity is now defined biologically by the presence of a specific neuropathological profile (Jack et al., 2018a): extracellular deposition of amyloid-β (Aβ) in the form of diffuse and neuritic plaques and the presence of intraneuronal neurofibrillary tangles and neuropil threads within dystrophic neurites consisting of aggregated hyperphosphorylated tau protein (Duyckaerts et al., 2009).
Dementia due to AD is associated with the onset of significant and progressive disability throughout the disease course with death an inevitable outcome, generally occurring within 5-12 years of symptom onset (Vermunt et al., 2019). The burden on caregivers and the public health sector are enormous (Alzheimer’s Association, 2019). There is a dire need for disease modifying therapies that may prevent or slow rate of disease progression, but unfortunately none are currently available. The history of pharmaceutical development for AD has been plagued by a seemingly endless parade of mid-to-late-stage clinical trial failures. Nonetheless, significant strides have been made in recent years in clarifying key aspects of the underlying pathobiology of AD. Though the therapeutic pipeline has faced struggles and some pharmaceutical companies have chosen to abandon their AD drug development divisions, novel therapeutic strategies are still being actively developed and tested. This review discusses recent advances in our understanding of the pathobiology of AD and summarizes treatment strategies and the challenges and opportunities on the path to development of truly disease-modifying treatments.
Clinical and Preclinical Disease
Symptomatic AD follows an insidious and progressive course. Typical amnestic cases are characterized by early impairment in learning and memory, followed by later impairments in complex attention, executive function, language, visuospatial function, praxis, gnosis and behavior/social comportment (McKhann et al., 2011). Symptomatic AD may also present as atypical clinical syndromes, in which there is early impairment in non-memory domains. Posterior cortical atrophy presents with early deficits in visuospatial function and praxis/gnosis (Tang-Wai et al., 2004). Logopenic variant of primary progressive aphasia is characterized by dysfluent language with prominent word-finding impairment and severe impairment in repetition (Gorno-Tempini et al., 2008). The behavioral/dysexecutive variant of AD presents with early executive dysfunction or behavioral impairment (especially apathy, hyperorality and perseveration) (Ossenkoppele et al., 2015). Clinical dementia severity can be graded by use of standardized instruments such as the Clinical Dementia Rating (CDR) (Burke et al., 1988; Morris, 1997), which grades disease severity based on composite level of dysfunction in domains of memory, orientation, judgement and problem solving, involvement in community affairs, function in home and hobbies, and self-care.
Antemortem AD neuropathologic diagnoses can now be made with reasonable validity using cerebrospinal fluid (CSF) or positron emission tomography (PET) imaging biomarkers as surrogate markers for cerebral Aβ and tau deposition (Brier et al., 2016; Fagan et al., 2006, 2007; Lowe et al., 2019; Morris et al., 2009). Recent studies demonstrate the ability to detect CNS Aβ deposition via the use of plasma assessment of Aβ species (Nakamura et al., 2018; Ovod et al., 2017; Palmqvist et al., 2019). Longitudinal studies of cognitive function and CSF and neuroimaging biomarker changes in ADAD and LOAD have identified a significant preclinical phase of disease preceding onset of clinical symptoms by at least 10-20 years (Vermunt et al., 2019), characterized by early deposition of Aβ in the precuneus and other cortical regions comprising the default mode network, followed sequentially by regional cortical hypometabolism, accumulation of tau pathology, hippocampal volume loss, and onset of symptomatic cognitive impairment (Figure 1) (Bateman et al., 2012; Fagan et al., 2006, 2007, 2014; Gordon et al., 2016, 2018; Hanseeuw et al., 2019; Jack and Holtzman, 2013; Morris et al., 2009; Vos et al., 2013). Synaptic and neuronal loss in the entorhinal cortex generally correlates well with onset of cognitive impairment (Gómez-Isla et al., 1996). CSF and plasma neurofilament light chain (NfL) is an emerging biomarker that appears to track the level of general neurodegeneration across all forms of neurodegenerative dementias (Bridel et al., 2019; Mielke et al., 2019). Studies of both ADAD and LOAD have demonstrated that rate of change in CSF and plasma NfL levels correlate with cortical thickness on structural MRI and cognitive performance (Mattsson et al., 2019; Preische et al., 2019).
Figure 1.
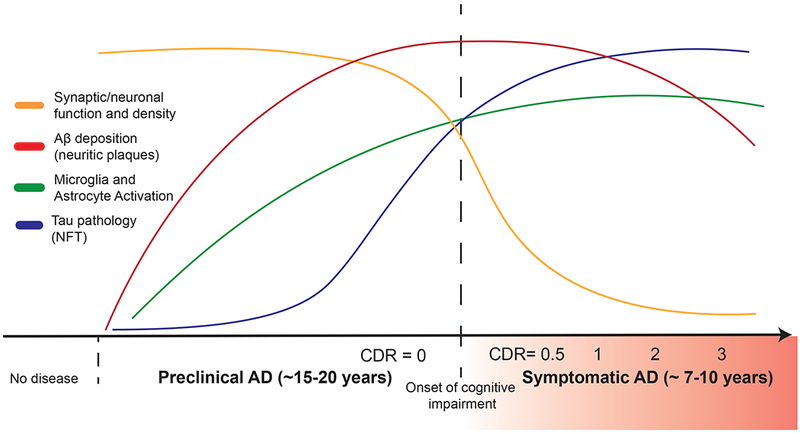
Timing of major AD pathophysiological events in relation to clinical course. A protracted preclinical phase of disease is characterized by the early onset of amyloid deposition. This is detected by a reduction in CSF and plasma levels of Aβ42 or increased global signal on amyloid PET imaging. Concurrently, there are early neuroinflammatory changes (such as microglial activation). Microgliosis can be detected longitudinally via use of PK11195 PET imaging though better agents are needed. This is followed by the spread of neurofibrillary tangle (NFT) tau pathology from the medial temporal lobes into neocortex. Increased signal on tau PET imaging and increased CSF phospho-tau levels mark this change in patients. Synaptic dysfunction, synapse loss and neurodegeneration accumulates with pathologic spread of tau aggregates. Imaging analysis of hippocampal and cortical volumes allows for longitudinal tracking of neurodegenerative changes. Onset and progression of cognitive impairment correlates with accumulation of tau and hippocampal volume loss but not amyloid deposition. Onset and severity of clinical symptoms in AD can be staged by use of the Clinical Dementia Rating (CDR) scale, where a score of 0 indicates normal cognition and scores of 0.5, 1, 2 and 3 indicate questionable, mild, moderate and severe dementia, respectively.
PATHOPHYSIOLOGY OF AD
Amyloid
Aβ peptide was first identified as the primary constituent of meningovascular amyloid in 1984 (Glenner and Wong, 1984) and subsequently as the main constituent in amyloid neuritic plaques (Masters et al., 1985). Over the ensuing decades, enormous research efforts were expended to clarify the underlying biology of this peptide and its role in AD pathophysiology. Aβ is produced by sequential cleavage of β-amyloid precursor protein (APP) by β-secretase and γ-secretase (Figure 2A) (for review see (Haass et al., 2012)). The β-secretase enzyme (BACE1) cleaves APP at the N-terminus of the Aβ sequence, releasing secreted APP-β and the membrane bound C99 fragment (Vassar et al., 1999). The γ-secretase complex consists of four protein subunits: presenilin (PSEN), presenilin enhancer (PEN), APH and Nicastrin. There are multiple isoforms of PSEN (PSEN1/PSEN2) and APH (APHA, APH B/C); up to four different gamma secretase complexes may exist in single cell (Voytyuk et al., 2018; Xia, 2019). Following cleavage by BACE1, the γ-secretase complex binds to N-terminally cleaved APP fragment (C99) and intramembranously cleaves at the epsilon site releasing C-terminal fragment (CTF) and Aβ48. The complex then processes along the remaining C-terminal end producing sequentially shorter peptides until the Aβ peptide is released from the complex (generally after producing peptides 38, 40 and 42 amino acids in length). Recent publication of high resolution substrate-enzyme cryo-EM molecular structures for γ-secretase-APP C83 fragment and γ-secretase-Notch 100 fragment (Yang et al., 2019; Zhou et al., 2019) has definitively determined substrate binding sites for γ-secretase and lends strong evidence for a helix-unwinding model of sequential substrate processing. BACE1, γ-secretase and APP form a large molecular complex in vivo, suggesting that Aβ production may be facilitated by directly shuttling APP from one processing enzyme to another (Liu et al., 2019). Aβ is produced predominantly in endosomes and its release from neurons is modulated by synaptic activity (Kamenetz et al., 2003; Wei et al., 2010) both presynaptically (Cirrito et al., 2005, 2008) and postsynaptically (Verges et al., 2011).
Figure 2.
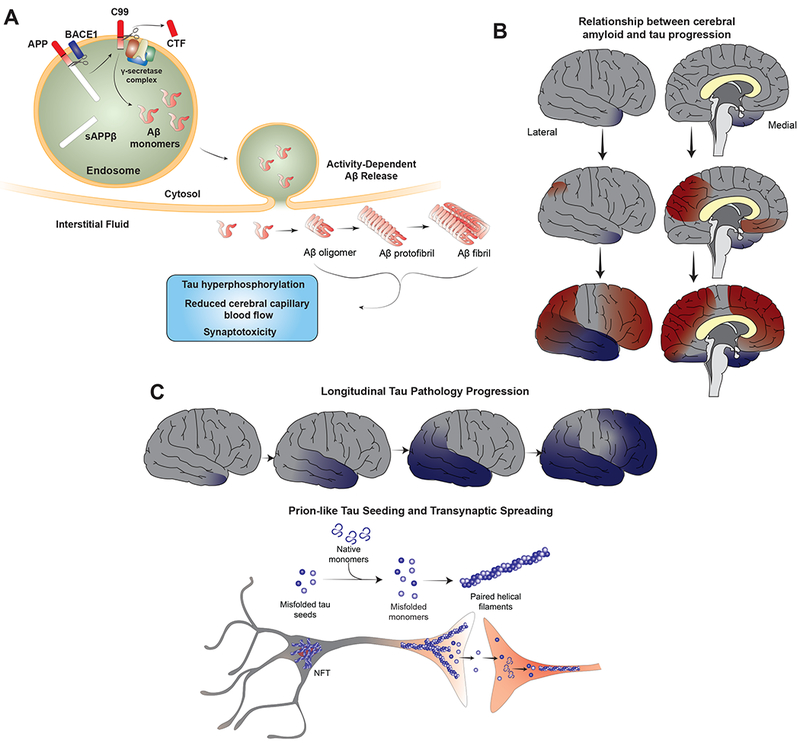
Selected roles of Aβ and tau in AD pathophysiology. (A) Aβ is derived from APP via the proteolytic functions of BACE1 and γ-secretase. BACE1 and APP are co-localized to endosomes, which is the location of intracellular Aβ production. Aβ is secreted into the interstitial fluid via a pathway that is enhanced in the setting of neuronal activity. Following secretion, Aβ aggregates into higher order oligomers and fibrils that have numerous effects on cellular function, including impaired synaptic activity and synapse loss, impaired cerebral capillary blood flow and direct promotion of tau pathology by stimulating tau hyperphosphorylation as well as other pathways. (B) Pathological tau aggregation (blue shading) in the medial temporal lobes occurs with aging and is not always associated with cognitive impairment (primary age-related tauopathy). The earliest accumulation of Aβ deposition (red shading) is in the precuneus and posterior cingulate. Longitudinal CSF and imaging biomarker studies suggest that global amyloid accumulation is required for the pathologic spread of tau from the medial temporal lobes to other cortical regions in AD. In this way, AD may represent an amyloid-facilitated tauopathy. (C) Pathologic spread of tau aggregates in AD usually occurs in a stereotyped fashion along neuroanatomically connected networks. Misfolded tau likely acts in a prion-like manner to promote templated misfolding of native monomers, leading to seeding of new pathological tau aggregates. Tau pathology can subsequently spread trans-synaptically to distant neurons, representing a molecular correlate for pathologic tau spreading noted in human AD brain.
Aβ peptides are prone to aggregate into beta-sheet conformations in the form of higher order oligomers, protofibrils, and fibrils, which are detectable in AD brain. Owing to increased hydrophobicity of its expanded C-terminus, Aβ42 has a greater propensity for aggregation. Recent cryo-EM experiments have elucidated the structure of synthetically-derived Aβ42 fibrils, demonstrating 7 nm diameter fibrils containing two twisted protofilaments consisting of Aβ42 monomers assuming a “LS” shape stacked in parallel with in-register cross-β structure (Gremer et al., 2017). 8 nm amyloid fibrils are present in the center of neuritic plaques, whereas oligomers appear to be detectable amongst the halo of dystrophic neurites surrounding neuritic plaques (Masters and Selkoe, 2012). There is evidence that the process of Aβ aggregation may require Aβ uptake by microglia followed by intracellular seeding and aggregation (Sosna et al., 2018)
Aβ fibrillization can be “seeded” in a prion-like manner by the presence of small assemblies of misfolded beta-sheet-containing Aβ seeds that template the formation of larger amyloid aggregates (Walker and Jucker, 2015). Brain extracts containing minute amounts of misfolded Aβ prepared from AD brain or APP transgenic mice, when injected into APP transgenic animals via intracerebral or intraperitoneal routes, will induce cerebral amyloidosis (Eisele et al., 2009, 2010; Ye et al., 2015). There is strong evidence that human-to-human transmission of amyloid pathology is possible. Patients who developed iatrogenic Creutzfeld-Jakob disease (CJD) following injections of prion-contaminated growth hormone derived from pooled cadaveric pituitary extracts prior to 1985 have been found in some cases to have significant cerebral and vascular amyloid pathology (congophilic amyloid angiopathy [CAA]) (Jaunmuktane et al., 2015). When injected intracerebrally into APP transgenic mice, these extracts are able to seed CAA pathology and cerebellar amyloid deposition (Purro et al., 2018). There is evidence that the process of Aβ aggregation may require Aβ uptake by microglia followed by intracellular seeding and aggregation (Sosna et al., 2018)
Based on a number of lines of evidence, Hardy and Higgins proposed the amyloid cascade hypothesis in 1992, positing that deposition of Aβ in the brain is the initiating step of AD pathogenesis, leading to subsequent tau deposition, neuron and synaptic loss and cognitive decline (Hardy and Higgins, 1992). This hypothesis has been the leading model of AD pathogenesis since it was first proposed, although portions have been revised or supplemented over time (Musiek and Holtzman, 2015; Selkoe and Hardy, 2016). The hypothesis is supported by the discovery that exclusively genetic forms of AD, such as in autosomal dominant AD (ADAD), Down syndrome (trisomy 21), or APP locus duplications, cause an increase in the Aβ42/40 ratio, increased total Aβ production, or increased Aβ fibrillinogenic properties and are sufficient to induce typical AD pathology (Tcw and Goate, 2017). Also, a rare APP mutation A673T that reduces risk of developing AD causes decreased Aβ production (Jonsson et al., 2012; Martiskainen et al., 2017). In addition, the strongest genetic risk factor for LOAD, apolipoprotein E, APOE, in large part appears to increase risk via influencing Aβ seeding and clearance (Bales et al., 1997; Castellano et al., 2011; Verghese et al., 2013). While the genetic evidence strongly supports the importance of Aβ aggregation in instigating the AD cascade, it seems clear that Aβ is necessary but not sufficient and that there are other downstream factors that play a key role. For example, there is minimal correlation between phases of amyloid deposition and degree of cognitive decline (Nelson et al., 2012). Also, patterns of regional cerebral amyloid deposition do not correlate with patterns of regional cerebral hypometabolism on functional neuroimaging (Altmann et al., 2015; Edison et al., 2007), although a recent study suggests that regional amyloid deposition does correlate with distant regional hypometabolism, suggesting that amyloid reduces the metabolic activity of distant neurons projecting to regions of amyloid deposition (Pascoal et al., 2019). Finally, despite decades of investment by the pharmaceutical industry in anti-amyloid therapies and numerous phase 3 clinical trials, no amyloid-targeting therapy has been successful in limiting progression of cognitive impairment in symptomatic AD. These data suggest that while amyloid accumulation may be key in beginning the pathological process, other downstream events such as neuroinflammation and tau accumulation may be the main drivers of neurodegeneration.
One extension of the amyloid hypothesis is the “cellular phase” of AD, proposed by Bart De Strooper and Eric Karran (De Strooper and Karran, 2016). This extension proposes that accumulation of cerebral amyloid and tau pathology (the “biochemical phase”) is a slow, gradual process that is tolerated by CNS cells early in the course of disease, serving as a risk factor for development of clinical disease, but that the disease is only manifest clinically when cellular homeostatic mechanisms fail, leading to impaired clearance of aggregated pathologic protein (proteopathy), increased cellular stress, and a complex breakdown of finely tuned intercellular physiologic functions that ultimately lead to neurodegeneration. Specifically, the cellular phase is characterized by dysfunction of the neurovascular unit, aberrant neuronal network activity, and impaired astrocyte and microglia homeostatic functions / possible gain-of-toxic functions.
, Several lines of evidence suggest Aβ deposition may be required for progression of tau pathology in AD (Figure 2B). Neuropathological studies in humanshave demonstrated that tau pathology generally does not progress from the entorhinal cortex into the neocortex in the absence of co-occurring amyloid pathology (Pontecorvo et al., 2019; Price and Morris, 1999; Price et al., 2009; Wang et al., 2016). Longitudinal assessment of amyloid and tau PET imaging suggests that rate of amyloid accumulation predicts onset of tau accumulation whereas rate of tau accumulation predicts onset of cognitive impairment (Hanseeuw et al., 2019). A study of tau kinetics in humans and cell culture using stable isotope labeling techniques demonstrates that tau production correlates with presence of amyloid suggesting a mechanistic link (Sato et al., 2018). A 3D in vitro culture system consisting of human neurons differentiated from induced pluripotent stem cells overexpressing human APP containing FAD mutations generates robust Aβ42 production and amyloid deposition. Importantly, this model also develops elevated phospho-tau levels and fibrillary tau aggregates, suggesting that elevated Aβ levels alone are sufficient to drive tau pathology in human neurons (Choi et al., 2014). Multiple studies have assessed the effect of combined Aβ deposition on local tau pathology by either crossing tau transgenic mice with APP transgenic mice or injecting Aβ42 fibrils into tau transgenic mice. In these studies, tau pathology and neurodegeneration is enhanced in mice with both pathologies, whereas amyloid pathology is generally unaffected(Bolmont et al., 2007; Götz et al., 2001; Hurtado et al., 2010; Lewis et al., 2001; Pooler et al., 2015). as demonstrated in a recent study where intracerebral injection of tau fibrils derived from human AD brain into Aβ plaque-bearing mouse models led to seeding of aggregated human tau in periplaque dystrophic neurites followed by development of neurofibrillary tangles (He et al., 2018).
Aβ may also lead to cognitive impairment in ways independent from its effects on tau. Soluble oligomers isolated from AD brain have been shown to potently inhibit long-term potentiation (LTP), enhance long-term depression (LTD) and reduce dendritic spine density in rodent hippocampal slice cultures (Kamenetz et al., 2003; Shankar et al., 2007; Wei et al., 2010). They also cause impairment in cognitive tasks when injected into the lateral ventricles of rodent models (Shankar et al., 2008). It is unclear whether soluble oligomers in human AD brain mediate synaptotoxic effects sufficient to cause cognitive impairment. One issue is that it is difficult to determine if soluble oligomers definitively exist in vivo due to technical challenges related to the biochemical extraction procedures required to detect them. A recent study shows that the presence of Aβ also causes capillary constriction in human cortical slices by acting on pericytes to generate reactive oxygen species (ROS) leading to endothelin-1 release (Nortley et al., 2019). Aβ may also lead to reduced cerebral blood flow by inducing neutrophils to occlude and stall capillary flow (Cruz Hernández et al., 2019). Hypoxia can induce increased Aβ production, possibly via increased BACE1 expression (Sun et al., 2006), suggesting the possibility of a pathological feedforward loop.
In summary, the available data still strongly supports the central role of pathologic Aβ accumulation in mediating AD pathogenesis as outlined in the original description of the amyloid cascade hypothesis, although its mechanism may be less direct than originally anticipated and requires further clarity via ongoing studies.
Tau
Multiple lines of evidence suggest that aggregated, hyperphosphorylated forms of tau may be a primary driver of neurodegeneration in AD. Clinico-neuropathologic correlation analyses have demonstrated that tau pathology propagates throughout the AD brain in a stereotyped fashion across neuroanatomically connected networks (Figure 2C) forming the basis of Braak staging (Braak and Braak, 1991), although recent cross-sectional and longitudinal tau-PET imaging studies in cognitively normal amyloid positive individuals demonstrate widely distributed and continuous accumulation of tau pathology outside of the entorhinal cortex suggesting that tau propagation may not be as spatially restricted as previously thought (Jack et al., 2018b; Schultz et al., 2018). Unlike Aβ, the stage of tau pathology correlates well with progression of cognitive impairment (Giannakopoulos et al., 2003; Nelson et al., 2012). With age, tau pathology accumulates in the entorhinal cortex and medial temporal lobes, even in the absence of cognitive decline, as so called primary age-related tauopathy (PART) (Crary et al., 2014). Cognitive impairment in AD is only noted when tau spreads from the entorhinal cortex into the neocortex in neuropathological studies (Price and Morris, 1999; Price et al., 2009). On longitudinal and cross-sectional studies of tau- and amyloid-PET imaging combined with structural MRI, only the presence or accumulation of tau was a predictor of cognitive impairment, whereas the presence or accumulation of amyloid was a predictor of more severe tau-associated cognitive impairment (Aschenbrenner et al., 2018; Hanseeuw et al., 2019). These findings have led to the hypothesis that cognitive decline and neurodegeneration in AD is primarily driven by onset and spread of tau pathology.
Tau protein is encoded by MAPT gene on chromosome 17, is primarily expressed by neurons in the brain, and is alternatively spliced at the N-terminal domain (N) and microtubulebinding repeat domain (R) to form 6 distinct isoforms (0N3R, 0N4R, 1N3R, 1N4R, 2N3R and 2N4R), which are differentially expressed during brain development. Although NFT in AD contain both 3R and 4R isoforms, different tau isoforms are over-represented in pathological aggregates in other human tauopathies (Guo et al., 2017). The physiological role of tau in the CNS is not entirely clear, although numerous in vitro experiments have documented important roles in microtubule assembly and stabilization of neuronal axons and regulation of microtubule transport (Dixit et al., 2008; Weingarten et al., 1975). However, tau knockout (KO) mice do not have a severe developmental phenotype (van Hummel et al., 2016) but do exhibit subtle deficits such as delayed neuronal maturation in cell culture (Dawson et al., 2001) and impaired synaptic plasticity (Ahmed et al., 2014).
Tau protein is subject to numerous post-translational modifications, including phosphorylation, acetylation, glycation, O-GlcNAcylation, nitration, SUMOylation, ubiquitination and truncation (Marcelli et al., 2018). Tau can be phosphorylated at 85 different residues (Guo et al., 2017). Pathological types and patterns of tau phosphorylation can occur even prior to development of NFT. In many cases, aberrant phosphorylation results in decreased binding affinity for microtubules (Biernat et al., 1993; Mandelkow et al., 2007). This disassembly increases the cytosolic pool of tau and is thought to promote aggregation and fibrillization. Hyperphosphorylated tau is also redirected from the axonal compartment to the somatodendritic compartment where it can impair synaptic function by inhibiting glutamate receptor trafficking or synaptic anchoring (Hoover et al., 2010). Tau acetylation has been shown to reduce degradation of phosphorylated tau and increase tau pathology (Min et al., 2010) but has also been shown to inhibit tau phosphorylation at certain residues and limit further aggregation (Cook et al., 2014). Tau acetylation has also been shown to result in axon initial segment cytoskeletal instability followed by mislocalization of tau to the somatodendritic compartment (Sohn et al., 2016). O-GlcNAc modification of tau inhibits toxic self-assembly (Ryan et al., 2019).
There are a number of tau kinases that mediate tau phosphorylation. They comprise three groups - proline-directed serine/threonine protein kinases, non-proline directed serine/threonine protein kinases and tyrosine protein kinases. Examples include glycogen synthase kinase (GSK) 3, cyclin-dependent kinase-5 (Cdk5), mitogen-activated protein kinase (MAPK), cAMP-dependent protein kinase A (PKA) and calcium/calmodulin-dependent protein kinase II (CaMKII), among many others (Guo et al., 2017). Various kinases have been proposed as putative drug targets for AD.
Tau is generally a soluble protein that is natively unfolded, but under the right conditions will aggregate into oligomers and fibrils. NFT and neuropil threads contain an insoluble form of tau aggregated into beta-sheet-containing amyloid fibrils, known as paired helical filaments (PHF) (Crowther and Wischik, 1985; Kidd, 1963; Mandelkow et al., 2007). These aggregated fibrils are known to be polymorphic (Frost et al., 2009) and differ in conformation when derived synthetically or from brains of patients with various tauopathies. (Guo et al., 2016; Sanders et al., 2014). Recent cryo-EM structures of tau fibrils derived from brains of AD patients (Fitzpatrick et al., 2017) and other tauopathies have demonstrated unexpected insights into fibril structures, and confirm that synthetic tau fibrils assume a vastly different folded structure as compared to AD brain-derived tau fibrils (for review see (Lippens and Gigant, 2019))
Since tau is normally highly soluble in its native monomeric form and does not have significant intrinsic hydrophobicity, the question arises as to what prompts tau aggregation in disease. Numerous studies over the last decade have demonstrated the “prion-like” ability of aggregated human tau fibrils to self-propagate, for both synthetically-prepared fibrils and those derived from human AD brain. Exogenously supplied fibrils will seed aggregation of transgenic human and endogenous mouse tau fibrils in mouse tauopathy models by enhancing nucleation of new fibrils. These aggregates then spread transsynaptically to remote anatomically connected brain regions inducing further seeding and aggregation, similar to the pathologic spread of tau noted in AD brain_(de Calignon et al., 2012; Clavaguera et al., 2009, 2013; Frost et al., 2009; Iba et al., 2013; Liu et al., 2012). Thus, prion-like seeding and spreading may represent a mechanism whereby tau pathology propagates from the entorhinal cortex to the neocortex in AD (DeVos et al., 2018).
ApoE
ApoE protein is an apolipoprotein whose major function is to serve as a lipid-binding protein in lipoprotein particles and participate in transport and delivery of lipids to target sites. ApoE is expressed at the highest levels in the liver and brain. In the brain, ApoE is expressed primarily in astrocytes and to a lesser degree in microglia.
APOE is the strongest genetic risk factor for LOAD (Corder et al., 1993; Strittmatter et al., 1993). The strength of this association has been confirmed in numerous clinical, pathological, epidemiological, genome-wide association (GWAS) and whole genome sequencing studies over the years (Kunkle et al., 2019). The APOE gene has three common alleles encoding three protein isoforms, ApoE2, ApoE3 and ApoE4, which differ in sequence by single amino acid substitutions at two different residues (E2 cys112/cys158, E3 cys112/arg158, and E4 arg112/arg158). A single inherited copy of the APOE ε4 allele increases risk of developing LOAD by approximately 3-4 fold, while two inherited copies increases risk by ~12-fold (Roses, 1996). Inheritance of the APOE ε2 allele is protective. Longitudinal CSF, MRI and PET imaging studies of the preclinical phase of AD have established that APOE ε4 allele carriers develop enhanced cerebral amyloid deposition with age, do so at an earlier age and accumulate amyloid at a more rapid rate relative to non-carriers (Bussy et al., 2019; Grimmer et al., 2010; Mishra et al., 2018; Morris et al., 2010; Risacher et al., 2015). The effect of APOE ε4 on measures of tau accumulation and hippocampal atrophy are less certain. ApoE has additionally been implicated in numerous AD-relevant neurobiological processes, many of which are mediated differentially by ApoE isoforms, likely contributing to its role in AD pathogenesis (Figure 2).
ApoE likely regulates AD risk in large part via effects on amyloid pathology (Figure 3A) (Huynh et al., 2017a). ApoE directly binds to Aβ present in plaques (Namba et al., 1991). APP transgenic animal models demonstrate markedly reduced fibrillar Aβ deposition and Aβ levels in the setting of ApoE KO, suggesting ApoE inhibits clearance and/or promotes Aβ seeding (Bales et al., 1997, 1999; Bien-Ly et al., 2012; Huynh et al., 2017b; Kim et al., 2011; Liu et al., 2017; Ulrich et al., 2018). In APP transgenic-ApoE targeted replacement (ApoE-TR) mice, where human apoE isoforms are expressed under the influence of endogenous murine ApoE regulatory sequences, ApoE isoforms have differential effects on amyloid pathology. ApoE4-TR mice have enhanced amyloid deposition and higher Aβ levels (Bales et al., 2009). Studies on the effects of ApoE on Aβ production are conflicting. In one study where iPSC-derived human neurons were co-cultured with mouse glia expressing human ApoE isoforms, neuronal Aβ production was significantly increased in ApoE4 glia co-cultures (Huang et al., 2017). However, this effect was not observed in other studies using different culture systems. (Biere et al., 1995; Cedazo-Minguez et al., 2001). Experiments where ApoE expression is controlled temporally, either via inducible expression or via the use of intrathecal antisense oligonucleotides (ASO), highlight that ApoE promotes the initial seeding of fibrillar Aβ deposition, whereas subsequent plaque growth after seeding seems to rely on other factors (Huynh et al., 2017b; Liu et al., 2017). Detailed in vivo microdialysis and stable isotope labeling kinetics experiments have demonstrated that ApoE regulates clearance of Aβ in the brain in an isoform-dependent manner (ApoE4-TR mice have diminished Aβ clearance relative to other TR mice), whereas Aβ production was not influenced by ApoE isoform (Castellano et al., 2011). Although it was initially postulated that ApoE regulates clearance via direct interactions with Aβ, in fact there is little direct binding of monomeric, soluble Aβ with ApoE in vivo. Instead, ApoE regulates clearance of Aβ by competitively binding to Aβ receptors, such as LDLR-related protein 1 (LRP1) on the surface of astrocytes, and blocking Aβ uptake. In this way, ApoE KO mice exhibit the highest rates of clearance since there is no competition with Aβ receptors. (Verghese et al., 2013). Possible routes of Aβ clearance include astrocytic uptake (Basak et al., 2012), microglial phagocytosis (Heckmann et al., 2019), blood-brain barrier transport (Castellano et al., 2012; Zlokovic, 2013), glymphatic clearance (Iliff et al., 2012) and meningeal lymphatics (Da Mesquita et al., 2018).
Figure 3.
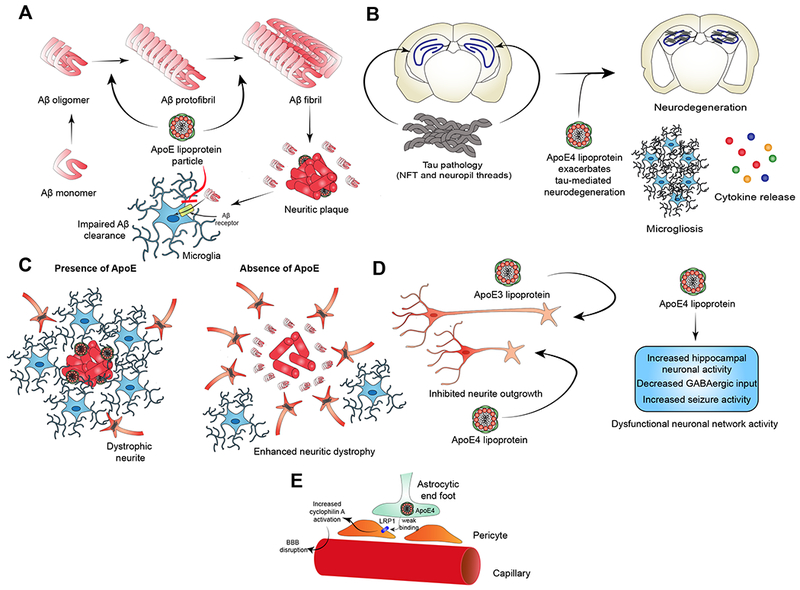
Postulated roles of ApoE in AD pathophysiology. (A) ApoE enhances seeding and fibrillization of Aβ leading to enhanced amyloid deposition. ApoE (especially E4) impedes Aβ clearance from brain parenchyma by competitively binding to Aβ receptors on glial cells. (B) ApoE (especially E4) enhances tau pathogenicity. Presence of ApoE leads to exacerbation of tau-mediated neurodegeneration, increased microgliosis and enhanced neuroinflammatory cytokine release from glial cells. (C) ApoE regulates the microglial response to amyloid plaques. In the presence of ApoE, phagocytically-active disease-associated microglia (DAM) and microglial neurodegenerative phenotype (MGnD) are located near plaques. Tight microglial clustering results in plaque compaction. In the absence of ApoE, periplaque microglia are sparser and amyloid plaques become larger and less compact. Neuritic dystrophy is significantly increased. On the other hand, if apoE levels are lower but not absent, neuritic dystrophy is lower. (D) ApoE may contribute to AD pathophysiology via direct effects on neurons and neuronal network activity. In cell culture, exogenously supplied ApoE4 inhibits neurite outgrowth relative to ApoE3. ApoE4 also contributes to dysfunctional neuronal network activity as evidenced by reduced hilar GABAergic interneurons, increase hippocampal network activity and propensity for seizures in ApoE4 target replacement mice. (E) ApoE4 may directly impair blood-brain barrier (BBB) function in AD by failing to efficiently bind to LRP1 expressed on cells such as pericytes. There is evidence that this leads to increased activation of cyclophilin A signaling pathways and ultimate breakdown of BBB, resulting in passage of serum proteins (such as fibrin) into the brain parenchyma and eventual neuronal death.
ApoE also has modulatory effects on tau pathology and tau-related neurodegeneration (Figure 3B) (Shi and Holtzman, 2018). Tau binds to ApoE3 but not ApoE4 in vitro (Strittmatter et al., 1994). In a recent study assessing the effect of ApoE-TR in the PS19 (tauP301S) tauopathy mouse model, ApoE4 dramatically exacerbated tau-mediated neurodegeneration (Shi et al., 2017). ApoE genetic deletion significantly reduced the extent of neuron and volume loss in PS19 mice and strongly attenuated microglial and astrocyte activation. A recent study failed to replicate the neurotoxic effect of ApoE4 in tauopathy mice. In this study, AAV viral transduction was used at postnatal day 0 to overexpress tauP301L in ApoE-TR mice. They found that ApoE2 mice, rather than ApoE4, had enhanced tau pathology, astrogliosis and behavioral impairment (Zhao et al., 2018). Of note, neurodegeneration was not observed in this model at 6 months of age, the time of endpoint analysis. Therefore, discrepancies between these studies are likely explained by differences in experimental models.
ApoE4 may mediate risk for AD though modulation of immune and microglial responses. APOE ε4 carriers have a stronger systemic inflammatory response to an intravenous LPS challenge, including more intense hyperthermia and higher levels of TNF secretion in whole blood (Gale et al., 2014). ApoE expression in microglia is required for the phenotypic expression of disease-associated microglia (see “Neuroimmune activation” below). ApoE also promotes microglial clustering around Aβ plaques and in the absence of ApoE expression, there is strong reduction in the total amount of fibrillar plaques and plaque size but an increase in overall Aβ-immunoreactive deposits (Figure 3C). Plaques appear less compact with fewer periplaque microglia and enhanced neuritic dystrophy (Ulrich et al., 2018). ApoE is also required for microglial intracellular endolytic degradation of Aβ by neprilysin and insulin-degrading enzyme (Jiang et al., 2008).This suggests that ApoE plays an important role in mediating the microglial response to Aβ plaques
ApoE4 may have direct pathologic effects on neurons and neuronal networks independent of effects on amyloid and tau pathology (Figure 3D) (Najm et al., 2019). In primary neuronal cell culture, exogenously applied ApoE4 directly inhibits neurite outgrowth, whereas ApoE3 stimulates neurite outgrowth (Holtzman et al., 1995; Nathan et al., 1994). In an ApoE transgenic model, ApoE3 expression in neurons was protective against kainic acid-induced excitotoxic neuronal damage, whereas neuronal ApoE4 expression had no protective effect (Buttini et al., 1999). When ApoE3 and ApoE4 were instead expressed in astrocytes, both isoforms were protective (Buttini et al., 2010). ApoE4-TR adult mice also have lower dendritic spine density and spine length in the cortex (but not hippocampus) relative to ApoE2- and ApoE3-TR mice (Dumanis et al., 2009).
ApoE4-TR mice have enhanced neural activity in the hippocampus and entorhinal cortex, as evidenced by hypermetabolism on fMRI, increased high frequency oscillations measured by in vivo electrophysiology, and reduced inhibitory input to neurons of entorhinal cortex (Nuriel et al., 2017). Along these lines, ApoE4-TR adult mice are prone to seizures (Hunter et al., 2012). In cognitively normal human APOE ε4 carriers, there is increased signal intensity and number of activated regions in the hippocampus on fMRI during memory-activation tasks. These data suggest that ApoE4 may cause neuronal dysfunction via direct neurotoxic effects and by promoting hyperactivity in hippocampal neuronal networks.
Neuronal hyperactivity may be mediated by impaired GABAergic input. GABAergic inhibitory networks are impaired in AD and may contribute to cognitive impairment (Najm et al., 2019). ApoE4-TR mice have decreased numbers of GABAergic interneurons in the hippocampus and this correlates with learning deficits. When given non-lethal doses of pentobarbital over 4 weeks to boost GABAergic pathways, learning deficits were reduced in ApoE4-TR mice. Conditional ApoE KO in all neurons or specifically in GABAergic neurons in ApoE4-TR mice was protective against neuron loss and memory deficits (Knoferle et al., 2014). Finally, neurons derived from iPSCs from E4/E4 patients as compared to E3/E3 or congenics had higher amounts of hyperphosphorylated tau and reduced GABAergic neurons, effects reduced after treating with a small molecule apoE structure corrector (Wang et al., 2018).
ApoE4 may also mediate neuropathological effects by directly modulating BBB integrity (Figure 3E). Studies in the Zlokovic lab have demonstrated that ApoE4 does not efficiently engage LRP1 on pericytes. Lack of ApoE-mediated signaling via this receptor results in activation of proinflammatory cyclophilin-A-NFκB-MMP-9 signaling cascade. Overactivation of this cascade leads to breakdown of BBB basement membrane and tight junctions, resulting in extravasation of serum proteins (e.g. fibrin) into the brain parenchyma. Exposure to serum proteins can then contribute to neurodegeneration (Bell et al., 2012; Zlokovic, 2013).
Neuroimmune activation
No area of AD research has experienced more intense investigation in the recent past than the role of the innate immune system in the pathophysiology of AD. Multiple lines of evidence suggest that activation of immune mediators is a critical regulator of AD pathology. Reactive astrogliosis and microgliosis are known to be prominent pathological features in AD brain. Numerous SNPs and rare coding variants in immune-related genes thought to be implicated in microglial function have been identified as risk factors for AD in whole genome sequencing and GWAS analyses (see for review (Efthymiou and Goate, 2017; Karch and Goate, 2015), including TREM2, CR1, SHIP1, BIN1, CD33, PICALM, CLU, and theMS4A gene cluster. A non-synonymous coding variant in PCLG2, a gene with suspected immune function, has been associated with decreased risk of AD (van der Lee et al., 2019). Of note, many of these immune-related AD risk factor SNPs or coding variants (Apoe, Trem2, Cd33, Ms4a6d) have been identified as genes differentially expressed in models of Aβ deposition as compared to tauopathy, suggesting they may represent amyloid response genes (Matarin et al., 2015). Systems-level analyses of gene regulatory networks in post-mortem human LOAD brain specimens have identified the immune/microglial molecular network as most highly associated with pathophysiology in LOAD (Zhang et al., 2013). A recent detailed morphological analysis of microglial activation state in brain specimens from two cohorts of cognitive aging found that the proportion of activated microglia (PAM) strongly correlated with presence of pathologic AD (Felsky et al., 2019). PAM was also strongly associated with total Aβ load and number of neuritic plaques and less strongly associated with amount of PHF tau. Causal mediation modeling generated a model consistent with a proposed pathologic sequence of increased PAM triggering increased PHF tau accumulation, leading to cognitive decline.
Single cell transcriptomic methods have been employed to identify unique microglial phenotypes associated with Aβ plaque deposition in animal models of amyloidosis (APP/PS1 and 5XFAD) (Figure 4A). Termed microglial neurodegenerative phenotype (MGnD) or disease-associated microglia (DAM), these cells are defined by a unique expression profile consisting of upregulation of certain inflammatory transcripts (e.g. Apoe, Trem2, Clec7a) and downregulation of homeostatic transcripts (e.g. P2ry12, Tmeml19). In both studies, these microglia were found to be in close association with neuritic plaques. In one study, MGnD could be induced in WT mouse brain by injection of apoptotic neurons, and required ApoE expression for induction (Keren-Shaul et al., 2017; Krasemann et al., 2017). Single nuclear transcriptomic techniques have recently been applied to human AD brain specimens and have identified microglial subtypes with increased Apoe expression but have failed to identify DAM or MGnD subpopulations similar to what has been seen in the mouse brain (Mathys et al., 2019). Further work to better understand similarities and difference between microglia in mouse models vs. the human brain are needed.
Figure 4.
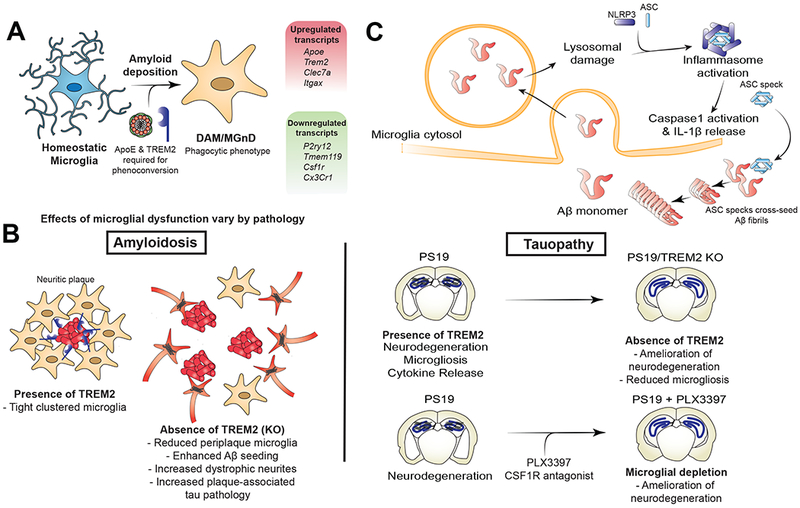
Selected roles of the innate immune system in AD pathophysiology. (A) Transcriptomic analyses of microglia isolated from mice with amyloid pathology have demonstrated a unique microglial subpopulation (DAM/MGnD) only found in diseased animals and defined by reduced expression of homeostatic genes and increased expression of genes involved in phagocytosis and microglial activation. Expression of ApoE and TREM2 is necessary for the development of this disease-specific subpopulation. (B) Microglial activation and phagocytosis have disparate effects based on the prominent pathology studied. TREM2 KO results in a microglial phenotype that is less activated and less phagocytic. In mouse models of amyloid deposition, TREM2 KO leads to reduced peri-plaque microglial clustering, increased Aβ seeding and deposition, and increased dystrophic neurites with enhanced plaque-associated tau pathology. Conversely, in PS19 tauopathy mice, TREM2 KO leads to reduced neurodegeneration and microgliosis. Similarly, near total microglial depletion in PS19 mice expressing ApoE4 leads to significant reduction in neurodegeneration and microgliosis. (C) Aβ phagocytosis leads to increased microglial activation. Lysosomal damage by Aβ can lead to activation of NLRP3 inflammasome, resulting in IL-1β secretion. Extracellular ASC specks released from activated microglial can then further cross-seed fibrillization of Aβ into fibrils.
Disparate Roles of Microglia in AD Pathogenesis
Although microglial activation is associated with disease pathology in AD, it is difficult to parse whether microglial activation in AD is damaging or protective. This may be because of disease stage specific effects with activation being protective in the setting of amyloid deposition and damaging in the setting of tau accumulation (Figure 4B).
Phagocytic microglia can limit amyloid-associated pathology
As an example, TREM2 is an AD risk gene with a rare variant allele R47H associated with significantly increased risk of AD (Gratuze et al., 2018). TREM2 protein is expressed on microglia, promotes microglial phagocytosis, modulates inflammatory signaling and promotes microglial survival. TREM2 also binds to soluble oligomers of Aβ and promotes phagocytosis of Aβ. R47H is thought to be a loss of function variant (Gratuze et al., 2018; Shi and Holtzman, 2018). In TREM2 deletion and haploinsufficiency studies using mouse models of Aβ deposition (APPPS1-21 and 5xFAD), levels of Aβ were decreased at 2 months of age (before plaque deposition) but increased at 8.5 months of age (after plaque deposition), suggesting an age or amyloid burden effect (Ulrich et al., 2014). Importantly, there was also a reduction in plaque-associated microglia with associated impairments in plaque compaction and increased levels of dystrophic neurites around plaques (Leyns et al., 2017; Song et al., 2018). Either TREM2 KO or TREM2 R47H transgene expression in mice that develop amyloid deposition results in enhanced amyloid seeding as well as enhanced tau seeding and spreading near neuritic plaques (Leyns et al., 2019; Parhizkar et al., 2019). This suggests a role of Trem2 function in suppressing amyloid-induced local toxicity that also inhibits tau seeding/spreading. CD33 is another AD susceptibility locus that encodes a transmembrane glycoprotein expressed on the surface of microglia. Increased surface CD33 expression results in decreased microglial activation. In the J20 amyloid mouse model, CD33 expression resulted in reduced microglial-mediated phagocytosis of Aβ, and increased amyloid pathology (Bradshaw et al., 2013; Griciuc et al., 2013). CD33 KO results in decreased amyloid pathology and improved cognition in 5xFAD mice. This is abrogated by additional TREM2 KO, however CD33 KO does not reciprocally abrogate effects of TREM2 KO, suggesting that CD33 acts upstream of TREM2 (Griciuc et al.,2019 . Microglia appear to clear Aβ through a process termed LC3-associated endocytosis (LANDO) that also recycles Aβ receptors (CD36, TREM2, TLR4) back to the cell surface; if this is genetically deleted in 5xFAD mice, there is increased Aβ accumulation, microgliosis with release of proinflammatory cytokines, tau hyperphosphorylation, synapse dysfunction, neuron loss and cognitive impairment (Heckmann et al., 2019). These data in models of Aβ deposition would suggest that dysfunctional microglia are less able to sequester Aβ pathology within compact plaques and less able to inhibit amyloid and tau seeding near plaques, resulting in neuronal damage.
Activated microglia can enhance amyloid pathology
Alternatively, phagocytosis of Aβ by microglia can lead to microglial activation. Following phagocytosis by microglia, Aβ activates the NLRP3 inflammasome leading to caspase-1 activation and IL-1β maturation and release (Halle et al., 2008). NLRP3 inflammasome is activated in AD and mild cognitive impairment (MCI) brains and in APPPS1 mice. NLRP3 and caspase-1 genetic deletion results in decreased Aβ deposition, reduced IL-1β release from microglia and improved cognitive performance in APPPS1 mice (Heneka et al., 2013). Following NLRP3 activation, monomers of the apoptosis associated speck-like protein containing a CARD domain (ASC) can form fibrils and recruit caspase-1 resulting in autoproteolysis and caspase-1 activation, leading to further assembly of ASC fibrils into paranuclear specks. Following inflammasome activation, ASC specks can be leaked extracellularly after which they can be taken up by neighboring microglia to sustain the innate immune response. Extracellular ASC specks have been found to bind to Aβ and can cross-seed Aβ oligomerization and plaque formation in APPPS1 mice (Figure 4C). This can be reversed by genetic deletion of ASC or use of an anti-ASC neutralizing antibody (Venegas et al., 2017). In support of a role for microglia in exacerbating amyloid pathology, a recent study using pharmacological depletion of microglia in amyloid mice during the period of plaque deposition demonstrated reduced parenchymal plaques following robust microglial depletion (Spangenberg et al., 2019). Therefore, phagocytosis of Aβ by microglia may be a double-edged sword: it may serve to limit spread of amyloid pathology in certain contexts or alternatively may promote spread of amyloid pathology via the mechanisms described above.
Activated microglia exacerbate tau-associated pathology
Different results have been observed in models of tau pathology that develop neurodegeneration. In the PS19 tauopathy mouse, TREM2 deletion significantly reduced tau-mediated neurodegeneration and astrogliosis (Leyns et al., 2017; Sayed et al., 2018)..These data would suggest that microglia contribute to neuron death in models of tau-mediated neurodegeneration. Microglial-derived exosomes appear necessary for tau propagation in a model of tau spreading using AAV viral transduced expression of P301Ltau to induce rapid tau spreading from the entorhinal cortex to the dentate gyrus of the hippocampus (Asai et al., 2015). Depletion of microglia or pharmacological inhibition of exosome secretion led to reduced tau spreading in this model.
Direct microglial effects on neuron viability
Activated microglia can directly secrete toxic proinflammatory cytokines (Colonna and Butovsky, 2017; Kinney et al., 2018) or secrete indirect mediators that stimulate astrocytes to secrete a neurotoxic substance (Liddelow et al., 2017). Microglia may also be directly synaptotoxic. Complement binding (C1q) and microglial phagocytosis leads to early synapse loss in a mouse model with Aβ plaque deposition (J20 model). C1q binds to synapses and, in the presence of soluble Aβ oligomers, promotes microglia phagocytosis of synaptic contents (Hong et al., 2016)
Role of Gut Microbiome in Innate Immunity and AD pathogenesis
There is increasing evidence that interactions between the gut microbiome and the CNS innate immune system (gut-brain axis) may modulate AD pathogenesis. Various aspects of CNS physiology are regulated by the microbiome, including microglial maturation and function (Abdel-Haq et al., 2019). Germ-free mice develop microglia with an immature phenotype, blunted response to inflammatory stimuli and defective immune response to infection (Erny et al., 2015). The mechanism may involve microbe-derived metabolites. Several studies have profiled the composition of the gut microbiota in both mouse models of amyloidosis (APP/PS1) and in human participants with dementia due to AD and have found significant differences in the overall abundance and diversity of microbial species relative to WT animals or non-demented control participants, respectively (Bäuerl et al., 2018; Saji et al., 2019; Vogt et al., 2017). These alterations may influence microglial responses to disease pathology. Indeed, one study demonstrated that APP/PS1ΔE9 mice chronically administered a cocktail of high dose antibiotics to modulate microbiome composition had significantly reduced Aβ plaque load, reduced plaque size, increased soluble Aβ42 levels and reduced plaque-associated microglia and reactive astrocytes (Minter et al., 2016). Further studies are required to validate these findings, explore the effect of microbiome modulation on other types of AD pathology (e.g. tau) and elucidate underlying mechanisms.
In summary, activation of the innate immune system undoubtedly plays a role in AD pathophysiology. However, data regarding role of activated microglia in animal models suggest possibly divergent effects depending on disease context. Gut microbiota may also modulate the microglial response to pathology. Future studies will need to characterize the expression profiles of microglia from both amyloid and tau-based animal models using single-cell techniques to determine if different microglial subpopulations with distinct pathology-associated phenotypes exist. The role of these various microglial-mediated mechanisms in modulating pathology in the human AD brain is unclear and is being actively investigated.
Infectious Hypothesis
Multiple recent studies have revived interest in a long-standing hypothesis that there may be an underlying infectious basis for AD. Dating back to 1991, studies have demonstrated the presence of herpesvirus in brains of patients with AD, as well as within amyloid plaques (Carbone et al., 2014; Jamieson et al., 1991, 1992; Wozniak et al., 2009). A recent systems-level molecular network analysis of preclinical AD brain identified a set of network driver genes noted to be enriched for viral susceptibility genes. Follow-up analyses in AD brain specimens found detectable human herpesvirus (HHV)-6 and herpes simplex virus (HSV)-1 DNA in 3 separate cohorts, and identified many putative viral-host interactions that regulate gene networks pertinent to AD biology, including innate immunity and APP processing (Readhead et al., 2018)
Aβ exhibits characteristics of an antimicrobial peptide. Synthetic and AD-brain derived Aβ peptide fibrils significantly inhibit the growth of gram positive and gram negative bacteria, fungus (Soscia et al., 2010) and herpesvirus, and inhibits the entry of herpesvirus into cells (Bourgade et al., 2015). Aβ also forms fibrils that entrap bacteria and fungal cells, leading to agglutination in vitro (Kumar et al., 2016; Spitzer et al., 2016). The presence of Aβ in brain parenchyma is linked with protection against infectious bacterial encephalomyelitis experimentally induced by intracerebral inoculation with Salmonella enterica typhimurium bacteria in 4-week old 5xFAD mice. 5xFAD mice survived longer with less severe disease than WT mice, while APP KO mice had slightly increased mortality compared to WT mice. At sites of bacterial deposition in the brain, significant Aβ deposition was also noted. (Kumar et al., 2016). Similarly, presence of parenchymal Aβ in 5xFAD mice results in decreased mortality following direct intracerebral HSV inoculation. Areas of brain parenchyma containing deposits of HSV-1 infection also demonstrated increased Aβ deposition (Eimer et al., 2018).
These experiments have led to the antimicrobial protection hypothesis of AD that suggests intracerebral infection by certain pathogens may induce Aβ fibrillization as an antimicrobial defense mechanism, leading to amyloid seeding and deposition, thereby initiating the amyloid cascade. (Moir et al., 2018). In support of this theory, HSV-1 viral particles have recently been shown to directly catalyze the fibrillization of Aβ42 in vitro by nucleating aggregation via contact with the viral surface (Ezzat et al., 2019).
Two recent Taiwanese-based nationwide, matched control, retrospective cohort studies have evaluated the association of HSV and varicella zoster virus (VZV) infection with risk of dementia. One study demonstrated a significantly increased risk of developing all-cause dementia (~3-fold increase) in patients greater than 50 years of age following a new HSV infection. This risk was nearly eliminated in patients that received antiherpetic treatment (Tzeng et al., 2018). A second study demonstrated a very slight increased risk of developing all-cause dementia after zoster infection (HR 1.12). Again, a significant decrease in dementia risk was observed in patients that received antiherpetic treatment (HR 0.47) (Chen et al., 2018). The results of these studies will need to be replicated, ideally in a large prospective longitudinal observational cohort study, before accepting herpesvirus infection as a bona fide risk factor for AD or related dementias.
Another recent study suggests that periodontal infections may contribute to AD pathogenesis. In this study, periodontal bacterial proteins (gingipains) and DNA were identified in human AD brain. Oral infection of WT mice with Porphyromsonas gingivalis resulted in elevated intraparenchymal Aβ levels, and inhibition of gingipain activity led to reduced intracerebral bacterial burden, decreased Aβ levels and reduced neuroinflammation (Dominy et al., 2019). Based on these results, a phase 2/3 double-blind, randomized control trial testing use of gingipain inhibitor COR388 is underway in mild to moderate AD (ClinicalTrials.gov Identifier: ).
Sleep
Sleep impairment can lead to impaired attention, concentration, and working memory but may also influence development of underlying AD pathology. Women with sleep disordered breathing have a higher risk of developing MCI or dementia relative to non-sleep disordered breathing patients (Yaffe et al., 2011). A single night of sleep deprivation leads to elevated CSF Aβ42 levels in healthy middle-aged men (Ooms et al., 2014). Reduced slow-wave sleep in cognitively normal elderly participants is also associated with increased CSF Aβ42 (Varga et al., 2016). A study exploring the effect of selective reduction of overnight slow-wave sleep via an automated detection-intervention method in healthy volunteers also demonstrated increased CSF Aβ levels in participants receiving the study intervention (Ju et al., 2017). However, in a recent study of partial sleep deprivation lasting 5-8 days, no changes in AD CSF biomarkers were noted although that study did not alter the quantity of slow-wave sleep (Olsson et al., 2018).
In animal microdialysis experiments, interstitial fluid (ISF) Aβ levels correlate with wakefulness and significantly increase following acute sleep deprivation or orexin infusion. In mice overexpressing Aβ, acute sleep deprivation caused a significant increase in amyloid plaque deposition. A decrease in amyloid plaque deposition was observed after chronic orexin receptor blockade resulting in increased sleep (Kang et al., 2009). In mice overexpressing Aβ, sleep-wake cycle and diurnal fluctuations in ISF and CSF Aβ levels are normal before an age when animals develop amyloid plaque formation. However, after Aβ plaque formation, there is an impaired sleep-wake cycle and loss of diurnal fluctuation in ISF/CSF Aβ levels (Roh et al., 2012). Two main mechanisms have been proposed as to how sleep deprivation or increased wakefulness increases extracellular Aβ in the brain. Studies from the lab of Maiken Nedergaard suggest that glymphatic clearance is slower during wakefulness vs. sleep (Iliff et al., 2012; Xie et al., 2013). Increased synaptic Aβ release due to elevated neuronal metabolism/activity during wakefulness vs. sleep is another mechanism Increased synaptic and network activity has been previously shown to stimulate release of extracellular Aβ and tau from neurons (Bero et al., 2011; Brody et al., 2008; Cirrito et al., 2005; Yamada et al., 2014). In a recent study of amyloid kinetics in human CSF using stable isotope labeling, there was an increase in Aβ levels following sleep deprivation but no change in rate of Aβ clearance. This suggests the mechanism for increased Aβ is increased production (Lucey et al., 2018). Similar findings are also noted with extracellular tau. Sleep deprivation in tauopathy mice and healthy human participants resulted in increased ISF/CSF tau levels (Holth et al., 2019). When seeded with recombinant synthetic tau fibrils, sleep deprived animals had more significant tau spreading relative to non-sleep deprived animals. In summary, these studies provide compelling evidence that sleep deprivation stimulates increased levels of Aβ and tau in human CSF and animal model brains and enhances intracerebral pathology.
THERAPEUTIC STRATEGIES FOR ALZHEIMER DISEASE
There are currently four FDA approved medications for the management of cognitive impairment and dysfunction in global activities in symptomatic AD. These include three cholinesterase inhibitors (ChEIs; donepezil, rivastigmine and galantamine) and memantine, an uncompetitive NMDA receptor modulator. Despite enormous efforts by the pharmaceutical industry, there remains no effective disease modifying therapy available today. More than 20 compounds have completed large phase 3 double-blind randomized control trials in cohorts of patients at various stages of AD and none has demonstrated any efficacy in slowing cognitive decline or improving global functioning (Table 1). These many trial failures highlight the need for different approaches to clinical trial design in AD.
Table 1.
Discontinued phase 3 disease modifying drug trials in AD (excluding ChEI and memantine trials)
| Drug (Study Name) | Study Population | Target/Mechanism | Type of molecule | Outcome | ClinicalTrials.gov NCT Identifier | Reference |
|---|---|---|---|---|---|---|
| Bapinezumab | Mild to moderate AD | Soluble and fibrillar Aβ | Monoclonal antibody | No effect on cognition or ADL | (Salloway et al., 2014) | |
| Solanezumab (EXPEDITION-1, 2 and 3) | Mild-to-moderate AD; mild AD | Soluble monomeric Aβ | Monoclonal antibody | No effect on cognition or ADL | (Doody et al., 2014; Honig et al., 2018) | |
| Crenezumab (CREAD-1/2) | Very mild to mild AD with amyloid positive biomarkers | Oligomeric, fibrillar and plaque-based Aβ | Monoclonal antibody | No effect on cognition or ADL on preliminary analysis | ||
| Aducanumab (ENGAGE; EMERGE) | Mild AD | Conformation-specific Aβ aggregates | Monoclonal Antibody | No change in rate of cognitive decline | (Selkoe, 2019) | |
| AN-1792 | Mild to moderate AD | Active immunization | Full length Aβ42 immunogen | Trial halted due to development of meningoencephalitis in 4 patients | ||
| Semagacestat (IDENTITY-1/2) | Mild to moderate AD | γ-secretase inhibitor | Small molecule | No effect on cognition or ADL; increased risk of skin cancer | (Doody et al., 2013) | |
| Tarenflurbil | Mild AD | γ-secretase modulator | Small molecule | No effect on cognition or ADL | (Green et al., 2009) | |
| CNP520 (Umibecestat) (API Generation) | Cognitively normal APOE ε4/ε4 carriers | BACE1 inhibitor | Small molecule | Worse cognitive performance, weight loss | (Lopez Lopez et al., 2019) | |
| Lanabecestat (AMARANTH; DAYBREAK-ALZ) | Very mild to mild AD | BACE1 inhibitor | Small molecule | No effect on cognition or ADL | ||
| Atabecestat | Preclinical AD; positive amyloid, normal cognition | BACE1 inhibitor | Small molecule | Worse performance on some cognitive tests; in some cases, prominent side effects | (Henley et al., 2019) | |
| Verubecestat (APECS) | Prodromal AD | BACE1 inhibitor | Small molecule | Worse performance on some cognitive tests and in ADL | (Egan et al., 2018, 2019) | |
| Intepirdine | Mild to moderate AD | 5-HT6 receptor antagonist | Small molecule | No effect on cognition or ADL | ||
| Idalopirdine (STARBEAM) | Mild to moderate AD | 5-HT6 receptor antagonist | Small molecule | No effect on cognition or ADL | (Atri et al., 2018) | |
| Dimebon | Mild to moderate AD | Pleiotropic: anti-histamine, mitochondria, others | Small molecule | No effect on cognition or ADL | (Cano-Cuenca et al., 2014) | |
| Nilvadipine | Mild to moderate AD | Calcium channel blocker | Small molecule | No effect on cognition or ADL | (Lawlor et al., 2018) | |
| Naproxen, celecoxib (ADAPT study) | Prevention study; Cognitively normal elderly with parent or sibling with dementia | Anti-inflammatory; COX inhibitor (NSAID) | Small molecule | No delay in time to incident cognitive impairment after 1-3 years treatment and 7 years follow-up | (ADAPT-FS Research Group, 2015) | |
| Indomethacin | Mild to moderate AD | Anti-inflammatory; COX inhibitor (NSAID) | Small molecule | No effect after 1 year treatment on cognition or ADL; study under-powered | (de Jong et al., 2008) | |
| Prednisone | Mild to moderate AD | Anti-inflammatory | Small molecule | No effect on cognition or ADL | (Aisen et al., 2000) | |
| Intravenous immunoglobulin (IVIg) | Mild to moderate AD | Immunomodulator | Pooled Ig | No effect on cognition or ADL | (Relkin et al., 2017) | |
| LMTM | Very mild to mild AD | Tau aggregation inhibitor; methylene blue derivative | Small molecule | No effect on cognition or ADL | (Gauthier et al., 2016) | |
| Simvastatin (CLASP and ESPRIT studies) | Mild to moderate AD | HMGCoA-reductase inhibitor; cholesterol lowering agent | Small molecule | Some improvement on a few cognitive tests but no effect on ADL | (Carlsson et al., 2008; Sano et al., 2011) | |
| Atorvastatin | Mild to moderate AD | HMGCoA-reductase inhibitor; cholesterol lowering agent | Small molecule | No effect on cognition or ADL | (Feldman et al., 2010) | |
| Rosiglitazone (REFLECT-2/3) | Mild to moderate AD | PPARγ agonist | Small molecule | No effect on cognition or ADL | (Harrington et al., 2011) | |
| Tricaprilin | Mild to moderate AD | Improve mitochondrial metabolism | Small molecule | No effect on cognition or ADL |
First, most of the failed phase 3 trials intervened on patients with mild-to-moderate symptomatic AD. Though this represents an intermediate phase of clinical disease, this is actually an advanced stage of the biological disease when considering that pathology accumulates in the AD brain 15-20 years prior to onset of clinical symptoms (Vermunt et al., 2019). At this stage of disease pathogenesis, significant and irreversible synaptic and neuronal loss has occurred and the pathological cascade would likely be very difficult to reverse (Gómez-Isla et al., 1996). Instead, disease modifying clinical interventions applied as early in the preclinical phase of disease as possible in a preventative study design might have a better chance of changing disease trajectory before the onset of frank neurodegeneration. Several recent large scale clinical trials have adopted a secondary or primary prevention paradigm: the Dominantly Inherited Alzheimer Network Trials Unit (DIAN-TU) drawing from the large DIAN cohort of ADAD patients (Bateman et al., 2017), the A4 trial enrolling participants with confirmed preclinical AD by biomarker assessment (Sperling et al., 2014), and the Alzheimer Prevention initiative (API) using a cohort of patients with ADAD as well as a separate cohort of older homozygous APOE ε4/ε4 carriers (Reiman et al., 2011). Importantly, each study is testing anti-amyloid therapies in cognitively normal patients who are at high risk for developing the disease.
Second, it is important to reconsider assumptions and underlying hypotheses with regards to disease pathogenesis. While the underlying evidence for the amyloid cascade hypothesis remains solid, new discoveries should continually inform and update our understanding of disease pathobiology, eventually leading to the development of novel treatment approaches.
The current AD clinical pipeline has over 100 different compounds currently being tested in various phases of clinical trials (Hara et al., 2019). We will now review available symptomatic treatments and developments on the path to identifying disease modifying therapies.
Symptomatic Treatment
Cholinesterase inhibitors
During the course of AD pathogenesis, cholinergic neurons in the nucleus basalis of Meynert and other septal nuclei that project widely throughout the cortex are lost, causing a general cholinergic deficit (Bartus et al., 1982). This loss of cholinergic input is thought to contribute to early attention and memory dysfunction in AD. Cholinesterase inhibitors (ChEI) work to reverse this deficiency by increasing synaptic levels of acetylcholine. Three ChEI are currently approved for use in mild-to-moderate AD: donepezil, rivastigmine and galantamine. They differ primarily in their pharmacokinetic profiles (donepezil has a much longer half-life than the others and is dosed once daily) and formulation (rivastigmine is available as a continuous release transdermal patch) but not in overall efficacy. Their symptomatic benefit in AD has been confirmed via meta-analyses assessing both cognitive performance and global functioning (Birks, 2006). However, the overall effect size is modest (mean difference in MiniMental State Examination (MMSE) of 1.37 points after 6 months treatment), and there is no effect on long-term disease progression. Available data does not support the use of ChEI in very mild AD (i.e. mild cognitive impairment), and in fact ChEI may worsen cognition at this early clinical stage (Han et al., 2019).
Anti-NMDA
Memantine is a uncompetitive NMDA receptor modulator that may act to inhibit glutamate-mediated neurotoxicity that develops as neurons die during AD progression, although the precise mechanism of its effect is unclear (Greenamyre et al., 1988; Livingston et al., 2017). Meta-analysis confirms the efficacy of memantine in moderate-to-severe AD on measures of cognition, activities of daily living (ADL) and neuropsychiatric symptoms (McShane et al., 2006). However, as with ChEI, the effect size is quite small and the medication has no effect on long-term disease progression. Effects in mild-to-moderate stage disease were marginal and therefore, it is not recommended that this medication be prescribed to patients with mild AD.
Disease Modifying Treatments
Aβ-directed therapeutics
The goal of many Aβ-directed therapies is to lower levels of parenchymal Aβ and amyloid deposits in the AD brain. The amyloid cascade hypothesis suggests that Aβ accumulation triggers disease pathogenesis, so therapies that lower parenchymal Aβ might be expected to slow disease progression. Since no integrated AD animal model exists in which to definitively test the validity of the amyloid cascade hypothesis, clinical trials using Aβ-directed therapies in humans have been considered one of the most direct ways to test this. Unfortunately, many clinical trials have been inadequately designed to robustly test the amyloid hypothesis, such that negative trial results cannot be necessarily interpreted as a rejection of the amyloid hypothesis (Karran and Hardy, 2014; Karran et al., 2011). Active and passive immunization strategies and use of secretase inhibitors have been the primary modes of targeting Aβ in clinical trials.
Active immunization
Aβ-directed immunotherapy has been explored as a disease-modifying therapy in AD for the last 20 years (Gallardo and Holtzman, 2017). The first successful demonstration of antibody-mediated clearance of Aβ was in 1999, when active immunization of PDAPP mice using full length human Aβ42 peptide resulted in a significant decrease in existing amyloid deposits in animals vaccinated at an older age and almost completely prevented the development of amyloid deposition in animals vaccinated at a younger age (Schenk et al., 1999). This subsequently led to the development of the first human Aβ immunotherapy trial using AN-1792, a mixture of synthetic full length human Aβ42 with adjuvant. In a phase 2a trial, four patients developed aseptic meningoencephalitis leading to the discontinuation of the trial, although ongoing neuroimaging and neuropathologic follow-up allowed for assessment of long-term treatment response. 25 of 129 patients were deemed to be antibody responders. Post-mortem evaluation determined that there was significant variability in degree of amyloid clearance but that some vaccinated patients had long-lasting nearly complete clearance of Aβ deposits. However, they still had prominent tau pathology with advanced Braak staging and most had severe dementia at time of death, even despite plaque clearance (Nicoll et al., 2019; Vellas et al., 2009). Active vaccination strategies using Aβ peptides as immunogens are still being pursued, with CAD106 in a phase 3 trial (Table 2) and ABVac40 currently in phase 2 trials (Lacosta et al., 2018; Vandenberghe et al., 2017). Both claim to avoid the risk of meningoencephalitis by excluding portions of the Aβ peptide responsible for inducing Th1 immune responses.
Table 2.
Active phase 3 disease-modifying drug trials in AD
| Drug | Study Population | Target | Type of molecule | Status | ClinicalTrials.gov NCT Identifier |
|---|---|---|---|---|---|
| Gantanerumab | Very mild to mild AD | Conformational epitope found only on fibrillar AD | Monoclonal antibody | Phase 3 enrolling | |
| BAN2401 (CLARITY) | Very mild to mild AD with positive amyloid biomarkers | Aβ protofibrils | Monoclonal antibody | Phase 3 enrolling | |
| Elenbecestat (MISSION-AD1/2) | Mild AD | BACE1 inhibitor | Small molecule | Phase 3 enrolling | |
| ALZT-OP1 (cromolyn sodium/ibuprofen) | Very mild AD with positive CSF amyloid | Anti-inflammatory | Small molecule | Phase 3 enrolling | |
| Levetiracetam | Very mild AD with positive amyloid PET imaging | SV2A | Small molecule | Phase 3 enrolling | |
| CAD106 | Cognitively normal ApoE ε4/ε4 homozygotes | Active immunization with Aβ1-6 peptide fused to virus-like particles | Immunogen | Phase 3 no longer enrolling; API Generation primary prevention study | |
| COR388 | Mild to moderate AD | Periodontal bacteria gingipain inhibitor | Small molecule | Phase 3 enrolling | |
| AVANEX 2-73 | Very early to mild AD with positive amyloid biomarkers | Sigma-1 protein agonist | Small molecule | Phase 3 enrolling |
Passive Immunization
Passive immunotherapy is another strategy for immunologically targeting Aβ clearance. With infusions of a monoclonal antibody, there is less variability in efficacy from patient to patient, and since titer levels can be more tightly controlled, there is less risk of adverse events. Six different monoclonal antibodies directed against Aβ have advanced to phase 3 clinical trials. The first monoclonal antibody to do so was bapinezumab. This is a humanized version of mouse 3D6 antibody that targets N-terminal Aβ peptide and binds to soluble and fibrillar Aβ, triggering microglial phagocytosis of Aβ deposits. This antibody was tested in two parallel phase 3 trials in mild-to-moderate AD patients and had no effect on cognition or ADL at the doses tested (Salloway et al., 2014). In high concentrations it was associated with amyloid-related imaging abnormalities (ARIA) edema and microhemorrhages. Solanezumab is a humanized version of mouse antibody m226 whose epitope lies within the Aβ mid-domain and binds only to soluble Aβ, not fibrillar or plaque-based Aβ. Preclinical studies demonstrated that peripheral administration sequestered peripheral Aβ and led to a dramatic and rapid rise in peripheral Aβ levels. It was hypothesized that the antibody might alter the equilibrium between plasma and CNS Aβ, the so-called “peripheral sink” hypothesis (DeMattos et al., 2001) as well as directly binding soluble Aβ in the CNS. Two phase 3 trials of solanezumab in mild-to-moderate AD failed to demonstrate a marked benefit on cognition or ADL and did not reduce cerebral amyloid deposition (Doody et al., 2014). A third phase 3 trial in mild AD also failed to demonstrate a significant benefit on the primary endpoint (Honig et al., 2018). Aducanumab is a fully human IgG1 antibody that binds a specific conformational epitope of Aβ. There was initial excitement for this antibody, as it was demonstrated to actively engage and clear amyloid plaques both in transgenic APP models and in human participants with prodromal or mild AD based on florbetapir-PET imaging. Further, after one year of monthly infusions, participants receiving aducanumab had slower decline in CDR-sum of boxes and MMSE in a phase 1b trial (Sevigny et al., 2016). Unfortunately, two phase 3 trials in mild AD were stopped early for futility in March 2019 after interim analysis suggested no change in cognitive decline (Selkoe, 2019). Crenezumab is a fully human IgG4 antibody that binds oligomeric, fibrillar and plaque-based Aβ. Two phase 3 trials were terminated early in January 2019 for futility after interim analysis failed to demonstrate any benefit. Gantenerumab is a fully human IgG1 antibody that binds to a conformational epitope on aggregated Aβ fibrils within plaques and triggers Fc-mediated microglial phagocytosis of Aβ. Gantenerumab entered a phase 3 clinical trial for mild AD in 2014 that ultimately stopped enrolling early due to futility. The study was continued as an open label extension at high dose for two years. Follow-up analyses demonstrated a dramatic decline in Aβ deposition in participants in the open extension, such that some became amyloid negative on imaging. Based on this, a new high dose phase 3 trial has been initiated in very mild to mild AD (Ostrowitzki et al., 2017). BAN2401 is a humanized version of murine antibody mAb158 that binds large soluble Aβ protofibrils in vitro. Phase 2 trials demonstrated significant engagement and clearance of cerebral amyloid deposits and possible reduction in cognitive decline. It has now entered a phase 3 clinical trial in very mild to mild AD patients (Logovinsky et al., 2016). Donanemab is a monoclonal antibody developed by Lilly that uniquely targets Aβ(p3-42), a pyroglutamate form of Aβ exclusively found in plaques, in which the goal is to directly engage plaque-based Aβ for clearance and is currently in a phase 2 trial.
Though the data for Aβ-directed monoclonal antibodies in symptomatic AD have been discouraging, several antibodies are currently being used in primary and secondary prevention studies, where dose administration at an earlier disease stage might provide a better opportunity for disease modification. DIAN-TU is using solanezumab and gantenerumab in a phase 2/3 adaptive design clinical trial in ADAD mutation carriers known to inevitably develop disease (Bateman et al., 2017). Crenezumab is being used in the API ADAD Colombia trial of known mutation carriers (Tariot et al., 2018), whereas active immunization with CAD106 is being used in the API Generation trial of APOE ε4 homozygotes (Lopez Lopez et al., 2019).
Secretase inhibitors
An alternative strategy for reducing Aβ levels is to reduce production via inhibition of β- and γ-secretase activities. The rate-limiting step for Aβ production is enzymatic cleavage by BACE1, so this would seem to be a particularly attractive target. In mouse amyloidosis model, BACE1 inhibition limits initiation of new plaque formation but does not prevent growth of established plaques (Peters et al., 2018). Thus, BACE1 inhibition might be expected to work synergistically in combination with Aβ-directed monoclonal antibodies to reduce Aβ deposition by addressing both clearance and production of Aβ. Ideally, BACE1 inhibition would be the preferred target for primary prevention studies, as blocking Aβ production prior to onset of pathology would be sufficient to prevent the development of pathology. The reality of BACE1 inhibitors in clinical testing has been more challenging.
A number of BACE1 inhibitors have entered clinical trials in recent years. Lilly inhibitors LY2886721 and LY3202626 failed in phase 2 trials due to hepatotoxicity and impaired cognitive performance, respectively. Four additional BACE1 inhibitors failed phase 3 clinical trials due to lack of efficacy and associated side effects (Table 1). Verubecestat failed to slow cognitive decline in a phase 3 trial of mild to moderate AD and resulted in worse cognition on some measures. (Egan et al., 2018, 2019). Lanabacestat was discontinued after interim analysis in biomarker-confirmed very mild to mild AD failed to detect any reduction in cognitive decline, despite evidence of target engagement (Clinicaltrials.gov ). Atabecestat was discontinued after a trial in preclinical AD demonstrated frequent hepatotoxicity and worse cognitive outcomes than placebo (Henley et al., 2019). CNP520 (umibecestat) was initially found to be safe and well-tolerated in early phase 1 and 2a studies. This inhibitor was then incorporated into one arm of the phase 2/3 API Generation study (along with CAD106; see above) for secondary prevention of AD in older APOE ε4/ε4 homozygotes (Lopez Lopez et al., 2019). Unfortunately study sponsors announced discontinuation of this arm in July 2019 when interim analysis demonstrated worse cognition, increased brain atrophy and more weight loss in the treatment group relative to placebo. Only one BACE1 inhibitor is now left standing in clinical trials. Elenbecestat is currently engaged in phase 3 clinical trials in biomarker positive MCI/prodromal AD and has not had issues with safety, tolerability or outcome futility to-date. In fact, it has been chosen by the Alzheimer’s Clinical Trials Consortium for use in upcoming primary and secondary prevention studies (along with BAN2401) (Panza et al., 2018).
At this point, it appears that worsening cognitive function and weight loss are possible class effects for BACE1 inhibitors, as they were observed as adverse effects across multiple trials. These may not be simply off-target effects. BACE1 has substrates other than APP that are likely important in neurodevelopment as evidenced by the fact that BACE1 KO mice develop seizures (Hitt et al., 2010), myelination deficits (Willem et al., 2006), cognitive impairment (Laird et al., 2005) and axon guidance defects (Rajapaksha et al., 2011). Conditional BACE1 KO in adulthood mitigates many of these side effects but there remains impaired axon guidance defects (Ou-Yang et al., 2018). Based on these data, it may be difficult to develop a BACE1 inhibitor without some expected on-target side effects.
Two γ-secretase inhibitors (GSI) have been tested clinically as disease modifying therapies for AD and each have failed due to lack of efficacy or worsening of cognition combined with dose-limiting adverse side effects. Avagacestat phase 2 trials in mild to moderate and prodromal AD were terminated after it failed to demonstrate any reduction in cognitive decline and caused adverse effects at higher doses, such as nausea, vomiting, diarrhea, rash, pruritis and increased incidence of non-melanoma skin cancer (Coric et al., 2015). Semagacestat was the first GSI to enter a phase 3 trial for AD. However, the trial was halted early due to concerns of worsening cognition, increased incidence of skin cancers and infection, and weight loss relative to placebo (Doody et al., 2013). Likely the broad-spectrum nature of these GSI contributed to their side effect profile, as Notch signaling and downstream pathways from other substrates would be inhibited (De Strooper, 2014). Alternatively, side effects might be attributable to on-target inhibition of APP processing, resulting in accumulating levels of membrane-bound N-terminally cleaved APP fragments. The same concern might also apply to BACE1 inhibitors.
An alternative means of inhibiting amyloidogenic processing of Aβ by γ-secretase is to use γ-secretase modulators (GSM) that alter processing of APP (lower Aβ42 and higher Aβ40 levels) without influencing processing of other substrates (Xia, 2019). The mechanism appears to be related to slowed dissociation of epsilon-cleaved C99 fragment thereby increasing enzyme processivity, leading to generation of shorter Aβ fragments (Okochi et al., 2013). The first GSMs were discovered by recognizing that ibuprofen, indomethacin and sulindac reduce Abeta42 levels (Weggen et al., 2001) through modulation of gamma-secretase activity (Eriksen et al., 2003). This eventually led to identification of R-flurbiprofen (tarenflurbil), a molecule having GSM activity but without inhibitory effects on cyclooxygenase activity. Despite promising preclinical data, unfortunately a phase 3 clinical trial did not demonstrate any benefits on cognition or ADLs over the course of 18 months (Green et al., 2009). However, significant CNS Aβ42 lowering was not likely achieved (Karran and Hardy, 2014), therefore more efficacious GSMs may still be a viable conceptual approach. Indeed, there are other promising GSMs now in development that may enter human trials in the near future (Wagner et al., 2017). With the recent publication of high resolution substrate-enzyme cryo-EM structures for γ-secretase-APP C83 fragment and γ-secretase-Notch 100 fragment (Yang et al., 2019; Zhou et al., 2019), it may now be possible to more easily develop substrate-specific γ-secretase inhibitors or modulators..
Tau-directed therapeutics
Tau is considered a critical target for developing disease modifying therapies. Multiple strategies have been proposed for reducing tau pathogenicity in AD. One of the first compounds tested in a clinical trial was methylene blue. Any putative benefit attributed to methylene blue has been thought to be secondary to reduced tau fibrillization and aggregation and induction of autophagy, which leads to reduced tau pathology and neuron death in mouse models of tauopathy (Congdon et al., 2012; Crowe et al., 2013; Wischik et al., 1996). A second-generation stabilized and reduced derivative of methylene blue called LMTM was created and moved to clinical trials. Unfortunately, in two different phase 3 trials testing LMTM in all-cause dementia and mild-to-moderate AD, there was no reduction in rate of decline of cognition or ADL compared to placebo (Gauthier et al., 2016). Whether LMTM at the doses administered prevented accumulation of aggregated tau or decreased existing deposits was not tested.
A second strategy for reducing tau pathogenicity is to inhibit abnormal hyperphosphorylation through use of tau kinase inhibitors. A GSK-3 inhibitor, tideglusib, was shown in a preclinical study using double transgenic mice expressing human tau and APP to reduce tau phosphorylation, decrease amyloid deposition, lead to less neuronal loss and improved cognition (Serenó et al., 2009). However, a phase 2 trial in mild to moderate AD did not demonstrate any reduction in rate of cognitive or functional decline as compared to placebo (Lovestone et al., 2015)
As with Aβ, a significant of amount of investment has been made in developing immunotherapies to facilitate tau pathology clearance from the brain, including both active and passive immunization. Two active immunization strategies are being investigated. AADvac-1 consists of tau peptide containing amino acids 294-305 conjugated to keyhole limpet hemocyanin with use of adjuvant. AADvac-1 recently completed phase 1 testing and is currently undergoing phase 2 trials (Novak et al., 2019). ACI-35 is another active vaccination attempt using 16 copies of synthetic phosphorylated tau fragment embedded in liposome. The goal is to generate an immune response against pathologic phosphorylated tau with no cross-reactivity against non-pathologic tau. This has completed phase 1 clinical testing and awaits further phase 2 testing in the future.
Numerous tau-directed monoclonal antibodies have been generated and are currently being tested in clinical trials. In preclinical studies, mouse anti-human tau monoclonal antibody HJ8.5 demonstrated reduced tau seeding, hyperphosphorylation and thioflavin S staining, reduced levels of detergent insoluble tau, and reduced brain atrophy and cognitive deficits in PS19 (P301S) mice, whether administered via intraventricular infusion (Yanamandra et al., 2013) or peripherally (Yanamandra et al., 2015) when administered prior to the development of significant tau pathology. This antibody has been humanized and in a single patient with progressive supranuclear palsy (PSP) led to increased plasma tau concentrations after intravenous injection. In PS19 mice, plasma tau was found to inversely correlate with brain soluble tau levels, suggesting that peripheral tau levels reflect changes occurring in the CNS (Yanamandra et al., 2017). The humanized version of HJ8.5 (ABBV-8E12) has been tested in a phase 1 trial and found to be safe and has now advanced to two phase 2 trials including patients with early AD and PSP (West et al., 2017). The PSP study was just terminated following a futility analysis. A separate Biogen humanized anti-tau monoclonal antibody (BIIB092) has also been tested in phase 1 study in PSP patients, passed safety parameters, and was demonstrated to reduce N-terminal tau in CSF of patients. This antibody has now advanced to phase 2 testing in PSP and AD (Boxer et al., 2019). Biogen and other companies have additional monoclonal antibodies targeting tau that are in the clinical pipeline: R07105705, BIIB076, JNJ-63733657, LY3303560, MC1-L, and UCB1017. A critical issue is to determine the extent to which pathological seeding and spreading of tau pathology occurs via extracellular forms of tau and whether such a mechanism occurs in humans. It is likely that if a peripherally administered anti-tau antibody is to be efficacious in a disease such as AD, such a mechanism would need to occur and an antibody would need to inhibit this process. If antibody based methods are developed to degrade and reduce intracellular, cytoplasmic forms of tau, this may prove to be a more effective way to not only prevent but clear existing tau pathology.
Finally, a separate strategy to limit tau-mediated pathology would be to reduce tau expression. One clinically-viable approach is the use of intrathecally-delivered ASO. ASO-based clinical treatments have demonstrated dramatic success in cases of spinal motor atrophy (Finkel et al., 2017) and are being tested in Huntington’s disease (Tabrizi et al., 2019). Preclinical studies using tau-overexpressing mice suggest this approach may be viable in tauopathies (DeVos et al., 2017). Ionis has initiated a phase 1/2 trial of anti-tau ASO (MAPTRx) in mild AD (Clinicatrials.gov ID# ).
ApoE-directed therapeutics
To date, no in-human AD clinical trials have targeted ApoE. However, a number of preclinical studies in relevant animal models have investigated ApoE-directed therapies that may eventually translate clinically.
Multiple studies employing ApoE genetic deletion (Bales et al., 1999; Verghese et al., 2013), conditional expression of ApoE isoforms (Liu et al., 2017) or ASO-mediated knockdown of ApoE expression (Huynh et al., 2017b) suggest that lowering ApoE levels reduces relevant pathology in both amyloidosis and tauopathy mouse models (Shi et al., 2017). Use of anti-ApoE ASO delivered via intracerebroventricular injection in ApoE-TR/APPPS1-21 mice demonstrated dramatically reduced Aβ plaque deposition if administered prior to plaque seeding but not after. Neuritic dystrophy was also significantly attenuated, independent of an effect on amyloid deposition even when administered after amyloid deposition (Huynh et al., 2017b). Therefore, targeting ApoE3 or ApoE4 expression via ASO delivery in AD may be beneficial, especially if administered in early preclinical phases. Since ApoE promotes plaque-associated microgliosis resulting in plaque compaction and reduced neuritic dystrophy (Ulrich et al., 2018), therapeutically reducing ApoE levels may also have unexpected effects on the innate immune response to established plaques. An alternative strategy for lowering Aβ levels via targeting a certain form of ApoE is the use of passive immunization. Monoclonal antibody HJ6.3 delivered peripherally to APP/PS1 mice either before or after plaque deposition resulted in reduced Aβ levels and fibrillary amyloid pathology, increased microglial activation, and improved spatial memory performance (Kim et al., 2012; Liao et al., 2014). HAE-4 is a monoclonal antibody that targets non-lipidated, aggregated ApoE3 and E4 preferentially located within amyloid plaques. Delivery of this antibody centrally or peripherally reduced fibrillary amyloid plaque load and soluble and insoluble Aβ levels without reducing cerebral or plasma ApoE levels. Instead, data suggests the antibody mediates its effect by binding apoE in plaques followed by antibody mediated plaque opsonization by microglia, likely promoting peri-plaque Aβ phagocytosis (Liao et al., 2018).
Another proposed strategy is to utilize gene therapy methods to overexpress ApoE2 in APOE ε4/ε4 carriers as means to overcome loss of function associated with ApoE4 (Rosenberg et al., 2018). This strategy would only be effective if ApoE4-mediated disease mechanisms are truly due to loss of function or, if secondary to ApoE4 toxic gain of function, that ApoE2 is able to act in dominant protective fashion to inhibit ApoE4 toxic effects. While there is data to support the concept that ApoE2 expression may be protective using a gene transfer approach (Hu et al., 2015; Hudry et al., 2013), it would be useful to validate protective effects and lack of toxicity in a number of different models. ApoE modulates AD pathobiology through various pathways (see above), so it will be important to confirm that APOE2 overexpression has only salutary effects on these many disease-relevant biological functions. This is especially critical after a recent study in the preclinical literature demonstrated APOE2 expression via viral delivery exacerbated pathology in tauopathy mice (Zhao et al., 2018). It is unclear whether ApoE2 gene delivery would be a superior strategy to simply lowering ApoE levels (discussed above). A recent study tested APOE2 gene delivery in non-human primates using AAVrh.10 hAPOE2-HA vector to deliver an APOE2 expression cassette. This study found that intracisternal viral injection led to the widest area of cerebral transduction with only minimal surgery (Rosenberg et al., 2018). A phase 1 clinical trial in humans using intracisternal injection of this vector is planned (ClinicalTrials.gov #: ).
Immune Modulation
Interest in anti-inflammatory treatments for AD were initially stoked by findings from multiple epidemiologic studies suggesting that chronic use of NSAIDs was associated with lower incidence of AD, with the most pronounced effect seen in patients taking chronic ibuprofen (in t’ Veld et al., 2001; Vlad et al., 2008). However, follow-up clinical trials testing the effect of low-dose prednisone (Aisen et al., 2000), low dose aspirin (AD2000 Collaborative Group et al., 2008), NSAIDS (Aisen et al., 2003; de Jong et al., 2008), and selective COX-2 inhibitors (Aisen et al., 2003; Thal et al., 2005) failed to show any reduction in rate of cognitive decline in mild to moderate AD. Prevention studies of naproxen and celecoxib in patients at risk for preclinical AD also failed to demonstrate any protective benefit (ADAPT-FS Research Group, 2015; Meyer et al., 2019). Therefore, a generic anti-inflammatory treatment approach for AD does not appear viable for secondary prevention or treatment of symptomatic patients.
In one study, patients with rheumatoid arthritis treated with etanercept, TNFα decoy receptor, had lower incidence of AD, but no effect was noted with use of other anti-rheumatic medications (prednisone, sulfasalazine, rituximab) or anti-TNF agents (infliximab, adalimumab) (Chou et al., 2016). A phase 2 trial of etanercept in mild to moderate AD established safety but saw no significant reduction in cognitive decline on analysis of secondary clinical outcomes (Butchart et al., 2015). It is notable that etanercept does not cross the BBB. One strategy to enhance etanercept delivery across the BBB is by fusing the molecule to transferrin receptor. This approach demonstrated reduced amyloid burden and improvement in recognition memory in preclinical studies using APP/PS1 mice (Chang et al., 2017). Preclinical models have demonstrated protection from synaptic deficits in rodent amyloidosis models following treatment with inflammasome inhibitors before onset of amyloid plaques (Qi et al., 2018)
Intravenous immunoglobulin (IVIg) is an immunomodulatory treatment approved for treatment of various neurologic and rheumatologic autoimmune diseases. However, a phase 3 trial of IVIg did not demonstrate any cognitive benefits in mild AD despite a favorable safety profile (Relkin et al., 2017).
A separate treatment approach is to boost microglial activation or phagocytic function, since several AD gene risk variants in TREM2 and CD33 act to decrease microglial activation and phagocytosis (see above). TREM2-activating monoclonal antibodies have entered phase I clinical trials (Clinicaltrials.gov Identifier #: ). CD33-blocking antibodies are also being planned for phase 1 clinical trials (Clinicaltrials.gov Identifier #: )
Young Blood
Recent heterochronic parabiosis experiments demonstrated that introduction of young blood in old mice led to increased neurogenesis and dendritic spine density, and improved age-related cognitive impairment, (Katsimpardi et al., 2014; Villeda et al., 2014). This effect was also seen after injection of human umbilical cord blood (Castellano et al., 2017). Serum from young mice promoted increased numbers of functional synapses, increased dendritic arborization, increased synaptic response to stimuli and increased NMDA synaptic responses in co-cultures of human neurons with mouse glia. Such effects were not seen with serum from old mice or FBS. Proteomic and functional validation experiments suggest that the effect is at least partly mediated by SPARC1L and thrombospondin-4 (Gan and Südhof, 2019). Heterochronic parabiosis and direct injection of young mouse plasma in APP amyloidosis mice resulted in increased levels of synaptic protein and improvement in cognitive performance on standard tests of spatial working memory and hippocampal-dependent associative learning (Middeldorp et al., 2016)
These studies have led to increased off-label clinical administration of young donor plasma to individuals for a list of age-related conditions, leading the FDA to release a statement recommending against this given lack of current evidence to support the practice (Commissioner, 2019)
This approach is being tested currently in clinical trials. A completed phase 1 trial of weekly infusions of 1 unit of young fresh frozen plasma from male donors (age 18-30) vs saline in patients with mild to moderate AD for total of 4 weeks demonstrated the intervention was mostly well tolerated (Sha et al., 2019). A concentrated plasma fraction derived from young donor plasma is now being tested in a phase 2 trial (Kheifets and Braithwaite, 2019). Plasma exchange may work to simply remove detrimental plasma component from old blood or mobilize CSF Aβ, rather than supply therapeutic factors from young blood. The AMBAR trial, a current phase 2b/3 trial, is testing this hypothesis by assessing the effect of monthly plasma exchange with albumin replacement on mild to moderate AD (Boada et al., 2019)
Combination Therapies
Using the example of treatment paradigms for other chronic diseases, such as hypertension, congestive heart failure, epilepsy, HIV and others, a combinatorial treatment strategy for AD seems more likely to yield clinical successes than monotherapy (Gauthier et al., 2019; Morris, 2019; Stephenson et al., 2015). Many of the diseases just listed are only effectively managed when multiple medications spanning different drug classes are employed. Given the multifactorial nature and underlying complexity of AD pathobiology, the expectation that a single agent targeting a single pathological pathway would be highly effective in slowing disease progression seems irrational once different pathologies of different types are present. In fact, combination strategies have begun to be employed in clinical trial design. The Lilly TRAILBLAZER phase 2 trial initially tested the combination of the BACE1 inhibitor LY3202626 with anti-pyroglutamate Aβ monoclonal antibody donanemab. Unfortunately, LY3202626 was found to be associated with slight cognitive worsening in dedicated phase 2 trials and so the combination treatment arm was ultimately dropped. Nonetheless, combination strategies in clinical trials should continue to be encouraged, especially those with combinations spanning different drug classes. Ideally, this type of treatment approach would also be applied to preventative or preclinical AD trials.
CONCLUSIONS
AD research is at a unique moment. Despite significant advances in our understanding of AD pathobiology, we have not yet identified a disease modifying therapy that has proven effective in humans. While biochemical analyses, in vitro and in vivo studies, genetic analyses and longitudinal imaging studies lend strong support to the role of Aβ aggregation in initiating disease pathogenesis, the clinical trials as conducted have not panned out. So far, amyloid-based therapeutics appear to be ineffective in modifying the disease course for symptomatic AD.Future clinical trial efforts should instead focus on applying these anti-amyloid treatment strategies to the preclinical disease, the earlier the better, with drugs shown to hit their target effectively. It is now more important than ever to also pursue non-amyloid based therapies. Tau- and ApoE-directed therapeutics remain at an early stage of development and both hold great potential going foward. There is also huge potential for therapies that modulate the neuroimmune or microglial response in AD, and this class of drug development is significantly underrepresented. Combination therapy is a strategy that should be employed more widely in AD clinical trial design. Finally, AD pathobiology is complex and the older one is, the greater likelihood that other age-related diseases contribute to cognitive decline together with AD pathology. Ongoing intensive investigation in this area will be critical to making key discoveries that will ultimately unveil novel therapeutic approaches leading to truly disease-modifying medications.
ACKNOWLEDGEMENTS
This work was supported by a grant from the Alzheimer’s Association to J.M.L (AACSF-18-564776), NIH grants NS090934, AG047644, NS074969 to D.M.H. and grants from the Tau consortium, the JPB Foundation, and Cure Alzheimer’s Fund to D.M. H.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATIONS OF INTEREST: J.M.L. reports serving as sub-investigator on the Lilly TRAILBLAZER trial but receives no financial compensation. D.M.H. reports being a cofounder of C2N Diagnostics, LLC; being on the scientific advisory board of C2N Diagnostics, Denali, and Genentech; and being a consultant for AbbVie and Idorsia. D.M.H is an inventor on a submitted patent #PCT/US2013/049333 “Antibodies to tau” that is licensed by Washington University to C2N Diagnostics. This patent was subsequently licensed to AbbVie.D.M.H. is an inventor on patent number 8,591,894 “Humanized antibodies that sequester amyloid beta” that was licensed by Washington University to Eli Lilly. D.M.H. is an inventor on patent number 8,232,107 “Methods for measuring the metabolism of neurally derived biomolecules in vivo” that was licensed by Washington University to C2N Diagnostics, LLC.
References
- Abdel-Haq R, Schlachetzki JCM, Glass CK, and Mazmanian SK (2019). Microbiome-microglia connections via the gut-brain axis. J. Exp. Med. 216, 41–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AD2000 Collaborative Group, Bentham P, Gray R, Sellwood E, Hills R, Crome P, and Raftery J (2008). Aspirin in Alzheimer’s disease (AD2000): a randomised open-label trial. Lancet Neurol 7, 41–49. [DOI] [PubMed] [Google Scholar]
- ADAPT-FS Research Group (2015). Follow-up evaluation of cognitive function in the randomized Alzheimer’s Disease Anti-inflammatory Prevention Trial and its Follow-up Study. Alzheimers Dement 11, 216–225. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed Z, Cooper J, Murray TK, Garn K, McNaughton E, Clarke H, Parhizkar S, Ward MA, Cavallini A, Jackson S, et al. (2014). A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 127, 667–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aisen PS, Davis KL, Berg JD, Schafer K, Campbell K, Thomas RG, Weiner MF, Farlow MR, Sano M, Grundman M, et al. (2000). A randomized controlled trial of prednisone in Alzheimer’s disease. Alzheimer’s Disease Cooperative Study. Neurology 54, 588–593. [DOI] [PubMed] [Google Scholar]
- Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, Farlow MR, Jin S, Thomas RG, Thal LJ, et al. (2003). Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA 289, 2819–2826. [DOI] [PubMed] [Google Scholar]
- Altmann A, Ng B, Landau SM, Jagust WJ, Greicius MD, and Alzheimer’s Disease Neuroimaging Initiative (2015). Regional brain hypometabolism is unrelated to regional amyloid plaque burden. Brain 138, 3734–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer’s Association (2019). 2019 Alzheimer’s disease facts and figures. Alzheimer Dement 15, 321–387. [DOI] [PubMed] [Google Scholar]
- Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kügler S, and Ikezu T (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 18, 1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschenbrenner AJ, Gordon BA, Benzinger TLS, Morris JC, and Hassenstab JJ (2018). Influence of tau PET, amyloid PET, and hippocampal volume on cognition in Alzheimer disease. Neurology 91, e859–e866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atri A, Frölich L, Ballard C, Tariot PN, Molinuevo JL, Boneva N, Windfeld K, Raket LL, and Cummings JL (2018). Effect of Idalopirdine as Adjunct to Cholinesterase Inhibitors on Change in Cognition in Patients With Alzheimer Disease: Three Randomized Clinical Trials. JAMA 319, 130–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ, et al. (1997). Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat. Genet. 17, 263–264. [DOI] [PubMed] [Google Scholar]
- Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, Fishman CE, DeLong CA, Piccardo P, Petegnief V, et al. (1999). Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 96, 15233–15238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Liu F, Wu S, Lin S, Koger D, DeLong C, Hansen JC, Sullivan PM, and Paul SM (2009). Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J. Neurosci. 29, 6771–6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartus RT, Dean RL, Beer B, and Lippa AS (1982). The cholinergic hypothesis of geriatric memory dysfunction. Science 217, 408–414. [DOI] [PubMed] [Google Scholar]
- Basak JM, Verghese PB, Yoon H, Kim J, and Holtzman DM (2012). Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Aβ uptake and degradation by astrocytes. J. Biol. Chem. 287, 13959–13971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Xiong C, Benzinger TLS, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, et al. (2012). Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. New England Journal of Medicine 367, 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Benzinger TL, Berry S, Clifford DB, Duggan C, Fagan AM, Fanning K, Farlow MR, Hassenstab J, McDade EM, et al. (2017). The DIAN-TU Next Generation Alzheimer’s prevention trial: Adaptive design and disease progression model. Alzheimers Dement 13, 8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäuerl C, Collado MC, Diaz Cuevas A, Viña J, and Pérez Martínez G (2018). Shifts in gut microbiota composition in an APP/PSS1 transgenic mouse model of Alzheimer’s disease during lifespan. Lett. Appl. Microbiol. 66, 464–471. [DOI] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, et al. (2012). Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485, 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee J-M, and Holtzman DM (2011). Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat. Neurosci. 14, 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bien-Ly N, Gillespie AK, Walker D, Yoon SY, and Huang Y (2012). Reducing human apolipoprotein E levels attenuates age-dependent Aβ accumulation in mutant human amyloid precursor protein transgenic mice. J. Neurosci. 32, 4803–4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biere AL, Ostaszewski B, Zhao H, Gillespie S, Younkin SG, and Selkoe DJ (1995). Co-expression of beta-amyloid precursor protein (betaAPP) and apolipoprotein E in cell culture: analysis of betaAPP processing. Neurobiol. Dis. 2, 177–187. [DOI] [PubMed] [Google Scholar]
- Biernat J, Gustke N, Drewes G, Mandelkow EM, and Mandelkow E (1993). Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron 11, 153–163. [DOI] [PubMed] [Google Scholar]
- Birks JS (2006). Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database of Systematic Reviews. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boada M, López O, Núñez L, Szczepiorkowski ZM, Torres M, Grifols C, and Páez A (2019). Plasma exchange for Alzheimer’s disease Management by Albumin Replacement (AMBAR) trial: Study design and progress. Alzheimers Dement (N Y) 5, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolmont T, Clavaguera F, Meyer-Luehmann M, Herzig MC, Radde R, Staufenbiel M, Lewis J, Hutton M, Tolnay M, and Jucker M (2007). Induction of tau pathology by intracerebral infusion of amyloid-beta -containing brain extract and by amyloid-beta deposition in APP x Tau transgenic mice. Am. J. Pathol. 171, 2012–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgade K, Garneau H, Giroux G, Le Page AY, Bocti C, Dupuis G, Frost EH, and Fülöp T (2015). β-Amyloid peptides display protective activity against the human Alzheimer’s disease-associated herpes simplex virus-1. Biogerontology 16, 85–98. [DOI] [PubMed] [Google Scholar]
- Boxer AL, Qureshi I, Ahlijanian M, Grundman M, Golbe LI, Litvan I, Honig LS, Tuite P, McFarland NR, O’Suilleabhain P, et al. (2019). Safety of the tau-directed monoclonal antibody BIIB092 in progressive supranuclear palsy: a randomised, placebo-controlled, multiple ascending dose phase 1b trial. Lancet Neurol 18, 549–558. [DOI] [PubMed] [Google Scholar]
- Braak H, and Braak E (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, Rosenkrantz LL, Imboywa S, Lee M, Von Korff A, et al. (2013). CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat. Neurosci. 16, 848–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridel C, van Wieringen WN, Zetterberg H, Tijms BM, Teunissen CE, and the NFL Group, Alvarez-Cermeño JC, Andreasson U, Axelsson M, Bäckström DC, et al. (2019). Diagnostic Value of Cerebrospinal Fluid Neurofilament Light Protein in Neurology: A Systematic Review and Meta-analysis. JAMA Neurol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brier MR, Day GS, Snyder AZ, Tanenbaum AB, and Ances BM (2016). N-methyl-D-aspartate receptor encephalitis mediates loss of intrinsic activity measured by functional MRI. J. Neurol. 263, 1083–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esparza TJ, Stocchetti N, Zipfel GJ, and Holtzman DM (2008). Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science 321, 1221–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke WJ, Miller JP, Rubin EH, Morris JC, Coben LA, Duchek J, Wittels IG, and Berg L (1988). Reliability of the Washington University Clinical Dementia Rating. Arch. Neurol. 45, 31–32. [DOI] [PubMed] [Google Scholar]
- Bussy A, Snider BJ, Coble D, Xiong C, Fagan AM, Cruchaga C, Benzinger TLS, Gordon BA, Hassenstab J, Bateman RJ, et al. (2019). Effect of apolipoprotein E4 on clinical, neuroimaging, and biomarker measures in noncarrier participants in the Dominantly Inherited Alzheimer Network. Neurobiol. Aging 75, 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butchart J, Brook L, Hopkins V, Teeling J, Püntener U, Culliford D, Sharples R, Sharif S, McFarlane B, Raybould R, et al. (2015). Etanercept in Alzheimer disease: A randomized, placebo-controlled, double-blind, phase 2 trial. Neurology 84, 2161–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, Mucke L, and Mahley RW (1999). Expression of human apolipoprotein E3 or E4 in the brains of Apoe-/-mice: isoform-specific effects on neurodegeneration. J. Neurosci. 19, 4867–4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttini M, Masliah E, Yu G-Q, Palop JJ, Chang S, Bernardo A, Lin C, Wyss-Coray T, Huang Y, and Mucke L (2010). Cellular source of apolipoprotein E4 determines neuronal susceptibility to excitotoxic injury in transgenic mice. Am. J. Pathol. 177, 563–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, et al. (2012). Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 73, 685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano-Cuenca N, Solis-Garcia del Pozo JE, and Jordan J (2014). Evidence for the efficacy of latrepirdine (Dimebon) treatment for improvement of cognitive function: a meta-analysis. J. Alzheimers Dis. 38, 155–164. [DOI] [PubMed] [Google Scholar]
- Carbone I, Lazzarotto T, Ianni M, Porcellini E, Forti P, Masliah E, Gabrielli L, and Licastro F (2014). Herpes virus in Alzheimer’s disease: relation to progression of the disease. Neurobiol. Aging 35, 122–129. [DOI] [PubMed] [Google Scholar]
- Carlsson CM, Gleason CE, Hess TM, Moreland KA, Blazel HM, Koscik RL, Schreiber NTN, Johnson SC, Atwood CS, Puglielli L, et al. (2008). Effects of simvastatin on cerebrospinal fluid biomarkers and cognition in middle-aged adults at risk for Alzheimer’s disease. J. Alzheimers Dis. 13, 187–197. [DOI] [PubMed] [Google Scholar]
- Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, et al. (2011). Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med 3, 89ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano JM, Deane R, Gottesdiener AJ, Verghese PB, Stewart FR, West T, Paoletti AC, Kasper TR, DeMattos RB, Zlokovic BV, et al. (2012). Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Aβ clearance in a mouse model of β-amyloidosis. Proc. Natl. Acad. Sci. U.S.A. 109, 15502–15507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano JM, Mosher KI, Abbey RJ, McBride AA, James ML, Berdnik D, Shen JC, Zou B, Xie XS, Tingle M, et al. (2017). Human umbilical cord plasma proteins revitalize hippocampal function in aged mice. Nature 544, 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedazo-Minguez A, Wiehager B, Winblad B, Hüttinger M, and Cowburn RF (2001). Effects of apolipoprotein E (apoE) isoforms, beta-amyloid (Abeta) and apoE/Abeta complexes on protein kinase C-alpha (PKC-alpha) translocation and amyloid precursor protein (APP) processing in human SH-SY5Y neuroblastoma cells and fibroblasts. Neurochem. Int. 38, 615–625. [DOI] [PubMed] [Google Scholar]
- Chang R, Knox J, Chang J, Derbedrossian A, Vasilevko V, Cribbs D, Boado RJ, Pardridge WM, and Sumbria RK (2017). Blood-Brain Barrier Penetrating Biologic TNF-α Inhibitor for Alzheimer’s Disease. Mol. Pharm. 14, 2340–2349. [DOI] [PubMed] [Google Scholar]
- Chen VC-H, Wu S-I, Huang K-Y, Yang Y-H, Kuo T-Y, Liang H-Y, Huang K-L, and Gossop M (2018). Herpes Zoster and Dementia: A Nationwide Population-Based Cohort Study. J Clin Psychiatry 79. [DOI] [PubMed] [Google Scholar]
- Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D’Avanzo C, Chen H, Hooli B, Asselin C, Muffat J, et al. (2014). A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515, 274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou RC, Kane M, Ghimire S, Gautam S, and Gui J (2016). Treatment for Rheumatoid Arthritis and Risk of Alzheimer’s Disease: A Nested Case-Control Analysis. CNS Drugs 30, 1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, and Holtzman DM (2005). Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48, 913–922. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Kang J-E, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, and Holtzman DM (2008). Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron 58, 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, et al. (2009). Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, Probst A, Winkler DT, Reichwald J, Staufenbiel M, et al. (2013). Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. U.S.A. 110, 9535–9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M, and Butovsky O (2017). Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 35, 441–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commissioner, O. of the (2019). Statement from FDA Commissioner Scott Gottlieb, M.D., and Director of FDA’s Center for Biologics Evaluation and Research Peter Marks, M.D., Ph.D., cautioning consumers against receiving young donor plasma infusions that are promoted as unproven treatment for varying conditions.
- Congdon EE, Wu JW, Myeku N, Figueroa YH, Herman M, Marinec PS, Gestwicki JE, Dickey CA, Yu WH, and Duff KE (2012). Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 8, 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C, Carlomagno Y, Gendron TF, Dunmore J, Scheffel K, Stetler C, Davis M, Dickson D, Jarpe M, DeTure M, et al. (2014). Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum. Mol. Genet. 23, 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, and Pericak-Vance MA (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. [DOI] [PubMed] [Google Scholar]
- Coric V, Salloway S, van Dyck CH, Dubois B, Andreasen N, Brody M, Curtis C, Soininen H, Thein S, Shiovitz T, et al. (2015). Targeting Prodromal Alzheimer Disease With Avagacestat: A Randomized Clinical Trial. JAMA Neurol 72, 1324–1333. [DOI] [PubMed] [Google Scholar]
- Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, Arnold SE, Attems J, Beach TG, Bigio EH, et al. (2014). Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 128, 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe A, James MJ, Lee VM-Y, Smith AB, Trojanowski JQ, Ballatore C, and Brunden KR (2013). Aminothienopyridazines and methylene blue affect Tau fibrillization via cysteine oxidation. J. Biol. Chem. 288, 11024–11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther RA, and Wischik CM (1985). Image reconstruction of the Alzheimer paired helical filament. EMBO J. 4, 3661–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz Hernández JC, Bracko O, Kersbergen CJ, Muse V, Haft-Javaherian M, Berg M, Park L, Vinarcsik LK, Ivasyk I, Rivera DA, et al. (2019). Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nat. Neurosci. 22, 413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Mesquita S, Louveau A, Vaccari A, Smirnov I, Cornelison RC, Kingsmore KM, Contarino C, Onengut-Gumuscu S, Farber E, Raper D, et al. (2018). Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature 560, 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, and Vitek MP (2001). Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell. Sci. 114, 1179–1187. [DOI] [PubMed] [Google Scholar]
- De Strooper B (2014). Lessons from a failed γ-secretase Alzheimer trial. Cell 159, 721–726. [DOI] [PubMed] [Google Scholar]
- De Strooper B, and Karran E (2016). The Cellular Phase of Alzheimer’s Disease. Cell 164, 603–615. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, and Holtzman DM (2001). Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 98, 8850–8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux CS, Wegener AJ, Chen G, Shen T, Tran H, Nichols B, et al. (2017). Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVos SL, Corjuc BT, Oakley DH, Nobuhara CK, Bannon RN, Chase A, Commins C, Gonzalez JA, Dooley PM, Frosch MP, et al. (2018). Synaptic Tau Seeding Precedes Tau Pathology in Human Alzheimer’s Disease Brain. Front Neurosci 12, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit R, Ross JL, Goldman YE, and Holzbaur ELF (2008). Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, Nguyen M, Haditsch U, Raha D, Griffin C, et al. (2019). Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv 5, eaau3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, et al. (2013). A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 369, 341–350. [DOI] [PubMed] [Google Scholar]
- Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, Raman R, Sun X, Aisen PS, et al. (2014). Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 311–321. [DOI] [PubMed] [Google Scholar]
- Dumanis SB, Tesoriero JA, Babus LW, Nguyen MT, Trotter JH, Ladu MJ, Weeber EJ, Turner RS, Xu B, Rebeck GW, et al. (2009). ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J. Neurosci. 29, 15317–15322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, Delatour B, and Potier M-C (2009). Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 118, 5–36. [DOI] [PubMed] [Google Scholar]
- Edison P, Archer HA, Hinz R, Hammers A, Pavese N, Tai YF, Hotton G, Cutler D, Fox N, Kennedy A, et al. (2007). Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology 68, 501–508. [DOI] [PubMed] [Google Scholar]
- Efthymiou AG, and Goate AM (2017). Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol Neurodegener 12, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan MF, Kost J, Tariot PN, Aisen PS, Cummings JL, Vellas B, Sur C, Mukai Y, Voss T, Furtek C, et al. (2018). Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 378, 1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan MF, Kost J, Voss T, Mukai Y, Aisen PS, Cummings JL, Tariot PN, Vellas B, van Dyck CH, Boada M, et al. (2019). Randomized Trial of Verubecestat for Prodromal Alzheimer’s Disease. N. Engl. J. Med. 380, 1408–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, György B, Breakefield XO, Tanzi RE, and Moir RD (2018). Alzheimer’s Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron 99, 56–63. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele YS, Bolmont T, Heikenwalder M, Langer F, Jacobson LH, Yan Z-X, Roth K, Aguzzi A, Staufenbiel M, Walker LC, et al. (2009). Induction of cerebral beta-amyloidosis: intracerebral versus systemic Abeta inoculation. Proc. Natl. Acad. Sci. U.S.A. 106, 12926–12931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele YS, Obermüller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, Walker LC, Staufenbiel M, Heikenwalder M, and Jucker M (2010). Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science 330, 980–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, Ozols VV, Jessing KW, Zavitz KH, Koo EH, et al. (2003). NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J. Clin. Invest. 112, 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erny D, Hrabe de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, Keren-Shaul H, Mahlakoiv T, Jakobshagen K, Buch T, et al. (2015). Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 18, 965–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzat K, Pernemalm M, Pålsson S, Roberts TC, Järver P, Dondalska A, Bestas B, Sobkowiak MJ, Levänen B, Sköld M, et al. (2019). The viral protein corona directs viral pathogenesis and amyloid aggregation. Nat Commun 10, 2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Mach RH, Lee S-Y, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, et al. (2006). Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann. Neurol. 59, 512–519. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, and Holtzman DM (2007). Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch. Neurol. 64, 343–349. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Xiong C, Jasielec MS, Bateman RJ, Goate AM, Benzinger TLS, Ghetti B, Martins RN, Masters CL, Mayeux R, et al. (2014). Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer’s disease. Sci Transl Med 6, 226ra30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman HH, Doody RS, Kivipelto M, Sparks DL, Waters DD, Jones RW, Schwam E, Schindler R, Hey-Hadavi J, DeMicco DA, et al. (2010). Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology 74, 956–964. [DOI] [PubMed] [Google Scholar]
- Felsky D, Roostaei T, Nho K, Risacher SL, Bradshaw EM, Petyuk V, Schneider JA, Saykin A, Bennett DA, and De Jager PL (2019). Neuropathological correlates and genetic architecture of microglial activation in elderly human brain. Nat Commun 10, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, Chiriboga CA, Saito K, Servais L, Tizzano E, et al. (2017). Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 377, 1723–1732. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, and Scheres SHW (2017). Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B, Ollesch J, Wille H, and Diamond MI (2009). Conformational diversity of wild-type Tau fibrils specified by templated conformation change. J. Biol. Chem. 284, 3546–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale SC, Gao L, Mikacenic C, Coyle SM, Rafaels N, Murray Dudenkov T, Madenspacher JH, Draper DW, Ge W, Aloor JJ, et al. (2014). APOε4 is associated with enhanced in vivo innate immune responses in human subjects. J. Allergy Clin. Immunol. 134, 127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo G, and Holtzman DM (2017). Antibody Therapeutics Targeting Aβ and Tau. Cold Spring Harb Perspect Med 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan KJ, and Südhof TC (2019). Specific factors in blood from young but not old mice directly promote synapse formation and NMDA-receptor recruitment. Proc. Natl. Acad. Sci. U.S.A. 116, 12524–12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier S, Feldman HH, Schneider LS, Wilcock GK, Frisoni GB, Hardlund JH, Moebius HJ, Bentham P, Kook KA, Wischik DJ, et al. (2016). Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 388, 2873–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier S, Alam J, Fillit H, Iwatsubo T, Liu-Seifert H, Sabbagh M, Salloway S, Sampaio C, Sims JR, Sperling B, et al. (2019). Combination Therapy for Alzheimer’s Disease: Perspectives of the EU/US CTAD Task Force. J Prev Alzheimers Dis 6, 164–168. [DOI] [PubMed] [Google Scholar]
- Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kövari E, Perl DP, Morrison JH, Gold G, and Hof PR (2003). Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60, 1495–1500. [DOI] [PubMed] [Google Scholar]
- Glenner GG, and Wong CW (1984). Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890. [DOI] [PubMed] [Google Scholar]
- Gómez-Isla T, Price JL, McKeel DW, Morris JC, Growdon JH, and Hyman BT (1996). Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J. Neurosci. 16, 4491–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon BA, Blazey T, Su Y, Fagan AM, Holtzman DM, Morris JC, and Benzinger TLS (2016). Longitudinal β-Amyloid Deposition and Hippocampal Volume in Preclinical Alzheimer Disease and Suspected Non-Alzheimer Disease Pathophysiology. JAMA Neurol 73, 1192–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon BA, Blazey TM, Su Y, Hari-Raj A, Dincer A, Flores S, Christensen J, McDade E, Wang G, Xiong C, et al. (2018). Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: a longitudinal study. Lancet Neurol 17, 241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorno-Tempini ML, Brambati SM, Ginex V, Ogar J, Dronkers NF, Marcone A, Perani D, Garibotto V, Cappa SF, and Miller BL (2008). The logopenic/phonological variant of primary progressive aphasia. Neurology 71, 1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz J, Chen F, van Dorpe J, and Nitsch RM (2001). Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 293, 1491–1495. [DOI] [PubMed] [Google Scholar]
- Gratuze M, Leyns CEG, and Holtzman DM (2018). New insights into the role of TREM2 in Alzheimer’s disease. Mol Neurodegener 13, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RC, Schneider LS, Amato DA, Beelen AP, Wilcock G, Swabb EA, Zavitz KH, and Tarenflurbil Phase 3 Study Group (2009). Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA 302, 2557–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenamyre JT, Maragos WF, Albin RL, Penney JB, and Young AB (1988). Glutamate transmission and toxicity in Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 12, 421–430. [DOI] [PubMed] [Google Scholar]
- Gremer L, Schölzel D, Schenk C, Reinartz E, Labahn J, Ravelli RBG, Tusche M, Lopez-Iglesias C, Hoyer W, Heise H, et al. (2017). Fibril structure of amyloid-β(1-42) by cryo-electron microscopy. Science 358, 116–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, and Tanzi RE (2013). Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78, 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griciuc A, Patel S, Federico AN, Choi SH, Innes BJ, Oram MK, Cereghetti G, McGinty D, Anselmo A, Sadreyev RI, et al. (2019). TREM2 Acts Downstream of CD33 in Modulating Microglial Pathology in Alzheimer’s Disease. Neuron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimmer T, Tholen S, Yousefi BH, Alexopoulos P, Forschler A, Forstl H, Henriksen G, Klunk WE, Mathis CA, Perneczky R, et al. (2010). Progression of cerebral amyloid load is associated with the apolipoprotein E ε4 genotype in Alzheimer’s disease. Biol. Psychiatry 68, 879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Narasimhan S, Changolkar L, He Z, Stieber A, Zhang B, Gathagan RJ, Iba M, McBride JD, Trojanowski JQ, et al. (2016). Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J. Exp. Med. 213, 2635–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Noble W, and Hanger DP (2017). Roles of tau protein in health and disease. Acta Neuropathol. 133, 665–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Kaether C, Thinakaran G, and Sisodia S (2012). Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med 2, a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, and Golenbock DT (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 9, 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J-Y, Besser LM, Xiong C, Kukull WA, and Morris JC (2019). Cholinesterase Inhibitors May Not Benefit Mild Cognitive Impairment and Mild Alzheimer Disease Dementia. Alzheimer Dis Assoc Disord 33, 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanseeuw BJ, Betensky RA, Jacobs HIL, Schultz AP, Sepulcre J, Becker JA, Cosio DMO, Farrell M, Quiroz YT, Mormino EC, et al. (2019). Association of Amyloid and Tau With Cognition in Preclinical Alzheimer Disease: A Longitudinal Study. JAMA Neurol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara Y, McKeehan N, and Fillit HM (2019). Translating the biology of aging into novel therapeutics for Alzheimer disease. Neurology 92, 84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy JA, and Higgins GA (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. [DOI] [PubMed] [Google Scholar]
- Harrington C, Sawchak S, Chiang C, Davies J, Donovan C, Saunders AM, Irizarry M, Jeter B, Zvartau-Hind M, van Dyck CH, et al. (2011). Rosiglitazone does not improve cognition or global function when used as adjunctive therapy to AChE inhibitors in mild-to-moderate Alzheimer’s disease: two phase 3 studies. Curr Alzheimer Res 8, 592–606. [DOI] [PubMed] [Google Scholar]
- He Z, Guo JL, McBride JD, Narasimhan S, Kim H, Changolkar L, Zhang B, Gathagan RJ, Yue C, Dengler C, et al. (2018). Amyloid-β plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 24, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckmann BL, Teubner BJW, Tummers B, Boada-Romero E, Harris L, Yang M, Guy CS, Zakharenko SS, and Green DR (2019). LC3-Associated Endocytosis Facilitates β-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer’s Disease. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng T-C, et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley D, Raghavan N, Sperling R, Aisen P, Raman R, and Romano G (2019). Preliminary Results of a Trial of Atabecestat in Preclinical Alzheimer’s Disease. N. Engl. J. Med. 380, 1483–1485. [DOI] [PubMed] [Google Scholar]
- Hitt BD, Jaramillo TC, Chetkovich DM, and Vassar R (2010). BACE1−/− mice exhibit seizure activity that does not correlate with sodium channel level or axonal localization. Mol Neurodegener 5, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holth JK, Fritschi SK, Wang C, Pedersen NP, Cirrito JR, Mahan TE, Finn MB, Manis M, Geerling JC, Fuller PM, et al. (2019). The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science eaav2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Pitas RE, Kilbridge J, Nathan B, Mahley RW, Bu G, and Schwartz A L. (1995). Low density lipoprotein receptor-related protein mediates apolipoprotein E-dependent neurite outgrowth in a central nervous system-derived neuronal cell line. Proc. Natl. Acad. Sci. U.S.A. 92, 9480–9484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, Hager K, Andreasen N, Scarpini E, Liu-Seifert H, et al. (2018). Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N. Engl. J. Med. 378, 321–330. [DOI] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan L-L, et al. (2010). Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Liu C-C, Chen X-F, Zhang Y-W, Xu H, and Bu G (2015). Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Aβ metabolism in apoE4-targeted replacement mice. Mol Neurodegener 10, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y-WA, Zhou B, Wernig M, and Südhof TC (2017). ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Aβ Secretion. Cell 168, 427–441. e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudry E, Dashkoff J, Roe AD, Takeda S, Koffie RM, Hashimoto T, Scheel M, Spires-Jones T, Arbel-Ornath M, Betensky R, et al. (2013). Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci Transl Med 5, 212ra161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hummel A, Bi M, Ippati S, van der Hoven J, Volkerling A, Lee WS, Tan DCS, Bongers A, Ittner A, Ke YD, et al. (2016). No Overt Deficits in Aged Tau-Deficient C57Bl/6.Mapttm1 (EGFP)Kit GFP Knockin Mice. PLoS ONE 11, e0163236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JM, Cirrito JR, Restivo JL, Kinley RD, Sullivan PM, Holtzman DM, Koger D, Delong C, Lin S, Zhao L, et al. (2012). Emergence of a seizure phenotype in aged apolipoprotein epsilon 4 targeted replacement mice. Brain Res. 1467, 120–132. [DOI] [PubMed] [Google Scholar]
- Hurtado DE, Molina-Porcel L, Iba M, Aboagye AK, Paul SM, Trojanowski JQ, and Lee VM-Y (2010). A{beta} accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am. J. Pathol. 177, 1977–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh T-PV, Davis AA, Ulrich JD, and Holtzman DM (2017a). Apolipoprotein E and Alzheimer’s disease: the influence of apolipoprotein E on amyloid-β and other amyloidogenic proteins. J. Lipid Res. 58, 824–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh T-PV, Liao F, Francis CM, Robinson GO, Serrano JR, Jiang H, Roh J, Finn MB, Sullivan PM, Esparza TJ, et al. (2017b). Age-Dependent Effects of apoE Reduction Using Antisense Oligonucleotides in a Model of β-amyloidosis. Neuron 96, 1013–1023. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, and Lee VM-Y (2013). Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J. Neurosci. 33, 1024–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, et al. (2012). A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med 4, 147ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, and Holtzman DM (2013). Biomarker modeling of Alzheimer’s disease. Neuron 80, 1347–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, et al. (2018a). NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14, 535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Wiste HJ, Schwarz CG, Lowe VJ, Senjem ML, Vemuri P, Weigand SD, Therneau TM, Knopman DS, Gunter JL, et al. (2018b). Longitudinal tau PET in ageing and Alzheimer’s disease. Brain 141, 1517–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson GA, Maitland NJ, Wilcock GK, Craske J, and Itzhaki RF (1991). Latent herpes simplex virus type 1 in normal and Alzheimer’s disease brains. J. Med. Virol. 33, 224–227. [DOI] [PubMed] [Google Scholar]
- Jamieson GA, Maitland NJ, Wilcock GK, Yates CM, and Itzhaki RF (1992). Herpes simplex virus type 1 DNA is present in specific regions of brain from aged people with and without senile dementia of the Alzheimer type. J. Pathol. 167, 365–368. [DOI] [PubMed] [Google Scholar]
- Jaunmuktane Z, Mead S, Ellis M, Wadsworth JDF, Nicoll AJ, Kenny J, Launchbury F, Linehan J, Richard-Loendt A, Walker AS, et al. (2015). Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 525, 247–250. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Lee CYD, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, et al. (2008). ApoE promotes the proteolytic degradation of Abeta. Neuron 58, 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong D, Jansen R, Hoefnagels W, Jellesma-Eggenkamp M, Verbeek M, Borm G, and Kremer B (2008). No effect of one-year treatment with indomethacin on Alzheimer’s disease progression: a randomized controlled trial. PLoS ONE 3, e1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, et al. (2012). A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488, 96–99. [DOI] [PubMed] [Google Scholar]
- Ju Y-ES, Ooms SJ, Sutphen C, Macauley SL, Zangrilli MA, Jerome G, Fagan AM, Mignot E, Zempel JM, Claassen JAHR, et al. (2017). Slow wave sleep disruption increases cerebrospinal fluid amyloid-β levels. Brain 140, 2104–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, and Malinow R (2003). APP processing and synaptic function. Neuron 37, 925–937. [DOI] [PubMed] [Google Scholar]
- Kang J-E, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, and Holtzman DM (2009). Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, and Goate AM (2015). Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran E, and Hardy J (2014). A Critique of the Drug Discovery and Phase 3 Clinical Programs Targeting the Amyloid Hypothesis for Alzheimer Disease. Ann Neurol 76, 185–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran E, Mercken M, and De Strooper B (2011). The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 10, 698–712. [DOI] [PubMed] [Google Scholar]
- Katsimpardi L, Litterman NK, Schein PA, Miller CM, Loffredo FS, Wojtkiewicz GR, Chen JW, Lee RT, Wagers AJ, and Rubin LL (2014). Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science 344, 630–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290. e17. [DOI] [PubMed] [Google Scholar]
- Kheifets V, and Braithwaite SP (2019). Plasma-Based Strategies for Therapeutic Modulation of Brain Aging. Neurotherapeutics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd M (1963). Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature 197, 192–193. [DOI] [PubMed] [Google Scholar]
- Kim J, Jiang H, Park S, Eltorai AEM, Stewart FR, Yoon H, Basak JM, Finn MB, and Holtzman DM (2011). Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-β amyloidosis. J. Neurosci. 31, 18007–18012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Eltorai AEM, Jiang H, Liao F, Verghese PB, Kim J, Stewart FR, Basak JM, and Holtzman DM (2012). Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Aβ amyloidosis. J. Exp. Med. 209, 2149–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, and Lamb BT (2018). Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement (N Y) 4, 575–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoferle J, Yoon SY, Walker D, Leung L, Gillespie AK, Tong LM, Bien-Ly N, and Huang Y (2014). Apolipoprotein E4 produced in GABAergic interneurons causes learning and memory deficits in mice. J. Neurosci. 34, 14069–14078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, et al. (2017). The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar DKV, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, Lefkowitz A, McColl G, Goldstein LE, Tanzi RE, et al. (2016). Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med 8, 340ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie-Wolf A, et al. (2019). Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacosta A-M, Pascual-Lucas M, Pesini P, Casabona D, Pérez-Grijalba V, Marcos-Campos I, Sarasa L, Canudas J, Badi H, Monleón I, et al. (2018). Safety, tolerability and immunogenicity of an active anti-Aβ40 vaccine (ABvac40) in patients with Alzheimer’s disease: a randomised, double-blind, placebo-controlled, phase I trial. Alzheimers Res Ther 10, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor B, Segurado R, Kennelly S, Olde Rikkert MGM, Howard R, Pasquier F, Börjesson-Hanson A, Tsolaki M, Lucca U, Molloy DW, et al. (2018). Nilvadipine in mild to moderate Alzheimer disease: A randomised controlled trial. PLoS Med. 15, e1002660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lee SJ, Conway OJ, Jansen I, Carrasquillo MM, Kleineidam L, van den Akker E, Hernández I, van Eijk KR, Stringa N, Chen JA, et al. (2019). A nonsynonymous mutation in PLCG2 reduces the risk of Alzheimer’s disease, dementia with Lewy bodies and frontotemporal dementia, and increases the likelihood of longevity. Acta Neuropathol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, et al. (2001). Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293, 1487–1491. [DOI] [PubMed] [Google Scholar]
- Leyns CEG, Ulrich JD, Finn MB, Stewart FR, Koscal LJ, Remolina Serrano J, Robinson GO, Anderson E, Colonna M, and Holtzman DM (2017). TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. U.S.A. 114, 11524–11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyns CEG, Gratuze M, Narasimhan S, Jain N, Koscal LJ, Jiang H, Manis M, Colonna M, Lee VMY, Ulrich JD, et al. (2019). TREM2 function impedes tau seeding in neuritic plaques. Nat. Neurosci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao F, Hori Y, Hudry E, Bauer AQ, Jiang H, Mahan TE, Lefton KB, Zhang TJ, Dearborn JT, Kim J, et al. (2014). Anti-ApoE antibody given after plaque onset decreases Aβ accumulation and improves brain function in a mouse model of Aβ amyloidosis. J. Neurosci. 34, 7281–7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao F, Li A, Xiong M, Bien-Ly N, Jiang H, Zhang Y, Finn MB, Hoyle R, Keyser J, Lefton KB, et al. (2018). Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J. Clin. Invest. 128, 2144–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung W-S, Peterson TC, et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippens G, and Gigant B (2019). Elucidating Tau function and dysfunction in the era of cryo-EM. J. Biol. Chem. 294, 9316–9325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C-C, Zhao N, Fu Y, Wang N, Linares C, Tsai C-W, and Bu G (2017). ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron 96, 1024–1032. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, and Duff K (2012). Trans-synaptic spread of tau pathology in vivo. PLoS ONE 7, e31302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Ding L, Rovere M, Wolfe MS, and Selkoe DJ (2019). A cellular complex of BACE1 and γ-secretase sequentially generates Aβ from its full-length precursor. J. Cell Biol. 218, 644–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston G, Sommerlad A, Orgeta V, Costafreda SG, Huntley J, Ames D, Ballard C, Banerjee S, Burns A, Cohen-Mansfield J, et al. (2017). Dementia prevention, intervention, and care. Lancet. [DOI] [PubMed] [Google Scholar]
- Logovinsky V, Satlin A, Lai R, Swanson C, Kaplow J, Osswald G, Basun H, and Lannfelt L (2016). Safety and tolerability of BAN2401--a clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimers Res Ther 8, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez Lopez C, Tariot PN, Caputo A, Langbaum JB, Liu F, Riviere M-E, Langlois C, Rouzade-Dominguez M-L, Zalesak M, Hendrix S, et al. (2019). The Alzheimer’s Prevention Initiative Generation Program: Study design of two randomized controlled trials for individuals at risk for clinical onset of Alzheimer’s disease. Alzheimers Dement (N Y) 5, 216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovestone S, Boada M, Dubois B, Hüll M, Rinne JO, Huppertz H-J, Calero M, Andrés MV, Gómez-Carrillo B, León T, et al. (2015). A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 45, 75–88. [DOI] [PubMed] [Google Scholar]
- Lowe VJ, Lundt ES, Albertson SM, Przybelski SA, Senjem ML, Parisi JE, Kantarci K, Boeve B, Jones DT, Knopman D, et al. (2019). Neuroimaging correlates with neuropathologic schemes in neurodegenerative disease. Alzheimers Dement. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucey BP, Hicks TJ, McLeland JS, Toedebusch CD, Boyd J, Elbert DL, Patterson BW, Baty J, Morris JC, Ovod V, et al. (2018). Effect of sleep on overnight cerebrospinal fluid amyloid β kinetics. Ann. Neurol. 83, 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow E, von Bergen M, Biernat J, and Mandelkow E-M (2007). Structural principles of tau and the paired helical filaments of Alzheimer’s disease. Brain Pathol. 17, 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelli S, Corbo M, Iannuzzi F, Negri L, Blandini F, Nistico R, and Feligioni M (2018). The Involvement of Post-Translational Modifications in Alzheimer’s Disease. Curr Alzheimer Res 15, 313–335. [DOI] [PubMed] [Google Scholar]
- Martiskainen H, Herukka S-K, Stančáková A, Paananen J, Soininen H, Kuusisto J, Laakso M, and Hiltunen M (2017). Decreased plasma β-amyloid in the Alzheimer’s disease APP A673T variant carriers. Ann. Neurol. 82, 128–132. [DOI] [PubMed] [Google Scholar]
- Masters CL, and Selkoe DJ (2012). Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb Perspect Med 2, a006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, and Beyreuther K (1985). Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 82, 4245–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matarin M, Salih DA, Yasvoina M, Cummings DM, Guelfi S, Liu W, Nahaboo Solim MA, Moens TG, Paublete RM, Ali SS, et al. (2015). A genome-wide gene-expression analysis and database in transgenic mice during development of amyloid or tau pathology. Cell Rep 10, 633–644. [DOI] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N, Cullen NC, Andreasson U, Zetterberg H, and Blennow K (2019). Association Between Longitudinal Plasma Neurofilament Light and Neurodegeneration in Patients With Alzheimer Disease. JAMA Neurol 76, 791–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, et al. (2011). The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShane R, Areosa Sastre A, and Minakaran N (2006). Memantine for dementia. Cochrane Database Syst Rev CD003154. [DOI] [PubMed] [Google Scholar]
- Meyer P-F, Tremblay-Mercier J, Leoutsakos J, Madjar C, Lafaille-Maignan M-É, Savard M, Rosa-Neto P, Poirier J, Etienne P, Breitner J, et al. (2019). INTREPAD: A randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology 92, e2070–e2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middeldorp J, Lehallier B, Villeda SA, Miedema SSM, Evans E, Czirr E, Zhang H, Luo J, Stan T, Mosher KI, et al. (2016). Preclinical Assessment of Young Blood Plasma for Alzheimer Disease. JAMA Neurol 73, 1325–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielke MM, Syrjanen JA, Blennow K, Zetterberg H, Vemuri P, Skoog I, Machulda MM, Kremers WK, Knopman DS, Jack C, et al. (2019). Plasma and CSF neurofilament light: Relation to longitudinal neuroimaging and cognitive measures. Neurology 93, e252–e260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min S-W, Cho S-H, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, et al. (2010). Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minter MR, Zhang C, Leone V, Ringus DL, Zhang X, Oyler-Castrillo P, Musch MW, Liao F, Ward JF, Holtzman DM, et al. (2016). Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci Rep 6, 30028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S, Blazey TM, Holtzman DM, Cruchaga C, Su Y, Morris JC, Benzinger TLS, and Gordon BA (2018). Longitudinal brain imaging in preclinical Alzheimer disease: impact of APOE ε4 genotype. Brain 141, 1828–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir RD, Lathe R, and Tanzi RE (2018). The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement 14, 1602–1614. [DOI] [PubMed] [Google Scholar]
- Morris JC (1997). Clinical dementia rating: a reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int Psychogeriatr 9 Suppl 1, 173–176; discussion 177-178. [DOI] [PubMed] [Google Scholar]
- Morris JC (2019). Editorial: Is Now the Time for Combination Therapies for Alzheimer Disease? J Prev Alzheimers Dis 6, 153–154. [DOI] [PubMed] [Google Scholar]
- Morris JC, Roe CM, Grant EA, Head D, Storandt M, Goate AM, Fagan AM, Holtzman DM, and Mintun MA (2009). Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch. Neurol. 66, 1469–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, and Mintun MA (2010). APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann. Neurol. 67, 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiek ES, and Holtzman DM (2015). Three dimensions of the amyloid hypothesis: time, space and “wingmen.” Nat. Neurosci. 18, 800–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najm R, Jones EA, and Huang Y (2019). Apolipoprotein E4, inhibitory network dysfunction, and Alzheimer’s disease. Mol Neurodegener 14, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Doré V, Fowler C, Li QX, Martins R, Rowe C, et al. (2018). High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 554, 249–254. [DOI] [PubMed] [Google Scholar]
- Namba Y, Tomonaga M, Kawasaki H, Otomo E, and Ikeda K (1991). Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 541, 163–166. [DOI] [PubMed] [Google Scholar]
- Nathan BP, Bellosta S, Sanan DA, Weisgraber KH, Mahley RW, and Pitas RE (1994). Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science 264, 850–852. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del Tredici K, et al. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71, 362–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll JAR, Buckland GR, Harrison CH, Page A, Harris S, Love S, Neal JW, Holmes C, and Boche D (2019). Persistent neuropathological effects 14 years following amyloid-β immunization in Alzheimer’s disease. Brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nortley R, Korte N, Izquierdo P, Hirunpattarasilp C, Mishra A, Jaunmuktane Z, Kyrargyri V, Pfeiffer T, Khennouf L, Madry C, et al. (2019). Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak P, Zilka N, Zilkova M, Kovacech B, Skrabana R, Ondrus M, Fialova L, Kontsekova E, Otto M, and Novak M (2019). AADvac1, an Active Immunotherapy for Alzheimer’s Disease and Non Alzheimer Tauopathies: An Overview of Preclinical and Clinical Development. J Prev Alzheimers Dis 6, 63–69. [DOI] [PubMed] [Google Scholar]
- Nuriel T, Angulo SL, Khan U, Ashok A, Chen Q, Figueroa HY, Emrani S, Liu L, Herman M, Barrett G, et al. (2017). Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nat Commun 8, 1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okochi M, Tagami S, Yanagida K, Takami M, Kodama TS, Mori K, Nakayama T, Ihara Y, and Takeda M (2013). γ-secretase modulators and presenilin 1 mutants act differently on presenilin/v-secretase function to cleave Aβ42 and Aβ43. Cell Rep 3, 42–51. [DOI] [PubMed] [Google Scholar]
- Olsson M, Ärlig J, Hedner J, Blennow K, and Zetterberg H (2018). Sleep deprivation and cerebrospinal fluid biomarkers for Alzheimer’s disease. Sleep 41. [DOI] [PubMed] [Google Scholar]
- Ooms S, Overeem S, Besse K, Rikkert MO, Verbeek M, and Claassen JAHR (2014). Effect of 1 night of total sleep deprivation on cerebrospinal fluid β-amyloid 42 in healthy middle-aged men: a randomized clinical trial. JAMA Neurol 71, 971–977. [DOI] [PubMed] [Google Scholar]
- Ossenkoppele R, Pijnenburg YAL, Perry DC, Cohn-Sheehy BI, Scheltens NME,Vogel JW, Kramer JH, van der Vlies AE, La Joie R, Rosen HJ, et al. (2015). The behavioural/dysexecutive variant of Alzheimer’s disease: clinical, neuroimaging and pathological features. Brain 138, 2732–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowitzki S, Lasser RA, Dorflinger E, Scheltens P, Barkhof F, Nikolcheva T, Ashford E, Retout S, Hofmann C, Delmar P, et al. (2017). A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther 9, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou-Yang MH Kurz JE, Nomura T, Popovic J, Rajapaksha TW, Dong H, Contractor A, Chetkovich DM, Tourtellotte WG, and Vassar R (2018). Axonal organization defects in the hippocampus of adult conditional BACE1 knockout mice. Sci Transl Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovod V, Ramsey KN, Mawuenyega KG, Bollinger JG, Hicks T, Schneider T, Sullivan M, Paumier K, Holtzman DM, Morris JC, et al. (2017). Amyloid β concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement 13, 841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmqvist S, Janelidze S, Stomrud E, Zetterberg H, Karl J, Zink K, Bittner T, Mattsson N, Eichenlaub U, Blennow K, et al. (2019). Performance of Fully Automated Plasma Assays as Screening Tests for Alzheimer Disease-Related β-Amyloid Status. JAMA Neurol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panza F, Lozupone M, Solfrizzi V, Sardone R, Piccininni C, Dibello V, Stallone R, Giannelli G, Bellomo A, Greco A, et al. (2018). BACE inhibitors in clinical development for the treatment of Alzheimer’s disease. Expert Rev Neurother 18, 847–857. [DOI] [PubMed] [Google Scholar]
- Parhizkar S, Arzberger T, Brendel M, Kleinberger G, Deussing M, Focke C, Nuscher B, Xiong M, Ghasemigharagoz A, Katzmarski N, et al. (2019). Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 22, 191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoal TA, Mathotaarachchi S, Kang MS, Mohaddes S, Shin M, Park AY, Parent MJ, Benedet AL, Chamoun M, Therriault J, et al. (2019). Aβ-induced vulnerability propagates via the brain’s default mode network. Nat Commun 10, 2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters F, Salihoglu H, Rodrigues E, Herzog E, Blume T, Filser S, Dorostkar M, Shimshek DR, Brose N, Neumann U, et al. (2018). BACE1 inhibition more effectively suppresses initiation than progression of β-amyloid pathology. Acta Neuropathol. 135, 695–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontecorvo MJ, Devous MD, Kennedy I, Navitsky M, Lu M, Galante N, Salloway S, Doraiswamy PM, Southekal S, Arora AK, et al. (2019). A multicentre longitudinal study of flortaucipir (18F) in normal ageing, mild cognitive impairment and Alzheimer’s disease dementia. Brain 142, 1723–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler AM, Polydoro M, Maury EA, Nicholls SB, Reddy SM, Wegmann S, William C, Saqran L, Cagsal-Getkin O, Pitstick R, et al. (2015). Amyloid accelerates tau propagation and toxicity in a model of early Alzheimer’s disease. Acta Neuropathol Commun 3, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preische O, Schultz SA, Apel A, Kuhle J, Kaeser SA, Barro C, Gräber S, Kuder-Buletta E, LaFougere C, Laske C, et al. (2019). Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat. Med. 25, 277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, and Morris JC (1999). Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann. Neurol. 45, 358–368. [DOI] [PubMed] [Google Scholar]
- Price JL, McKeel DW, Buckles VD, Roe CM, Xiong C, Grundman M, Hansen LA, Petersen RC, Parisi JE, Dickson DW, et al. (2009). Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol. Aging 30, 1026–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purro SA, Farrow MA, Linehan J, Nazari T, Thomas DX, Chen Z, Mengel D, Saito T, Saido T, Rudge P, et al. (2018). Transmission of amyloid-β protein pathology from cadaveric pituitary growth hormone. Nature 564, 415–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Klyubin I, Cuello AC, and Rowan MJ (2018). NLRP3-dependent synaptic plasticity deficit in an Alzheimer’s disease amyloidosis model in vivo. Neurobiol. Dis. 114, 24–30. [DOI] [PubMed] [Google Scholar]
- Rajapaksha TW, Eimer WA, Bozza TC, and Vassar R (2011). The Alzheimer’s β-secretase enzyme BACE1 is required for accurate axon guidance of olfactory sensory neurons and normal glomerulus formation in the olfactory bulb. Mol Neurodegener 6, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Readhead B, Haure-Mirande J-V, Funk CC, Richards MA, Shannon P, Haroutunian V, Sano M, Liang WS, Beckmann ND, Price ND, et al. (2018). Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron 99, 64–82. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Langbaum JBS, Fleisher AS, Caselli RJ, Chen K, Ayutyanont N, Quiroz YT, Kosik KS, Lopera F, and Tariot PN (2011). Alzheimer’s Prevention Initiative: a plan to accelerate the evaluation of presymptomatic treatments. J. Alzheimers Dis. 26 Suppl 3, 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relkin NR, Thomas RG, Rissman RA, Brewer JB, Rafii MS, van Dyck CH, Jack CR, Sano M, Knopman DS, Raman R, et al. (2017). A phase 3 trial of IV immunoglobulin for Alzheimer disease. Neurology 88, 1768–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risacher SL, Kim S, Nho K, Foroud T, Shen L, Petersen RC, Jack CR, Beckett LA, Aisen PS, Koeppe RA, et al. (2015). APOE effect on Alzheimer’s disease biomarkers in older adults with significant memory concern. Alzheimers Dement 11, 1417–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh JH, Huang Y, Bero AW, Kasten T, Stewart FR, Bateman RJ, and Holtzman DM (2012). Disruption of the sleep-wake cycle and diurnal fluctuation of β-amyloid in mice with Alzheimer’s disease pathology. Sci Transl Med 4, 150ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg JB, Kaplitt MG, De BP, Chen A, Flagiello T, Salami C, Pey E, Zhao L, Ricart Arbona RJ, Monette S, et al. (2018). AAVrh.10-Mediated APOE2 Central Nervous System Gene Therapy for APOE4-Associated Alzheimer’s Disease. Hum Gene Ther Clin Dev 29, 24–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roses AD (1996). Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Annu. Rev. Med. 47, 387–400. [DOI] [PubMed] [Google Scholar]
- Ryan P, Xu M, Davey AK, Danon JJ, Mellick GD, Kassiou M, and Rudrawar S (2019). O-GlcNAc Modification Protects against Protein Misfolding and Aggregation in Neurodegenerative Disease. ACS Chem Neurosci 10, 2209–2221. [DOI] [PubMed] [Google Scholar]
- Saji N, Niida S, Murotani K, Hisada T, Tsuduki T, Sugimoto T, Kimura A, Toba K, and Sakurai T (2019). Analysis of the relationship between the gut microbiome and dementia: a cross-sectional study conducted in Japan. Sci Rep 9, 1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Ferris S, et al. (2014). Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley A, Thorpe JR, Serpell LC, et al. (2014). Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82, 1271–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano M, Bell KL, Galasko D, Galvin JE, Thomas RG, van Dyck CH, and Aisen PS (2011). A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology 77, 556–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato C, Barthélemy NR, Mawuenyega KG, Patterson BW, Gordon BA, Jockel-Balsarotti J, Sullivan M, Crisp MJ, Kasten T, Kirmess KM, et al. (2018). Tau Kinetics in Neurons and the Human Central Nervous System. Neuron 97, 1284–1298. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayed FA, Telpoukhovskaia M, Kodama L, Li Y, Zhou Y, Le D, Hauduc A, Ludwig C, Gao F, Clelland C, et al. (2018). Differential effects of partial and complete loss of TREM2 on microglial injury response and tauopathy. Proc. Natl. Acad. Sci. U.S.A. 115, 10172–10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, et al. (1999). Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177. [DOI] [PubMed] [Google Scholar]
- Schultz SA, Gordon BA, Mishra S, Su Y, Perrin RJ, Cairns NJ, Morris JC, Ances BM, and Benzinger TLS (2018). Widespread distribution of tauopathy in preclinical Alzheimer’s disease. Neurobiol. Aging 72, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (2019). Alzheimer disease and aducanumab: adjusting our approach. Nat Rev Neurol. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, and Hardy J (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serenó L, Coma M, Rodríguez M, Sánchez-Ferrer P, Sánchez MB, Gich I, Agulló JM, Pérez M, Avila J, Guardia-Laguarta C, et al. (2009). A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 35, 359–367. [DOI] [PubMed] [Google Scholar]
- Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, et al. (2016). The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537, 50–56. [DOI] [PubMed] [Google Scholar]
- Sha SJ, Deutsch GK, Tian L, Richardson K, Coburn M, Gaudioso JL, Marcal T, Solomon E, Boumis A, Bet A, et al. (2019). Safety, Tolerability, and Feasibility of Young Plasma Infusion in the Plasma for Alzheimer Symptom Amelioration Study: A Randomized Clinical Trial. JAMA Neurol 76, 35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, and Sabatini BL (2007). Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, et al. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, and Holtzman DM (2018). Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 18, 759–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, et al. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn PD, Tracy TE, Son H-I, Zhou Y, Leite REP, Miller BL, Seeley WW, Grinberg LT, and Gan L (2016). Acetylated tau destabilizes the cytoskeleton in the axon initial segment and is mislocalized to the somatodendritic compartment. Mol Neurodegener 11, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WM, Joshita S, Zhou Y, Ulland TK, Gilfillan S, and Colonna M (2018). Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J. Exp. Med. 215, 745–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, et al. (2010). The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS ONE 5, e9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosna J, Philipp S, Albay R, Reyes-Ruiz JM, Baglietto-Vargas D, LaFerla FM, and Glabe CG (2018). Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol Neurodegener 13, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA, Zhang Y, Spevak W, Lin J, Phan NY, et al. (2019). Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat Commun 10, 3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Rentz DM, Johnson KA, Karlawish J, Donohue M, Salmon DP, and Aisen P (2014). The A4 study: stopping AD before symptoms begin? Sci Transl Med 6, 228fs13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer P, Condic M, Herrmann M, Oberstein TJ, Scharin-Mehlmann M, Gilbert DF, Friedrich O, Grömer T, Kornhuber J, Lang R, et al. (2016). Amyloidogenic amyloid-β-peptide variants induce microbial agglutination and exert antimicrobial activity. Sci Rep 6, 32228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson D, Perry D, Bens C, Bain LJ, Berry D, Krams M, Sperling R, Dilts D, Luthman J, Hanna D, et al. (2015). Charting a path toward combination therapy for Alzheimer’s disease. Expert Rev Neurother 15, 107–113. [DOI] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, and Roses AD (1993). Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 1977–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Goedert M, Weisgraber KH, Dong LM, Jakes R, Huang DY, Pericak-Vance M, Schmechel D, and Roses AD (1994). Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 91, 11183–11186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, He G, Qing H, Zhou W, Dobie F, Cai F, Staufenbiel M, Huang LE, and Song W (2006). Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. U.S.A. 103, 18727–18732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, Wild EJ, Saft C, Barker RA, Blair NF, Craufurd D, Priller J, Rickards H, et al. (2019). Targeting Huntingtin Expression in Patients with Huntington’s Disease. N. Engl. J. Med. 380, 2307–2316. [DOI] [PubMed] [Google Scholar]
- Tang-Wai DF, Graff-Radford NR, Boeve BF, Dickson DW, Parisi JE, Crook R, Caselli RJ, Knopman DS, and Petersen RC (2004). Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology 63, 1168–1174. [DOI] [PubMed] [Google Scholar]
- Tariot PN, Lopera F, Langbaum JB, Thomas RG, Hendrix S, Schneider LS, Rios-Romenets S, Giraldo M, Acosta N, Tobon C, et al. (2018). The Alzheimer’s Prevention Initiative Autosomal-Dominant Alzheimer’s Disease Trial: A study of crenezumab versus placebo in preclinical PSEN1 E280A mutation carriers to evaluate efficacy and safety in the treatment of autosomal-dominant Alzheimer’s disease, including a placebo-treated noncarrier cohort. Alzheimers Dement (N Y) 4, 150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcw J, and Goate AM (2017). Genetics of β-Amyloid Precursor Protein in Alzheimer’s Disease. Cold Spring Harb Perspect Med 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal LJ, Ferris SH, Kirby L, Block GA, Lines CR, Yuen E, Assaid C, Nessly ML, Norman BA, Baranak CC, et al. (2005). A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology 30, 1204–1215. [DOI] [PubMed] [Google Scholar]
- Tzeng N-S, Chung C-H, Lin F-H, Chiang C-P, Yeh C-B, Huang S-Y, Lu R-B, Chang H-A, Kao Y-C, Yeh H-W, et al. (2018). Anti-herpetic Medications and Reduced Risk of Dementia in Patients with Herpes Simplex Virus Infections-a Nationwide, Population-Based Cohort Study in Taiwan. Neurotherapeutics 15, 417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, Stewart FR, Piccio L, Colonna M, and Holtzman DM (2014). Altered microglial response to Aβ plaques in APPPS1–21 mice heterozygous for TREM2. Mol Neurodegener 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich JD, Ulland TK, Mahan TE, Nyström S, Nilsson KP, Song WM, Zhou Y, Reinartz M, Choi S, Jiang H, et al. (2018). ApoE facilitates the microglial response to amyloid plaque pathology. J. Exp. Med. 215, 1047–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe R, Riviere M-E, Caputo A, Sovago J, Maguire RP, Farlow M, Marotta G, Sanchez-Valle R, Scheltens P, Ryan JM, et al. (2017). Active Aβ immunotherapy CAD106 in Alzheimer’s disease: A phase 2b study. Alzheimers Dement (N Y) 3, 10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga AW, Wohlleber ME, Giménez S, Romero S, Alonso JF, Ducca EL, Kam K, Lewis C, Tanzi EB, Tweardy S, et al. (2016). Reduced Slow-Wave Sleep Is Associated with High Cerebrospinal Fluid Aβ42 Levels in Cognitively Normal Elderly. Sleep 39, 2041–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, et al. (1999). Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741. [DOI] [PubMed] [Google Scholar]
- in t’ Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, Breteler MM, and Stricker BH (2001). Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N. Engl. J. Med. 345, 1515–1521. [DOI] [PubMed] [Google Scholar]
- Vellas B, Black R, Thal LJ, Fox NC, Daniels M, McLennan G, Tompkins C, Leibman C, Pomfret M, Grundman M, et al. (2009). Long-term follow-up of patients immunized with AN1792: reduced functional decline in antibody responders. Curr Alzheimer Res 6, 144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, Vieira-Saecker A, Schwartz S, Santarelli F, Kummer MP, et al. (2017). Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 552, 355–361. [DOI] [PubMed] [Google Scholar]
- Verges DK, Restivo JL, Goebel WD, Holtzman DM, and Cirrito JR (2011). Opposing synaptic regulation of amyloid-β metabolism by NMDA receptors in vivo. J. Neurosci. 31, 11328–11337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, Bu G, Frieden C, and Holtzman DM (2013). ApoE influences amyloid-β (Aβ) clearance despite minimal apoEM-β association in physiological conditions. Proc. Natl. Acad. Sci. U.S.A. 110, E1807–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermunt L, Sikkes SAM, van den Hout A, Handels R, Bos I, van der Flier WM, Kern S, Ousset P-J, Maruff P, Skoog I, et al. (2019). Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimers Dement. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeda SA, Plambeck KE, Middeldorp J, Castellano JM, Mosher KI, Luo J, Smith LK, Bieri G, Lin K, Berdnik D, et al. (2014). Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat. Med. 20, 659–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlad SC, Miller DR, Kowall NW, and Felson DT (2008). Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 70, 1672–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, Carlsson CM, Asthana S, Zetterberg H, Blennow K, et al. (2017). Gut microbiome alterations in Alzheimer’s disease. Sci Rep 7, 13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos SJ, Xiong C, Visser PJ, Jasielec MS, Hassenstab J, Grant EA, Cairns NJ, Morris JC, Holtzman DM, and Fagan AM (2013). Preclinical Alzheimer’s disease and its outcome: a longitudinal cohort study. Lancet Neurol 12, 957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voytyuk I, De Strooper B, and Chavez-Gutierrez L (2018). Modulation of γ- and β-Secretases as Early Prevention Against Alzheimer’s Disease. Biol. Psychiatry 83, 320–327. [DOI] [PubMed] [Google Scholar]
- Wagner SL, Rynearson KD, Duddy SK, Zhang C, Nguyen PD, Becker A, Vo U, Masliah D, Monte L, Klee JB, et al. (2017). Pharmacological and Toxicological Properties of the Potent Oral γ-Secretase Modulator BPN-15606. J. Pharmacol. Exp. Ther. 362, 31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LC, and Jucker M (2015). Neurodegenerative diseases: expanding the prion concept. Annu. Rev. Neurosci. 38, 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Najm R, Xu Q, Jeong D-E, Walker D, Balestra ME, Yoon SY, Yuan H, Li G, Miller ZA, et al. (2018). Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 24, 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Benzinger TL, Su Y, Christensen J, Friedrichsen K, Aldea P, McConathy J, Cairns NJ, Fagan AM, Morris JC, et al. (2016). Evaluation of Tau Imaging in Staging Alzheimer Disease and Revealing Interactions Between β-Amyloid and Tauopathy. JAMA Neurol 73, 1070–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, et al. (2001). A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 414, 212–216. [DOI] [PubMed] [Google Scholar]
- Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, and Malinow R (2010). Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci. 13, 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingarten MD, Lockwood AH, Hwo SY, and Kirschner MW (1975). A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U.S.A. 72, 1858–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West T, Hu Y, Verghese PB, Bateman RJ, Braunstein JB, Fogelman I, Budur K, Florian H, Mendonca N, and Holtzman DM (2017). Preclinical and Clinical Development of ABBV-8E12, a Humanized Anti-Tau Antibody, for Treatment of Alzheimer’s Disease and Other Tauopathies. J Prev Alzheimers Dis 4, 236–241. [DOI] [PubMed] [Google Scholar]
- Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, and Haass C (2006). Control of peripheral nerve myelination by the beta-secretase BACE1. Science 314, 664–666. [DOI] [PubMed] [Google Scholar]
- Wischik CM, Edwards PC, Lai RY, Roth M, and Harrington CR (1996). Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. U.S.A. 93, 11213–11218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak MA, Mee AP, and Itzhaki RF (2009). Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J. Pathol. 217, 131–138. [DOI] [PubMed] [Google Scholar]
- Xia W (2019). γ-Secretase and its modulators: Twenty years and beyond. Neurosci. Lett. 701, 162–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O’Donnell J, Christensen DJ, Nicholson C, Iliff JJ, et al. (2013). Sleep drives metabolite clearance from the adult brain. Science 342, 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe K, Laffan AM, Harrison SL, Redline S, Spira AP, Ensrud KE, Ancoli-Israel S, and Stone KL (2011). Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA 306, 613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Holth JK, Liao F, Stewart FR, Mahan TE, Jiang H, Cirrito JR, Patel TK, Hochgräfe K, Mandelkow E-M, et al. (2014). Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 211, 387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, Wozniak DF, Diamond MI, and Holtzman DM (2013). Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 80, 402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanamandra K, Jiang H, Mahan TE, Maloney SE, Wozniak DF, Diamond MI, and Holtzman DM (2015). Anti-tau antibody reduces insoluble tau and decreases brain atrophy. Ann Clin Transl Neurol 2, 278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanamandra K, Patel TK, Jiang H, Schindler S, Ulrich JD, Boxer AL, Miller BL, Kerwin DR, Gallardo G, Stewart F, et al. (2017). Anti-tau antibody administration increases plasma tau in transgenic mice and patients with tauopathy. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Zhou R, Zhou Q, Guo X, Yan C, Ke M, Lei J, and Shi Y (2019). Structural basis of Notch recognition by human γ-secretase. Nature 565, 192–197. [DOI] [PubMed] [Google Scholar]
- Ye L, Fritschi SK, Schelle J, Obermüller U, Degenhardt K, Kaeser SA, Eisele YS, Walker LC, Baumann F, Staufenbiel M, et al. (2015). Persistence of Aβ seeds in APP null mouse brain. Nat. Neurosci. 18, 1559–1561. [DOI] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea L-G, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, et al. (2013). Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N, Liu C-C, Van Ingelgom AJ, Linares C, Kurti A, Knight JA, Heckman MG, Diehl NN, Shinohara M, Martens YA, et al. (2018). APOE ε2 is associated with increased tau pathology in primary tauopathy. Nat Commun 9, 4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yang G, Guo X, Zhou Q, Lei J, and Shi Y (2019). Recognition of the amyloid precursor protein by human γ-secretase. Science 363. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV (2013). Cerebrovascular effects of apolipoprotein E: implications for Alzheimer disease. JAMA Neurol 70, 440–444. [DOI] [PMC free article] [PubMed] [Google Scholar]