

SUMMARY
Although cellular reprogramming enables the generation of new cell types for disease modeling and regenerative therapies, reprogramming remains a rare cellular event. By examining reprogramming of fibroblasts into motor neurons and multiple other somatic lineages, we find that epigenetic barriers to conversion can be overcome by endowing cells with the ability to mitigate an inherent antagonism between transcription and DNA replication. We show that transcription factor overexpression induces unusually high rates of transcription and that sustaining hypertranscription and transgene expression in hyperproliferative cells early in reprogramming is critical for successful lineage conversion. However, hypertranscription impedes DNA replication and cell proliferation, processes that facilitate reprogramming. We identify a chemical and genetic cocktail that dramatically increases the number of cells capable of simultaneous hypertranscription and hyperproliferation by activating topoisomerases. Further, we show that hypertranscribing, hyperproliferating cells reprogram at 100-fold higher, near-deterministic rates. Therefore, relaxing biophysical constraints overcomes molecular barriers to cellular reprogramming.
In Brief
Privileged populations of hypertranscribing, hyperproliferating cells (HHCs) reprogram at near-deterministic rates. By reducing sources of genomic stress, such as R-loops and DNA supercoiling, topoisomerases support HHCs to facilitate rapid cellular reprogramming of mouse and human fibroblasts to neural cells types with increased functional maturity.
Graphical Abstract
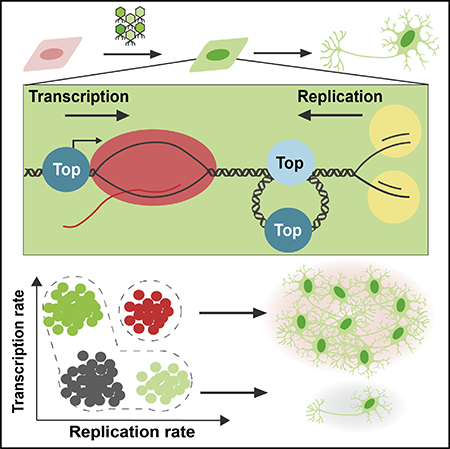
INTRODUCTION
Cellular reprogramming redirects the transcriptional state of a cell to a new fate (Xu et al., 2015). By supplying inaccessible somatic cell types in unique genomic contexts, transcription-factor-mediated reprogramming massively expands the potential for in vitro disease modeling (Ma et al., 2018; Shi et al., 2018, 2019; Wainger et al., 2014; Wen et al., 2014; Zhao et al., 2015). However, epigenetic barriers limit reprogramming between somatic lineages to rare events (Guo et al., 2014b; Lee et al., 2018; Son et al., 2011; Wapinski et al., 2013; Yoo et al., 2011; Zhou et al., 2008, 2016) and cause incomplete conversion of gene regulatory networks (GRNs) (Cahan et al., 2014). Efforts to identify epigenetic factors limiting reprogramming have focused primarily on induced pluripotent stem cell (iPSC) generation, and many of these findings are specific to iPSC reprogramming (dos Santos et al., 2014; Mor et al., 2018; Papp and Plath, 2013; Rais et al., 2013; Soufi et al., 2012).
We sought to identify universal roadblocks to reprogramming that extend beyond iPSCs into other lineages and define strategies to overcome them. To this end, we examined systems-level constraints limiting the conversion of fibroblasts into motor neurons, as well as other paradigms. We find that addition of the reprogramming factors sharply increases the transcription rate in cells and reduces the rate of DNA synthesis and cell division, highlighting the existence of trade-offs between transcription and cell replication during the conversion process. Most cells display either a high rate of transcription and limited proliferation or a high rate of proliferation and limited transcription, with both cell states being refractory to reprogramming. However, we identify a privileged population of cells capable of both high proliferation and high transcription rates that contribute to the majority of reprogramming events. This indicates that a high rate of proliferation is not sufficient for efficient reprogramming and that it must be coupled with high rates of transcription. Using genetic and chemical factors, we expand the hypertranscribing, hyperproliferating cell (HHC) population and achieve induced motor neuron reprogramming at near-deterministic rates. Importantly, this approach is effective across all starting and target cell types we tested. Transcription and DNA synthesis interfere directly through collisions of transcription and replication machinery, as well as indirectly by generating inhibitory DNA structures and topologies (e.g., R-loops and supercoiling). We identify topoisomerases as key regulators supporting the emergence and expansion of these privileged HHCs. By expanding the population of HHCs, we accelerate the maturation and reduce the heterogeneity of the resulting cells. Thus, relieving biophysical constraints governing transcription and replication overcomes the molecular barriers to reprogramming.
RESULTS
Transcription Factor Overexpression Induces Genomic Stress
We focused on the motor neuron lineage because it is a well-defined neuronal subtype with established markers. Utilizing mouse embryonic fibroblasts (MEFs) isolated from Hb9::GFP transgenic mice, we generated induced motor neurons (iMNs) by viral overexpression of six transcription factors (Ascl1, Brn2, Myt1l, Ngn2, Isl1, and Lhx3 [6F]; Son et al., 2011). We observed a large number of binucleated iMNs (~10%; Figure 1A), suggesting cell division and incomplete cytokinesis during reprogramming. Using longitudinal tracking from 1 to 8 days post-infection (dpi), we found that cells activated Hb9::GFP following division, definitively indicating that reprogramming cells do divide (Figure S1A; Video S1).
Figure 1. Genetic and Chemical Factors Relieve Genomic Stress and Reprogramming Block.

(A) Binucleated iMN at 14 dpi. Scale bar represents 10 μm.
(B) Mitotic cell with a micronucleus at 2 dpi. Arrow denotes micronucleus. Scale bar represents 5 μm. In Figure 1, mitotic cells were identified based on morphology of DAPI+ nuclei as described (Slattery et al., 2012).
(C) Percentage of mitotic anaphase-telophase cells with a micronucleus at 2 dpi. Anaphase-telophase cells with a non-integrated DNA fragment were scored as having micronuclei. n = 150–175 cells from 3–6 independent conversions per condition. Percentage ± 95% confidence interval is shown; chi-square test.
(D) Mitotic cell with a chromatin bridge at 4 dpi. Arrow denotes bridge. Scale bar represents 10 μm.
(E) Percent of mitotic anaphase-telophase cells with a chromatin bridge at 4 dpi. Anaphase-telophase cells with a DNA strand between daughter cells were scored as having a bridge. n = 63–100 cells from 3–6 independent conversions per condition. Percentage ± 95% confidence interval per condition is shown; chi-square test.
(F) Legend of genetic and chemical combinations used in conversion. 6F, 6 transcription factors only; 6FDD, 6 transcription factors and p53DD, a p53 mutant; 6FDDRR, 6 transcription factors and p53DD, hRasG12V, and RepSox.
(G) 6F iMNs, 14 dpi. Scale bar represents 100 μm.
(H) 6FDDRR-iMNs, 14 dpi. Scale bar represents 100 μm.
(I) iMN yield in 6F, 6FDD, or 6FDDRR conditions at 14 dpi. Conversion yield determined by counting Hb9::GFP+ cells with neuronal morphology divided by number of cells seeded is shown. n = 10–20 independent conversions per condition. Mean ± SEM; one-way ANOVA.
(J) Percentage of mitotic anaphase-telophase cells with a micronucleus at 2 dpi. n = 100 cells from 3 independent conversions per condition. Percentage ± 95% confidence interval is shown; chi-square test.
(K) Percentage of mitotic anaphase-telophase cells with a chromatin bridge at 4 dpi. n = 100 cells from 3 independent conversions per condition. Percentage ± 95% confidence interval is shown; chi-square test.
(L) Percentage of binucleated iMNs at 14 dpi; n = 6 independent conversions; mean ± SEM; unpaired t test.
Significance summary: p > 0.05 (ns); *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; and ****p ≤ 0.0001.
Impaired DNA replication can cause failed cytokinesis, chromatin bridges between separating nuclei, and micronuclei as the chromatin bridges resolve (Keszthelyi et al., 2016; Wang et al., 2010). Transduction with the iMN factors, but not a puromycin resistance gene (Control-Puro), induced DAPI+ micronuclei and chromatin bridges in ~30% of the mitotic anaphase-telophase cells at 2 and 4 dpi, respectively (Figures 1B–1E and S1B). Thus, cell division occurs during iMN reprogramming and transcription factor overexpression induces DNA replication stress.
Identification of a Genetic and Chemical Cocktail that Massively Increases Reprogramming
To identify factors that promote lineage conversion into somatic cell types, we screened small molecule kinase inhibitors, epigenetic modifiers, and oncogenes for the ability to increase the efficiency of MEF-to-iMN reprogramming. Suppression of Gatad2a-Mbd3/NuRD enables deterministic iPSC reprogramming (Rais et al., 2013; Mor et al., 2018). In partial agreement, Mbd3 suppression modestly increased iMN reprogramming (Figures S1C–S1E). However, unlike in iPSC studies, Gatad2a suppression did not increase iMN reprogramming (Figures S1F and S1G). Thus, Gatad2a-Mbd3/NuRD does not regulate iMN reprogramming as strongly as it regulates iPSC reprogramming.
A combination of RepSox, a transforming growth factor β (TGF-β) inhibitor (Ichida et al., 2009), a Ras mutant (hRasG12V; Kotsantis et al., 2016), and p53DD (DD), a p53 mutant lacking a DNA-binding domain (Bowman et al., 1996; 6FDDRR; Figure 1F), increased iMN reprogramming by 100-fold (Figures 1G–1I). In reprogramming cultures, DD, RepSox, and hRasG12V significantly reduced micronuclei, chromatin bridges, and binucleated iMNs (Figures 1J–1L). This suggests a strong correlation between reducing genomic stress and increased iMN formation.
Hypertranscription and Hyperproliferation Drive Neuronal Reprogramming
Transcription and DNA replication antagonize each other by increasing torsional strain and steric interference on genomic DNA (Keszthelyi et al., 2016; Kotsantis et al., 2016; Merrikh et al., 2011; Tuduri et al., 2009). Measuring 5-ethynyl uridine (EU) incorporation by fluorescence-activated cell sorting (FACS) at 2 dpi (Figure S2A) revealed that 6F transduction induced a significant increase in mean EU intensity in MEFs (Figure 2A). We defined the relative transcription rate as the mean EU incorporation of 6F-transduced MEFs relative to non-transduced MEFs on the same day. The transcription rate in 6Ftransduced MEFs increased 50% from 1 to 2 dpi (Figure 2B). Nuclear EU signal in 6F MEFs increased within and outside of nucleoli at 2 dpi compared to 1 dpi (Figures S2A and S2B), suggesting that both RNA-polymerase-I- and II-dependent transcription are elevated. Because this reflected an increased overall transcription rate in MEFs, we termed this a state of “hypertranscription.”
Figure 2. Hypertranscribing and Hyperproliferating Cells Drive Reprogramming.

(A) FACS plot showing relative EU incorporation in viable cells from 6F-infected MEF cultures at 1 and 2 dpi. Cell viability was determined by forward scatter (FSC) and side scatter (SSC) profiles in FACS analysis.
(B) Relative transcription rate measured by EU incorporation via flow cytometry at 1 and 2 dpi in 6F-infected MEFs compared to uninfected control. Mean EU intensity of non-transduced MEFs = 1. Only viable cells, determined by FSC and SSC profile via FACS, were analyzed. n = 5 independent transductions per condition. Mean ± SEM; unpaired t test.
(C) Schematic of CSFE-based flow sorting and replating of populations for reprogramming assays.
(D) CFSE intensity measured by flow cytometry at 4 dpi. “HyperP,” hyperproliferating cells, defined as cells showing a two-fold increase in division rate (an 8-fold decrease in CFSE intensity) compared to the average of Control-Puro MEFs.
(E) CFSE intensity measured by flow cytometry for Puro-infected cells (control), Ascl1-infected cells, or Brn2+Ascl1+Myt1l-infected cells (BAM) at 4 dpi.
(F) Effect of addition of Ascl1 or neuronal reprogramming factors BAM on the percentage of hyperproliferating cells measured by flow cytometry at 4 dpi. n = 3–6 independent transductions per condition. Mean ± SEM; one-way ANOVA.
(G) CFSE intensity measured by flow cytometry at 4 dpi with gates showing CFSE-low (HyperP) and CFSE-high. (H) Yield of iMNs from reprogramming populations sorted by CFSE intensity (CFSE-low and CFSE-high) at 4 dpi. Percent yield determined by counting total iMNs normalized by total number of cells counted per population at 4 dpi is shown. n = 4–23 independent conversions per condition. For 6F and 6FDD, median ± interquartile range and Mann-Whitney test between CFSE high and low groups in each transduction condition are shown. For 6FDDRR, mean ± SEM. Unpaired t test between CFSE high and low groups is shown.
(I) Schematic of CFSE-EU assay for measuring transcription and proliferation rates via flow cytometry at 4 dpi.
(J) Dot plot of CFSE intensity and fluorescently labeled EU for Control-Puro (gray), 6F (green), and 6FDDRR (red). Histograms of CFSE and EU intensity adjacent to dot plot are shown. Quadrant to demark HHCs set by reference to 6F condition is shown. Hyperproliferating and slow cycling cells set by selecting CFSE value in 6F condition to allow the dimmest 15% are shown. High EU values set by top half of 6F condition are shown, resulting in ~7% HHCs in 6F.
K) Relative transcription rate measured by EU incorporation via flow cytometry at 4 dpi of the whole population (all cells) of 6F-infected cells compared to hyperproliferative cells measured in 6F-infected MEFs. n = 10 independent transductions per condition. Mean ± SEM; one-way ANOVA.
(L) Percent relative transcription rate increase upon inhibition of DNA synthesis with aphidicolin treatment at 4 dpi. Relative transcription rate determined by difference between rates with and without aphidicolin treatment normalized to without for each transduction condition. n = 3 independent transductions per condition. Mean ± SEM; unpaired t test between with and without aphidicolin treatment for each transduction condition.
(M) Percentage of HHCs. n = 11–16 independent conversions per condition. Median ± interquartile range is shown; Kruskal-Wallis test.
(N) Yield of Hb9::GFP+ cells counted via flow cytometry at 17 dpi normalized to number of seeded cells. n = 7–8 independent conversions per condition. Mean ± SEM; unpaired t test.
(O) Yield of Hb9::GFP+ cells normalized to total cell number at 17 dpi. Cells were quantified via flow cytometry at 17 dpi. n = 7 or 8 independent conversions per condition. Mean ± SEM; unpaired t test.
(P) Schematic of CFSE-EU-pulse label assay to sort and label HHCs at 4 dpi followed by evaluation of Hb9::GFP intensity at 8 dpi.
(Q) Percentage of Hb9::GFP+ cells in 6FDDDR conditions for various gated populations. Cells gated for low EU intensity are shown. EU-low, cells with EU intensity in the lowest three quartiles, and EU-high, cells with EU intensity in the top quartile, at 8 dpi compared to all viable cells are shown, both EU-high and EU-low (all cells). Viable cells defined based on FSC and SSC profiles via FACS are shown. n = 7 or 8 independent conversions. Mean ± SEM; one-way ANOVA.
(R) Percentage of replated hyperproliferating cells in 6FDDRR conditions gated for high EU intensity (cells with EU intensity in the top quartile as measured by FACS) at 8 dpi. By definition, the whole viable population (all) contained 25% EU-Hi cells and Hb9::GFP+ and Hb9::GFP+ Bright cells (Hb9::GFP intensity in the top half of all viable Hb9+ cells) displayed enrichment of EU-high cells. n = 4–8 independent conversions. Median ± interquartile range is shown; Kruskal-Wallis test. Significance summary: p > 0.05 (ns); *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; and ****p ≤ 0.0001.
We next evaluated the impact of 6F transduction on cell proliferation. We measured cell proliferation by labeling MEFs with the stable dye CFSE (carboxyfluorescein succinimidyl ester) 24 h after transduction and flow sorting 72 h later (Figure 2C). Similar to published studies, we defined “hyperproliferation” or “fast-cycling” cells as those showing a two-fold increase in division rate (an eight-fold decrease in CFSE intensity) compared to control MEFs (Guo et al., 2014a). 6F transduction reduced the percentage of hyperproliferating cells ten-fold (Figure 2D). Staining for Ki67 at 4 dpi confirmed that 6F reduced the proliferative population and DDRR restored it (Figure S2C). DsRed retrovirus did not reduce proliferation, suggesting the effect was specific to transcription factors (Figure S2D). Ascl1 alone or Brn2, Ascl1, and Myt1l (BAM) also significantly reduced hyperproliferative cells (Figures 2E and 2F). Thus, transcription factor overexpression reduces cell proliferation.
DDRR greatly increased the number of hyperproliferating cells during iMN reprogramming (Figure 2G). We labeled reprogramming cultures with CFSE at 1 dpi, prospectively isolated hyperproliferative cells by FACS 72 h after CFSE labeling (at 4 dpi), and measured their ability to form iMNs by 14–17 dpi (Figure 2C). In 6F, 6F + DD (6FDD), and 6FDDRR conditions, cells that hyperproliferated from 1 to 4 dpi formed iMNs at higher rates (Figure 2H). Reducing cell division by MEF passaging, mitomycin C treatment, or p21 overexpression impaired iMN conversion (Figures S2E–S2H). Mitomycin C treatment at different time points indicated that cell division early in conversion promotes reprogramming (Figure S2G). DDRR did not reduce apoptosis during reprogramming (Figure S2I). Importantly, hyperproliferative cells only reprogrammed with substantially greater efficiency in the 6FDD and 6FDDRR conditions, suggesting that DDRR provided hyperproliferative cells with additional properties that enabled efficient reprogramming (Figure 2H).
We next measured cellular proliferation and transcription rates during reprogramming with DDRR (Figure 2I). We defined hypertranscribing cells as cells in the top half of EU intensity within the hyperproliferating population in the 6F condition (Figure 2J). Hyperproliferating cells displayed significantly reduced transcription levels when transduced with the six iMN factors (Figures 2J and 2K). Aphidicolin dramatically reduced the percent of cells in S phase as measured by EdU incorporation at 4 dpi while only slightly reducing cell count (Figures S2J–S2L), and this increased the transcription rates in Control-Puro and 6FDDRR cells (Figure 2L). Thus, transcription and DNA synthesis oppose each other during reprogramming.
DDRR increased the transcription rate of 6F-infected hyperproliferative cells, resulting in a larger population of HHCs (Figures 2J and 2M; hypertranscribing and hyperproliferating cells defined as above in Figures 2J and 2D, respectively). α-amanitin treatment at 4 dpi to reduce the average transcription rate in 6FDDRR cells did not reduce viability but significantly impaired reprogramming (Figures S2M–S2O). Conversely, overexpressing TATA-binding protein (TBP), which increases transcription in fibroblasts (Kotsantis et al., 2016), significantly increased iMN reprogramming in the 6F condition (Figure S2P). Thus, there is a strong correlation between hypertranscription, hyperproliferation, and the rate of iMN conversion, and DDRR increases HHCs after 6F infection.
Given the high density of Hb9::GFP+ cells in 6FDDRR conditions (Figures 1G and 1H), we improved our quantification of reprogramming efficiency by generating 6F or 6FDDRR iMNs without primary glia and exhaustively quantifying cell number by flow cytometry. Although 6F resulted in less than 10 iMNs per 100 MEFs plated, 6FDDRR yielded ~300 iMNs (Figure 2N). Without DDRR, 90% of cells failed to activate Hb9::GFP. With DDRR, 30% of cells activated Hb9::GFP (Figure 2O). Because HHCs represent 20%–30% of 6FDDRR cells (Figure 2M) and comprise the majority of reprogrammable cells (see Figures 2P–2R below), 30% of MEFs activating Hb9::GFP represent near-deterministic reprogramming of the HHC population. Thus, DDRR boosts reprogramming by increasing the number of cells capable of maintaining hypertranscription and hyperproliferation early in conversion.
To test whether HHCs identified at 4 dpi possess greater reprogramming potential relative to hyperproliferative cells with lower transcription rates, we prospectively isolated HHCs and non-HHCs (Figure 2P). We CFSE-labeled Hb9::GFP MEFs at 1 dpi, pulse-labeled cells with EU at 4 dpi prior to isolating CFSE-low, hyperproliferative cells by FACS, and replated these cells from 4 to 8 dpi. From 4 to 8 dpi, CFSE dimmed 10-fold and EU signal only dropped 10% (Figures S2Q and S2R). Using FACS at 8 dpi, we identified HHCs by high EU levels and analyzed them for Hb9::GFP expression (Figure 2P). We used cells with EU intensity in the top quartile of hyperproliferative cells to stringently examine hypertranscribing cells.
Over 40% of HHCs expressed Hb9::GFP at 8 dpi, although only 13% of non-hypertranscribing cells were Hb9::GFP+ (Figure 2Q). Thus, HHCs were 3 times more likely to activate Hb9::GFP relative to hyperproliferative but non-hypertranscribing cells. 90% of bright Hb9::GFP+ cells, which display better neuronal morphology and gene expression than low Hb9::GFP+ cells (Figures S2S, S2T, and 3D), had an EU intensity in the top quartile of all cells. Thus, of the Hb9::GFP+ cells that advanced to the terminal iMN stage, most originated from HHCs (Figure 2R). These prospective isolation studies indicate that HHCs possess significantly greater reprogramming potential than non-HHCs, and the inability of most cells to sustain hypertranscription and hyperproliferation early in conversion limits reprogramming to rare cells. Increasing the population of cells capable of mediating both processes improves reprogramming to near-deterministic rates.
Figure 3. Sustained Transgene Expression Differentiates Complete from Partial Reprogramming.

(A) Hb9::GFP+ cells with fibroblast (top) or neuronal (bottom) morphology at 17 dpi. Scale bars represent 20 μm.
(B) Percentage of Hb9::GFP+ cells of all viable cells measured by flow cytometry at 8 dpi. n = 6 independent conversions per condition. Mean ± SEM; one-way ANOVA.
(C) Percentage of Hb9::GFP+ cells with neuronal morphology of total Hb9::GFP+ cells at 17 dpi. n = 9 independent conversions per condition. Mean ± SEM; one-way ANOVA.
(D) Relative gene expression of cells collected at 14 dpi sorted based on No, Low, or Bright Hb9::GFP expression. Bright Hb9::GFP, cells in the top 50% of Hb9::GFP in the 6F condition. Gene expression was calculated based on qRT-PCR data. The expression level that was highest among the three conditions was set to 1 and used to normalize levels for the other two conditions. n = 2 independent experiments for each condition.
(E) Relative expression for single cells with either fibroblast (n = 16) or neuronal (n = 39) morphology for qPCR assays for endogenous Ngn2 and Isl1 and viral Isl1 (vIsl1). Median ± interquartile range is shown; Mann-Whitney test.
(F) Relative Isl1-GFP intensity in all viable cells (all) and HyperP infected with Isl1-GFP and 6F or 6FDDRR measured by flow cytometry at 4 dpi. n = 4–6 independent transductions per condition. Mean ± SEM; one-way ANOVA.
(G) Percentage of Isl1-GFP+ cells in all viable cells (all) and HyperP measured by flow cytometry at 4 dpi. Isl1-GFP+ determined by expression exceeding fluorescein isothiocyanate (FITC) values for untransfected cells is shown. n = 6 independent transductions per condition. Mean ± SEM; one-way ANOVA.
(H) Relative integrations of Isl1-GFP and NIL viruses in cells collected at 4 dpi. Relative integrations determined by qPCR are shown. Delta Ct of transgene calculated by difference of Ct between transgene and endogenous genomic region is shown. Relative integrations calculated by normalizing to NIL condition are shown. n = 3 independent transductions per condition. Mean ± SEM; one-way ANOVA.
Significance summary: p > 0.05 (ns); *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; and ****p ≤ 0.0001.
Sustained Transgene Expression Differentiates Complete from Partial Reprogramming
Previous work showed that components of the fibroblast GRN remain active within induced neurons (iNs) (Cahan et al., 2014; Morris et al., 2014). Potentially, mechanisms limiting reprogramming may arrest cells at intermediate states, leading to heterogeneous cultures composed of fully and partially neuronal cells.
Using live imaging, we identified a post-mitotic intermediate state characterized by Hb9::GFP reporter activation and retention of a fibroblast morphology (Figure 3A, top panel). This state frequently preceded Hb9::GFP+ iMN formation (Figure S3A), and 50% of Hb9::GFP+ cells remained trapped in this state with 6F alone (Figure S3B). Longitudinal tracking showed that, in the presence of DD, Hb9::GFP+ intermediates were four times more likely to fully convert into iMNs (Figure S3C; n = 65–80 cells in 6F and n = 1,200–1,400 in 6FDD). FACS purification of Hb9::GFP+ cells at 8 dpi showed that less than 1% of 6F cells activated Hb9::GFP, and 8% and 40% of 6FDD and 6FDDRR cells activated Hb9::GFP, respectively (Figure 3B). Additionally, although 50% of 6F Hb9::GFP+ cells remained trapped in the fibroblastic intermediate state, 90% of 6FDDRR Hb9::GFP+ cells became iMNs by 17 dpi (Figure 3C).
To identify transcriptional patterns that differentiate successful from unsuccessful reprogramming, we collected cells at 14 dpi, flow sorted based on Hb9::GFP+ into No, Low, and Bright Hb9::GFP populations, and performed qRT-PCR analysis. Cells lacking Hb9::GFP (No, top, gray) expressed high levels of a cluster enriched with fibroblast genes (cluster 1, left, gray; Figure 3D). Cluster 3 (left, bright green) genes were enriched with transgenes and neuronal markers and highly expressed in the Bright Hb9::GFP population (Bright, top, bright green; Figure 3D). Hb9::GFP Bright cells were more neuronal and showed sustained transgene expression compared to No and Low Hb9::GFP cells (Figures 3D and S3D), suggesting that the ability to sustain exogenous reprogramming transcription factor expression until 14 dpi is critical for reaching the Bright Hb9::GFP+ iMN state.
Single-cell qRT-PCR showed that iMNs (Figure 3A, bottom) displayed increased expression of neuronal markers relative to Hb9::GFP+ fibroblast-like intermediates (Figure S3E). Hb9::GFP+ fibroblasts did not show substantially more fibroblast gene expression than Hb9::GFP+ neurons, suggesting that activating neuronal gene expression rather than suppressing fibroblast expression was the limiting step in the Hb9::GFP+ intermediate stage (Figure S3E). In particular, high expression of transgenic and endogenous Isl1 differentiated neuronal from fibroblast morphologies (Figures 3E and S3F).
To examine transgene expression during reprogramming, we constructed an Isl1-GFP fusion construct. Isl1-GFP was insufficient to replace Isl1 in reprogramming, suggesting the fusion impacted Isl1 function (Figure S3G). Although Isl1-GFP intensity dropped in hyperproliferative cells in both 6F and 6FDDRR conditions (Figure 3F), DDRR doubled the percentage of hyperproliferative cells with detectable Isl1-GFP expression, suggesting DDRR could sustain transgene activation in hyperproliferative cells (Figure 3G). To evaluate how multiple virus expression varied in 6F and 6FDDRR, we used YFP- and DsRed-labeled viruses (Figures S3H and S3I). Although individual viruses showed high expression efficiency in 6F and 6FDDRR after single-virus infections (80%–90%; Figure S3H), the percentage of cells exhibiting detectable expression of both fluorescent proteins upon double infection was significantly higher in 6FDDRR than 6F (Figure S3I).
To measure transgenic integrations in a relevant context but eliminate the complexity of 6 individual transcription factors, we constructed a polycistronic cassette of Ngn2, Isl1, and Lhx3 (NIL). These factors reprogram embryonic stem cells (ESCs) to motor neurons (Mazzoni et al., 2013). NIL is sufficient to mediate reprogramming, and DDRR increased HHCs and reprogramming in this system (Figures S3J and S3K). At 4 dpi, NIL+DDRR did not have more integrations of the NIL cassette than NIL-alone cells (Figure 3H). Similarly, NIL+DDRR cells transduced with Isl1-GFP from the previous experiments did not contain more Isl1-GFP transgenes than NIL cells (Figure 3H). Thus, DDRR does not enable higher transgene expression by increasing the number of transgene integrations. Instead, DDRR enables higher levels of transgene expression in hyperproliferative cells, leading to efficient activation of Hb9::GFP and transition of partially reprogrammed intermediates to the neuronal state. Therefore, transgene expression levels in proliferating cells, rather than viral transduction rates, limit reprogramming.
Topoisomerases Enable Simultaneous Hypertranscription and Hyperproliferation in HHCs
To determine how DDRR enables combined hypertranscription and hyperproliferation, we performed RNA sequencing (RNA-seq) on single cells on a successful reprogramming trajectory by profiling hyperproliferative cells at 4 dpi (CFSE-low) and Hb9::GFP+ cells at 8 and 14 dpi (Figure 4A). 6F and 6FDDRR iMNs were similar to each other relative to MEFs and reprogramming cells, suggesting cells take similar trajectories to the iMN state in either condition (Figures 4A and 4B). At 4 dpi, 6F and 6FDDRR cells mapped to similar locations (Figure 4B). At 8 dpi, pseudotime analysis indicated more 6FDDRR cells were proximal to the iMN state than 6F cells (Figure 4C; note color scheme is consistent among Figures 4C and 4E–4H and distinct from that in Figure 4B). Thus, 6F and 6FDDRR cells traverse through a conserved trajectory, but DDRR increases reprogramming speed and efficiency.
Figure 4. Topoisomerase Expression Enables Cells to Exhibit Both Hyperproliferation and Hypertranscription.

(A) Schematic of populations collected across conversion and profiled via single-cell RNA-seq. Individual libraries were prepared for MEFs (1,357 cells), hyperproliferating cells (CFSE-low) for 6F (1,174 cells) and 6FDDRR (1,189 cells) collected at 4 dpi (6F 4 dpi and 6FDDRR 4 dpi), and Hb9::GFP+ cells for 6F (259 cells) and 6FDDRR (406 cells) at 8 dpi (6F 8 dpi and 6FDDRR 8 dpi) and 6F iMNs (1,863 cells) and 6FDDRR iMNs (2,869 cells) at 14 dpi (iMNs).
(B) t-distributed stochastic neighbor embedding (tSNE) projection of all cells mapped during reprogramming colored by condition.
(C) Distribution of pseudotime across cells in each condition.
(D) Relative UMI distribution across cells.
(E) Clustering of three cellular states across the tSNE projection.
(F) Relative expression of Col1a1, Mki67, Top2a, Top1, and Map2 over pseudotime. Colors correspond to states identified in (E).
(G) Violin plot of UMI (top, unique molecular identifiers) and relative Mki67 expression (bottom) for clusters identified in (E).
(H) Violin plot of relative expression of Top1 (top) and Top2a (bottom) for clusters identified in (E).
(I) Reads from Top1 and Top2a quantified by cell number normalized (CNN) RNA-seq at 4 dpi.
(J) Percentage of mitotic anaphase-telophase cells with a chromatin bridge at 4 dpi for 6FDDRR conditions. n = 3 independent conversions per condition,n = 50–70 cells per condition. Percentage ± 95% confidence interval; chi-square test.
(K) Percentage of HHCs in 6FDDRR conditions. n = 4–6 independent conversions per condition. Mean ± SEM; one-way ANOVA.
(L) Percentage of HHCs in 6FDDRR conditions treated for 18 h with camptothecin (Cpt) or doxorubicin (Doxo) prior to 4 dpi compared to DMSO control. n = 4 or 5 independent conversions per condition. Mean ± SEM; one-way ANOVA.
(M) Yield of iMNs in 6DDDRR conditions at 14 dpi. n = 7–9 independent conversions per condition. Mean ± SEM; one-way ANOVA.
(N) Yield of iMNs at 14 dpi in 6FDDRR conditions treated for 18 h with Cpt or Doxo prior to 4 dpi compared to DMSO control. n = 3 or 4 independent conversions per condition. Mean ± SEM; one-way ANOVA.
(O) Yield of iMNs at 14 dpi in 6FDD condition treated with RepSox with or without Top1 overexpression. n = 7–9 independent conversions per condition. Mean ± SEM; unpaired t test.
Significance summary: p > 0.05 (ns); *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; and ****p ≤ 0.0001.
We next examined the different single-cell states to identify transcriptional programs enabling combined hypertranscription and hyperproliferation (Figures 4D–4H). As expected, converting cells decreased collagen gene expression during transit to iMNs and increased Map2, a marker of post-mitotic neurons (Figure 4F). Monocle 2 clustered cells based on differentially expressed genes and aligned cells along a reprogramming trajectory. Most state 1 cells remained close to the starting fibroblasts, although some 6F iMN cells clustered into state 1 most likely based on sustained Col1a1 expression (Figure 4E). In contrast, state 2 cells possessed a proliferative signature and high expression of Mki67 (Figures 4F and 4G). Because about 80% of MEFs were Ki67+ by immunostaining (Figure S2B), the low level of Mki67 in state 1 is due to the limited sensitivity of single-cell RNA-seq and indicates that Mki67 increases in state 2 above levels observed in proliferating MEFs. State 2 was also enriched in unique molecular identifiers (UMIs), a proxy of total mRNAs (Figures 4D and 4G), signifying a putative HHC population.
State 2 cells showed increased expression of two topoisomerases (Figure 4H). Top1 expression increased at early stages and was sustained throughout reprogramming, and Top2a peaked as cells transitioned from fibroblasts (high Col1a1; low Map2) to iMNs (low Col1a1; high Map2; Figure 4F). Bulk RNA-seq at 4 dpi confirmed that DDRR increased Top1 and Top2a (Figure 4I).
Short hairpin RNAs (shRNAs) targeting either Top1 or Top2a (Figure S4A) increased genomic stress and reduced HHCs in 6FDDRR conditions (Figures 4J, 4K, S4B, and S4C). Transient inhibition of TOP1 or TOP2A by camptothecin or doxorubicin treatment, respectively, decreased the percentage of HHCs in 6FDDRR conditions (Figure 4L). Doxorubicin reduced active DNA synthesis to levels of irradiated MEFs (Figure S4D) and only impacted cell viability modestly relative to the drop in HHCs and proliferating cells (Figures 4L, S4D, and S4E). Consistent with HHCs comprising most of the reprogramming-competent cells, shRNA knockdown or chemical inhibition of either TOP1 or TOP2A resulted in a significant drop in iMN yield with 6FDDRR (Figures 4M, 4N, and S4F). Although overexpression of Top2a did not increase conversion (Figures S4G and S4H), Top2a is only transiently induced during 6FDDRR reprogramming (Figure 4F), and constitutive overexpression may prohibit iMN formation. Top1 overexpression significantly increased iMN conversion (Figures 4O and S4I). Thus, DDRR upregulates topoisomerases to promote HHC formation and enable highly efficient reprogramming.
DDRR and Topoisomerases Reduce Negative DNA Supercoiling and R-Loop Formation and Sustain Transcription in S Phase
Transcription and DNA replication increase positive and negative supercoiling in the genome (Ma and Wang, 2016). Negative supercoiling promotes R-loop formation, which in turn can stall DNA replication forks (Manzo et al., 2018; Gan et al., 2011). To investigate whether reprogramming perturbed supercoiling, we incubated cells with trimethylpsoralen (TMP), which preferentially intercalates into negatively supercoiled DNA (i.e., underwound) and signifies the amount of negative supercoiling in the genome (Teves and Henikoff, 2014; Naughton et al., 2013). Because DNA synthesis can influence DNA supercoiling (Kurth et al., 2013), we normalized DNA synthesis in Control-Puro, 6F, and 6FDDRR conditions before detecting supercoiling by using aphidicolin to inhibit DNA polymerases.
Bleomycin treatment, which causes DNA double-strand breaks and decreases DNA supercoiling (Naughton et al., 2013), decreased biotinylated TMP intercalation (Figures S5A and S5B). At 4 dpi, 6F MEFs showed increased biotinylated TMP intercalation compared to Control-Puro MEFs, indicating that transcription factor overexpression increased negative supercoiling (Figures 5A and 5B). Addition of DDRR to 6F normalized TMP incorporation (Figures 5A and 5B), and shRNA knockdown of Top1 or Top2a in 6FDDRR cells blocked this reduction (Figures 5C and S5C).
Figure 5. DDRR and Topoisomerase Expression Reduces Negative DNA Supercoiling and R-Loop Formation and Sustains Transcription in S Phase.

(A) Psoralen incorporation at 4 dpi. Scale bars represent 10 μm. Dotted white lines outline the nucleus.
(B) Mean intensity of biotinylated psoralen conjugated streptavidin-Alexa Fluor 594 at 4 dpi. Cultures treated with 1 μM aphidicolin for 2 h prior to collection at 4 dpi are shown. n = 42–130 cells from 3 independent conversions per condition. Median ± interquartile range is shown; Kruskal-Wallis test.
(C) Mean intensity of biotinylated psoralen conjugated streptavidin-Alexa Fluor 594 at 4 dpi in 6FDDRR conditions at 4 dpi. n = 99–162 cells from 3 independent conversions per condition. Median ± interquartile range is shown; Kruskal-Wallis test.
(D) Relative amount of DNA protected by exonuclease digestion in regions 500 bp upstream of transcription start sites for listed genes at 4 dpi. n = 4 independent transductions per condition per gene. Mean ± SEM; unpaired t test.
(E) R-loop immunostaining (S9.6) at 4 dpi. Scale bars represent 10 μm. Dotted white lines outline the nucleus.
(F) R-loop intensity per area at 4 dpi. n = 101–158 cells from 3 independent conversions per condition. Median ± interquartile range is shown; Kruskal-Wallis test.(G) R-loop intensity per area at 4 dpi in 6FDDRR conditions. n = 119–135 cells from 3 independent conversions per condition. Median ± interquartile range is shown; Kruskal-Wallis test.
(H) DNA fiber labeling scheme to identify progressing replication forks (red-green), stalled forks (red only), and new origins (green only).
(I) Relative number of stalled replication forks at 4 dpi. Stalled replication forks were quantified and normalized to all replicative fiber species to generate the percentage of stalled replication forks. n = 1,000 fibers per condition from 4 independent transductions. Percentage ± 95% confidence interval is shown; Fisher’s exact test.
(J) Relative number of new origins at 4 dpi. New origins were quantified and normalized to all replicative fiber species to generate the percentage of stalled replication forks. n = 1,000 fibers per condition from 4 independent transductions. Percentage ± 95% confidence interval is shown; Fisher’s exact test.
(K) Dot plot of EdU and active RNA polymerase II intensity at 4 dpi for Control-Puro (gray), 6F (green), and 6FDDRR (red). Gating to demark S phase cells with high active RNAPII (RNAPII Ser2p) is shown. S phase determined by intensity above EdU incorporation in non-proliferative, irradiated MEFs is shown (Figure S4H). High RNAPIISer2p, the top quartile of RNAPIISer2p intensity in Control-Puro infected cells in S phase cells.
(L) Percentage of cells in S phase with high RNAPII activity from area gated in (K) measured via flow cytometry at 4 dpi. Percentage relative to total viable cell population based on FSC and SSC profile via FACS is shown. n = 4 independent conversions per condition. Mean ± SEM; one-way ANOVA.
(M) Relative DNA synthesis rate of S phase cells at 4 dpi. Relative DNA synthesis rate determined by EdU intensity of S phase population normalized to EdU intensity of S phase population in Control-Puro condition is shown. n = 4 independent conversions per condition. Mean ± SEM; one-way ANOVA.
(N) Relative active RNAPII of S phase cells at 4 dpi. Relative active RNAPII rate in S phase cells determined by intensity of RNAPII Ser2p in S phase population normalized to intensity of RNAPII Ser2p in S phase population in Control-Puro condition is shown. n = 4 independent conversions per condition. Mean ± SEM; one-way ANOVA.
(O) Dot plot of EdU and active RNA polymerase II intensity at 4 dpi for 6FDDRR+Scrambled shRNA (gray), 6FDDRR+shTop2a (blue), and 6FDDRR+shTop1 (red) shRNAs. Gating to demark S phase cells with high active RNAPII (RNAPII Ser2p) is shown.
(P) Percentage of cells in S phase with high RNAPII activity from area gated in (O) at 4 dpi in 6FDDRR conditions. n = 4 independent conversions per condition. Mean ± SEM; one-way ANOVA.
Significance summary: p > 0.05 (ns); *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; and ****p ≤ 0.0001.
Transcription bubbles induce negative supercoiling upstream of the transcription start site (TSS) (Ma and Wang, 2016; Ma et al., 2013). TMP cross-linking protects negatively supercoiled genomic regions against digestion with exonuclease I and enables their quantification by qPCR (Teves and Henikoff, 2014). Indeed, in Control-Puro-infected cells at 4 dpi, the promoter region upstream of the Actb transcription start site was significantly more protected from exonuclease I digestion with TMP cross-linking than without (Figure S5D). At 4 dpi, 6FDDRR cells had significantly less negative supercoiling than 6F cells at the promoters of three genes with similar expression in 6F and 6FDDRR cells (Gapdh, Actb, and Sod1; Figures S5E and 5D). Chromatin immunoprecipitation sequencing (ChIP-seq) showed that RNAPII bound a greater fraction of TSS-proximal peaks in 6F compared to 6FDDRR (Figure S5F), suggesting that DDRR reduces RNAPII pausing. Thus, 6F transduction increases negative DNA supercoiling and DDRR reduces this in a topoisomerase-dependent manner.
Negatively supercoiled DNA and high transcription rates promote R-loops, hybrid structures formed between genomic DNA, and nascent transcripts that can impair DNA replication (Thomas et al., 1976; Aguilera and García-Muse 2012; Gan et al., 2011). Using an R-loop-specific antibody (S9.6; Boguslawski et al., 1986), we employed dot blot analysis (Figures S5G and S5H) and immunofluorescence (Figures 5E, 5F, and S5I–S5K) to quantify R-loops at 4 dpi in Control-Puro, 6F, and 6FDDRR conditions. As expected, RNase H reduced S9.6 signal intensity in lysates (Figure S5G) and non-nucleolar nuclear regions (Figures S5I and S5J). 6F cells showed increased R-loop formation compared to Control-Puro cells (Figures 5E, 5F, S5G, S5H, and S5K). DDRR reduced R-loop formation compared to 6F (Figures 5E, 5F, S5G, S5H, and S5K), and shRNA knockdown of Top1 or Top2a increased R-loops in 6FDDRR (Figures 5G and S5L).
To determine whether increased DNA supercoiling and R-loops after 6F transduction impedes DNA replication, we used DNA fiber labeling. Pulse labeling of IdU for 20 min followed by CldU for 30 min yields patterns of IdU and CldU marking progressing forks (IdU and CldU labeling), stalled forks (only IdU labeling), and new origins (only CldU labeling; Kotsantis et al., 2016; Nieminuszcy et al., 2016; Figure 5H). 6F increased stalled replication forks at 4 dpi, and DDRR mitigated this effect (Figures 5I and S5M, top). Additionally, 6FDDRR MEFs initiated more new replication origins than 6F cells (Figures 5J and S5M).
To measure transcriptional activity in S phase cells, we examined RNAPIISer2p levels by immunolabeling and DNA synthesis by EdU (Figure 5K). Aphidicolin-treated cells did not incorporate EdU and provided a negative control to gate S phase cells (Figure S5N). 6FDDRR cultures had 3-fold more high RNAPIISer2p S phase cells than Control-Puro and 6F (Figure 5L). Additionally, EdU and RNAPIISer2p intensity were higher in 6FDDRR compared to 6F (Figures 5M, 5N, S5O, and S5P). Knockdown of Top1, but not Top2a, reduced the percentage of S phase cells with high RNAPIISer2p (Figures 5O, 5P, and S5Q). Thus, 6F expression induces genomic stress by increasing negative DNA supercoiling, R-loop formation, and DNA replication fork stalling. DDRR rescues these stresses by activating topoisomerases.
Converting HHCs Adopt the iMN Transcriptional Program, Accelerating Maturation
To determine whether DDRR affects the resulting iMNs, we analyzed 6F, 6FDD, and 6FDDRR Hb9::GFP+ cells by RNA-seq. 6F and 6FDDRR Hb9::GFP+ cells were similar, although small variations differentiated them (Figures 6A–6C). Compared to 6F, 6FDDRR Hb9::GFP+ cells downregulated fibroblast genes and upregulated genes involved in neuron projection development and processes modulated by hRasG12V, such as apoptosis, cell cycle, and migration (Figures 6B and 6C). Thus, DDRR accelerates the shift toward the iMN profile generated by 6F.
Figure 6. Converting HHCs Adopt the Induced Motor Neuron Transcriptional Program and Accelerate Morphological Maturation.

(A) RNA-seq heatmap for Hb9::GFP+ cells at 17 dpi from different conditions compared to starting MEFs across the 1,186 genes that are differentially expressed between MEFs and Hb9::GFP+ cells. n = 3 independent conversions per condition.
(B) Volcano plot comparison of genes up- (blue) or downregulated (red) in Hb9::GFP+ cells at 17 dpi.
(C) List of gene ontology (GO) terms for genes upregulated (top, blue) or downregulated (bottom, red) in 6FDDRR cells compared to 6F at 17 dpi.
(D) tSNE projection of Hb9::GFP+ embryonic motor neurons (embMNs) collected at 12.5 dpi and iMNs generated by three different cocktails (6F, 6FDDRR, and 6FDDRR+Top1) colored by individual condition. embMNs were bioinformatically identified by Isl1 expression to distinguish from other Hb9::GFP+ populations.
(E) Relative expression colored by intensity of Col1a1, Isl1, Map2, and Chat over the populations in the tSNE in (D).
(F) Hb9::GFP+ iMNs immunostained for Map2 at 17 dpi. Scale bars represent 5 μm.
(G) Percentage of the Hb9::GFP+ cell population with neuronal gene expression profile at 17 dpi.
(H) Relative expression of neurosignaling genes (i.e., Scg2, Chgb, Sncg, and Snca) colored by intensity over the populations in the tSNE in (D).
(I) List of gene ontology (GO) terms for marker genes upregulated in iMN clusters.
(J) Percentage of multipolar iMNs derived from MEFs at 14 dpi. n = 6 or 7 independent conversions per condition. Mean ± SEM; unpaired t test.
(K) SFA ratio evoked action potentials (Aps) of mouse iMNs at 14 dpi. n = 7 or 8 cells from 3 independent conversions per condition. Median ± interquartile range is shown; Mann-Whitney test.
(L and M) Representative action potentials evoked in mouse iMNs by a positive current injection (indicated by solid bar across bottom) illustrating SFA over the course of the stimulus of iMNs in 6FDD (M) and 6F (L) conditions at 14 dpi.
Significance summary: p > 0.05 (ns); *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; and ****p ≤ 0.0001.
In single-cell RNA-seq analysis of primary embryonic motor neurons at embryonic day 12.5 (E12.5) and 6F, 6FDDRR, and 6FDDRR+Top1 iMNs, each iMN condition grouped into multiple clusters, each with a larger Map2+ population and a smaller Col1a1+ cluster (Figures 6D, 6E, S6A, and S6B). Immunostaining confirmed high MAP2 levels in Hb9::GFP+ cells (Figure 6F) and most cells grouped into the Map2+, neuronal population for each condition (Figure 6G). Small gene expression differences distinguished iMN clusters, including variations in neurosignaling and cell cycle (Figures 6H and 6I).
To determine whether expanding HHCs accelerates maturation, we examined morphological and electrophysiological properties. Mature spinal motor neurons are multipolar (Takazawa et al., 2012; Vrieseling and Arber, 2006). DD significantly increased the percentage of multipolar iMNs (Figures 6J and S6C). Upon repetitive stimulation, mature neurons display spike-frequency adaptation (SFA), increasing the time interval between spikes (Takazawa et al., 2012). Unlike 6F iMNs, several 6FDD iMNs achieved SFA, with an SFA ratio several-fold higher than 6F iMNs (Figures 6K–6M) and iPSC motor neurons with prolonged culture (Takazawa et al., 2012). Thus, expanding HHCs improves maturation.
Chemical and Genetic Factors that Increase HHCs Promote Reprogramming across Cell Types and Species
We next assessed the generality of inducing this HHC population in other reprogramming schemes. DD or DDRR increased reprogramming of MEFs into induced neurons via Ascl1, Brn2, and Mytl1L (Vierbuchen et al., 2010); induced dopaminergic neurons (iDANs) via Ascl1, Brn2, Mytl1L, Lmx1A, and FoxA2 (Pfisterer et al., 2011); and induced hair cells (iHCs) via Atoh1, Gata3, and Brn3C (Costa et al., 2015; Figures 7A–7D). The reprogramming increase into iMNs extended across age and species in the starting cells to include mouse adult tail tip fibroblasts and myoblasts (Figures 7E and 7F) and human adult fibroblasts (Son et al., 2011; Figure 7G). Although human fibroblasts reprogrammed less efficiently than mouse fibroblasts (compare Figure 7E versus 7G), the rates of HHC and iMN formation in the 6FDDRR condition were similar between mouse embryonic and adult fibroblasts (Figure 2M versus S7A and Figure 1I versus 7E). DD increased the percentage of multipolar human iMNs (Figures 7H and S7B) and resulted in faster sodium and potassium currents (Figures 7I, 7J, S7C, and S7D) and tighter, more mature action potentials (Figures 7K and 7L). Thus, HHCs promote reprogramming into post-mitotic lineages across age and species.
Figure 7. The DDRR Cocktail Boosts Reprogramming across Multiple Cell Types and Species.

(A) Yield of induced neurons for different conditions, including control with 3 factors (3F [Brn2, Ascl1, and Myt1l]), 3FDD, and 3FDDRR counted by MAP2+ cells at 17 dpi over number of cells seeded. n = 6 or 7 independent conversions per condition. Mean ± SEM; one-way ANOVA.
(B) Yield of induced dopaminergic neurons (iDANs) for different conditions, including control with 5 factors (5F [Brn2, Ascl1, Myt1l, Lmx1A, and FoxA2]), 5FDD, and 5FDDRR counted by MAP2+ cells at 17 dpi. n = 6–8 independent conversions per condition. Mean ± SEM; one-way ANOVA.
(C) Yield of induced inner ear hair cells (iHCs) for different conditions, including control with 3 factors (3F [Brn3C, Atoh1, and Gfi1]), 3FDD, and 3FDDRR counted by Atoh1::nGFP+ cells at 17 dpi. n = 3–16 independent conversions per condition. Mean ± SEM; one-way ANOVA.
(D) 3F-iHCs and 3FDD-iHCs immunostained with Myosin VIIa at 17 dpi. Scale bars represent 100 μm.
(E) Yield of iMNs generated from adult tail tip fibroblasts with 6F, 6FDD (both conditions with RepSox), and 6FDDRR at 28 dpi. n = 4–9 independent conversions per condition. Mean ± SEM; one-way ANOVA.
(F) Yield of iMNs generated from Hb9::GFP+ adult mouse muscle explants at 28 dpi. n = 4 or 5 independent conversions per condition. Mean ± SEM; unpaired t test.
(G) Yield of iMNs generated from human fibroblasts with factors alone (7F) or 7FDD (both conditions with RepSox), counted by MAP2+ cells at 35 dpi. n = 4–6 independent conversions per condition. Median ± interquartile range is shown; Mann-Whitney test.
(H) Percentage of multipolar iMNs derived from primary human fibroblasts at 35 dpi. n = 3 independent conversions per condition. Mean ± SEM; unpaired t test. (I and J) Step voltage depolarizations result in functional sodium and potassium channels in human 7F (I) or 7FDD-iMNs (J) at 35 dpi.
(K and L) Action potentials evoked by step current injection in current-clamp configuration for human 7F (K) or 7FDD-iMNs at 35 dpi (L).
(L) Model of topoisomerase-mediated reprogramming through hypertranscribing, hyperproliferating cells.
Significance summary: p > 0.05 (ns); *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; and ****p ≤ 0.0001.
DISCUSSION
We have identified combined hypertranscription and hyperproliferation as a central driver of reprogramming that is able to overcome molecular barriers to lineage conversion across multiple species and somatic cell states. Combined hypertranscription and hyperproliferation is rare because transcription and proliferation antagonize each other during reprogramming. Forced expression of the reprogramming transcription factors increases genomic stress in the form of R-loops, DNA torsion, and reduced processivity of DNA replication forks. Consequently, reprogramming remains restricted to rare cells with high transcriptional and proliferative capacity that reprogram at near-deterministic rates. By introducing chemical and genetic perturbations that mitigate antagonism by activating topoisomerases, we expand capacity for high rates of coincident transcription and proliferation and extend conversion to otherwise unreprogrammable cells (Figure 7M). Cells exhibiting combined hypertranscription and hyperproliferation are also capable of achieving greater functional maturity in the reprogrammed state, indicating that increasing the cell’s capacity to balance trade-offs during conversion can surmount maturity barriers. Our work suggests that the enhanced design of reprogramming vectors to account for limitations in cellular hardware may improve the predictability and determinism of reprogramming.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents used in this study should be directed to the Lead Contact, Dr. Justin Ichida (ichida@usc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines and Tissue Culture
HEK293, Plat-E, mouse embryonic fibroblasts, and primary human fibroblasts were cultured in DMEM supplemented with 10% FBS at 37°C with 5% CO2. Mouse tailtip fibroblasts were cultured in DMEM supplemented with 40% FBS at 37°C with 5% CO2. The following are the sex of primary human fibroblasts used in this study: Foreskin fibroblasts (BJ) – male, adult fibroblasts (GM05116) – female.
METHOD DETAILS
Isolating fibroblasts and cell culture
Hb9::GFP-transgenic mice (Jackson Laboratories) were mated with C57BL/6 mice (Jackson Laboratories) and MEFs were harvested from Hb9::GFP E12.5 – E13.5 embryos under a dissection microscope (Nikon SMZ 1500). To eliminate contaminating neurons, the head and spinal cord were removed. The fibroblasts were passaged at least once before being used for experiments. Neonatal mesenchymal cells were harvested by collagenase I digestion of hindlimb muscle. Human adult fibroblasts were obtained from Coriell (GM05116). Irradiated MEFs were obtained from GlobalStem (Cat. No: GSC-6001G).
Plasmid construction
Retroviral and lentiviral plasmids were constructed by Gateway and Gibson cloning into entry vectors pDONR221 or pENTR4-DS. Entry clones were recombined into destination vectors via LR reaction into the pMXS-DEST (retro) FUWO-tetO-DEST (lenti). For complete list of plasmids see Table of Reagents.
Viral transduction and iMN reprogramming
Retroviral transduction and iMN reprogramming from MEFs was performed as described. Briefly, retroviral transduction was performed using Plat-E retroviral packaging cells (Cell Biolabs, Inc., RV-101). MEFs were transduced twice with Ascl1, Brn2, Isl1, Lhx3, Myt1l, and Ngn2 or a polycistronic NIL construct at 48 and 72 hours after transfection. For human iMN experiments, retroviruses were generated in 293T cells and co-transfected with pLK and pHDMG packaging plasmids, and NEUROD1 was added to the reprogramming cocktail. In experiments in which iMN formation was quantified by microscopy or iMNs were functionally evaluated using electrophysiology, we added mixed glia isolated from P2 ICR mouse pups to infected fibroblasts 2 days after transduction. It is important to note that in other experiments, including those in which the number of hyperproliferating or hypertranscribing cells were analyzed, CFSE labeling was used, FACS sorting was used prior to iMN formation, or FACS sorting was used to quantify the number of iMNs out of total viable cells at the end of reprogramming, we omitted glia from the glia media added to the reprogramming cultures in order to avoid confounding our results. The day after glia media addition, medium was switched to complete N3 neuronal medium (DMEM/F-12 (VWR) with N2 and B27 (ThermoFisher) supplements and 1% glutamax (ThermoFisher)). Medium was supplemented with neurotrophics, GDNF, BDNF, and CNTF (R&D Systems) and FGF-Basic (Peprotech), each at 10 ng/ml. When included for reprogramming, RepSox (Selleck) was added to N3 media at a final concentration of 7.5uM.
iHC reprogramming
Retroviral transduction of MEFs was performed using Plat-E retroviral packaging cells (Cell Biolabs, Inc., RV-101). Atoh1::nGFP MEFs were transduced with Atoh1, Brn3c, and Gfi1 at 48 and 72 hours post-transfection. Two days after transduction, media was changed to induced hair cell media (DMEM/F-12 + N2 + B27) supplemented with FGF-Basic (Peprotech) and HB-EGF (Peprotech) and final concentrations of 2.5 ng/uL and 5 ng/uL, respectively.
iDAN and iN reprogramming
Retroviral transduction of non-transgenic or Tau::GFP MEFs was performed using Plat-E. MEFs were transduced with Ascl1, Brn2, Myt1l, FoxA2, and Lmx1a for iDAN reprogramming. Alternately, MEFs were transduced with Ascl1, Brn2, and Myt1l for iN reprogramming. Two days after transduction, mixed glia isolated from P2 ICR mouse pups were added to converting cultures. The next day, complete neuronal N3 media with neurotrophic factors (FGF, GDNF, CNTF, and BDNF at each at 10 ng/ml) was added to converting cultures.
shRNA-mediated knockdown of Mbd3, Gatad2a, Top1, Top2a
Mbd3, Gatad2a, Chd4, Top1 and Top2a shRNA lentiviral constructs were obtained from Sigma. Lentiviruses were generated in 293T cells and packaged via co-transfection with pPax2 as well as VSVG envelope using PEI transfection reagent. Hb9::GFP+ MEFs were co-transduced with scrambled, Mbd3, Gatad2a, Top1, or Top2a shRNAs during the second day of PlatE transduction with the motor neuron factors.
qRT-PCR quantification of shRNA-mediated Mbd3, Gatad2a, Top1, Top2a knockdown
For experiments measuring knockdown of Mbd3, Gatad2a, Top1 or Top2a, cells were collected 4 days after transduction with motor neuron factors and shRNAs. RNA isolation was performed using TRIzol LS reagent according to the manufacturer’s instructions (ThermoFisher Cat. No: 10296010). Reverse transcription of purified RNA was performed using random hexamer primers and New England Biolabs protoscript first strand cDNA synthesis (VWR Cat. No: 101640–908). qPCR was performed using primers for Mbd3, Gatad2a, Top1 and Top2a and iTaq universal SYBR green (Bio-Rad Cat. No: 1725125). The following primer sequences for endogenous Mbd3, Gatad2a, Top1 and Top2a genes were used:
Mbd3 (5′ – TCCAGGTCTCAGTGCAGGGA and 5′ – TGACTTCCTGGTGGGCTGCT), Gatad2a (5′ – AATAACGGGTCCTCACTACAG and 5′ – GTATTCTCGCTGTCGATCCA), Top1 (5′ – TCTCTAGTCCGCCACGAATTA and 5′ – CATCTCGAAGCCTCTTCAATGG) and Top2a (5′ – GCTCCTCGAGCCAAATCTGA and 5′ – CTACCTATAAAACTGGCTCCGT).
Quantification of Conversion Yield
All reprogrammed cultures were imaged using either the Biostation CT or Molecular Devices Image Express and manually quantified using Fiji. Yield of converted cells was calculated as the number of cells with the proper morphology and marker(s) on the final day of conversion over the number of cells seeded for conversion. For iMNs, the number of Hb9::GFP+ MEF- or explant-derived cells with neuronal morphologies was quantified between 14–17 dpi. In the single cell RNA sequencing experiments in Figure 4, iMNs were collected at 14 dpi. For iDANS and iNs, Tau::GFP+ or Map2+ cells with neuronal morphologies were manually quantified between 17dpi. For iHCs, the number of Atoh1::nGFP+ cells was used to quantify percent iHCs between 17 dpi. For iMNs, adult human fibroblast-derived Map2+/DsRed or p53DD-T2A-RFP+ double-positive cells with neuronal morphologies were manually quantified between 35 dpi.
Whole cell patch clamp electrophysiology
Whole cell membrane potential and current recordings in voltage- and current-clamp configurations were made using an EPC9 patch clamp amplifier controlled with PatchMaster software (HEKA Electronics). Voltage-and current-clamp data was acquired at 50kHz and 20kHz, respectively, with a 2.9kHz low-pass Bessel filter. For experiments, culture media was exchanged with warm extracellular solution consisting of (in mM): 140 NaCl, 2.8 KCl, 10 HEPES, 1 MgCl2, 2 CaCl2, and 10 glucose, with pH adjusted to 7.3 and osmolarity adjusted to 310 mOsm. Glass patch pipettes were pulled on a Narishige PC-10 puller and polished to 5–7 MΩ resistance. Pipettes were also coated with Sylgard 184 (Dow Corning) to reduce pipette capacitance. The pipette solution consisted of (in mM): 130 K-gluconate, 2 KCl, 1CaCl2, 4 MgATP, 0.3 GTP, 8 phosphocreatine, 10 HEPES, 11 EGTA, adjusted to pH 7.25 and 300 mOsm. Pipettes were sealed to cells in GΩ-resistance whole cell configuration, with access resistances typically between 10–20 MΩ, and leakage currents less than 100 pA. Capacitance transients were compensated automatically through software control. For current-voltage (IV) curves, cells were held in voltage clamp configuration at −70 mV and stepped through depolarizing voltages from −70 to 100 mV. A P/4 algorithm was used to subtract leakage currents from the traces. For action potential measurements, cells were held in current clamp configuration at 0 pA and action potentials were evoked by injecting positive depolarizing currents for 1 s. SFA ratios were calculated as the time interval between the first two APs evoked to the time interval between the last two APs evoked (35) using the lowest current injection that generated APs. Measurements were taken at room temperature (approximately 20–25°C). Data was analyzed and plotted in Igor Pro (WaveMetrics).
CFSE cell labeling to measure cellular proliferation
One day after retroviral infection, fibroblasts were labeled with CellTrace CFSE Cell Proliferation Kit GFP (Invitrogen, Cat. No: C34554) or Far Red (Invitrogen, Cat. No: C34572) at a final concentration of 10uM. Briefly, media was removed, CFSE added to the cells, and incubated at 37°C for 30 minutes. After incubation cells were washed once with PBS, then replaced with fresh media. Generally, cells were harvested for FACS sorting 72 hr following labeling without addition of glial cultures. Fast cycling cells were determined by examining the distribution from cells infected with reprogramming factors. During reprogramming, the dimmest 15% of cells in 6F conditions at 4 dpi were used to set the gate for fast-cycling cells. Cells with lower CFSE intensity were gated as fast-cycling. For all replating experiments, gates were set using the dimmest 15% of cells in 6F conditions. Generally, we found that the absolute CFSE intensity of the fast-cycling cells was 8-fold lower than mean CFSE of the entire population, indicating three more division over 72 hr. With a putative average 24 hr cell cycle, cells divide 3 times over 72 hours, while fast cells divide 6 times or more, suggesting a < 12 hr cell cycle.
Chromatin Immunoprecipitation (ChIP)-sequencing for RNA PoIII
One day after addition of N3 media (and without addition of glial cultures), cells were fixed by adding fresh formaldehyde to culture media at a 1:10 volume (11% final concentration) and fixed for 15 minutes at room temperature with agitation. Formaldehyde was quenched with by adding glycine solution to cells at 1:20 volume (2.5M final concentration) and incubated at room temperature for 5 minutes. Cells were then scraped from cell dish, collected into 1.5mL Eppendorf tubes, and kept on ice for the remainder of processing. Cells were spun down at 800×g for 10 minutes at 4°C. After pelleting, supernatant was removed and cells were resuspended in 1mL chilled 0.5% Igepal in PBS, triturating each cell sample by pipetting up and down several times. Samples were then spun down for 10 minutes at 800×g at 4°C. After spinning down, cells were again resuspended in 1mL 0.5% Igepal in PBS and 1uL PMSF was added (final concentration of 100mM). Cell pellets were then snap-frozen and stored at −80°C. Cells were processed by Active Motif via a standard ChIPseq protocol to enrich for RNAPII bound regions of DNA. Replicate RNA Pol II ChIP reactions were performed using 25 μg of primary MEF, 6F, and DDRR chromatin and 4 μg of Abflex RNAPII antibody (Active Motif, cat # 91151). Libraries were generated via standard Illumina protocols and sequenced to generate 30M reads per sample. The 75-nt sequence reads generated by Illumina sequencing (using NextSeq 500) are mapped to the mm10 genome using the BWA algorithm with default settings. Sicer was used to call peaks of enrichment resulting in 20,000 peaks per sample. Peaks called within 500 bp of a transcription start site were deemed “TSS-proximal peaks.”
Cleaved caspase-3, mKi67, RNA PolII immunolabeling for FACS sorting
One day after addition of N3 media (and without addition of glial cultures), cleaved caspase-3 and mKi67 labeling and subsequent FACS sorting for analysis was performed. Cells were trypsinized with 0.25% Trypsin-EDTA (Genesee Scientific), resuspended, and then spun down. Cells were then fixed in 4% paraformaldehyde for 15 minutes at room temperature in the dark. Cells were washed with PBS, pelleted, and permeabilized with 0.5% Triton X-100 for 15 minutes at room temperature in the dark. After permeabilization, cells were blocked in 3% FBS in PBS block solution for 30 minutes at room temperature in the dark with rotation. After being spun down, cells were then incubated in primary antibodies (1:200 dilution in 3% block solution) for 45 minutes at room temperature with rotation. Cells were washed with block solution, spun down, and then incubated in secondary antibodies (1:200 dilution in 3% block solution) for 30 minutes at room temperature in the dark with rotation. Cells were then washed in block solution, spun down, and resuspended in 150–200 μL PBS containing DAPI (100×) prior to FACS sorting and analysis. The following primary antibodies were used: rabbit anti-cleaved caspase-3 (abcam Cat No: ab13847), rabbit anti-Ki67 (GeneTex GTX16667 Cat. No: 89351–224) and rabbit anti-RNA polymerase II CTD repeat YSPTSPS (phospho S2) (abcam Cat No: ab5095).
DNA Fiber Assay
One day after addition of N3 media (and without glial cultures), cells were pulse-labeled with IdU (50 μM) and CIdU (100 μM final concentration) for 20 and 30 minutes, respectively at 37°C. Cells were washed with PBS, trypsinized with 0.25% Trypsin-EDTA and spun down. Cells were resuspended in 50 μL, put on ice, and resuspended to a concentration of 400 cells/μL in PBS. Three, 2 μL aliquots of each cell sample was spotted onto silane-coated slides and tilted to allow the cells to streak across the slide lengthwise. The cell preparations were dried for ~15–20 minutes and then lysed (1M Tris pH 7.4 + 0.5M EDTA + 10% SDS in ddH2O). DNA spreads were air-dried for 12 hours at room temperature and then fixed in methanol: acetic acid (3:1) for 2 minutes at room temperature. Slides were dried overnight at room temperature protected from light and then stored at −20°C for at least 24 hours before antibody labeling. The fiber spreads were treated with 2.5M HCl for 30 minutes and then blocked in 5% BSA for 30 minutes in “humidified chamber.” Fiber spreads were incubated with mouse α-BrdU (1:500, to detect IdU) and rat α-BrdU (1:500, to detect CIdU) primary antibodies for 1 hour at room temperature and then incubated for 15 minutes in stringency buffer (1M Tris pH 7.4 + 5M NaCl + 10% Tween + 10% NP40 in ddH2O). Slides were blocked again for 30 minutes and then incubated with rabbit α-mouse 594 (1:1000) and chicken α-rat 488 (1:750) secondary antibodies for 30 minutes at room temperature. After washes in 0.1% Tween in PBS, slides were blocked again at room temperature and then incubated with goat α-rabbit 594 (1:1000) and goat α-chicken 488 (1:750) tertiary antibodies for 30 minutes at room temperature. After a wash with 0.1% Tween in PBS followed by PBS washes, glass coverslips were mounted onto the silane slides using Antifade. The following primary antibodies were used: Monoclonal anti-IdU antibody produced in mouse (Sigma Cat. No: SAB3701448–100UG) and anti-BrdU antibody [BU1/75 (ICR1)] detects CIdU (abcam Cat. No: ab6326).
DNA-RNA Hybrid R-loop Staining and RNase Treatment
One day after addition of N3 media (and without addition of glial cultures), cells were fixed in 4% paraformaldehyde for one hour at 4°C in the dark. Cells were then permeabilized in 0.2% Triton X-100 in PBS for one hour in the dark. Coverslips were then split into two and 1 half was used for RNase H treatment. Briefly, coverslip halves were treated with 250 μL of 1X buffer + 2 μL RNase H at 37°C for 36 hours prior to proceeding with antibody labeling. Then, all coverslips were incubated in 2% BSA in PBS block solution for 1 hour at room temperature. Cells were then incubated in primary antibodies (1:1000 nucleolin to label nucleoli + 1:200 S9.6 to label DNA-RNA R-loops in 2% block solution) for 1 hour at room temperature followed by two PBS washes. Then, cells were stained with secondary antibodies (1:500 dilution in 2% block solution) for 2 hours at room temperature in the dark. After two PBS washes, cells were stained with Hoescht (1:1000) for 10 minutes at room temperature in the dark, washed again, and mounted onto glass slides using ImmuMount (ThermoFisher). The following primary antibodies were used: DNA-RNA R-loop S9.6 antibody (Kerafast Cat. No: ENH001), and nucleolin (abcam Cat. No: ab22758).
Dot Blot for R-loop Analysis
For each sample, genomic DNA was purified from one well of a 6-well dish using the DNeasy Kit from QIAGEN. Samples were eluted using 150 uls of elution buffer. Samples were then ethanol precipitated and resuspended in 7–10 uls of water. 1 ul of each sample was spotted onto a positively charged nylon membrane (GE Healtcare) and dried for 10 minutes before cross-linking by exposure to 254 nm light for 3 minutes. Membranes were then blocked with 5% milk/TBST (20 mM Tris-Hcl, 150 mM NaCl, 0.05% Tween 20, pH 7.5) for 1 hr at room temperature. If RNase H treatment was performed, the membrane was incubated in 11 mls of 1× RNase H buffer with 44 uls of RNase H (New England Biolabs, Cat. No: M0297L) at 37°C for 36 hours. Membranes were then washed twice with 5% milk/TBST. S9.6 (1:1000, Kerafast Cat. No: ENH001) or single-stranded DNA (1:10,000, Millipore Cat. No: MAB3868) antibodies were added in 1% BSA/TBST and incubated at 4°C overnight. For DNA that was going to be probed with the single-stranded DNA antibody, samples were heat denatured at 95°C for 10 minutes and snap-cooled on ice for 2 minutes prior to spotting on the membrane. Membranes were then washed twice with TBST and probed with an anti-mouse horseradish peroxidase-linked antibody (1:5,000, Cell Signaling Cat. No: 7076S) for one hour at room temperature. Membranes were exposed using either the Amersham ECL Western Blotting Detection Kit (GE Healthcare, Cat. No: RPN2108) or the SuperSignal West Femto Maximum Sensitivity Substrate (Thermofisher Scientific Cat. No: 34577).
EU Incorporation for FACS Sorting
One day after addition of N3 media (and without addition of glial cultures), EU incorporation assays were performed according to manufacturer’s instructions modified for FACS sorting (Invitrogen, Cat. No: C10330). Cells were incubated with 1mM EU for 1 hour at 37°C, washed once with PBS, dissociated with 0.25% Trypsin-EDTA (Genesee Scientific), resuspended, and then spun down. Cells were fixed with 3.7% PFA for 15 minutes at room temperature in the dark. Cells were then washed with PBS, pelleted, and then permeabilized with 0.5% Triton X-100 for 15 minutes at room temperature in the dark. After permeabilization, Click-iT reaction mix was added to each sample proceeded by incubation for 30 minutes at room temperature with rotation in the dark. Cells were then washed with Click-iT Reaction Rinse Buffer (Component F), pelleted, washed once with PBS, and then pelleted again. Cells were resuspended in N3 neuronal media containing DAPI (100×) and then FACS sorted.
EdU Incorporation for FACS Sorting
One day after addition of N3 media (and always omitting glia), EdU incorporation assays were performed according to manufacturer’s instructions (Invitrogen, Cat. No: C10424). Cells were incubated with 10uM EdU for 1 hour at 37°C, washed once with PBS, dissociated with 0.25% Trypsin-EDTA (Genesee Scientific), resuspended, and spun down. Cells were fixed in 100uL Click-iT fixative (Component D) and incubated for 15 minutes at room temperature in the dark. Cells were washed with 1% BSA in PBS, pelleted, and resuspended in 100uL of 1X Click-iT saponin-based permeabilization and wash reagent (Component E) for 15 minutes at room temperature in the dark. Cells were then incubated with Click-iT reaction cocktail for 30 minutes at room temperature in the dark with shaking. Cells were washed with 1X Click-iT saponin based permeabilization and wash reagent (Component E) and then pelleted. Cells were resuspended in N3 neuronal media or PBS containing DAPI (100×) for FACS sorting.
Flow cytometry and FACS analysis
Cells were harvested as previously described for each cell type with trypsin processing for MEFs and 4 dpi samples and DNaseI/Papain (Worthington Biochemical) processing for 8 dpi and iMN samples. Sorting of cells for analysis or collection was performed on an Aria I or Aria II (BD). Live single cells were identified by SSC and FSC gating and as DAPI negative. For fixed cells processed for CFSE-EU assays, cells were identified by SSC and FSC gating and DAPI staining was used to identify positive stained cells. Nonfluorescent controls were included to identify fluorescent populations. For multiple fluorophore experiments, single-labeled cell populations were included to allow proper compensation (e.g., EU-only, EdU-only, CFSE-only controls, primary antibody-only controls, non-labeled cells for CFSE-EU/EdU assays). Sample compensation was performed prior to other analyses. For all CFSE-EU assays, fast cycling cells were determined by gating the dimmest 15% of cells in 6F conditions at 4 dpi. Cells with lower CFSE intensity were gated as fast-cycling. From the fast-cycling population of cells, hypertranscribing cells were identified as the top 50% of the 6F only conditions.
Alpha-amanitin treatment for FACS and conversion
Converting cultures were treated with complete N3 media supplemented with water control or α-amanitin (1 μg/mL) at 3 dpi and transcription rate was measured by flow cytometry at 4 dpi using EU incorporation. For iMN conversion, cultures were treated complete N3 media supplemented with water control or α-amanitin (1 μg/mL) from 3–7 dpi, at which point cultures were maintained in complete N3 without water control or α-amanitin until 14–17 dpi.
Aphidicolin, camptothecin, doxorubicin treatment for FACS and conversion
Converting cultures were treated with complete N3 media supplemented with DMSO control, aphidicolin (1 μM) or doxorubicin (0.25 μM) at 3 dpi for 18 hours. Transcription rate was measured by flow cytometry using EU incorporation or DNA synthesis rate was measured by flow cytometry using EdU incorporation at 4 dpi. For iMN conversion, cultures were treated with complete N3 media supplemented with DMSO control, aphidicolin (1 μM), camptothecin (1 μM), or doxorubicin (0.25 μM) at 3 dpi for 18 hours, at which point cultures were maintained in complete N3 media without DMSO or small molecules until 14–17 dpi.
Quantification of anaphase-telophase chromatin bridges, micronuclei
For quantification of anaphase-telophase micronuclei or bridges, converting cultures grown on plastic coverslips were fixed with 4% paraformaldehyde at 2 or 4 dpi, respectively. Cells were then stained with DAPI (1:1000) for 10 minutes at room temperature in the dark. After mounting onto glass slides using ImmuMount (Thermo Scientific), cells were acquired on the Zeiss LSM 800 confocal microscope using a 40X objective. Anaphase-telophases with chromatin bridges or micronuclei were identified based on their DAPI profile as has been previously reported (Slattery et al., 2012; Broderick et al., 2015; Dykhuizen et al., 2013; Kotsantis et al., 2016). Anaphase-telophase cells with one or more non-integrated DNA fragments were determined as having micronuclei. Anaphase-telophase cells with one or more DNA strands between the separating/separated daughter cells were determined as having a bridge. The number of anaphase-telophase mitotic cells with chromatin bridges or micronuclei over all anaphase-telophases was recorded.
Quantification of multipolar neurons
Converted iMN cultures were imaged using the Molecular Devices Image Express at 14 dpi for mouse or at 35 dpi for human and manually quantified using Fiji. Cells expressing the proper marker(s), neuronal morphology, and at least 3 or more neurite processes were included in the quantification of percent multipolar neurons.
RNA Polymerase II + CFSE + EdU labeling for FACS analysis
For CFSE labeling, one day after retroviral infection, fibroblasts were labeled with CellTrace CFSE Cell Proliferation Kit Far Red (Invitrogen, Cat. No: C34572) at a final concentration of 10 μM as described above. For EdU labeling, cells were then incubated with EdU one day after addition of N3 media (without addition of glial cultures) also as described above. After a 30 minute incubation with Click-iT reaction mixture (using Alexa Fluor 594) followed by the wash with 1X Click-iT saponin based permeabilization and wash reagent (Component E), cells were then incubated in 3% FBS in PBS block solution for 30 minutes at room temperature with shaking. After spinning down and resuspending, cells were then incubated with primary antibody (1:200 dilution in 3% block solution) for 45 minutes at room temperature with rotation. Cells were washed with block solution, spun down, and incubated in secondary antibody (1:200 dilution of Alexa Fluor 488 in 3% block solution) for 30 minutes at room temperature in the dark with rotation. Cells were then washed in block solution, spun down, and resuspended in 150–200 μL PBS containing DAPI (100×) prior to FACS sorting and analysis. The following primary antibody was used: rabbit anti-RNA polymerase II CTD repeat YSPTSPS (phospho S2) (abcam Cat No: ab5095). The following secondary antibody was used: donkey anti-rabbit IgG highly cross-adsorbed secondary antibody, Alexa Fluor 488 (Thermo Fisher Cat. No: A-21206).
Genomic analysis of viral integrations
To analyze integration of viral constructs into cells during reprogramming, we collected three replicates of 40,000 cells at 4 dpi by trypsinization. To gather cells based on Isl1-GFP expression, populations were collected via FACS for high and low Isl1-GFP as well as gated for CFSE intensity (e.g., CFSE-Low for hyperproliferative populations, CFSE-High for slowly dividing cells). Following isolation, cells were pelleted and responded in Direct Lysis Buffer (Viagen) with 1 mg/mL Proteinase K (Viagen) and processed per manufacturer’s instructions. Briefly, cell solutions were incubated at 55°C for 45 minutes, followed by 85°C for 1 hour to inactivate Proteinase K. Cell extracts were diluted 1:3 in water. Relative number of integrations were analyzed by qPCR with iTaq Universal SYBR Green Supermix (Biorad) and primers specific for the native MALAT1 genomic region (primers: MALAT1-FWD: GGTTTCTCTCTCCCCTCCCT, MALAT1-REV: TTCGCATACGTGTGTCTGCT), Isl1-GFP transgene (primers: Isl1-GFP-FWD: AACA GCATGGTAGCCAGTCC, Isl1-GFP-REV: GCTGAACTTGTGGCCGTTTA), and Ngn2-F2A-Isl1 transgene (primers: Ngn2-F2A-Isl1 - FWD: GAGAAGCATCGTTATGCGCC, Ngn2-F2A-Isl1 -REV: TCCCATTGGACCTGGATTGC). Relative integrations were determined by calculating each samples delta CT for the transgenes relative to the native MALAT1 region and calculating 2 raised to the negative delta CT.
Single cell qPCR
Single iMNs of different morphologies were identified and isolated using an inverted microscope equipped with micromanipulator and micropipette. Cells were collected directly into 5 uL of CellsDirect 2XBuffer (Cells Direct One-step qRT-PCR kit, Thermo). Cells were processed using the manufacturer’s protocol for reverse transcription (RT) and specific target amplification (STA). cDNA was synthesized and pre-amplified from single-cell lysate. Single-cell qPCR was performed using the Fluidigm BioMark HD system on amplified cDNA templates, with primer and SsoFast EvaGreen supermix. Primers were validated in-house to yield efficient PCR amplification. A matrix of CTs and quality metrics was generated and extracted for each cell. Cells and genes were excluded for low-quality scores. In all, expression across 17 genes for fibroblast and neuronal markers was performed for 25 fibroblast-like cells and 36 neuronal cells. A profile of expression was generated for each cell using delta CTs normalized across total expression of the panel of genes. A heatmap was generated to visualize the profile of expression across the different gene sets and morphologies.
Single cell RNA-sequencing
Cells were harvested at different points in conversion. Specific populations were identified and collected via FACS and all cells were sorted to obtain viable single cell suspensions. Fast-cycling cells were identified by low CFSE intensity at 4 dpi. Hb9::GFP+ at 8 dpi and 14 dpi cells were identified relative to Hb9::GFP negative control. Cell suspensions were loaded into a chip and processed with the Chromium Single Cell Controller (10× Genomics). To generate single-cell gel beads in emulsion (GEMs), individual populations were assigned individual libraries using Single Cell 3′ Library and Gel Bead Kit V2 (10× Genomics, 120237). For each population, the target population size was between 1000–1500 cells. Cell suspensions were calibrated to capture the target number of cells. Fewer cells were captured at 8 dpi due to limited Hb9::GFP+ cells in 6F condition. RNA from lysed cells was barcoded through reverse transcription in individual GEMs. Barcoded cDNAs were pooled and cleanup by using DynaBeads® MyOne Silane Beads (Invitrogen, 37002D). Single-cell RNA-seq libraries were prepared using Single Cell 3′ Library Gel Bead Kit V2 (10× Genomics, 120237). Sequencing was performed with using multiple NextSeq 500/550 High Output Kit v2 on an Illumina NextSeq with pair end 150bp (PE150). On average, sequencing generated 100–200K reads per cell on average over the libraries.
Single Cell RNA-seq Analysis
Cluster analysis via Seurat
Analysis of embryonic motor neurons and induced motor neurons from various conditions was performed using Seurat 2.2. Following alignment and processing in CellRanger, variable genes were identified using FindVariableGenes. Clustering was performed using FindClusters based on the number of PCs identified through the PCElbow plot function. Cluster markers were identified for each cluster using the FindMarkers function.
Cluster analysis and pseudotemporal ordering via Monocle
We used the cellranger count pipeline (10× Genomics) to align and quantify single cell expression for each library. Software details available (Table S1). Samples were combined into a single matrix via the aggr pipeline and normalized by read depth across the libraries. scRNaseq datasets were imported into Monocle using cellrangerRkit in R to create a cellDataSet. Data were normalized using estimateSizeFactors. Outliers were removed based on variance using estimateDispersion to remove 108 outlier cells. Clustering was performed using 10,300 genes with high dispersion and mean gene expression > = 0.1 on the first 10 PCs. Clusters of varying number were examined and clustering via 3 primary clusters was chosen to capture different populations (e. g. MEFs, converting cells, and iMNs). Pseudotemporal ordering was performed using identified clusters. Pseudotemporal ordering was rooted in the identified iMN endpoint. To generate the pseudotime trajectory corresponding to reprogramming time, pseudotime was reversed to generate trajectory spanning MEFs at t = 0 and iMNs at t = 30 (end time). All subsequent pseudotime analyses were performed with the resulting cellDataSet.
Bulk RNA-sequencing and analysis
For cultures at 17 dpi, cells were harvest by DNase/papain (Worthington Chemical) treatment to dissociate cells. Cells were washed three times in DMEM-F12 media and resuspended in N3 neuronal media for sorting. Cells in replicates of 50K were collected based on gates set to identify viable, single Hb9::GFP+ cells. Following sorting, cells were spun and resuspended in 100 uL RLT buffer from the RNAeasy micro kit (QIAGEN). RNA in RLT and RNA extracted via RNAeasy kit were sequenced by Amaryllis (Emryville, CA) via single-end sequencing to generate 30M reads per sample. Additionally, Fastq files for previously acquired data for MEFs, embryonic motor neurons (embMNs), iPSC-derived MNs, and ES-derived MNs samples in duplicate were acquired and processed with newly generated datasets. Sequencing reads from triplicate or more replicates were trimmed and aligned to mm10 reference transcriptome with STAR aligner 2.5.3a. Gene counts quantified using annotation model (Partek E/M). Differentially expressed genes were identified using DEseq2 for with genome wide false discovery rate (FDR) of less than 0.05 and log2 fold change greater than 1. Comparison of MEFs with all MN samples generated 1186 DEGs. Heatmap analysis of MEFs and iMNs from different conditions was generated using this DEG set. Direct comparison of iMNs from 6F and DDRR conditions generated 756 DEGs. Metascape analysis (http://www.metascape.org) was used to generate GO terms for up and downregulated genes.
Cell number normalized (CNN) RNA-sequencing and analysis
For cultures at 4 dpi, cells were harvest by trypsin treatment to dissociate cells. Cells were washed three times in DMEM-F12 media and resuspended in N3 neuronal media for sorting. Cells in replicates of 50K were collected based on gates set to identify viable, fastcycling cells (e.g., CFSE-lo) or each condition (e.g., 6F and DDRR). Following sorting, cells were spun and resuspended in 100 uL RLT buffer from the RNAeasy micro kit (QIAGEN). To normalize to a standard number of cells, ERCC spike-in mix (Thermo-Fisher, 1ul at 1:100 dilution) was added to 50K cells in RLT. RNA in RLT and RNA extracted via RNAeasy kit, libraries were prepared by DNAlink (San Diego, CA) using SMARTer Stranded Total RNA-Seq Kit-Pico Input Mammalian (Clontech) and were sequenced using NextSeq 500 Mid-output 75PE (Illumina) to generate 30M reads per sample. Sequencing reads from triplicate or more replicates were trimmed and aligned to mm10 reference transcriptome with STAR aligner 2.5.3a. Gene counts quantified using annotation model (Partek E/M). Samples were aligned to ERCC spike-in reference to quantify total spike-in reads per sample. Sample reads were normalized by spike-in reads to generate cell number normalized reads per sample.
Biotinylated-trimethylpsoralen (bTMP) Immunofluorescence
One day after addition of N3 media (and without addition of glial cultures), cells were treated with 1 μM aphidicolin for 1.5–2 hours in N3 media. For control experiments, MEFs were treated with or without 100 μM bleomycin prior to incubation with psoralen. Then cells were incubated with 0.3 mg/mL EZ-Link Psoralen-PEG3-Biotin (Thermo Cat. No: 29986) for 15 minutes. Cultures were then exposed to 3kJ m-2 of 365nM light (Fotodyne UV Transilluminator 3–3000 with 15W bulbs) for 15 minutes at room temperature in the dark followed by 3 washes in PBS. Then cells were fixed with cold 70% ethanol for 30 minutes at 4°C followed by another 3 washes in PBS. Cells were then incubated with Alexa Fluor 594 Streptavidin (Thermo Cat. No: S32356) for one hour at room temperature in the dark, washed with PBS 3 times, and then stained with Hoescht (1:1000) for 10 minutes at room temperature in the dark. Coverslips were mounted onto glass slides using ImmuMount and imaged using the Zeiss LSM 800 confocal microscope.
Trimethylpsoralen-qPCR
Cell Harvest and DNA Extraction
One day after addition of N3 media (and without addition of glial cultures), cells were treated with 1 μM aphidicolin for 1.5–2 hours in N3 media. Cells were then trypsinized in 0.25% Trypsin-EDTA, spun down, and resuspended in complete N3 media + 1 μM aphidicolin + 2 μg/mL trimethylpsoralen (Sigma). 500 μL of control-puro was removed and saved for the no UV crosslinking control. Each 1mL of the remaining samples were added to individual wells of a 24-well plate and then exposed to 3kJ m-2 of 365nM light (Fotodyne UV Transilluminator 3–3000 with 15W bulbs) for 15 minutes at room temperature in the dark. Cells were then re-collected, spun at 1000 × g for 5 minutes, washed with 1mL PBS, and spun down again. Then cells were resuspended in 200 μL PBS and purified using QIAGEN DNeasy Blood and Tissue Kit with inclusion of an RNase A digestion (QIAGEN Cat. No: 69504). Samples were eluted once in 200 μL followed by a second elution in 100 μL of Buffer AE and eluates were then combined.
Sonication, Quantification, and Exonuclease Digestion
To achieve fragment sized of 100–500bp, each sample was sonicated in a Bioruptor for 30 s on/30 s off for 45 minutes on High. To ensure the same amount of DNA was then used for Exonuclease digestion, sample concentrations were quantified with a qPCR reaction. Briefly, samples were heat denatured at 95°C for 10 minutes, put on ice for 2 minutes, and then spun down briefly. For the qPCR reaction, 2 μL DNA for each sample was used in a 20 μL iTaq Universal SYBR Green Supermix (Biorad Cat. No: 1725125) reaction using primers −500bp upstream of the TSS for Actb. The qPCR results were used to determine the relative concentrations of each sample, using the least concentrated sample as the reference to adjust all other sample concentrations to. Samples were brought to a total volume of 280 μL after adjustment for DNA concentration and then heat denatured at 95°C for 10 minutes followed by 2 minute recovery on ice. Then, 240 μL of each sample was put into a new tube, saving the remaining undigested 40 μL of DNA at 4°C. The 240 μL samples were then heat denatured at 95°C for 10 minutes, incubated on ice for 2 minutes, and then briefly spun down. To each 240 μL sample, the following was added: 29 μL 10X ExonucleaseI buffer + 1 μL ExonucleaseI and samples were incubated at 37°C for one hour. Samples were then heat denatured at 95°C again, put on ice for 2 minutes, spun down, and another 10 μL of ExonucleaseI was added. After another 1 hour incubation at 37°C, samples were heated at 95°C for 10 minutes and put on ice for 2 minutes to stop the exonuclease reaction.
TMP-qPCR
The non-exonuclease digested samples were diluted 1:8 in Milli-Q water to a total volume of 320 μL. A qPCR reaction was then performed on both exonuclease digested and non-exonuclease digested samples with the upstream primers (−500bp from TSS) for several genes. Inclusion of non-exonuclease digested samples were used to normalize input levels for each exonuclease treated sample. Each biological sample was run in technical triplicate using 4 μL DNA per well in a 20 μL iTaq Universal SYBR Green Supermix reaction using the ViiA 7 Software. The following primers were used for qPCR quantification:
Actb
5′-GTCTCGGTTACTAGGCCTGC-3′
5′- ATCCACGTGACATCCACACC-3′
Gapdh
5′-GGTGAGATCAGTGAGGGGAG-3′
5′- CAAGAGGCTAGGGGCTTCC-3′
Sod1
5′-TCCGCATTTCCAGACACAGT-3′
5′- GAGCGGGGAAAGTCGCTATT-3′
Live imaging
Live imaging was carried out using a Nikon Biostation CT.
QUANTIFICATION AND STATISTICAL ANALYSIS
Sample numbers and experimental repeats are indicated in figure legends. Unless otherwise stated, data presented as mean ± SEM of at least three biological replicates. Significance determined by one-way ANOVA for multiple comparisons while an unpaired t test was used when comparing two datasets. If a dataset was non-normally distributed according to the D’Agostino & Pearson omnibus normality test, Kruskal-Wallis or Mann-Whitney testing was used for multiple comparisons or when comparing two datasets, respectively. Significance summary: p > 0.05 (ns), *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001.
DATA AND CODE AVAILABILITY
The datasets for RNA sequencing and RNAPII ChIP generated during this study are available at NCBI GEO: GSE134732.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Map2 Polyclonal Antibody | ThermoFisher | PA5–24589; RRID:AB_2542089 |
| p-Histone H3 antibody (C-2) | Santa Cruz | sc-374669; RRID:AB_11150094 |
| Cleaved Caspase-3 antibody | Abcam | ab13847; RRID:AB_443014 |
| Ki67 antibody | GeneTex | 89351–224 |
| RNA polymerase II CTD repeat YSPTSPS (phospho S2) | Abcam | ab5095; RRID:AB_304749 |
| DNA-RNA Hybrid [S9.6] antibody | Kerafast | ENH001; RRID:AB_2687463 |
| Nucleolin antibody | Abcam | ab22758; RRID:AB_776878 |
| Monoclonal anti-IdU antibody produced in mouse | Sigma | SAB3701448 |
| anti-BrdU antibody [BU1/75 (ICR1)] | Abcam | ab6326; RRID:AB_305426 |
| Single-stranded DNA antibody | Millipore | MAB3868; RRID:AB_570342 |
| Anti-mouse horseradish peroxidase-linked antibody | Cell Signaling | 7076S; RRID:AB_330924 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| RepSox | Selleck | S7223 |
| FGF-Basic (AA 1–155) Recombinant Human Protein | Peprotech | 100–18B |
| Recombinant Human GDNF | R&D Systems | 212-GD |
| Recombinant Human CNTF | R&D Systems | 257-NT |
| Recombinant Human BDNF | R&D Systems | 248-BD |
| Recombinant Human HB-EGF | Peprotech | 100–47 |
| a-Amanitin | Cayman Chemicals | CAS No 23109–05–9 |
| (+)-Aphidicolin | Cayman Chemicals | CAS No 38966–21–1 |
| Camptothecin | Cayman Chemicals | 11694 |
| Doxorubicin (hydrochloride) | Cayman Chemicals | CAS No 25316–40–9 |
| Trioxsalen (TMP) | Sigma | T6137 |
| Critical Commercial Assays | ||
| CellTrace CFSE Cell Proliferation Kit | ThermoFisher | Cat#C34570 |
| CellTrace Far Red Cell Proliferation Kit | ThermoFisher | Cat#C34572 |
| Click-iT® RNA Alexa Fluor® 594 Imaging Kit | ThermoFisher | Cat#C10330 |
| Click-iT® EdU Alexa Fluor® 647 Flow cytometry Kit | ThermoFisher | C10424 |
| Single Cell 3′ Library and Gel Bead Kit V2 | 10× Genomics | Cat#120237 |
| Chromium single cell chip kit V2 | 10× Genomics | Cat#120236 |
| RNeasy Micro Kit | QIAGEN | Cat#74004 |
| EZ-Link Psoralen-PEG3-Biotin | ThermoFisher | Cat#29976 |
| Amersham ECL Western Blotting Detection Kit | GE Healthcare | RPN2108 |
| SuperSignal West Femto Maximum Sensitivity Substrate | ThermoFisher | 34577 |
| Deposited Data | ||
| RNaseq and RNAPII ChIP | This study | GEO: GSE134732 |
| Experimental Models: Cell Lines | ||
| GM05116, Fibroblast from skin | Coriell Institute | GM05116 |
| BJ, Fibroblast from foreskin | ATCC | ATCC CRL-2522 |
| ND39024, Fibroblast from skin | NINDS Human Cell and Data Repository | ND39023 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J– Tg(Hb9::GFP) | Jackson Laboratory | stock # 005029 |
| Oligonucleotides | ||
| Top2a shRNA-B | Sigma-Aldrich | SHCLNG-NM_011623, TRCN0000070986, pJI.248 |
| Top2a shRNA-A | Sigma-Aldrich | SHCLNG-NM_011623, TRCN0000070987, pJI.245 |
| Top1 shRNA-B | Sigma-Aldrich | SHCLNG-NM_009408, TRCN0000011883, pJI.281 |
| Top1 shRNA-A | Sigma-Aldrich | SHCLNG-NM_009408, TRCN0000435820, pJI.285 |
| Mbd3 shRNA-A | Gift of Kevin Eggan | |
| Mbd3 shRNA-B | Sigma-Aldrich | SHCLNG-NM_013595, TRCN0000301772 |
| Gatad2a shRNA-A | Sigma-Aldrich | SHCLNG-NM_145598, TRCN0000376883 |
| Gatad2a shRNA-B | Sigma-Aldrich | SHCLNG-NM_145600, TRCN0000348687 |
| pLKO.1-TRC Control | Addgene | Addgene #10879, pJI.273 |
| Recombinant DNA | ||
| pMXS-p53DD-T2A-RFP | This paper | pJI.66 |
| pMXS-hRASV12 | This paper | pJI.271 |
| pMXS-Ascl1 | Gift courtesy of Kevin Eggan | pJI.175 |
| pMXS-Brn2 | Addgene | Addgene #32925, pJI.10 |
| pMXS-Myt1l | Addgene | Addgene #32926, pJI.3 |
| pMXS-Ngn2 | Addgene | Addgene #32928, pJI.6 |
| pMXS-Isl1 | Addgene | Addgene #32929, pJI.9 |
| pMXS-Lhx3 | Addgene | Addgene #32927, pJI.8 |
| pMXS-NeuroD1 | Gift courtesy of Kevin Eggan | pJI.2 |
| pMXS-p53DD | Addgene | Addgene #22729, pJI.333 |
| pSLIK-NEO+p53DD-T2A-mRFP | This paper | pJI.219 |
| pMXS-dsRed | This paper | pJI.99 |
| pMXS-Atoh1 | This paper | |
| pMXS-Brn3C | This paper | |
| pMXS-Gfi1 | This paper | |
| pMXS-FoxA2 | This paper | |
| pMXS-Lmx1a | This paper | |
| Hb9::RFP Lenti | Addgene | Addgene #37081, pJI.46 |
| pMXs-NIL | This paper | pJI.100 |
| ORF expression clone for TOP2A (Lentiviral Plasmid), 10ug purified, Ef1a promoter, IRES2-mcherry-IRES-puromyocin | GeneCopoeia | EX-E0236-Lv224 |
| ORF expression clone for TOP2A (Lentiviral Plasmid), 10ug purified, UBC promoter, IRES2-mcherry-IRES-puromyocin | GeneCopoeia | EX-E0236-Lv247 |
| Software and Algorithms | ||
| Cell Ranger 2.0.1 | 10× Genomics | https://support.10xgenomics.com/ |
| R 3.4.1 | R Project | https://www.r-project.org |
| Monocle 2.4.0 | Qiu et al., 2017 | https://github.com/cole-trapnell-lab/monocle-release |
| TopHat 2.1.1 | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat |
| Cufflinks 2.2.1 | Trapnell et al., 2013 | https://github.com/cole-trapnell-lab/cufflinks |
| STAR 2.5.1b | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
Highlights.
Chemical, genetic cocktail reduces genomic stress induced by TF reprogramming
DDRR cocktail expands population of hypertranscribing, hyperproliferating cells (HHCs)
Supported by topoisomerases, HHCs reprogram at near-deterministic rates
Topoisomerase expression reduces negative DNA supercoiling and R-loop formation
ACKNOWLEDGMENTS
We thank the NINDS Biorepository at Coriell Institute for the ND39023 cell line, members of the Ichida lab for useful feedback, the Choi Family Therapeutic Screening Facility for imaging, the USC Flow Cytometry Core, Dave Ruble and Gigi Ostrow of Children’s Hospital LA Center for Personalized Medicine for single-cell RNA-seq, and Paul Chastain of University of Illinois for assistance with DNA fiber analyses. K.N.B. was supported by CIRM Predoctoral Training Grant TG2–01161. K.E.G. was supported by Kirschstein-NRSA Postdoctoral Fellowship 5F32NS092417. This work was supported by NIH grants R00NS077435, R01NS097850, and 5R01DC015530; US Department of Defense grant W81XWH-15–1-0187; and grants from the Donald E. and Delia B. Baxter Foundation, the Alzheimer’s Drug Discovery Foundation and the Association for Frontotemporal Degeneration, the Harrington Discovery Institute, the Tau Consortium, the Pape Adams Foundation, the Frick Foundation for ALS Research, the Muscular Dystrophy Association, the John Douglas French Alzheimer’s Foundation, the Merkin Family Foundation, the New York Stem Cell Foundation, the USC Keck School of Medicine, the USC Broad Innovation Award, and the SC Clinical and Translational Science Institute to J.K.I. J.K.I. is a New York Stem Cell Foundation-Robertson Investigator and a Richard N. Merkin Scholar. K.K. and B.Z. are supported by the NIH grants NS090904 and NS100459 and the Foundation Leducq Transatlantic Network of Excellence for the Study of Perivascular Spaces in Small Vessel Disease reference no. 16 CVD 05.
Footnotes
DECLARATION OF INTERESTS
J.K.I. is a co-founder of AcuraStem and is bound by confidentiality agreements that prevent him from disclosing details of his financial interests in this work. Other authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.stem.2019.08.005.
REFERENCES
- Aguilera A, and García-Muse T (2012). R loops: from transcription byproducts to threats to genome stability. Mol. Cell 46, 115–124. [DOI] [PubMed] [Google Scholar]
- Boguslawski SJ, Smith DE, Michalak MA, Mickelson KE, Yehle CO, Patterson WL, and Carrico RJ (1986). Characterization of monoclonal antibody to DNA.RNA and its application to immunodetection of hybrids. J. Immunol. Methods 89, 123–130. [DOI] [PubMed] [Google Scholar]
- Bowman T, Symonds H, Gu L, Yin C, Oren M, and Van Dyke T (1996). Tissue-specific inactivation of p53 tumor suppression in the mouse. Genes Dev. 10, 826–835. [DOI] [PubMed] [Google Scholar]
- Broderick R, Nieminuszczy J, Blackford AN, Winczura A, and Niedzwiedz W (2015). TOPBP1 recruits TOP2A to ultra-fine anaphase bridges to aid in their resolution. Nat. Commun 6, 6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahan P, Li H, Morris SA, Lummertz da Rocha E, Daley GQ, and Collins JJ (2014). CellNet: network biology applied to stem cell engineering. Cell 158, 903–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A, Sanchez-Guardado L, Juniat S, Gale JE, Daudet N, and Henrique D (2015). Generation of sensory hair cells by genetic programming with a combination of transcription factors. Development 142, 1948–1959. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Santos RL, Tosti L, Radzisheuskaya A, Caballero IM, Kaji K, Hendrich B, and Silva JC (2014). MBD3/NuRD facilitates induction of pluripotency in a context-dependent manner. Cell Stem Cell 15, 102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykhuizen I, Georgescu RE, and O’Donnell ME (2013). A solution to release twisted DNA during chromosome replication by coupled DNA polymerases. Nature 496, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan W, Guan Z, Liu J, Gui T, Shen K, Manley JL, and Li X (2011). R-loop-mediated genomic instability is caused by impairment of replicatinon fork progression. Genes Dev. 25, 2041–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Zi X, Schulz VP, Cheng J, Zhong M, Koochaki SH, Megyola CM, Pan X, Heydari K, Weissman SM, et al. (2014a). Nonstochastic reprogramming from a privileged somatic cell state. Cell 156, 649–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Zhang L, Wu Z, Chen Y, Wang F, and Chen G (2014b). In vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an Alzheimer’s disease model. Cell Stem Cell 14, 188–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichida JK, Blanchard J, Lam K, Son EY, Chung JE, Egli D, Loh KM, Carter AC, Di Giorgio FP, Koszka K, et al. (2009). A small-molecule inhibitor of tgf-Beta signaling replaces sox2 in reprogramming by inducing nanog. Cell Stem Cell 5, 491–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keszthelyi A, Minchell NE, and Baxter J (2016). The causes and consequences of topological stress during DNA replication. Genes (Basel) 7, E134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzber SL (2013). TopHat2: accurate alignment of transcriptomes in the presecent of insertions, deletions and gene fusions. Genome Biology 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotsantis P, Silva LM, Irmscher S, Jones RM, Folkes L, Gromak N, and Petermann E (2016). Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun 7, 13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurth I, Georgescu RE, and O’Donnell ME (2013). A solution to release twisted DNA during chromosome replication by coupled DNA polymerases. Nature 496, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SW, Oh YM, Lu YL, Kim WK, and Yoo AS (2018). MicroRNAs overcome cell fate barrier by reducing EZH2-controlled REST stability during neuronal conversion of human adult fibroblasts. Dev. Cell 46, 73–84.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, and Wang MD (2016). DNA supercoiling during transcription. Biophys. Rev 8, 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Xie M, Laurent T, and Ding S (2013). Progress in the reprogramming of somatic cells. Circ. Res 112, 562–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Wert KJ, Shvartsman D, Melton DA, and Jaenisch R (2018). Establishment of human pluripotent stem cell-derived pancreatic β-like cells in the mouse pancreas. Proc. Natl. Acad. Sci. USA 115, 3924–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzo SG, Hartono SR, Sanz LA, Marinello J, De Biasi S, Cossarizza A, Capranico G, and Chedin F (2018). DNA topoisomerase I differentially modulated R-loops across the human genome. Genome Biol. 19, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni EO, Mahony S, Closser M, Morrison CA, Nedelec S, Williams DJ, An D, Gifford DK, and Wichterle H (2013). Synergistic binding of transcription factors to cell-specific enhancers programs motor neuron identity. Nat. Neurosci 16, 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrikh H, Machón C, Grainger WH, Grossman AD, and Soultanas P (2011). Co-directional replication-transcription conflicts lead to replication restart. Nature 470, 554–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mor N, Rais Y, Sheban D, Peles S, Aguilera-Castrejon A, Zviran A, Elinger D, Viukov S, Geula S, Krupalnik V, et al. (2018). Neutralizing Gatad2a-Chd4-Mbd3/NuRD complex facilitates deterministic induction of naive pluripotency. Cell Stem Cell 23, 412–425.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SA, Cahan P, Li H, Zhao AM, San Roman AK, Shivdasani RA, Collins JJ, and Daley GQ (2014). Dissecting engineered cell types and enhancing cell fate conversion via CellNet. Cell 158, 889–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naughton C, Avlonitis N, Corless S, Prendergast JG, Mati IK, Eijk PP, Cockroft SL, Bradley M, Ylstra B, and Gilbert N (2013). Transcription forms and remodels supercoiling domains unfolding large-scale chromatin structures. Nat. Struct. Mol. Biol 20, 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieminuszcy J, Schwab RB, and Niedzwiedz W (2016). The DNA fibre technique – tracking helicases at work. Methods 108, 92–98. [DOI] [PubMed] [Google Scholar]
- Papp B, and Plath K (2013). Epigenetics of reprogramming to induced pluripotency. Cell 152, 1324–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfisterer U, Kirkeby A, Torper O, Wood J, Nelander J, Dufour A, Björklund A, Lindvall O, Jakobsson J, and Parmar M (2011). Direct conversion of human fibroblasts to dopaminergic neurons. Proc. Natl. Acad. Sci. USA 108, 10343–10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rais Y, Zviran A, Geula S, Gafni O, Chomsky E, Viukov S, Mansour AA, Caspi I, Krupalnik V, Zerbib M, et al. (2013). Deterministic direct reprogramming of somatic cells to pluripotency. Nature 502, 65–70. [DOI] [PubMed] [Google Scholar]
- Shi Y, Lin S, Staats KA, Li Y, Chang WH, Hung ST, Hendricks E, Linares GR, Wang Y, Son EY, et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med 24, 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Hung ST, Rocha G, Lin S, Linares GR, Staats KA, Seah C, Wang Y, Chickering M, Lai J, et al. (2019). Identification and therapeutic rescue of autophagosome and glutamate receptor defects in C9ORF72 and sporadic ALS neurons. JCI Insight 5, 127736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery SD, Newberg JY, Szafran AT, Hall RM, Brinkley BR, and Mancini MA (2012). A framework for image-based classification of mitotic cells in asynchronous populations. Assay Drug Dev. Technol 10, 161–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son EY, Ichida JK, Wainger BJ, Toma JS, Rafuse VF, Woolf CJ, and Eggan K (2011). Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell 9, 205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A, Donahue G, and Zaret KS (2012). Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 151, 994–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takazawa T, Croft GF, Amoroso MW, Studer L, Wichterle H, and Macdermott AB (2012). Maturation of spinal motor neurons derived from human embryonic stem cells. PLoS ONE 7, e40154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves S, and Henikoff S (2014). Transcription-generated torsional stress destabilizes nucleosomes. Nat. Struct. Mol. Biol 21, 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M, White RL, and Davis RW (1976). Hybridization of RNA to double-stranded DNA: formation of R-loops. PNSA 73, 2294–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, and Pachter L (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol 31, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuduri S, Crabbé L, Conti C, Tourriére H, Holtgreve-Grez H, Jauch A, Pantesco V, De Vos J, Thomas A, Theillet C, et al. (2009). Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol 11, 1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Südhof TC, and Wernig M (2010). Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrieseling E, and Arber S (2006). Target-induced transcriptional control of dendritic patterning and connectivity in motor neurons by the ETS gene Pea3. Cell 127, 1439–1452. [DOI] [PubMed] [Google Scholar]
- Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SS, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, et al. (2014). Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LH, Mayer B, Stemmann O, and Nigg EA (2010). Centromere DNA decatenation depends on cohesin removal and is required for mammalian cell division. J. Cell Sci 123, 806–813. [DOI] [PubMed] [Google Scholar]
- Wapinski OL, Vierbuchen T, Qu K, Lee QY, Chanda S, Fuentes DR, Giresi PG, Ng YH, Marro S, Neff NF, et al. (2013). Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell 155, 621–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, Lin S, Shneider NA, Monaghan J, Pandey UB, et al. (2014). Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84, 1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Du Y, and Deng H (2015). Direct lineage reprogramming: strategies, mechanisms, and applications. Cell Stem Cell 16, 119–134. [DOI] [PubMed] [Google Scholar]
- Yoo AS, Sun AX, Li L, Shcheglovitov A, Portmann T, Li Y, Lee-Messer C, Dolmetsch RE, Tsien RW, and Crabtree GR (2011). MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 476, 228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Sagare AP, Ma Q, Halliday MR, Kong P, Kisler K, Winkler EA, Ramanathan A, Kanekiyo T, Bu G, et al. (2015). Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat. Neurosci 18, 978–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Brown J, Kanarek A, Rajagopal J, and Melton DA (2008). In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 455, 627–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang L, Vaseghi HR, Liu Z, Lu R, Alimohamadi S, Yin C, Fu JD, Wang GG, Liu J, and Qian L (2016). Bmi1 is a key epigenetic barrier to direct cardiac reprogramming. Cell Stem Cell 18, 382–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets for RNA sequencing and RNAPII ChIP generated during this study are available at NCBI GEO: GSE134732.