

Abstract
The sRNA Yfr1 and members of the Yfr2 sRNA family are almost universally present within cyanobacteria. The conserved motifs of these sRNAs are nearly complementary to each other, suggesting their ability to participate in crosstalk. The conserved motif of Yfr1 is shared by members of the Yfr10 sRNA family, members of which are otherwise less conserved in sequence, structure, and synteny compared to Yfr1. The different structural properties enable the discrimination of unique targets of Yfr1 and Yfr10. Unlike most studied regulatory sRNAs, Yfr1 gene expression only slightly changes under the tested stress conditions and is present at high levels at all times. In contrast, cellular levels of Yfr10 increase during the course of acclimation to darkness, and levels of Yfr2 increase when cells are shifted to high light or nitrogen limitation conditions. In this study, we investigated the targetomes of Yfr2, Yfr1, and Yfr10 in Prochlorococcus MED4, establishing CRAFD-Seq as a new method for identifying direct targets of these sRNAs that is applicable to all bacteria, including those that are not amenable to genetic modification. The results suggest that these sRNAs are integrated within a regulatory network of unprecedented complexity in the adjustment of carbon and nitrogen-related primary metabolism.
Subject terms: Small RNAs, Marine biology
Introduction
Small non-coding RNAs (sRNAs) are present within all domains of life and modulate the expression of target genes1. Compared to other regulators of gene expression, such as transcription factors, sRNAs exhibit some unique features with respect to regulatory dynamics, i.e., regulation by an sRNA leads to a faster decrease in the level of the protein product of the target gene than does regulation by a transcription factor2. Studies of sRNAs in bacteria and archaea show that they can regulate gene expression at the post-transcriptional level1, either by occluding or exposing ribosomal binding sites or ribonuclease cleavage sites1. Other distinct modes of sRNA action have been described that often require RNA chaperones such as Hfq or ProQ for their full functionality1,3,4.
In the most abundant photosynthetic organism on Earth5, the marine picocyanobacterium Prochlorococcus, sRNAs have been investigated since 20056 and until today more than twenty were discovered in the model strain Prochlorococcus marinus MED4 (Prochlorococcus MED4)7,8. In contrast to the reduction in the number of protein regulators associated with the genome streamlining observed in this clade, the number of sRNAs is not reduced in Prochlorococcus MED47. The relatively high number of sRNAs present in this bacterium may represent an adaptation to the extremely oligotrophic environment it inhabits, where nutrients are scarce and the synthesis of an RNA regulator requires fewer resources than that of a protein regulator7. Of the completely sequenced Prochlorococcus genomes, those of the low-light II/III ecotype are the only ones that do not contain yfr1 genes encoding cyanobacterial functional RNA 1 (Yfr1), which are otherwise ubiquitous in cyanobacteria9. Most yfr1 homologs are located between the genes encoding inosine-5′-monophosphate dehydrogenase (guaB) and thioredoxin (trxA)10. To date, only two porins, which originated from a gene duplication event, have been shown to be targets of Yfr1 in Prochlorococcus MED411. In addition to yfr1, the yfr2 sRNA family is similarly conserved and is present in all analysed cyanobacteria12. Unlike yfr1, yfr2 genes are present in multiple copies in cyanobacterial genomes and occur at unconserved loci12. Many Prochlorococcus strains possess two copies of yfr2, although four gene copies are present in Prochlorococcus MED4 (yfr2-5) and Synechococcus CC9611 harbours nine12. With the exception of tmRNA, the 6 S and 4.5 S sRNAs and RNase P RNA, which are universally conserved throughout all bacterial phyla, all other sRNAs identified in Prochlorococcus are restricted to the marine picocyanobacteria (Prochlorococcus and marine Synechococcus) and do not occur outside this group7,8.
While it appears that Prochlorococcus relies heavily on sRNAs to modulate gene expression, the transcription of sRNAs is also coordinated by sigma factors and transcription factors. Despite the reduced set of regulatory proteins harboured by Prochlorococcus MED4, this bacterium encodes representatives of a variety of regulatory proteins. To activate regulatory pathways, Prochlorococcus MED4 encodes five sigma factors from groups 1 and 2, two Fur-type transcriptional regulators, three Crp-type transcriptional regulators (including NtcA), the LysR-type transcriptional regulator RbcR, the global response regulator LexA, one ArsR-type transcriptional regulator, two AbrB-type transcriptional regulators, the GntR-type transcriptional regulator PMM1637, and six two component response regulators (including RpaA, RpaB, and PhoB)9.
Studies of sRNA targets have developed from the prediction and verification of one or a few targets towards sequencing of the entire targetome of an sRNA, for which various approaches have been developed. RNA chaperones can be immunoprecipitated to allow for subsequent sequencing of the bound RNA (RIP-Seq)13. MAPS technology is based on the affinity purification of MS2-tagged sRNA and its bound targets followed by RNA-Seq, and this method can also be used to study sRNAs that function without an RNA chaperone14. Another approach, called GRIL-Seq, is based on the recombinant expression of T4 ligase and the sRNA of interest, taking advantage of proximity ligation and the subsequent enrichment of hybrid sRNA-target fragments followed by sequencing15. Target-enrichment sequencing of sRNAs (TEsR) uses a biotinylated in vitro transcript of an sRNA of interest to enrich targets that were reverse transcribed into cDNA16.
In this study, we established a new method for sRNA target enrichment that uses a hybrid sRNA-target pulldown approach we call CRAFD-Seq (cell-free RNA affinity pull-down followed by sequencing), a technique that is also applicable to organisms for which no genetic tools are available. Our CRAFD-Seq results suggest that Yfr2 interacts with both Yfr1 and the highly conserved sRNA Yfr10, which is less conserved in position and structure but contains the same conserved motif as Yfr1. Although Yfr1 and Yfr10 share a set of targets, these sRNAs also appear to have unique targets. The distinct targetomes of Yfr2, Yfr1 and Yfr10 coupled with their interdependency constitute a complex regulatory network comprising hundreds of genes in the Prochlorococcus MED4 genome.
Results
Use of CRAFD-Seq to identify the Yfr2 targetome
We recently showed that the GntR family transcriptional regulator PMM1637, which is common to all cyanobacteria, binds to promotor regions of yfr2 homologs that contain the marine picocyanobacterial CGRE1 motif17. Because the CGRE1 motif is not present anywhere else in the Prochlorococcus MED4 genome, we investigated the regulon of Yfr2, resulting in an elucidation of the extended regulon of PMM1637.
Because tools for the genetic manipulation of Prochlorococcus do not currently exist, we developed a pipeline for the genome-wide enrichment of sRNA targets that is based on coupling in vitro-synthesized bait RNA to magnetic beads that are then incubated with total cell lysate (Fig. S1). Subsequently, a strand-specific cDNA library of specifically enriched target RNAs is prepared and amplified by PCR18 followed by sequencing. To identify enriched peaks, we developed a peak caller (Fig. S2) that is available to be downloaded (refer to the Material and Methods section for more details). We termed that method CRAFD-Seq for cell-free RNA affinity pull-down followed by sequencing. The advantage of this approach is that the targetome of any sRNA in any bacterium can be analysed, even in organisms such as Prochlorococcus that are recalcitrant to genetic manipulation.
We applied the CRAFD-Seq method using Yfr2 as bait with Prochlorococcus cells cultivated under standard growth conditions and identified 404 enriched peaks that could be assigned to 302 mRNAs (Table S1). For the mRNAs that bound on Yfr2, we observed a functional enrichment in the categories of translation, photosynthesis and respiration, regulatory functions and energy metabolism (Fig. 1A). Overall, the enriched peaks for the Yfr2 affinity pull-down assay were primarily located at sites that were antisense to a CDS (33%), inside of a CDS (23%), or within a 5′ UTR (17%) (Fig. 1B).
Figure 1.
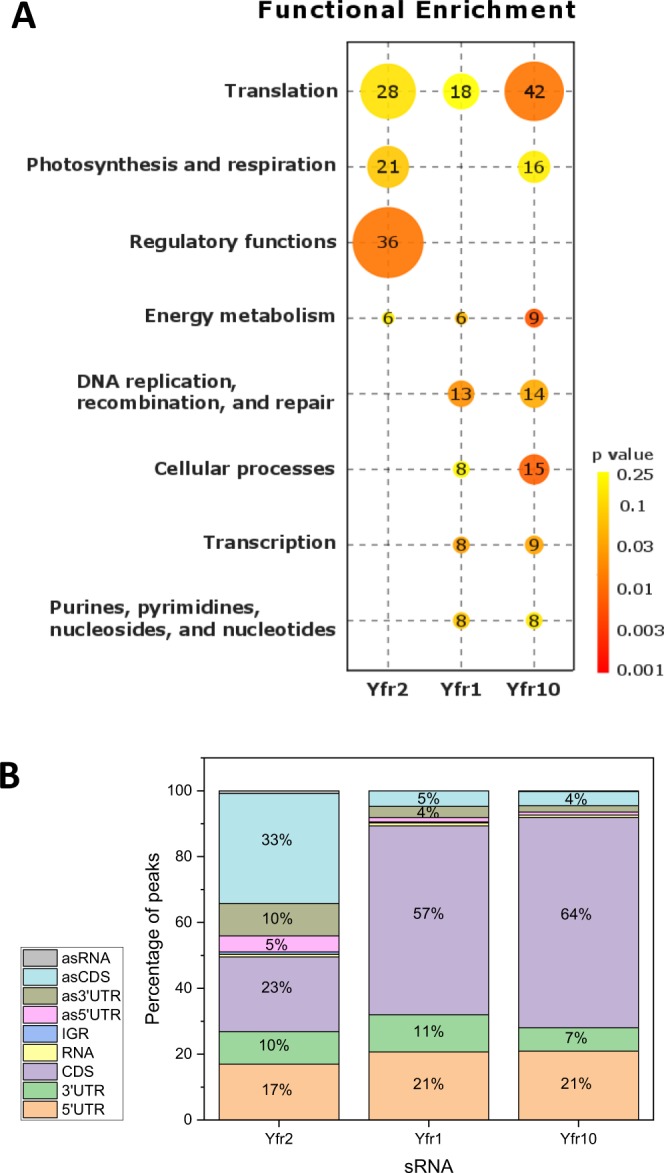
Overview on biological processes and sRNA-target RNA interaction regions regulated by Yfr2, Yfr1 or Yfr10. (A) Functional enrichment analysis based on CyanoBase functional categories. Only functional classes with a p-value of < 0.05 in at least one of the RNA affinity pull-down libraries are shown. (B) Relative distribution of enriched peaks located in gene coding regions (CDS), 5′ and 3′ untranslated regions (5′UTR and 3′UTR), untranslated genes (RNA) or antisense to CDS (asCDS), 5′/3′UTR (as5′UTR and as3′UTR) or RNA (asRNA).
Among the genes, those belonging to the category photosynthesis and respiration were essential genes of PSII, such as psbA, which encodes the PSII reaction centre protein D1, and psbD, which encodes the PSII core antenna protein CP43 (Fig. 2). The enrichment of targets for regulatory functions expands the Yfr2 regulatory network even further. For example, the transcriptional regulator RbcR, which is believed to control genes of the Calvin-Benson-Bassham cycle and RubisCO19, as well as the two-component sensor histidine kinase NblS, which controls acclimation to high light via the response regulator RpaB20, were enriched targets of Yfr2 (Fig. 2). Intriguingly, we also observed enrichment of the GntR family transcriptional regulator PMM1637, which is a regulator of Yfr2 (Fig. 2). Furthermore, we detected ribonuclease D (encoded by rnd), which is responsible for the 3′ processing of tRNAs, in the Yfr2 dataset (Fig. 2). The mRNA of the E. coli rnd homologue is undetectable during stationary phase21, a time during which nutrients such as nitrogen are limiting. The induction of Yfr2 under nitrogen starvation may also lead to decreased levels of Prochlorococcus MED4 rnd mRNA, which would lead to an increase in its substrates during nitrogen deprivation.
Figure 2.

Coverage plots of selected Yfr2 targets discovered using the CRAFD-Seq approach. Mapped read regions of the Yfr2 affinity pull-down and the control libraries are coloured in dark and light grey, respectively. The orange boxes correspond to the gene positions of psbA and psbC (encoding the PSII reaction centre proteins D1 and D2, respectively), psbD (encoding the PSII core antenna protein CP43), PMM1637 (encoding the transcription factor GntR), rnd (encoding ribonuclease D), rbcR (encoding the Rubisco transcriptional regulator), nblS (encoding a two-component sensor histidine kinase) and the sRNAs yfr1 and yfr10. The numbers in the grey boxes correspond to peak IDs of the Yfr2 enrichment library listed in Table S1. White boxes correspond to called peak regions that were not enriched. Genome coordinates for genes located on the reverse strand (rbcR and yfr1) are displayed with respect to the forward strand.
Yfr2 interferes with the interactions between Yfr1 and Yfr10 and their targets and is controlled by RNase E
Interestingly, two well-known sRNAs, Yfr1 and Yfr10, were also enriched in the Yfr2 affinity purification library (Table S1, Fig. 2). This result validates the earlier presumption of a physical interaction between the complementary regions of Yfr2 and Yfr1/107 (Fig. 3A). In a previous study, we demonstrated the interaction of Yfr1 with two of the top in silico-predicted targets, the homologs PMM1119 and PMM112111. To evaluate the ability of Yfr2 to interfere with interactions between Yfr1 and its targets, we modified a heterologous GFP reporter system to evaluate sRNA-mRNA interactions in E. coli22 by inserting both the sRNA genes yfr1 and yfr2, each under the control of their own PLlacO promoter, in the sRNA plasmid (for more details see the Materials & Methods section). The sRNA plasmids carrying yfr1, yfr2, yfr1 + yfr2, a non-interacting control RNA (control) or yfr1/yfr2+ control were cotransformed with the plasmid carrying the PMM1121 5′ UTR fused to sfgfp. In the presence of Yfr1 or Yfr1+ control, a pronounced repression in GFP fluorescence (4.7-fold ±0.8 and 3.8-fold ±0.9, respectively) was observed (Fig. 3C). GFP fluorescence was restored to the initial levels when both the Yfr1 and Yfr2 sRNAs were coexpressed, demonstrating that Yfr2 counteracts the inhibition of translation initiation in the PMM1121 5′UTR caused by Yfr1 in E. coli (Fig. 3C).
Figure 3.

Effectors of Yfr2. (A) Co-folded structures of Yfr1-Yfr2 and Yfr10-Yfr2. (B) Read coverage plots of the Yfr1-enriched (dark grey) and the control library (light grey). The positions of genes are indicated by the orange boxes, and the numbers in the grey boxes correspond to peak IDs for the Yfr1 and Yfr10 enrichment libraries listed in Tables S2 and S3, respectively. The genome coordinates of PMM1121 are displayed with respect to the forward strand. (C) Verification of Yfr1-Yfr2 interaction by GFP reporter assay. Average florescence values were calculated for 50,000 events (cells), and the fold repression and respective error were calculated from twelve independent clones for each strain. Each transformed E. coli strain contains a plasmid carrying sfgfp fused to the PMM1121 5′UTR and a second plasmid expressing Yfr1 or Yfr2 alone or in combination with each other or a non-sense RNA (control). pXG0+ control does not express GFP and serves as negative control. The horizontal dashed line indicates background fluorescence. (D) Cleavage of Cy3-labelled Yfr2 (indicated by pink coloured text) by RNase E in the presence of equal amounts of unlabelled Yfr2 D1, Yfr1, or Yfr10. The black arrow indicates the full-length Yfr2 transcript and the grey arrow indicates the Yfr2 cleavage product.
Because endoribonuclease E (RNase E) frequently plays a role in sRNA-target interactions in bacteria23, we characterized the interaction between Yfr2 with Yfr1 or Yfr10 through RNase E in vitro cleavage assays. The results showed that Yfr2 is cleaved by RNase E and that this cleavage is abolished when Yfr2 binds to Yfr1 or Yfr10 (Fig. 3D). To exclude the impact of titration effects, we performed the RNase E cleavage assay in the presence of Yfr2 D1, an Yfr2 mutant missing the RNase E recognition site and the Yfr1 interaction region (Fig. S3). Cleavage assays conducted with Yfr1, Yfr10 or Yfr2 D1 alone in vitro showed that none of these RNAs is a substrate of RNase E (Fig. S3).
Yfr1 and Yfr10 have overlapping but distinct target sets
Because of the unique interaction between the sRNAs Yfr2 and Yfr1 or Yfr10, we also used the CRAFD-Seq protocol to elucidate the targetomes of Yfr1 and Yfr10 by using these sRNAs as bait. The reciprocal fishing approach, using Yfr1 or Yfr10 as bait, largely confirmed that Yfr2 and its three homologs interact with Yfr1 or Yfr10 (Fig. 3B, Tables S2 and S3). For unknown reasons, Yfr2 was only slightly enriched in the Yfr10 library, but enrichment for the remaining Yfr2 homologs Yfr3, Yfr4, and Yfr5 was clearly above the set threshold. In total, we identified 288 and 384 enriched peaks associated with 206 and 253 mRNAs for Yfr1 and Yfr10, respectively.
Overall, the majority of peaks were located within a CDS (57 and 64% for Yfr1 and Yfr10, respectively), in a 5′ UTR (21% for Yfr1 and Yfr10), or in a 3′ UTR (11 and 7% for Yfr1 and Yfr10, respectively) (Fig. 1B). The functional enrichment of Yfr1 and Yfr10 targets was comparable, except for the photosynthesis- and respiration-associated targets, which were only enriched for Yfr10 (Fig. 1A). We compared the rank of the best intaRNA11-predicted interaction site for full-length genes of Yfr1, Yfr2 and Yfr10 with the lists of enriched/non-enriched CRAFD-Seq identified peaks (Tables S1–3, Fig. S4) and performed a non-parametric Kruskal-Wallis ANOVA test to show that intaRNA ranks are significantly better for enriched compared to non-enriched peaks with p-values of 1.69 × 10−3 (Yfr1), 1.74 × 10−5 (Yfr10) and 2.73 × 10−7 (Yfr2) (Fig. S4).
The CRAFD-Seq results also verified that the mRNA of PMM1121 is a target of Yfr1 (Fig. 3B) as well as those of the previously suggested Yfr1 targets11 PMM1119 (the homolog of PMM1121), inorganic pyrophosphatase (ppa, PMM0494) and the bifunctional ornithine acetyltransferase/N-acetylglutamate synthetase (argJ, PMM0050) (Table S2). The latter two targets could not be previously verified using the GFP reporter assay because of the poor translation efficiency of their UTRs in E. coli11. Inorganic pyrophosphatase plays an important role in the oxidative phosphorylation pathway by splitting pyrophosphate into two molecules of inorganic phosphate that can subsequently be added to ADP by the ATP synthase complex. The peak areas located in the 5′ region of ppa (Fig. 4B) were 6- and 3-fold enriched in Yfr1 and Yfr10 pulldown libraries, respectively (Tables S2 and S3). We further validated these interactions by primer extension and showed that the termination signals of ppa vary between Yfr1 and Yfr10, which might be explained by different structures that are formed during the interaction of Yfr1-ppa and Yfr10-ppa complexes, respectively (Fig. 4A). However, our data indicate that the interaction sites of both Yfr1 and Yfr10 with ppa are at the same position, the same position that was predicted by intaRNA11 (Fig. 4C,D). The formation of RNA-RNA complexes is stronger for ppa-Yfr10 than for ppa-Yfr1, which might be explained by structural differences in the complexes that are also suggested by primer extension results. We exchanged UCCU of the Yfr1/Yfr10 conserved motif with AAAA (M1) or UGGU (M2) (Fig. 4D). Neither of the mutants were able to interact with ppa as indicated by the loss of the footprint (Fig. 4C). The affinity of the sRNAs Yfr1 or Yfr10 to Yfr2 is stronger compared to the mRNA target ppa as the interaction of Yfr1 or Yfr10 with ppa was completely inhibited by Yfr2, even when the latter sRNA was present in very small amounts (Fig. 4A). Despite the identical conserved motifs in Yfr1 and Yfr10, we observed distinct sets of enriched mRNAs for the Yfr1 and Yfr10 targetomes. Almost half of all the Yfr1 targets were not present in the Yfr10 library, and two thirds of the targets were unique to Yfr10 (Fig. S5). In contrast to ppa, argJ and phosphoglucomutase (pgm, PMM0076) are targeted by Yfr1 but not Yfr10 (Fig. 5). Amongst the specific targets of Yfr10 are atpA and atpC, which encode the α and γ subunits of the F1 region of ATP synthase, as well as csoS1 and csoS2, which encode carboxysome shell polypeptides (Fig. 5). In both cases, the genes are organized within operons, and all of the other genes within these operons are not targeted by Yfr10 (Fig. 5).
Figure 4.

Yfr2 circumvents the interaction of Yfr1 and Yfr10 with their shared target inorganic pyrophosphatase (ppa). (A) Primer extension of 0.2 pmol in vitro-synthesized 5′ region ppa mRNA in the absence of any synthesized sRNA or in the presence of varying amounts of Yfr1 (left panel) or Yfr10 (right panel) alone or with Yfr1 (left panel) or Yfr10 (right panel) combined with Yfr2. Primer extension termination signals of ppa in presence of Yfr1 or Yfr10 are denoted in blue and grey lines, respectively. The interaction region predicted by intaRNA11 is shown in green lines. (B) Read coverage plots of the Yfr10- (dark grey), Yfr1-enriched (blue) and the control library (light grey). The position of ppa is indicated by the blue box, and the numbers in red boxes correspond to the peak IDs of the Yfr1 and Yfr10 enrichment libraries shown in Tables S2 and S3, respectively. The orange box marks the region of template DNA used for primer extension. The blue and grey boxes within the orange box correspond to the observed termination sites, and the green box corresponds to the predicted interaction site in A. The vertical dashed lines mark the start and end of the termination sites. The sequence of the 5′ UTR with Yfr1 (blue) and Yfr10 (grey) primer extension termination sites, the predicted interaction site (green) and the first ATG codon of the ORF are shown. (C) RNA footprinting assays of 0.1 pmol in vitro-synthesized 5′ region ppa mRNA in the presence of increasing amounts of Yfr1, Yfr1 M1, Yfr1 M2, Yfr10, Yfr10 M1 or Yfr10 M2, respectively. The first three lanes correspond to RNA – untreated control, OH – alkaline RNA ladder and T1 – RNase T1 treated ppa in vitro transcript. (D) Interactions between ppa and Yfr1/Yfr10 predicted by intaRNA11. The arrows indicate mutations M1 and M2 (red letters).
Figure 5.

Coverage plots of selected Yfr1 and Yfr10 targets discovered using the CRAFD-Seq approach. Mapped read regions of the Yfr1 and Yfr10 affinity pull-down libraries are coloured in blue and dark grey, respectively, and those of the control library are shown in light grey. The orange boxes correspond to gene positions of parts of the ATP synthase operon, the carboxysome shell polypeptides encoded by csoS1 and csoS2, the small and large subunits of Rubisco (encoded by rbcS and rbcL, respectively), argJ (encoding a bifunctional ornithine acetyltransferase/N-acetylglutamate synthetase) and pgm (encoding phosphoglucomutase). The numbers in the grey or blue boxes correspond to the peak IDs of Yfr1 and Yfr10, respectively, listed in Tables S2 and S3, respectively. White boxes correspond to called peak regions that were not enriched.
Complex post-transcriptional regulation of carbon primary metabolism
We observed enrichment for several mRNAs encoding enzymes involved in carbon primary metabolism in both libraries of Yfr1 and Yfr10. In addition, these mRNAs possess asRNAs that were enriched in the Yfr2 library. The glucose-1-phosphate adenyltransferase (glgC, PMM0769) mRNA appears to be under the control of a highly complex regulatory network of non-coding RNAs, including Yfr1/10, Yfr2 and the cis-encoded antisense RNA (asRNA) asGlgC (Fig. 6). The glgC mRNA is internally targeted by Yfr1 and Yfr10 as well as by asGlgC (Fig. 6), which we first observed in a previous study8. Peak 1328 corresponds to asGlgC and was enriched 2.6-fold in the Yfr2 affinity-purified library (Table S1), indicating that asGlgC is regulated by Yfr2. A similarly complex regulatory circuit can be anticipated for gap2 (glyceraldehyde-3-phosphate dehydrogenase, PMM0023), pgk (phosphoglycerate kinase, PMM0195) and pgmI (phosphoglycerate mutase, PMM1434), which appear to be controlled by Yfr1/Yfr10 and corresponding asRNAs, the latter of which seems to be under the control of Yfr2 (Fig. 6). Regulation of asRNAs by Yfr2 is a common feature, as almost half of all enriched peaks (48%) are located antisense to RNAs (peaks antisense to the CDS and the 5′ and 3′ UTRs combined), whereas this fraction makes up less than 10% for Yfr1 and Yfr10 (Fig. 1B).
Figure 6.

Coverage plots of selected Yfr2, Yfr1 and Yfr10 targets discovered using the CRAFD-Seq approach. Mapped read regions of the Yfr2, Yfr1 and Yfr10 affinity pull-down libraries are coloured in green, blue and dark grey, respectively, and those of the control library are shown in light grey. The orange boxes correspond to gene positions of glgC (encoding glucose-1-phosphate adenyltransferase) and asglgC, gap2 (encoding glyceraldehyde-3-phosphate dehydrogenase), pgk (encoding phosphoglycerate kinase) and pgmI (encoding phosphoglycerate mutase). The numbers in the grey, blue and green boxes correspond to the peak IDs of Yfr10, Yfr1 and Yfr2 listed in Tables S1, S2 and S3, respectively. Boxes coloured in green and white stripes correspond to peak regions where only one of the duplicate libraries was more than 2-fold enriched. White boxes correspond to called peak regions that were not enriched.
Differential expression and conservation of Yf1, Yfr2 and Yfr10
To improve our understanding of the interaction between Yfr2 and Yfr1 or Yfr10 and of their distinct targetomes, we investigated the expression of Yfr2, Yfr1, and Yfr10. Yfr2 was previously shown to be induced during late nitrogen starvation and early high light stress17. The abundance of Yfr1 and especially Yfr10 declines during high light acclimation (Fig. 7A), prompting us to investigate the effect of darkness on Yfr2, Yfr1, and Yfr10 expression. While Yfr1 and Yfr2 are rather stable, Yfr10 was 2.4-fold upregulated after one hour of darkness (Fig. 7B). Yfr10 is especially downregulated during elongated nitrogen starvation, whereas Yfr1 is at least 2.6-fold induced after 48 h (Fig. 7C).
Figure 7.
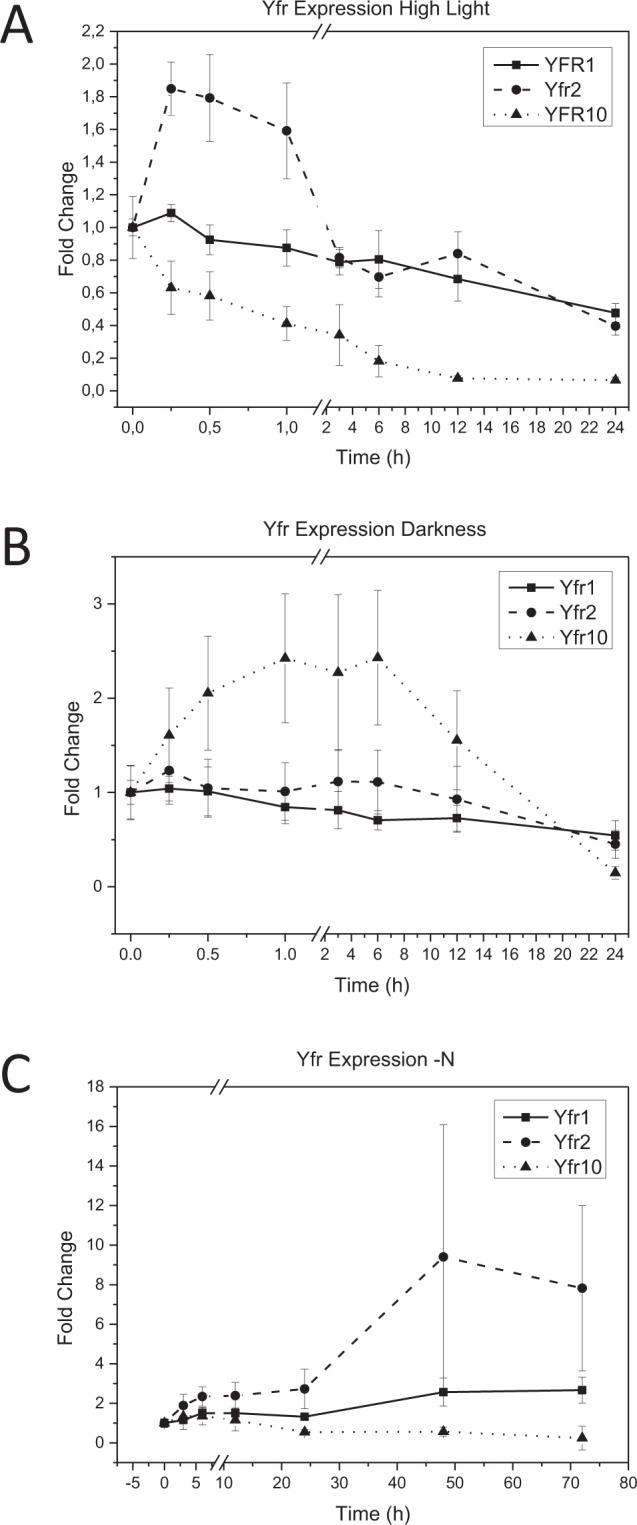
Temporal expression profiles of Yfr1, Yfr2 and Yfr10 during nitrogen depletion, high light-, or darkness stress. (A) Prochlorococcus MED4 cells were shifted from 30 µE to 300 µE, and samples were taken at 0, 0.25, 0.5, 1, 3, 6, 12, and 24 h after stress application and were probed against Yfr1 and Yfr10. (B) Acclimation of Prochlorococcus MED4 to darkness. Samples were taken at 0, 0.25, 0.5, 1, 3, 6, 12, and 24 h after stress application and were probed against Yfr1, Yfr2, and Yfr10. (C) Yfr1 and Yfr10 expression profiles of Prochlorococcus MED4 samples after 0, 3, 6, 12, 24, 48, and 72 h of nitrogen starvation. Data for Yfr2 during high light stress (A) and nitrogen starvation (C) were replotted from17. All stress experiments were carried out in biological triplicates. Extracted RNA samples were separated on 7 M urea-10% PAA gels, transferred on Hybond N+ membranes and hybridized with the respective probe.
Until this study, it was assumed that Yfr1 and the Yfr2 sRNA family are present in almost all cyanobacteria, whereas Yfr10 appeared to be restricted to a few Prochlorococcus strains despite harbouring the same motif as Yfr17. We showed that Yfr10-like sRNAs are more common than previously believed. For example, we verified the presence of Yfr10 in the marine picocyanobacterium Synechococcus WH8102 (Fig. S6). Furthermore, other species within the cyanobacterial phylum possess sRNAs that encode the conserved Yfr1 motif in addition to Yfr1, such as Syr5 in Synechocystis sp. PCC 680324, TSS 2515196r and TSS 3750139 f in Nostoc sp. PCC 712025 and Yfr104 in Prochlorococcus MIT93138 (Fig. S7). Intriguingly, we also detected Yfr10 in Prochlorococcus SS120 (Fig. S6), which was previously considered to be devoid of a Yfr1 homolog9 but is now known to at least possess an sRNA with the Yfr1 motif.
Discussion
We previously showed that two Yfr2 homologs in Prochlorococcus MED4 are regulated by the cyanobacterial GntR transcriptional regulator PMM163717. Studies on homologous GntR proteins in other cyanobacteria, such as Synechocystis sp. PCC 6803, showed that GntR mutants fail to establish correct ratios of PSII/PSI during acclimation to high light26 and fail to degrade phycobilisomes during nitrogen starvation27. Consistent with these findings, we observed increased expression of Yfr2 during both conditions17. Our findings that Yfr2 possibly targets genes involved in photosynthesis and respiration are therefore in good agreement with the results of these previous studies. Changing light conditions and nitrogen availability are presumably the most important factors that cyanobacteria, especially Prochlorococcus, have to cope with in the oligotrophic oceans. Therefore, it is reasonable that Yfr2 also targets other regulatory factors, such as the transcriptional regulator rbcR, the sensor histidine kinase nblS or asRNAs, which together comprise 48% of the enriched peaks. Yfr2 thereby integrates the perceived multiple environmental signals into other regulatory circuits. Because we can assume that at least one function of antisense transcripts is the regulation of cis-encoded genes, asRNAs can be added to the regulatory functions category of protein-coding genes. Furthermore, the high number of enriched antisense peaks highlights the importance of antisense transcription for gene regulation. These findings emphasize the utility of Yfr2 as a modulator of gene expression of regulatory elements.
We previously showed that PMM1637 does not act as an autoregulator17. However, the results from this study indicate that a feedback loop occurs via Yfr2. Modulation of gene expression of an sRNA by a transcription factor has been previously described. For instance, the sRNAs NsiR4 in Synechocystis sp. PCC 6803 and NsrR1 in Nostoc sp. PCC 7120 are regulated by NtcA28,29. However, feedback control of an sRNA on its associated transcription factor has only been described in a few instances in bacteria. In Vibrio harveyi, the quorum-sensing related Qrr sRNAs posttranscriptionally repress LuxO, which functions as their transcriptional repressor30. Our data suggest the existence of feedback control of Yfr2 on its transcriptional regulator PMM1637, adding another example to this class of control mechanisms. We further showed that the activity of Yfr2 can be neutralized either by its pairing with Yfr1/10 or through its RNase E-mediated cleavage. It is unclear if the cleavage of Yfr2 by RNase E initiates its degradation. However, in addition to the generally very high stability of Yfr26, the Yfr2 cleavage product possibly represents a modified version of the sRNA that is unable to interact with Yfr1 or Yfr10 via its conserved 5′ motif and can only facilitate gene regulation via the conserved motif in the first stem loop12.
The CRAFD-Seq results obtained in our study revealed a relatively high occurrence of enriched peaks within CDS regions. This result is in contrast to those obtained in RIL-Seq experiments, where an almost equal distribution of chimeric sRNA fragments with CDS and 5′ UTRs was observed31. These differences may be explained by differences in the experimental setup or a by a different mode of action of sRNAs in Prochlorococcus. The results of recent ribosomal profiling studies suggest that translational inhibition via blocking of the Shine-Dalgarno sequence (SD) is possibly not as established as previously thought for cyanobacterial sRNAs and their targets32. The SD sequence does not occur more often than by chance in Prochlorococcus MED4 and MIT93138, and in Synechocystis sp. PCC 6803 it is only detected in 26% of all genes33. The latter number of SD sequences in Synechocystis sp. PCC 6803 correlates well with the 27% of genes that were observed to have a higher than average intergenic coverage in the ribosomal profiling experiment for which SD sequences were detected32. The genes containing SD sequences were enriched in the categories of photosynthesis and respiration and for translation, possibly because genes in these categories are highly translated32. However, this does not appear to be the case for Prochlorococcus, and further studies are required to examine the detailed influence of internally binding sRNAs in Prochlorococcus on their targets.
During nitrogen starvation, cells undergo chlorosis and reduce the levels of their photosynthetic apparatus, since phycobiliproteins and chlorophyll are rich in nitrogen that can be recycled. This process is abolished in mutants of the cyanobacterial GntR transcriptional regulator in Synechocystis sp. PCC 6803 despite the presence of elevated nblA levels27. Furthermore, nitrogen starvation is also indicative as an excess of carbon supply34. The metabolic fluxes of fixed carbon are therefore rerouted from amino acid synthesis, which cannot be sustained, towards gluconeogenesis35,36. The genes encoding enzymes that metabolize fixed carbon in the form of 3-phosphoglycerate, either in the direction of gluconeogenesis (such as pgk and gap2) or towards the citrate cycle (such as pgmI) are regulated by Yfr1 and/or Yfr10. However, because Yfr2 can interact with Yfr1/10, the stimulus of late nitrogen or high light stress can be integrated into these metabolic pathways via Yfr1/10 and through the Yfr2-controlled asRNAs of pgk, gap2, and pgmI. The pathway from 3-phosphoglycerate towards the citrate cycle appears to be indirectly controlled by Yfr2, because pgmI is also a target of Yfr2, possibly enhancing the shutdown of this route for fixed carbon. A highly complex regulatory circuit involving Yfr2, Yfr1/10 and asglgC can be similarly anticipated for glgC, which catalyses the first committed step in glycogen synthesis. Another target of Yfr1 is the important metabolic branch point enzyme phosphoglucomutase (encoded by pgm), which links glycolysis, the oxidative pentose phosphate pathway and glycogen metabolism, making it directly involved in the processes of polyglucan storage (glycogen synthesis) and utilization (e.g., respiration via the glycolysis and OPP pathways)37. Unfortunately, in this study we only determined the targetomes of Yfr1, Yfr2, and Yfr10, and a functional analysis of the mode of action of the assayed sRNAs requires further investigations.
Comparing the expression profiles of an sRNA and its targets may provide useful information on the mode of interaction. Previous studies showed that ppa expression is downregulated during high light stress38 and nitrogen depletion39, both conditions where Yfr2 is upregulated. This result may suggest that Yfr1 and Yfr10 have stabilizing effects on ppa mRNA. A similar observation was made for the Yfr10-specific targets of the carboxysome-associated genes csoS1 and csoS2 and for atpA and atpC, which are downregulated during nitrogen stress39. However, because multiple regulatory inputs affect one gene, and the mode of action for sRNAs in Prochlorococcus is enigmatic, we could not detect this effect in general for the sRNA targets presented here.
The importance of Yfr1, beside its interaction with targets involved in central pathways such as energy metabolism, is supported by its almost omnipresent occurrence in members of the cyanobacterial phylum10. This idea is further confirmed with the discovery of a second sRNA (Yfr10) that shares the same conserved motif as Yfr17. Unlike Yfr1, synteny and sequence conservation, except from the Yfr1 motif, is not conserved, resulting in secondary structure conservation among Yfr10 homologs being very low (Fig. S7). These features could be the reason that the context of additionally encoded Yfr1 motifs remained undiscovered by computational methods. In HL-adapted Prochlorococcus strains, Yfr10 homologs are encoded upstream of Yfr2 homologs17. The proximity of expression of both sRNAs possibly enhances their interaction potential. Previous studies suggested that Yfr1 is required for growth during various stresses40 and may explain why Prochlorococcus SS120, which lacks Yfr19, is especially susceptible to abiotic stresses. Thus, Prochlorococcus SS120 may only be viable because it still possesses Yfr10, which is able to partially fulfil the functions of Yfr1, a theory that is supported by the observed overlap between the Yfr1 and Yfr10 regulons (Fig. S5). However, we were surprised to observe that half of the enriched genes for Yfr1 and two thirds of those for Yfr10 were sRNA-specific (Fig. S5), which clearly shows that RNA-RNA interactions are very complex and cannot be limited to simple complementarity. The conserved motif in Yfr1 is a single-stranded region between two stem loops10,40, whereas the conserved motif in Yfr10 is present in the single-stranded loop on top of a stem7. The different accessibility and surroundings of the conserved motif appears to enable the discrimination of targets.
Collectively, our data suggest the existence of a complex, interconnected network of interactions among Yfr2, Yfr1 and Yfr10 and their targets (Fig. 8). We identified late nitrogen starvation and high light stress as important factors that trigger the induction of Yfr2 mediated by the GntR transcription factor PMM1637. A change in the light regime (high light and darkness) regulates the expression of Yfr10, although the regulators involved in the modulation of yfr10 gene expression during varying light conditions are unknown. However, yfr1 is constitutively expressed under all stress conditions assayed in this study. In addition to the reciprocal influence of Yfr2 and Yfr1/10, the cleavage of Yfr2 by RNase E is another layer of regulation in this system that impedes the interaction between Yfr2 and Yfr1/10 and presumably of Yfr2 with many of its targets.
Figure 8.
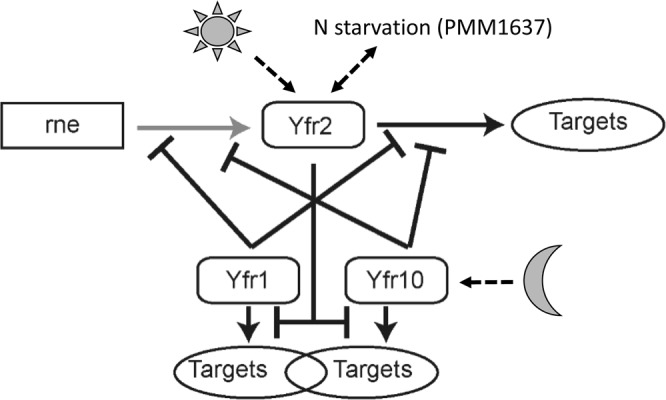
Schematic representation of the interplay between Yfr2, Yfr1, Yfr10, RNase E and their targets. Central to this model is the capability for titration between Yfr2 and Yf1/Yfr10 sRNAs. Therefore, changes in the concentration of any of these sRNAs will also affect the possible outcome by any of the other sRNAs. Interactions drawn in black represent RNA-RNA interactions and those in grey represent RNA-protein interactions. Arrowheads on the tips indicate an unknown mode of action, while bars on the tips indicate inhibition. Dashed black arrows indicate abiotic factors that modulate the gene expression of Yfr2 and Yfr10.
Materials and Methods
Culturing and RNA preparation
Prochlorococcus MED4 cultures were grown at 22 °C in AMP1 medium41 under 30 µmol quanta m−2 s−1 of continuous white cool light to cell densities of 1–3 × 108 cells per ml. Stress experiments for high-light and nitrogen starvation were performed as described previously17. For darkness experiments, cells were transferred from 30 µmol quanta m−2 s−1 of continuous white cool light to complete darkness for 0, 15, or 30 min or for 1, 3, 6, 12, or 24 h, with the cells subsequently harvested in darkness by filtration as previously described17. RNA extraction and northern hybridization were performed as described previously6,17. Prochlorococcus SS120 and Synechococcus sp. WH8102 cultures were grown at 22 °C in AMP1 medium41 under 15 µmol quanta m−2 s−1 of continuous white cool light, and nitrogen depletion experiments were performed as described preveiously17. Yfr10 homologs were probed with primers #1–3 (Table S4).
In vitro RNA synthesis and biotinylation of RNA
Transcript templates for in vitro RNA synthesis were generated from purified PCR products or annealed complementary oligonucleotides using primers #4–13 (Table S4). The desired RNAs were transcribed using a MegaShort script Kit (ThermoFisher Scientific), and residual DNA was removed by TURBO DNase I treatment, with both steps performed according to the manufacturer’s instructions. RNA was purified and concentrated by phenol-chloroform extraction and ethanol precipitation or using RNA Clean & Concentrator columns (Zymo Research) following the manufacturer’s instructions. For the primer extension assay, if required, in vitro-transcribed RNA was separated on 7 M urea-10% polyacrylamide gels or on 2% non-denaturing agarose gels, and full length fragments were excised and purified using either a ZR small-RNA PAGE Recovery Kit (Zymo Research) or a NucleoSpin Gel and PCR-Clean-up kit (Macherey-Nagel) according to manufacturer’s instructions. For biotinylation, two volumes of KIO4 (6 mM) were added to 1.5 nmol of RNA and was incubated at room temperature, in the dark for one hour. Subsequently, one volume of ethylene glycol/H2O (1:1) was added, and the RNA was precipitated by adding 2.9 volumes of ethanol (100%) and 0.1 volumes of NaCl (3.3 M) at −20 °C for a maximum of 60 min. The RNA was centrifuged at 13,000 g for 30 min at 4 °C, the supernatant was removed, and the pellet was washed with 70% ethanol. The air-dried pellet was then resuspended in 24 µl 10 × PBS (pH 7.4), 6 µl H2O, and 20 µl EZ-Link Alkoxyamine-PEG12-Biotin (50 mM in DMSO). The solution was incubated at 37 °C in the dark for 3 h and was periodically mixed, after which 50 µl of NaBH4 (20 mM) and 100 µl of Tris-HCl (0.1 M, pH 7.5 at room temperature) were added and the mixture was incubated in the dark at 4 °C for 30 min. After the RNA was precipitated overnight as described above, free EZ-link-PEG12-alkoxyamine was removed by washing the RNA with water in Amicon ultracel 10-K columns (Merck).
Affinity purification of target RNAs
Prochlorococcus MED4 cultures (500 ml for each sRNA to be analysed) were pelleted at 15,000 g for 10 min at 22 °C. The cell pellets were washed with a solution containing 1.3 M betaine and 10 mM HEPES (pH 8) and were centrifuged again. The pellets were resuspended in lysis buffer (100 mM Tris-HCl (pH 7.5), 500 mM LiCl, 10 mM EDTA, 5 mM DTT, and 1% LiDS) and lysed using a OneShot constant pressure system (Constant Systems Limited) at 1.5 kBar. Cell debris was separated from the cell lysate by centrifugation for 30 min, 13,000 g, at 4 °C. For immobilization of the bait RNA, 250 µl of Dynabeads Streptavidin MyOne C1 (ThermoFisher Scientific) per sample were prepared following the manufacturer’s instructions. The beads were washed once with binding buffer (20 mM Tris-HCl (pH 7.5), 1 M LiCl, and 2 mM EDTA). Biotinylated RNA was then loaded onto the beads in binding buffer for 10 min at room temperature with gentle agitation. Subsequently, the beads were washed first with binding buffer then with lysis buffer. To reduce the binding of nonspecific RNA to the beads, the cell lysate was first incubated with unloaded beads, which were prepared as described above, for 10 min at room temperature with gentle agitation. The unbound cell lysate incubated with the empty beads was then incubated with beads charged with the sRNA of interest for 10 min at room temperature with gentle agitation. The beads were washed with 1 ml buffer A (10 mM Tris-HCl (pH 7.5), 0.3 M LiCl, 1 mM EDTA, and 0.1% LiDS), 1 ml buffer B (10 mM Tris-HCl (pH 7.5), 0.3 M LiCl, and 1 mM EDTA), and 0.5 ml buffer C (10 mM Tris-HCl (pH 7.5), 0.15 M LiCl, and 1 mM MgCl2). Bound RNA was eluted in 150 µl of elution buffer (10 mM Tris-HCl, pH 7.5) by incubating the sample for 2 min at 65 °C, and the beads were immediately removed by placing the tube on a magnet and transferring the supernatant to a new tube.
Primer extension
The primers were labelled as described previously42. Annealing mixtures containing 0.2 pmol of in vitro-synthesized target RNA, 2 pmol of the 5′ end-labelled primer ppa_RT (Table S4) without or with 40/80/160/320 pmol of in vitro-synthesized sRNAs (Yfr1, Yfr2, or Yfr10) were heated for 10 min at 70 °C and then chilled on ice for at least 5 min. cDNA synthesis was performed for 2 h at 30 °C using SuperScript III Reverse Transcriptase (ThermoFisher Scientific) according to the manufacturer’s instructions. The reaction was inactivated by incubation for 15 min at 70 °C, which was followed by RNase H treatment for 20 min at 37 °C and a final heat inactivation for 5 min at 95 °C in RNA loading buffer. DNA sequencing ladder reactions were performed with the same 5′ end-labelled primer used for cDNA synthesis and the same template DNA used for target RNA in vitro synthesis using a USB Thermo Sequenase Cycle Sequencing Kit (Affymetrix). Primer extension products and sequencing reactions were separated on 8.3 M urea-6% polyacrylamide sequencing gels, and the vacuum-dried gels were exposed to imaging plates. Signals were visualized using a Thyphoon FLA 9500 instrument (GE Healthcare) with Quantity One software (Bio-Rad).
RNA footprinting
In vitro-synthesized RNA was produced from annealed primers #4, 5, 23–34 (Table S4) using the HighScribe T7 Quick High Yield RNA Synthesis Kit (NEB). For removal of 5′ triphosphates, 20 pmol ppa in vitro-synthesized RNA was incubated with 20 units FastAP (Thermo Fischer Scientific) in a 200 µl reaction volume at 37 °C for one hour. Dephosphorylated in vitro-synthesized RNA was labeled with [γ-32P] ATP by T4 Polynucleotide Kinase (Thermo Fischer Scientific). RNA footprinting was performed using Ambion RNase T1 according to manufacturer’s description. Briefly, 0.1 pmol of labeled ppa in vitro-synthesized RNA was mixed with 20, 40, or 80 pmol of unlabeled Yfr1, Yfr10, or mutant in vitro-synthesized RNA, denatured for 1 min at 95 °C and cooled to room temperature for 5 min. Subsequently, 1 µl of 1 µg/µl yeast RNA and 1 µl of 10 x structure buffer was added and samples were incubated at room temperature for 15 min. RNase T1 treatment was performed by adding 1 µl of 0.2 U/µl RNase T1 and the samples were incubated for 15 min at room temperature. Reactions were stopped by adding 10 µl of denaturing formamide loading dye. Alkaline ladder was obtained by incubating 0.1 pmol of labeled ppa in vitro-synthesized RNA at 95 °C for 5 min in 7.5 µl of alkaline hydrolysis buffer containing 1.5 µg of yeast RNA. Reactions were stopped by adding 10 µl of denaturing formamide loading buffer. RNase T1 G ladders was obtained by incubating 0.1 pmol of labeled ppa in vitro-synthesized RNA and 1 µl of 1 µg/µl yeast RNA in 9 µl sequencing buffer for 10 min at 50 °C, followed by the addition of 1 µl of 0.2 U/µl RNase T1 and incubation at room temperature for 15 min. Reactions were stopped by adding 12 µl of denaturing formamide loading dye. The samples were separated on 8.3 M urea-10% polyacrylamide sequencing gels, and the vacuum-dried gels were exposed to imaging plates. Signals were visualized using a Thyphoon FLA 9500 instrument (GE Healthcare) with Quantity One software (Bio-Rad).
Library preparation and data analysis
Libraries were prepared following the whole transcriptome protocol as previously described18. In brief, 2 µg of affinity-purified RNA samples and 4 µg of control RNA samples (unbound cell lysate of empty beads) were treated with 4 units of TURBO DNase (Life Technologies) in 2 consecutive incubation steps, each at 37 °C for 15 min. Next, RNA clean-up and separation into small (17–200 nt) and large ( > 200 nt) RNA fractions was performed using RNA Clean & Concentrator columns (Zymo Research). The large RNA fraction was fragmented according to the protocol18 and was recombined with the small RNA fraction after RNA Clean & Concentrator column purification (Zymo Research). All subsequent follow-up steps were performed according to the protocol18 with the following modifications: the clean-up of RNA 5′-pholyphosphatase (Epicentre)-treated samples was performed using Clean & Concentrator column purification (Zymo Research). For RNA adapter ligation, the UGA linker (Supplementary Table S4) was used. After RNA adapter ligation and cDNA synthesis, the samples were gel excised from 2% agarose gels and purified using a NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel) with the optional NTC buffer to solubilize the gel slices. After PCR amplification, residual primers were removed by adding 10 µl of ExoSAP-IT (USB) to 50 µl of PCR, and with samples incubated for 15 min at 37 °C followed by heat inactivation of the enzyme for 15 min at 85 °C. The samples were cleaned-up using a NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel), and the cDNA libraries were analysed on an Illumina HiSeq 2500 or 3000 sequencer. Sequencing data were analysed using the Galaxy43 Platform https://usegalaxy.eu/. Reads were mapped to the Prochlorococcus MED4 genome with BWA-MEM (Galaxy Version 0.7.17.1), unmapped reads were removed with BAM filter (Galaxy Version 0.5.9) and that data was converted to the wig format using BAM to Wiggle (Galaxy Version 2.6.4). To merge different wig files, missing nucleotide positions were added and the read number was set 0. Read numbers were normalized to library size and merged grp files for each strand were generated. Grp files are available as supplementary files 1 and 2.
Peak calling
The start and end of a peak were set if the difference between the average read coverage over a defined base range separated by a spacer exceeded the threshold (Fig. S2A). In addition, the coverage between the defined start and endpoints had to pass the coverage threshold. In this study, we compared the coverage over 5 nucleotides separated by 10 nucleotides, which had to exhibit a fold change of 1.5 to be considered as the start- or endpoint. The coverage of a peak had to be at least 20% of the average coverage of a nucleotide in the dataset. A schematic overview on the peak calling procedure is presented in Fig. S2A. The input for peak calling was a grp file and the Python script for data analysis is available at https://github.com/SJLambrecht/GRP_Peakcall.
RNase E cleavage assays and RNase E purification
RNase E purification and cleavage assays were essentially performed as previously described42. Yfr2 transcripts (25 pmol) labelled with Cy3 and equal amounts of unlabelled Yfr2 D1, Yfr1, or Yfr10 were incubated with RNase E. Aliquots were withdrawn immediately after reaction assembly and after 20 min of incubation at 30 °C. Prior to analysis of RNA cleavage on 10% urea PAGE, cleavage reactions were purified using an RNA Clean and Concentrator Kit (Zymo).
Construction of plasmids and GFP reporter assay
To introduce a second sRNA gene (yfr1) or the control RNA under its own PLlacO promoter in the pZE12-luc-yfr2 plasmid, PCRs were performed with primers PromFw-AvrII and Yfr1PromRv-AvrII or primers PromFw-AvrII and pJV300PromRv-AvrII (Table S4) using the pZE12-luc-yfr2 or pJV300 control plasmid, respectively, as template. The PCR products, pZE12-luc-yfr2 and pJV300 were digested with AvrII, and the amplified yfr1 gene with the PLlacO promoter was introduced into pZE12-luc-yfr2 and pJV300. For higher GFP signals, the 5′ UTR of PMM1121 was generated by annealing primers PMM1121_aqua_sense and PMM1121_aqua_as (Table S4). The backbone of pXG10 (expressing sfGFP) was amplified by inverted PCR using the primers pXG10_sfGFP_aqua_righ and pXG10_sfGFP_aqua_left (Tables S4) and a plasmid containing the 5′ UTR of PMM1121 fused to sfGFP was generated by AQUA cloning44.
In general, GFP assays were performed as previously described11. Briefly, E. coli Top10 cells were transformed with the plasmids encoding the PMM1121 UTR fused to sfGFP and one of the sRNA encoding plasmids. The colonies were inoculated into 200 µl antibiotics-containing LB medium and grown overnight at 37 °C in a 96-well plate. Cells were diluted 1:10 into fresh LB medium and fixed in 1% HistoFix (Roth). Single-cell fluorescence was determined by flow cytometry using an Accuri C6 flow cytometer (BD Bioscience). Cell fluorescence was measured with an excitation wavelength of 488 nm and the emission was detected at 533/30 nm. The mean fluorescence per plasmid combination was calculated from 50,000 events (cells) of 12 individual clones.
Supplementary information
Acknowledgements
We thank Matthias Futschik for providing an R script for functional enrichment analyses and Steffen C. Lott for providing a Python script for the visualization of functional enrichment results. We also thank Wolfgang R. Hess for helpful discussions and critical reading of the manuscript. The research was supported by Deutsche Forschungsgemeinschaft grant STE1119/4-2 to C.S. and by the MEXT-supported program for strategic research foundation at private universities 2013–2017 (S1311017) to Y.K.
Author Contributions
S.J.L. and C.S. conceived the project, performed biochemical experiments, and analyzed and interpreted RNA-Seq data. S.J.L. Developed CRAFD-Seq and the peak calling tool and performed CRAFD-Seq experiments. Y.K., J.F., B.H. and R.R. performed RNA sequencing. S.J.L. and C.S. wrote the manuscript.
Data Availability
The sequencing data have been deposited in NCBI-SRA under accession numbers SRR8280994, SRR8280995, SRR8280996, SRR8280997, SRR8280998 and SRR8280999.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-49881-9.
References
- 1.Wagner, E. G. H. & Romby, P. Small RNAs in Bacteria and Archaea. In Advances in Genetics90, 133–208 (Elsevier, 2015). [DOI] [PubMed]
- 2.Nitzan M, Rehani R, Margalit H. Integration of Bacterial Small RNAs in Regulatory Networks. Annu. Rev. Biophys. 2017;46:131–148. doi: 10.1146/annurev-biophys-070816-034058. [DOI] [PubMed] [Google Scholar]
- 3.Chaulk SG, et al. ProQ is an RNA chaperone that controls ProP levels in Escherichia coli. Biochemistry. 2011;50:3095–3106. doi: 10.1021/bi101683a. [DOI] [PubMed] [Google Scholar]
- 4.Smirnov A, et al. Grad-seq guides the discovery of ProQ as a major small RNA-binding protein. Proc. Natl. Acad. Sci. 2016;113:11591–11596. doi: 10.1073/pnas.1609981113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biller SJ, Berube PM, Lindell D, Chisholm SW. Prochlorococcus: the structure and function of collective diversity. Nat. Rev. Microbiol. 2015;13:13–27. doi: 10.1038/nrmicro3378. [DOI] [PubMed] [Google Scholar]
- 6.Axmann IM, et al. Identification of cyanobacterial non-coding RNAs by comparative genome analysis. Genome Biol. 2005;6:R73. doi: 10.1186/gb-2005-6-9-r73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steglich C, et al. The challenge of regulation in a minimal photoautotroph: non-coding RNAs in Prochlorococcus. PLoS Genet. 2008;4:e1000173. doi: 10.1371/journal.pgen.1000173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voigt K, et al. Comparative transcriptomics of two environmentally relevant cyanobacteria reveals unexpected transcriptome diversity. ISME J. 2014;8:2056–2068. doi: 10.1038/ismej.2014.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scanlan DJ, et al. Ecological Genomics of Marine Picocyanobacteria. Microbiol. Mol. Biol. Rev. 2009;73:249–299. doi: 10.1128/MMBR.00035-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voss B, Gierga G, Axmann IM, Hess WR. A motif-based search in bacterial genomes identifies the ortholog of the small RNA Yfr1 in all lineages of cyanobacteria. BMC Genomics. 2007;8:375. doi: 10.1186/1471-2164-8-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richter AS, Schleberger C, Backofen R, Steglich C. Seed-based IntaRNA prediction combined with GFP-reporter system identifies mRNA targets of the small RNA Yfr1. Bioinformatics. 2010;26:1–5. doi: 10.1093/bioinformatics/btp609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gierga, G., Voss, B. & Hess, W. R. The Yfr2 ncRNA family, a group of abundant RNA molecules widely conserved in cyanobacteria. RNA Biol. 222–227 (2009). [DOI] [PubMed]
- 13.Dugar G, et al. The CsrA-FliW network controls polar localization of the dual-function flagellin mRNA in Campylobacter jejuni. Nat. Commun. 2016;7:11667. doi: 10.1038/ncomms11667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lalaouna D, Massé E. Identification of sRNA interacting with a transcript of interest using MS2-affinity purification coupled with RNA sequencing (MAPS) technology. Genomics. Data. 2015;5:136–138. doi: 10.1016/j.gdata.2015.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han K, Tjaden B, Lory S. GRIL-Seq, a method for identifying direct targets of bacterial small regulatory RNA by in vivo proximity ligation. Nat. Microbiol. 2016;2:16239. doi: 10.1038/nmicrobiol.2016.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nguyen Q, et al. Target-enrichment sequencing for detailed characterization of small RNAs. Nat. Protoc. 2018;13:768–786. doi: 10.1038/nprot.2018.001. [DOI] [PubMed] [Google Scholar]
- 17.Lambrecht SJ, Wahlig JML, Steglich C. The GntR family transcriptional regulator PMM1637 regulates the highly conserved cyanobacterial sRNA Yfr2 in marine picocyanobacteria. DNA Res. 2018;25:489–497. doi: 10.1093/dnares/dsy019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pfeifer-Sancar K, Mentz A, Rückert C, Kalinowski J. Comprehensive analysis of the Corynebacterium glutamicum transcriptome using an improved RNAseq technique. BMC Genomics. 2013;14:888. doi: 10.1186/1471-2164-14-888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burnap R, Hagemann M, Kaplan A. Regulation of CO2 Concentrating Mechanism in Cyanobacteria. Life. 2015;5:348–371. doi: 10.3390/life5010348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moronta-Barrios F, Espinosa J, Contreras A. In vivo features of signal transduction by the essential response regulator RpaB from Synechococcus elongatus PCC 7942. Microbiology. 2012;158:1229–1237. doi: 10.1099/mic.0.057679-0. [DOI] [PubMed] [Google Scholar]
- 21.Deutscher MP. How bacterial cells keep ribonucleases under control. FEMS Microbiol. Rev. 2015;39:350–361. doi: 10.1093/femsre/fuv012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corcoran CP, et al. Superfolder GFP reporters validate diverse new mRNA targets of the classic porin regulator, MicF RNA. Mol. Microbiol. 2012;84:428–445. doi: 10.1111/j.1365-2958.2012.08031.x. [DOI] [PubMed] [Google Scholar]
- 23.Saramago M, et al. The role of RNases in the regulation of small RNAs. Curr. Opin. Microbiol. 2014;18:105–115. doi: 10.1016/j.mib.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 24.Georg J, et al. Evidence for a major role of antisense RNAs in cyanobacterial gene regulation. Mol. Syst. Biol. 2009;5:305. doi: 10.1038/msb.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brenes-Álvarez, M., Olmedo-Verd, E., Vioque, A. & Muro-Pastor, A. M. Identification of Conserved and Potentially Regulatory Small RNAs in Heterocystous Cyanobacteria. Front. Microbiol. 7 (2016). [DOI] [PMC free article] [PubMed]
- 26.Fujimori T, et al. The Mutant of sll1961, Which Encodes a Putative Transcriptional Regulator, Has a Defect in Regulation of Photosystem Stoichiometry in the Cyanobacterium Synechocystis sp. PCC 6803. PLANT Physiol. 2005;139:408–416. doi: 10.1104/pp.105.064782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sato H, Fujimori T, Sonoike K. sll1961 is a novel regulator of phycobilisome degradation during nitrogen starvation in the cyanobacterium Synechocystis sp. PCC 6803. FEBS Lett. 2008;582:1093–1096. doi: 10.1016/j.febslet.2008.02.063. [DOI] [PubMed] [Google Scholar]
- 28.Klähn S, et al. The sRNA NsiR4 is involved in nitrogen assimilation control in cyanobacteria by targeting glutamine synthetase inactivating factor IF7. Proc. Natl. Acad. Sci. 2015;112:E6243–E6252. doi: 10.1073/pnas.1508412112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Álvarez-Escribano, I., Vioque, A. & Muro-Pastor, A. M. NsrR1, a Nitrogen Stress-Repressed sRNA, Contributes to the Regulation of nblA in Nostoc sp. PCC 7120. Front. Microbiol. 9 (2018). [DOI] [PMC free article] [PubMed]
- 30.Tu KC, Long T, Svenningsen SL, Wingreen NS, Bassler BL. Negative Feedback Loops Involving Small Regulatory RNAs Precisely Control the Vibrio harveyi Quorum-Sensing Response. Mol. Cell. 2010;37:567–579. doi: 10.1016/j.molcel.2010.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melamed S, et al. Global Mapping of Small RNA-Target Interactions in Bacteria. Mol. Cell. 2016;63:884–897. doi: 10.1016/j.molcel.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karlsen, J., Asplund-Samuelsson, J., Thomas, Q., Jahn, M. & Hudson, E. P. Ribosome Profiling of Synechocystis Reveals Altered Ribosome Allocation at Carbon Starvation. mSystems3 (2018).
- 33.Ma J, Campbell A, Karlin S. Correlations between Shine-Dalgarno Sequences and Gene Features Such as Predicted Expression Levels and Operon Structures. J. Bacteriol. 2002;184:5733–5745. doi: 10.1128/JB.184.20.5733-5745.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang, C.-C., Zhou, C.-Z., Burnap, R. L. & Peng, L. Carbon/Nitrogen Metabolic Balance: Lessons from Cyanobacteria. Trends Plant Sci. 0 (2018). [DOI] [PubMed]
- 35.Gründel M, Scheunemann R, Lockau W, Zilliges Y. Impaired glycogen synthesis causes metabolic overflow reactions and affects stress responses in the cyanobacterium Synechocystis sp. PCC 6803. Microbiology. 2012;158:3032–3043. doi: 10.1099/mic.0.062950-0. [DOI] [PubMed] [Google Scholar]
- 36.Joseph A, et al. Rre37 stimulates accumulation of 2-oxoglutarate and glycogen under nitrogen starvation in Synechocystis sp. PCC 6803. FEBS Lett. 2014;588:466–471. doi: 10.1016/j.febslet.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 37.Zilliges, Y. Glycogen, a dynamic cellular sink and reservoir for carbon. in The cell biology of cyanobacteria (eds Flores, E. & Herrero, A.) 189–210 (Caister Academic Press, 2014).
- 38.Steglich C, Futschik M, Rector T, Steen R, Chisholm SW. Genome-wide analysis of light sensing in Prochlorococcus. J Bacteriol. 2006;188:7796–7806. doi: 10.1128/JB.01097-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tolonen AC, et al. Global gene expression of Prochlorococcus ecotypes in response to changes in nitrogen availability. Mol Syst Biol. 2006;2:53. doi: 10.1038/msb4100087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakamura T, Naito K, Yokota N, Sugita C, Sugita M. A cyanobacterial non-coding RNA, Yfr1, is required for growth under multiple stress conditions. Plant Cell Physiol. 2007;48:1309–1318. doi: 10.1093/pcp/pcm098. [DOI] [PubMed] [Google Scholar]
- 41.Moore LR, et al. Culturing the marine cyanobacterium Prochlorococcus: Prochlorococcus culturing. Limnol. Oceanogr. Methods. 2007;5:353–362. doi: 10.4319/lom.2007.5.353. [DOI] [Google Scholar]
- 42.Stazic D, Lindell D, Steglich C, Antisense RNA. protects mRNA from RNase E degradation by RNA-RNA duplex formation during phage infection. Nucleic Acids Res. 2011;39:4890–4899. doi: 10.1093/nar/gkr037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Afgan E, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018;46:W537–W544. doi: 10.1093/nar/gky379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beyer HM, et al. AQUA Cloning: A Versatile and Simple Enzyme-Free Cloning Approach. PLOS ONE. 2015;10:e0137652. doi: 10.1371/journal.pone.0137652. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data have been deposited in NCBI-SRA under accession numbers SRR8280994, SRR8280995, SRR8280996, SRR8280997, SRR8280998 and SRR8280999.