

Key Points
LL-37 antimicrobial peptides promote macrophage uptake of NETs.
NETs modulate LPS-induced cytokine responses in macrophages and DCs.
NETs are digested intra- and extracellularly by TREX1 and DNase1L3.
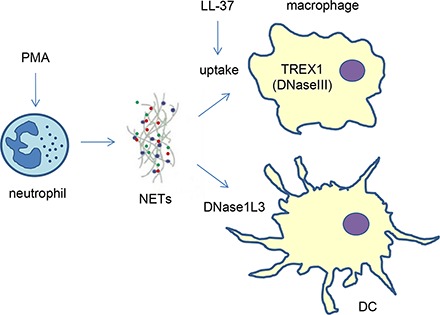
Visual Abstract
Abstract
Neutrophil extracellular traps (NETs) composed of nuclear DNA associated with histones and granule proteins are involved in the extracellular killing of pathogens. Excessive NET formation has been implicated in several noninfectious pathological conditions. The disposal of NETs is, therefore, important to prevent inadvertent effects resulting from the continued presence of NETs in the extracellular environment. In this study, we investigated the interaction of NETs released by freshly isolated, PMA-stimulated primary human neutrophils with primary human monocyte–derived macrophages or dendritic cells (DCs). NETs were internalized by macrophages, and removal of the protein component prevented engulfment of NETs, whereas complexation with LL-37 restored the uptake of “naked” (protein-free) NETs. NETs were also found to dampen the bacterial LPS-induced maturation of DCs. Cytokine profiling was conducted by using a multiplex array following the interaction of NETs with macrophages or DCs, and NETs alone were found to be noninflammatory, whereas immunomodulatory effects were noted in the presence of LPS with significant upregulation of IL-1β secretion, and a marked suppression of other LPS-induced factors including vascular endothelial growth factor (VEGF) in both cell types. Moreover, macrophage digestion of NETs was dependent on TREX1 (also known as DNaseIII), but not DNaseII, whereas extracellular DNase1L3-mediated degradation of NETs was observed for DCs. Collectively, these findings shed light on the interactions between NETs and phagocytic cells and provide new insights regarding the clearance of NETs, double-edged swords of innate immunity.
Introduction
Neutrophil extracellular traps (NETs) have attracted considerable attention in the past decade, with numerous studies aiming to disentangle the mechanisms of NET formation, the antimicrobial function of NETs, and the impact of NET formation in the pathogenesis of various human diseases (1, 2). Hence, whereas NETs are known to capture and/or destroy offending microorganisms, including bacteria and fungi (3), NET formation in the circulation may also promote coagulation, vascular occlusion, and thrombosis (4, 5). NETs were also shown to sequester circulating tumor cells and promote metastasis (6) and to contribute to cancer-associated thrombosis (7). Moreover, NETs triggered by cholesterol were suggested to promote atherosclerosis by priming macrophages for cytokine release, illustrating that danger signals may drive sterile inflammation through their interactions with neutrophils (8). Interestingly, whereas NETs may promote inflammation, aggregated NETs could also limit inflammation through the degradation of cytokines and chemokines (9). Systemic lupus erythematosus (SLE) is a multisystemic autoimmune disease in which patients develop autoantibodies to DNA, histones, and other host structures. Previous studies have shown that a subset of SLE patients degraded NETs poorly because of an impairment of serum endonuclease DNaseI function (10). The latter findings suggested that defective dismantling of NETs could contribute to the occurrence of autoantibodies in SLE patients. Circulating neutrophils from SLE patients also released more NETs than those from healthy donors (11), and NETs extruded by SLE patient neutrophils activated IFN‐producing plasmacytoid dendritic cells (pDCs), one of the main drivers of inflammation and damage in SLE (12). However, whereas an impairment in DNaseI-mediated degradation of NETs was suggested to be associated with an exacerbation of SLE (10, 13), physiological concentrations of extracellular DNaseI are not sufficient to fully degrade NETs (14), indicating that other mechanisms of degradation and/or clearance of NETs are required to prevent inadvertent immune responses (15). Indeed, we have shown in a previous study that human monocyte–derived macrophages (HMDMs) readily internalized and digested NETs released from neutrophils from healthy donors without any proinflammatory effects (14). In this study, we investigated the interactions of PMA-triggered NETs with primary human macrophages and dendritic cells (DCs) and examined the subsequent cytokine responses as well as the role of different intra- and extracellular DNases for their potential involvement in the degradation of NETs. We also asked whether the antibacterial peptide, LL-37 is involved in macrophage uptake of NETs. Previous studies suggested that LL-37 may promote the formation of NETs and demonstrated that the presence of LL-37 in NETs stabilizes the neutrophil-derived DNA against degradation by bacterial nucleases (16, 17).
Materials and Methods
Neutrophil isolation and the purification of NETs
Isolation of neutrophils from buffy coat of healthy human volunteers (Karolinska University Hospital, Stockholm, Sweden) was performed as described earlier (14). Briefly, after density gradient centrifugation with Lymphoprep (Axis-Shield Diagnostics), neutrophils were further separated from erythrocytes via density gradient sedimentation in a 5% dextran solution, followed by hypotonic lysis of the RBCs and washes with PBS. Neutrophils were cultured in phenol red-free RPMI 1640 medium (Sigma-Aldrich) with 2 mM l-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (Life Technologies) in the absence of serum, as FCS contains heat-stable nucleases capable of degrading NETs (18). To trigger NETs, neutrophils were incubated with 25 nM PMA (Sigma-Aldrich) for 2 h at 37°C in 5% CO2. The PMA-containing medium was then removed, and NETs were collected by thorough and repetitive pipetting in fresh medium or HBSS (Life Technologies) and the mixture was centrifuged for 5 min at 1500 rpm. The supernatants containing purified NETs were subsequently retrieved and either used immediately or kept frozen at −20°C for further use.
Monocyte-derived macrophages and DCs
Primary human monocytes were isolated from buffy coat of healthy human donors (Karolinska University Hospital) as previously described (19). In brief, density gradient centrifugation with Lymphoprep (Axis-Shield Diagnostics) was performed to obtain PBMCs, followed by positive selection of CD14+ monocytes using CD14 MACS magnetic beads (Miltenyi Biotec). The cells were then cultured in RPMI 1640 medium (Sigma-Aldrich) supplemented with 10% heat-inactivated FCS (Life Technologies), 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (Life Technologies). To obtain HMDMs, cells were cultured for 3 d with 50 ng/ml human rM-CSF (PeproTech) at 37°C in 5% CO2. To obtain DCs, the medium was supplemented instead with 16 ng/ml human rGM-CSF (PeproTech) and 50 ng/ml human rIL-4 (Life Technologies), and the cells were cultured for 6 d at 37°C in 5% CO2, substituting the culture medium with fresh medium after 3 d.
Cell viability assessment
To evaluate cell viability of HMDMs and monocyte-derived DCs (MDDCs) exposed to NETs, both cell types were incubated with NETs for 1 h at 37°C in 5% CO2, then supernatants were removed and cells cultured in the relevant medium for 24 h, at which time cell viability was evaluated using trypan blue staining, and cells were counted with a TC20 automated cell counter (Bio-Rad Laboratories). Cell viability values are expressed as the percentage of live cells in each sample.
Phenotypic assessment of macrophages and DCs
Bright field microscopy.
To assess morphological changes in DCs exposed to NETs, cells were seeded at a density of 0.5 × 106 cells per well in a 24-well plate and exposed to purified NETs in RPMI 1640 medium, or 100 ng/ml bacterial LPS (Sigma-Aldrich), NETs plus LPS, or RPMI 1640 medium alone for 1 h. Then, cells were washed and the cell medium replaced with fresh medium for 24 h. Cells were then imaged using the bright field option on a Nikon ECLIPSE TE2000-S Inverted Fluorescence Microscope.
Flow cytometric analysis.
For analysis of surface markers, cells from three different donors were isolated and differentiated into MDDC, as indicated above. Cells were then exposed to the respective different stimuli for 1 h, and the medium was removed and replaced with fresh medium for a further 24 h. Samples were then collected, washed with PBS, and resuspended in 1% BSA in PBS for blocking. Each sample was aliquoted and stained separately for 30 min with either FITC-conjugated mouse anti-human CD80 (560926), mouse anti-human CD83 (560929), mouse anti-human CD86 (560958), or mouse IgG1 κ isotype control (555748), all from BD Pharmingen, and used according to the manufacturer’s protocol. The samples were analyzed on a BD Accuri C6 flow cytometer using BD Accuri C6 Software.
Immunofluorescence staining and confocal microscopy
For immunofluorescence staining, neutrophils, macrophages, or DCs were seeded on coverslips at a density of 0.5 × 106 cells per well. For visualization of NETs following a 2 h incubation with 25 nM PMA (Sigma-Aldrich), neutrophils were washed and fixed using 2% paraformaldehyde (PFA) solution in PBS for 30 min, followed by blocking with 2% BSA (Sigma-Aldrich) in PBS for 30 min at room temperature. Incubation with rabbit anti-human cathelicidin (LL-37) primary Ab (ab69484; Abcam) and mouse anti-human myeloperoxidase (MPO) primary Ab (M0748; DAKO) was carried out at 1: 300 dilution in 2% BSA in PBS at room temperature for 1 h, followed by staining with FITC-conjugated sheep anti-rabbit (F7512; Sigma-Aldrich) and Alexa 594–conjugated goat anti-mouse (A11005; Invitrogen) secondary Abs at 1:500 dilution in 2% BSA in PBS for 45 min at room temperature. Coverslips were mounted on microscope glass slides using ProLong Gold Antifade Mountant with DAPI (Molecular Probes). For localization of nucleases, HMDMs were allowed to attach to the glass coverslips for 3 h, and MDDCs were seeded on poly-l-lysine–coated coverslips, then cells were fixed with 2% PFA in PBS for 30 min. Permeabilization with 0.2% Triton X-100 in PBS was performed for 5 min at room temperature, followed by blocking with 2% BSA in PBS for 30 min. Staining with primary Abs was performed at 1:300 dilution in 2% BSA in PBS for 1 h at room temperature. The primary Abs used were the following: mouse anti-human LAMP1 (sc-20011; Santa Cruz Biotechnology), mouse anti-human calreticulin (ab22683; Abcam), goat anti-human EEA1 (sc-6515; Santa Cruz Biotechnology), mouse polyclonal anti-DNaseII (ab168782; Abcam), rabbit polyclonal anti-DnaseII (ab8119; Abcam), rabbit anti-human TREX1 (ab185228; Abcam), and rabbit anti-human DNASE1L3 Ab (ab152118; Abcam). The samples were further incubated with secondary Abs at 1:500 dilution in 2% BSA in PBS at room temperature for 45 min. Secondary Abs used were the following: Alexa 594–conjugated goat anti-mouse (A11005; Invitrogen), FITC-conjugated sheep anti-rabbit (F7512; Sigma-Aldrich), and Alexa 594–conjugated donkey anti-goat (A-11058; Invitrogen). Samples mounted on glass slides were visualized with a ZEISS LSM 510 META or ZEISS LSM 880 confocal microscope (Carl Zeiss, Oberkochen, Germany).
Cytokine and chemokine multiplex measurements
To quantify the release of soluble mediators, cell culture medium from HMDMs on day 3 of differentiation or MDDCs on day 6 of differentiation were removed, and cells were exposed to either control RPMI 1640 medium, RPMI 1640 medium containing purified NETs, LPS (100 ng/ml), or NETs plus LPS for 1 h. Then, cells were washed and incubated in fresh cell culture medium for 24 h. Cells from three different donors were used. After 24 h, supernatants were collected and saved at −80°C for further analysis using the Luminex Bio-Plex Pro Human Cytokine Panel 27-plex (M500KCAF0Y; Bio-Rad Laboratories) according to the manufacturer’s protocol. To perform cluster analysis and generate heatmaps, data were processed with the R software using complete linkage and Euclidean distances as metrics for the dendrograms after quantile normalization of the data (20).
Silencing of DNASEII, TREX1, and DNASE1L3
Gene silencing in HMDMs was conducted using the Amaxa Nucleofector 2B system (Lonza, Basel, Switzerland) and the Amaxa Human Monocyte Nucleofector Kit (VPA-1007; Lonza) according to the manufacturer’s protocol. HMDMs were transfected 2 d after isolation from buffy coats with 3 μM of total small interfering RNA (siRNA) and either Silencer Select Negative Control No.1 siRNA (4390843; Ambion), DNASEII-specific ON-TARGETplus siRNA (5′-UGGUCACAGUGAACUAUGAdTdT-3′), TREX1-specific ON-TARGETplus siRNA pool (5′-ACAAUGGUGACCGCUACGAUU-3′ and 5′-CCAAGACCAUCUGCUGUCAUU-3′) (Dharmacon), or a mixture of DNASEII and TREX1 siRNAs in a 1:1 ratio. After transfection, HMDMs were cultured for another 2 d in complete-cell culture medium supplemented with M-CSF, and silencing efficiency was evaluated by RT-PCR as described below. Experiments were carried out with cells isolated from three different donors. For silencing of DNASE1L3, the Lipofectamine 3000 Reagent (Thermo Fisher Scientific) was used to transfect monocytes, stimulated with GM-CSF and IL-4 for 3 d, with either nontargeting Silencer Negative Control No. 2 siRNA (AM4613; Ambion) or specific ON-TARGETplus DNASE1L3 siRNA pool (L-012694-00; Dharmacon) at a concentration of 75 nM. In brief, cells were seeded in RPMI 1640 medium at a density of 0.3 × 106 cells per well in a 24-well plate, 100 μl/well, and siRNA/Lipofectamine 3000 complexes were incubated for 30 min prior to addition to the cells (100 μl). After 5 h, 200 μl of complete cell medium including GM-CSF and IL-4 was added, and cells were kept in culture for an additional 3 d, then used for further experiments.
Quantitative real-time PCR analysis
Total RNA was extracted from freshly isolated monocytes, HMDMs on days 3 or 4 after isolation, and MDDCs using the QIAGEN RNeasy Mini Kit following the manufacturer’s protocol. Quantification of RNA was performed using NanoDrop (Thermo Fisher Scientific, Whaltham, MA), and 1 μg of total RNA was used for reverse transcription using the RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). Analysis of transcripts of the three different nucleases, as well as downregulation of TREX1, DNASEII, and DNASE1L3, was performed with an Applied Biosystems 7500 Real-Time PCR System using the Power SYBR Green PCR Master Mix (Applied Biosystems); analysis of the data were carried out with the Applied Biosystems 7500 Real-Time PCR software v.2.3. Primers used were from Sigma-Aldrich and were as follows: GAPDH forward 5′-CCCCTTCATTGACCTCAACTAC-3′; GAPDH reverse 5′-GAGTCCTTCCACGATACCAAAG-3′; DNASEII forward 5′-TTCCTGCTCTACAATGACCAAC-3′; DNASEII reverse 5′-GGAAGTTAGGTACACTGTGGACC-3′; TREX1 forward 5′-GCATCTGTCAGTGGAGACCA-3′; TREX1 reverse 5′-AGATCCTTGGTACCCCTGCT-3′; DNASE1L3 forward 5′-GTGCATATGACAGGATTGTG-3′; and DNASE1L3 reverse 5′-AATTCAACTGGAAAGTGGTC-3′. Data were normalized on the endogenous level of GAPDH mRNA. Each experiment was performed with cells derived from three donors.
Western blot analysis
On day 6 of differentiation, culture media was removed, and MDDCs were seeded at a density of 1.0 × 106 cells/ml in either Ca2+- and Mg2+-free HBSS or HBSS containing NETs and cultured for the indicated time-points. At each time-point, supernatants were collected, and pellets were lysed in radioimmunoprecipitation assay buffer. Total protein content was assessed using the BCA assay (Thermo Fisher Scientific); equal amounts of protein from the lysates or equal volumes from the supernatants were loaded in each well on a NuPAGE 4–12% SDS-PAGE Gel (Invitrogen), and proteins were separated on the gel under denaturing conditions. Transfer on a Hybond low-fluorescence 0.2 μm PVDF membrane (Amersham) was performed, and the LI-COR Biosciences REVERT Total Protein Stain was used according to the manufacturer’s protocol to ease normalization on total protein content of the samples. Incubation with primary anti-human DNASE1L3 rabbit Ab (Abcam) was carried out at a 1:1000 dilution overnight at 4°C. After washing, the membrane was probed with the IRDye 800CW goat anti-rabbit secondary Ab for 1 h at room temperature, and detection was performed on an Odyssey CLx LI-COR Biosciences scanner.
Confocal microscopy of cellular uptake of NETs
To assess cellular uptake of NETs, HMDMs after 3 d of differentiation or MDDCs after 6 d of differentiation were seeded on glass coverslips at a density of 0.5 × 106 cells per well in a 24-well plate. In experiments in which the expression of nucleases had been silenced, HMDMs were used 2 d after transfection as described above. Cells were incubated with CellTracker Orange (Molecular Probes) at a final concentration of 0.5 μM for 30 min, then Hoechst 33342 (Sigma-Aldrich) at a final concentration of 5 μg/ml was added for 15 min to stain nuclear DNA. Cells were then washed to remove the dyes, and purified NETs, prestained with the cell-impermeable SYTOX Green dye (Molecular Probes) at a concentration of 5 μM in HBSS for 15 min, were added for 1 h. For the study of intracellular localization of NETs, no CellTracker was added; instead, 30 min prior to fixation, LysoTracker Red (Molecular Probes) at a concentration of 50 nM was added to visualize lysosomes. Samples were washed three times with PBS and fixed with 2% PFA in PBS for 30 min. Coverslips were mounted on microscope glass slides using ProLong Gold Antifade Mountant without DAPI, and samples were analyzed using a ZEISS LSM 880 confocal microscope. For evaluation of uptake of NETs, extranuclear green DNA dots were considered as phagocytosed NETs, and cells containing these dots were counted as positive. At least 200 cells per condition were scored, and the experiment was repeated with three different donors.
Mechanism of NET internalization
To evaluate the role of proteins contained in NETs for the cellular uptake of NETs, HMDMs, isolated as described above, were seeded on coverslips and allowed to attach for 3 h. Staining with CellTracker Orange (red) and Hoechst (blue) was carried out for 30 and 15 min, respectively, and cells were then washed and incubated with SYTOX Green–labeled NETs for 1 h. In some cases, NETs were subjected to protein digestion with proteinase K (200 μg/ml, AM2548; Ambion) for 1 h at 50°C, with subsequent inactivation of proteinase K for 20 min at 65°C prior to incubation with HMDMs. NETs, with or without pretreatment with proteinase K, were also incubated with synthetic LL-37 peptide (10 μM, SP-LL37-1; Innovagen, Lund, Sweden) for 30 min to allow for NET-LL-37 complex formation prior to staining with SYTOX Green. After incubation for 1 h with NETs, the cells were washed and fixed with 2% PFA in PBS and mounted on microscope glass slides using ProLong Gold Antifade Mountant (Molecular Probes). Immunofluorescence staining with LAMP1 was performed as described above. The samples were visualized using a ZEISS LSM880 confocal microscope.
Extracellular degradation of NETs
To assess extracellular degradation of NETs, we analyzed samples using agarose gel electrophoresis. To this end, 1 ml of purified NETs in HBSS without Ca2+ and Mg2+ were added to 1.0 × 106 MDDCs on day 6 of differentiation or to MDDCs transfected with control siRNA or specific DNASE1L3 siRNA, as described above, and incubated for the indicated time-points. Supernatants were collected, and enzymatic activity of Dnase1L3 was promoted by addition of 2 mM CaCl2 and 2 mM MgCl2 as described (21). The degradation was carried out for 1 h at 37°C, and the samples were subjected to a 1% Tris-acetic acid-EDTA-agarose gel electrophoresis, followed by staining of the gel using SYBR Green I dye at 1:10,000 concentration and visualization with a Molecular Imager scanner (Bio-Rad Laboratories).
Public microarray database analysis
Data on the expression of DNASE1L3 across different human tissues was extracted from the public in silico transcriptomics database (IST Online) that contains data from almost 10.000 microarray gene expression analyses of human tissues (http://ist.medisapiens.com) (22).
Statistical analysis
Statistical analysis was performed using the GraphPad Prism software for Windows version 5.02 (GraphPad Software, San Diego, CA). One-way ANOVA was applied for the comparison of multiple groups, and Tukey posttest was employed to assess statistical significance comparing each group against all the other groups. Where specified, comparison between two groups was carried out using two-tailed Student t test. Statistical significance is indicated in each figure as the following: *p < 0.05, **p < 0.01, ***p < 0.001.
Results
Uptake of NETs by primary HMDMs
NET release by primary human neutrophils is a well-studied phenomenon, and a variety of different stimuli have been applied to trigger the production of NETs (23, 24). In this study, we triggered NETs in freshly isolated neutrophils by stimulation with 25 nM PMA for 2 h, and we were able to consistently detect fiber-like NETs as evidenced based on staining for DNA and the neutrophil granule protein, MPO (Fig. 1A). The antimicrobial peptide LL-37, a key component of NETs, was also found on PMA-triggered NETs (Fig. 1A). Uptake of NETs in HMDMs has previously been reported, and visualization of NETs in macrophages was performed by staining with the cell-permeable dye, DAPI (14). In the current study, we used the more sensitive, cell-impermeable SYTOX Green dye for the detection of NETs following a 1 h incubation with cells. LysoTracker Red was added to visualize lysosomes, and cell nuclei were stained by using Hoechst 33342. In this manner, internalized NETs were visualized as extranuclear dots in HMDMs and, to a lesser extent, in MDDCs (refer to Fig. 1B for representative images). NET-positive HMDMs were typically 5% of the whole population, but this varied depending upon the donor. NETs were not colocalized with lysosomes in HMDMs or MDDCs, as evidenced by the absence of colocalization with LysoTracker Red (Fig. 1B). To investigate this further, we also performed colocalization studies using specific Abs against EEA1 and LAMP1, markers of early and late endosomes, respectively, but we could not detect any NETs in these compartments (Supplemental Fig. 1A, 1B).
FIGURE 1.

NETs are engulfed by monocyte-derived phagocytes. (A) Freshly isolated primary human neutrophils from healthy adult donors were exposed to 25 nM PMA for 2 h. NETs were detected by immunofluorescence staining of the following components: MPO (red), LL-37 (green), and DNA (blue). Scale bars are 20 μm for the upper and 10 μm for the lower panels. (B) HMDMs and MDDCs were incubated with Hoechst 33342 for 15 min to visualize nuclear DNA, whereas purified NETs were prestained with the cell-impermeable DNA dye SYTOX Green for 15 min. HMDMs or MDDCs were then washed to remove the Hoechst staining and incubated with SYTOX Green–labeled NETs for 1 h. Thirty minutes prior to the end of incubation, LysoTracker Red was added to visualize lysosomes. This dual DNA staining allows for the differentiation between internalized NETs (arrows) and the cell nuclei of the phagocytic cells. Confocal images are representative of three independent experiments. Scale bars in (B) are 5 μm for all panels. Refer to Supplemental Fig. 1 for results on costaining with EEA1 and LAMP1. (C and D) Cell viability of HMDMs (C) and MDDCs (D) exposed to NETs or control media for 1 h, then cultured in the relevant culture medium for 24 h prior to the assessment of viability. Data shown are mean values ± SD of two independent experiments.
Previous studies have shown that prolonged exposure to NETs can compromise cell viability of macrophages and DCs (25). Indeed, when HMDMs (Fig. 1C) or MDDCs (Fig. 1D) were exposed to purified NETs for 1 h, followed by cell culture in normal medium for 24 h, we observed a significant reduction in cell viability in both cell types.
Contribution of antimicrobial peptides to the cellular uptake of NETs
We previously reported that macrophage clearance of NETs could be reduced to some extent by pretreatment of the cells with cytochalasin D, suggesting the involvement of an actin cytoskeleton-dependent endocytosis/phagocytosis mechanism (14). To further probe the mechanism of uptake of NETs, and to address the potential role of proteins contained in the NETs, we subjected purified NETs to proteinase K treatment. NETs were then stained with SYTOX Green and incubated with HMDMs for 1 h. CellTracker Orange was used to uniformly stain the macrophages, and cell nuclei were stained by using Hoechst 33342. As depicted in Fig. 2A, the uptake of NETs, as evidenced by the presence of extranuclear dots, was abrogated when protein-free NETs were added. However, when LL-37 was allowed to complex with the protein-free NETs prior to incubation with macrophages, uptake of NETs was noted (Fig. 2A). Moreover, LL-37 significantly promoted the uptake of NETs that had not been pretreated with proteinase K, and the nuclear SYTOX Green staining was intensified, suggesting that NETs complexed with LL-37 may have translocated to the nucleus (Fig. 2A, 2B). We also found that cytochalasin D blocked uptake of NETs by HMDMs, as shown before (14), and that the uptake was partially restored by LL-37 (Fig. 2C). We confirmed the extralysosomal localization of internalized NETs following their complexation with LL-37 by costaining of the cells with LAMP1 (Supplemental Fig. 2A). Interestingly, IFN-β mRNA expression was induced when HMDMs were exposed to NETs complexed with exogenous LL-37, but not in the presence of LL-37 alone or NETs alone (Supplemental Fig. 2B).
FIGURE 2.

Macrophage uptake of NETs is facilitated by LL-37. (A) Representative confocal images of HMDMs prestained with Cell Tracker Orange (red) for 30 min and counterstained with Hoechst 33342 (blue) for 15 min to identify the perimeter of the cell and cell nuclei, respectively. Purified NETs were subjected to pretreatment with proteinase K (200 μg/ml) or untreated, and LL-37 (10 μM) was added as indicated prior to staining with SYTOX Green for 15 min, followed by incubation with HMDMs for 1 h. Scale bar, 10 μm. Refer to Supplemental Fig. 2 for results on costaining with LAMP1. (B) Quantification of macrophage uptake of NETs. HMDMs were exposed as described above. For each condition, at least 200 cells were scored and normalized to the samples transfected with control siRNA. (C) Quantification of macrophage uptake of NETs following pretreatment of cells with cytochalasin D (10 μg/ml), followed by addition of LL-37 (10 μM). The results in (B) and (C) are presented as average values of three independent experiments performed with cells from three different donors ± SEM. One-way ANOVA with Tukey posttest was used to compare the groups (where not specified, the differences did not achieve significance). *p < 0.05, **p < 0.01.
Phenotypic changes in DCs incubated with NETs with/without LPS
We then investigated how HMDMs and MDDCs respond to purified NETs. We also sought to investigate whether NETs modulated the response of HMDMs and MDDC to endotoxin. Following exposure of HMDMs and MDDC to NETs, we could observe morphological changes in both cell types. HMDMs exposed to NETs presented with cytosolic vacuoles, compatible with vesicular trafficking because of uptake of NETs, whereas this morphology was not observed in cells exposed to LPS (100 ng/ml) (data not shown). Untreated MDDCs were nonadherent or loosely adherent to plastic, and the cells displayed short dendrites, but when exposed to NETs, MDDCs became strongly adherent and produced long dendrites (Fig. 3A). LPS treatment did not elicit obvious morphological changes in MDDCs, and the cells maintained a nonadherent phenotype and were partially clustered. In the presence of both LPS and purified NETs, a combined phenotype was observed in MDDCs, with cells organizing in clusters of adherent cells and dendrites were also seen (Fig. 3A). We also studied surface markers of activation and maturation on MDDCs. LPS induced significant upregulation of all markers (CD80, CD83, and CD86), whereas NETs decreased the expression of CD80 and seemed not to affect CD83 and CD86 (Fig. 3B, 3C). Notably, when MDDCs were exposed to both NETs and LPS (100 ng/ml), the LPS-induced upregulation of all the examined markers was significantly dampened (Fig. 3B, 3C). To exclude any effects of residual PMA, HMDMs, or MDDCs were exposed to a range of concentrations of PMA (0.025–25 nM), but we did not detect any loss of cell viability, nor did we observe changes in the expression of CD80, CD83, or CD86 in MDDCs (data not shown).
FIGURE 3.

NETs induce phenotypic changes in DCs stimulated with LPS. (A) Representative bright field images at original magnification ×20 of MDDCs exposed to control medium, or medium containing purified NETs for 1 h, followed by 24 h of culture in the relevant medium. (B) Flow cytometry analysis of MDDCs exposed for 1 h to either LPS (100 ng/ml), control RPMI medium, RPMI medium containing purified NETs or NETs plus LPS, followed by 24 h culture in the relevant media. Histograms display representative results from one experiment, with the black line indicating isotype control and colored lines indicating CD80 (blue), CD83 (green), and CD86 (purple), respectively. (C) Quantification of three independent experiments performed as described above. Data shown as average mean fluorescence intensity (MFI) values ± SEM of three experiments performed with different human donors. One-way ANOVA with Tukey posttest for comparison of all groups was carried out to evaluate statistical significance (where not specified, the differences between groups were NS). *p < 0.05, **p < 0.01, ***p < 0.001.
Immunomodulatory effects of NETs in cells coexposed to LPS
We then performed profiling of cytokines, chemokines, and growth factors in both HMDMs and MDDCs exposed to purified NETs in the presence or absence of LPS for 1 h, followed by continued culture for 24 h in fresh culture medium. Cells derived from three different donors were used for these studies, and profiling was performed on the supernatants of exposed cells using the Luminex multiplex assay platform. Overall, NETs alone did not generally induce any cytokine production in HMDMs and MDDCs in relation to control, with the exception of the IL-1R antagonist (IL-1ra), for which the secretion was increased, albeit not to a statistically significant level (Fig. 4A, 4B). Interestingly, coexposure to NETs and LPS (100 ng/ml) triggered a pronounced release of IL-1β in both cells types (Fig. 4A, 4B), suggestive of inflammasome activation (26). Furthermore, in cells exposed to NETs plus LPS, NETs reduced the LPS-triggered release of IL-10, an immunomodulatory cytokine, and IL-12, a T cell stimulatory factor, both in HMDMs and MDDCs (Fig. 4A, 4B). With respect to chemokines, we noted that NETs alone triggered the secretion of IL-8 (also known as CXCL8) in both HMDMs and MDDCs (Fig. 4A, 4B). NETs also induced the release of MIP-1α (CCL3) and MIP-1β (CCL4) by HMDMs and MDDCs, whereas the LPS-triggered secretion of IP-10 (also known as CXCL10) was abolished by coincubation with NETs in the case of HMDMs and significantly reduced in the case of MDDCs (Supplemental Fig. 3). Finally, with regards to growth factors, purified NETs alone did not elicit the release of any of the growth factors included in the panel, whereas incubation with NETs plus LPS triggered the secretion of G-CSF and GM-CSF in HMDMs and the secretion of G-CSF in MDDCs, although not to a statistically significant degree (Supplemental Fig. 3). However, we noted that NETs completely abolished the LPS-triggered release of vascular endothelial growth factor (VEGF; originally known as a vascular permeability factor) in both cell types (Fig. 4A, 4B). To highlight differences or similarities between the different conditions, we performed hierarchical cluster analysis (20). For both cell types, exposure to NETs alone clustered together with the medium control, in line with the notion that purified NETs are, in general, immunologically “inert” (14), whereas exposures to LPS or NETs plus LPS were more closely related (refer to vertical dendrograms in Fig. 5A, 5B). For HMDMs, NETs plus LPS shifted the cytokine secretion profile when compared with LPS (horizontal dendrogram, Fig. 5A).
FIGURE 4.

Cytokine profiling of HMDMs and MDDCs exposed to NETs and/or LPS. Profiling of HMDMs (A) and MDDCs (B) exposed to LPS (100 ng/ml), purified NETs, LPS plus NETs, or control RPMI medium for 1 h, and then to the relevant culture media for 24 h. After 24 h, supernatants were collected and used for quantification with the Luminex platform. Data are displayed as average values of three independent experiments with cells from three different blood donors ± SEM. One-way ANOVA with Tukey posttest for comparison of all groups was carried out to evaluate statistical significance (where not specified, the differences between groups were NS). Refer to Supplemental Fig. 3 for additional results. *p < 0.05, **p < 0.01, ***p < 0.001.
FIGURE 5.

Hierarchical clustering of cytokine responses to LPS or NETs. Heatmaps depicting the hierarchical cluster analysis of chemokines, cytokines, and growth factors released by HMDMs (A) and MDDCs (B) in response to cell medium alone, purified NETs, LPS (100 ng/ml), or NETs plus LPS. Cells were exposed for 1 h, and cells were then washed and cultured for a further 24 h in fresh medium followed by analysis. The results were processed as described in Materials and Methods. Horizontal and vertical dendrograms in (A) and (B) indicate the degree of similarity between the samples/conditions, with shorter branches designating a higher similarity.
Expression of different DNases in phagocytic cells
We then asked which nucleases are involved in the degradation of NETs. Macrophage-internalized NETs were previously presumed to be localized partly in lysosomes (14). However, using SYTOX Green for the detection of NETs instead of DAPI, we observed that NETs, visible as extranuclear dots, were present outside lysosomes (Fig. 1B). We therefore sought to study nucleases that are known to be active in nonlysosomal compartments. DNaseII is a lysosomal nuclease involved in erythropoiesis and apoptosis, whereas TREX1 (DNaseIII), in contrast, is an endoplasmic reticulum–bound nuclease with cytosolic activity (27). DNase1L3 (also known as DNase γ), in turn, is produced and secreted extracellularly mainly by tissue resident macrophages and DCs (28). We examined the mRNA expression of DNASEII, TREX1, and DNASE1L3 in primary human monocytes, M-CSF–stimulated monocyte-derived macrophages (HMDMs), and IL-4– and GM-CSF–MDDCs (Fig. 6A–C). The expression of DNASEII in MDDCs was found to be much lower than in HMDMs, and TREX1 expression was also expressed mainly in HMDMs, but not in MDDCs. However, DNASE1L3 was prominently expressed in MDDCs in comparison with HMDMs or monocytes (Fig. 6C), suggesting that this nuclease may be especially important in MDDCs. Next, we employed confocal microscopy to study the cellular expression and localization of DNaseII (Fig. 6D) and TREX1 (Fig. 6E) in HMDMs, and DNase1L3 in MDDCs (Fig. 6F). We confirmed the expression of both DNaseII and TREX1 in HMDMs. However, contrary to our expectations, DNaseII was not colocalized with LAMP1. We used two different Abs, a mouse polyclonal and a rabbit polyclonal, and we obtained similar results (data not shown). It remains possible that DNaseII is trafficked to lysosomes upon cellular activation, as suggested in a recent study (29). TREX1 was not colocalized with the lysosomal marker, LAMP1 (Fig. 6E), or with calreticulin (data not shown). Interestingly, DNase1L3 was found to have both a nuclear and extranuclear localization in MDDCs, in line with the fact that this enzyme possesses nuclear localization signal motifs (30), whereas it is also a secreted protein (28).
FIGURE 6.

Expression and localization of DNases in phagocytes. (A–C) mRNA expression of DNASEII (A), TREX1 (B), and DNASE1L3 (C) in freshly isolated monocytes and monocytes differentiated with M-CSF for 3 or 4 d (HMDMs) or GM-CSF plus IL-4 for 6 d (MDDCs). Target mRNA was normalized to GAPDH, and mRNA relative quantity of freshly isolated monocytes was set to one, and normalization was carried out to simplify the comparison between the different treatments. The data are displayed as average values of independent experiments with samples from three adult donors ± SEM. One-way ANOVA with Tukey posttest for comparison of all groups was carried out to evaluate statistical significance (where not specified, the differences between groups were NS). **p < 0.01. (D–F) Representative confocal images from three independent experiments showing the expression and subcellular localization of the respective nucleases. HMDMs on day 3 of differentiation were stained for DNaseII (green) (D) or TREX1 (E) (green), LAMP1 (red) for lysosomes, and counterstained with DAPI (blue) to visualize cell nuclei. Scale bar, 10 μm. Results with isotype-matched control Abs were negative (data not shown). (F) MDDCs on day 6 of differentiation were stained for DNASE1L3 (green), LAMP1 (red) (lysosomes), and counterstained with DAPI (blue). Scale bar, 10 μm.
TREX1, but not DNaseII, involved in degradation of NETs in macrophages
With the purpose to assess the involvement of these nucleases in the degradation of NETs, we optimized a model in which expression of DNASEII and/or TREX1 is transiently downregulated in differentiated primary human macrophages by the use of specific siRNAs. HMDMs were transfected with control siRNA, specific DNASEII siRNA or specific TREX1 siRNA, or a pool of siRNAs targeting both DNASEII and TREX1 simultaneously. We successfully achieved downregulation of DNASEII using both the specific siRNA and the siRNA pool (Fig. 7A) as well as relevant downregulation of TREX1 with both the specific siRNA and the siRNA pool (Fig. 7B). Using this model, we performed phagocytosis assays of purified NETs prelabeled with SYTOX Green and noticed that TREX1-deficient HMDMs displayed a greater number of undigested NETs compared with HMDMs transfected with control siRNA or DNASEII siRNA (Fig. 7C, Supplemental Fig. 4). To quantify these results, we performed the same experiment using cells obtained from three different donors and scored all the samples (Fig. 7D). We observed that silencing of DNASEII did not affect degradation of NETs, whereas silencing of TREX1 alone, or in combination with DNASEII, resulted in approximately twice as many cells with undigested NETs (Fig. 8D).
FIGURE 7.

TREX1 is involved in intracellular degradation of NETs. (A) mRNA expression of DNASEII in HMDMs transfected with control siRNA, DNASEII siRNA, or DNASEII plus TREX1 siRNA simultaneously as described in Materials and Methods. Data are expressed as mRNA quantity normalized to GAPDH transcripts and values for the control siRNA sample were arbitrarily set to 100 and normalization carried out to simplify the comparison. Results are presented as mean values of three independent experiments carried out with cells from three adult donors ± SEM. One-way ANOVA with Tukey posttest was used to compare all the groups and evaluate statistical significance (where not specified, the differences between groups were found to be NS). (B) mRNA quantification of TREX1 transcripts in HMDMs transfected with TREX1 siRNA, DNASEII plus TREX1 siRNA simultaneously, or control siRNA. Data were reported as described in (A). (C) Representative confocal images of HMDMs transfected with either TREX1 siRNA or control siRNA cocultured with NETs carried for 1 h to evaluate internalization of NETs. HMDMs were stained with CellTracker Orange (red), NETs with SYTOX Green, and cell nuclei are shown in blue (Hoechst 33342). Arrows in the upper panels at lower magnification indicate cells presenting SYTOX Green–positive extranuclear NETs, whereas in the lower panels, arrows are pointing at undigested intracellular NETs (green dots). Scale bars are 20 μm for the upper and 10 μm for the lower panels. Refer to Supplemental Fig. 4 for DNASEII siRNA and DNASEII plus TREX1 siRNA results. (D) Quantification of the internalization of NETs by HMDMs transfected as indicated. For each condition, at least 200 cells were scored and normalized to the samples transfected with control siRNA. The results are presented as average values of three independent experiments performed with cells from three donors ± SEM. One-way ANOVA with Tukey posttest was used to compare all the groups (where not specified, the differences were NS). *p < 0.05, **p < 0.01, ***p < 0.001.
FIGURE 8.

Secreted DNase1L3 degrades NETs extracellularly. Western blots of cell lysates from pellets (A) and supernatants (B) of MDDCs cultured in HBSS or exposed to HBSS containing purified NETs for 1, 2, 6, and 24 h. Membranes were probed with Abs against human DNase1L3 (predicted molecular mass = 36 kDa). Note that two bands were observed in cells and only one band in the supernatants. Equal loading was verified by using a total protein normalization protocol (data not shown). (C) Expression of DNASE1L3 in MDDCs transfected with control siRNA or DNASE1L3 siRNA. Data are expressed as mRNA normalized to GAPDH, and the control siRNA values were arbitrarily set to 100, and normalization was carried out to simplify the comparison. Results shown are mean values ± SEM. **p < 0.01. (D) NET-containing supernatants retrieved from the same experiments as in (B), or supernatants retrieved from MDDCs transfected with either control siRNA or DNASE1L3 siRNA as in (C) containing NETs were run on a 1% agarose gel after promoting DNase1L3 enzymatic activity by addition of 2 mM CaCl2 and 2 mM MgCl2 for 1 h. NETs alone were loaded as a control (left gel). The bands just below the loading wells indicate nondigested NETs. The results shown are representative of three similar experiments using cell supernatants from different donors. (E) Expression of human DNASE1L3 was derived from the IST microarray database (22). Each box represents the quartile distribution (25–75%) range with the median value indicated with a horizontal line. The 95% range including individual outlier samples is also displayed. The y-axis indicates the relative gene expression levels.
DNase1L3 is secreted by human DCs and is capable of degrading NETs
As we identified DNase1L3 to be the relevant DNase expressed by MDDCs, we sought to confirm its role in the digestion of NETs in our model. For this purpose, MDDCs on day 6 of differentiation were kept in culture in HBSS for up to 24 h, with sampling at 1, 2, 6, and 24 h. Both pellets and supernatants were collected at these time-points, and protein extracts were used for Western blot analysis of DNase1L3. For supernatants, equal volumes of samples were loaded onto the gel, and assessment of total loaded protein was performed for both pellets and supernatant samples using the LI-COR Biosciences REVERT Total Protein Stain normalization protocol (data not shown). DNase1L3 was detected at all time-points in the cell pellets (Fig. 8A), and a slight increase over time was seen in the protein expression in the supernatants (Fig. 8B). We also evaluated the effect of HBSS containing purified NETs and observed a decrease of DNase1L3 expression in the cell pellets, and an increase of the protein in the supernatants, compared with supernatants from cells maintained in HBSS alone (Fig. 8A, 8B), suggesting that NETs induced the secretion of DNase1L3 in this model. Interestingly, when examining the cell pellets, we could detect two separate bands (Fig. 8A), possibly indicating that two forms of DNase1L3 are expressed in MDDCs, whereas for the supernatants, only one band was present, suggesting that only one of the two (putative) isoforms of DNase1L3 is released (Fig. 8B).
Having established that MDDCs are capable of releasing DNase1L3, we monitored the degradation of NETs in supernatants of these cells. Hence, we observed a time-dependent degradation of NETs in the supernatants. The time-dependent disappearance of the high-m.w. band at the top of the gel, corresponding to nondegraded DNA, indicated that NET degradation had taken place (Fig. 8D, left panel). To confirm a role for DNase1L3 in this model, MDDCs were transfected with specific siRNA–targeting DNASE1L3 versus control siRNA, and silencing was confirmed by RT-PCR (Fig. 8C). We then performed experiments with purified NETs and noted that the DNA was degraded by supernatants from MDDCs transfected with control siRNA, whereas supernatants from MDDCs with diminished DNASE1L3 expression were ineffective (Fig. 8D, right panel). Finally, we have shown that DNASE1L3 is expressed in MDDCs, but not in HMDMs (Fig. 6C), whereas other reports have indicated that DNase1L3 is also produced by macrophages (28). To shed light on this, we queried a public microarray database (22) and found that DNASE1L3 mRNA is abundantly expressed in DCs when compared with other blood cells (Fig. 8E). High expression of DNASE1L3 was also noted in liver and spleen, in line with a previous study showing expression of DNASE1L3 in DCs and in tissue resident macrophages (28).
Discussion
Nucleic acid degradation is required to prevent inadvertent autoimmune responses, as evidenced by the fact that mutations in various nucleases, including DNaseI, DNase1L3, and TREX1 (DNaseIII), are associated with inherited forms of SLE (31–33). Mice deficient for these nucleases also develop lupus-like disease apparently because of the persistence of nucleic acids (28, 34, 35). Mice deficient for DNaseII, a lysosomal nuclease, display defective erythropoiesis, and DNaseII in macrophages was suggested to be responsible for destroying the nuclear DNA that is expelled from erythroid precursors (36). Previous work has shown that nuclear chromatin present in microparticles released from apoptotic cells serves as a potential source of autoantigen in SLE (28). However, less is known about the degradation of nuclear DNA present in NETs. DNaseI present in serum is capable of digesting NETs, and several pathogens deploy similar nucleases to escape NETs and may even repurpose NETs to trigger cell death (37). We have previously reported that preprocessing of NETs with DNaseI facilitates their clearance by macrophages; however, DNaseI is not sufficient to degrade NETs (14), suggesting that other complementary systems are needed to prevent deleterious effects of NETs. In the current study, we show that TREX1, but not DNaseII, is required for degradation of NETs in HMDMs. These results expand on the repertoire of potential substrates for TREX1 (38, 39). We also provide evidence that DNase1L3 released from DCs is involved in the extracellular degradation of NETs (Fig. 8D). During the preparation of the current manuscript, it was reported that DNaseI and DNase1L3 cooperated in the degradation of NETs in circulation, thereby preventing vascular occlusion by NETs during severe bacterial infections (40). Taken together, DNase1L3 emerges as an important extracellular NET-degrading nuclease, alongside DNaseI, whereas TREX1, but not DNaseII, is implicated in the intracellular degradation of NETs following internalization of NETs by macrophages. DNase1L3 may also participate in the intracellular degradation of DNA from apoptotic cells (41), whereas DNaseI has been implicated in the disposal of necrotic cells (42), both potential sources of autoantigens. The fact that so many endo- and exonucleases participate in DNA degradation testifies to the importance of avoiding unscheduled immune responses to this ubiquitous self-antigen.
We successfully demonstrated the expression of DNaseII in the current model, but DNaseII failed to colocalize with the lysosomal marker, LAMP1. DNaseII, also known as acid DNase, functions optimally at acidic pH and is believed to be present in lysosomes. However, Chan et al. (29) recently reported that a majority of DNaseII was expressed outside the lysosomes, whereas DNaseII trafficking into lysosomes was induced by CpG-A stimulation. We believe that further studies are needed to (re)evaluate DNaseII localization and its potential trafficking in human macrophages. Nevertheless, our data showed that silencing of DNaseII did not affect degradation of NETs.
We found that DNASE1L3 was highly expressed in MDDCs, but not in macrophages, in line with the recent report by Sisirak et al. (28), who demonstrated expression of DNase1L3 in DCs in humans and mice by microarray analysis, whereas expression in macrophages was apparent only in select tissues (liver, spleen, and intestines). The authors concluded that circulating DNase1L3 is predominantly produced by DCs and by certain tissue macrophages (28). Indeed, our results showed that DNase1L3 can digest the DNA component of NETs (i.e., nuclear DNA associated with histones), and we found that exposure of DCs to NETs promoted the release of DNase1L3 from these cells. Unlike other DNase I family members, DNase1L3 contains nuclear localization signals (30). Based on the present observation regarding the localization of DNase1L3 in the nucleus and cytosol of DCs, one may speculate that two isoforms of DNase1L3 exist, and that the nuclear isoform is devoted to chromatin fragmentation in cells undergoing apoptosis (43), whereas the fraction present in the cytosol is released constitutively or in response to relevant stimuli, such as NETs, to digest DNA extracellularly. Alternatively, deletion of the N-terminal signal peptide may allow the protein to be distributed in the nucleus and cytosol, as shown for murine DNase1L3 (21).
Lande et al. (11) reported that the antibacterial peptide LL-37 protected NETs from degradation by DNaseI, thus contributing to the exacerbation of SLE. In addition, it has been demonstrated that mitochondrial DNA complexed with LL-37 is resistant to degradation by DNaseII, leading to activation of TLR9-mediated inflammatory responses (44). Furthermore, LL-37 was suggested to transfer genomic DNA from the extracellular environment to the cytosol (45) and may also shuttle plasmid DNA into the cell nucleus (46). These findings are intriguing in light of the fact that we observed a nonlysosomal localization of NETs in macrophages in our study, raising the possibility that NETs are “transfected” into the cytoplasm of macrophages by LL-37. In fact, our data show that macrophage uptake is dependent on the protein component of NETs purified from primary human neutrophils, and we were able to demonstrate that the addition of LL-37 restored uptake of “naked” NETs (i.e., purified, protein-free NETs). LL-37 also enhanced the uptake of NETs in cells exposed to cytochalasin D. It is instructive to note that previous mass spectrometry studies have shown that that the uptake of naked DNA is aided by the presence of LL-37, as well as HMGB1 and histones, all known components of NETs (47). However, citrullination of LL-37 may abolish its ability to bind DNA, and this may influence the inflammatory potential of NETs (48). Nevertheless, the available data suggest that LL-37 not only facilitates the internalization of NETs into cells, but may also protect NETs against degradation by bacterial and cellular nucleases. We have also noticed that cell nuclei in TREX1 siRNA-treated cells displayed a stronger SYTOX Green signal upon uptake of NETs. Although we cannot exclude that this could be an artifact, it is interesting to speculate that the downregulation of TREX1 and/or the complexation of NETs with LL-37 leading to the persistence of undigested DNA in the cytosol may favor the translocation of NETs into the nucleus. For comparison, horizontal gene transfer via apoptotic bodies has been reported (49), and DNaseII together with the Chk2, p53, and p21 were suggested to form a genetic “barrier” blocking the replication of potentially harmful DNA introduced via such apoptotic bodies (50). Perhaps TREX1 serves a similar function with respect to NETs.
Carmona-Rivera et al. (51) recently reported that synovial fibroblasts are capable of internalizing NETs into EEA1-positive compartments in an actin cytoskeleton-independent manner (i.e., insensitive to cytochalasin D). The authors suggested that fibroblasts internalized NETs through a RAGE/TLR9 pathway, promoting an inflammatory phenotype and upregulation of MHC class II in these cells (51). Our studies, using primary human macrophages, did not disclose any colocalization of internalized NETs with endosomes or lysosomes, and silencing of DNaseII did not have an impact on the intracellular degradation of NETs. We found, instead, that silencing of TREX1 resulted in a marked reduction in NET degradation. The cellular localization of TREX1 was previously shown to be determined by a transmembrane domain in the C terminus of the protein, and mutations in this region have been shown to affect its localization, but not the nuclease activity of the protein (32, 52). Mislocalization would, nevertheless, deprive immune-competent cells of a cytosolic DNA digestion machinery. The discovery that TREX1 is involved in the degradation of NETs was somewhat unexpected, as a previous study has shown that oxidative damage of DNA purified from NETs conferred resistance to TREX1 degradation and potentiated stimulator of IFN genes (STING)–dependent immune sensing (53). However, in our study, we used “pristine” NETs purified from PMA-triggered neutrophils, without DNA extraction of the DNA component, as oxidative damage may arise from purification methods (54), and we could show that TREX1 is required for intracellular degradation of NETs in macrophages. These results do not exclude that oxidized genomic DNA (53) or mitochondrial DNA (55) may serve as critical danger signals in chronic inflammatory or autoimmune diseases.
Beyond their antimicrobial function (56), it is also important to understand the potential immunomodulatory role of NETs. We previously reported that macrophages are capable of internalizing NETs in a “silent” manner [i.e., without a proinflammatory response (14)], whereas others have found that NETs generated after neutrophil activation by Leishmania promastigotes interfered with IL-4/GM-CSF–driven differentiation of monocytes, resulting in reprogramming into anti-inflammatory macrophages (57). In contrast, previous work has shown that NETs trigger proinflammatory IL-1β secretion in macrophages primed with LPS, and this was enhanced in macrophages from patients with SLE (26, 58). Furthermore, macrophage-like THP-1 cells were shown to display a phenotype-dependent response after degradation of NETs insofar as M2 macrophages (i.e., IL-4–stimulated cells) displayed a proinflammatory response, whereas M1 macrophages (i.e., IFN-γ plus LPS–stimulated cells) underwent cell death (59). In the current study, we confirmed IL-1β secretion in monocyte-derived, M-CSF–stimulated macrophages upon coexposure to LPS, and we also showed that DCs derived from IL-4/GM-CSF–stimulated monocytes produced IL-1β when coexposed to NETs and LPS. Furthermore, we could detect a significant release of the chemokine, IL-8/CXCL8, as well as MIP-1α (CCL3) and MIP-1β (CCL4), but not RANTES (CCL5) or IP-10/CXCL10, in response to NETs, both in HMDMs and MDDCs. These findings suggest that NETs have the potential to elicit production of chemotactic factors that could attract neutrophils and other phagocytes to the site of infection. Thus, to clarify, our previous work (14) demonstrated that the clearance of NETs by macrophages is immunologically “silent” in the sense that this process does not trigger proinflammatory cytokines, and the present study, using both HMDMs and MDDCs, also supports the view that NETs are not proinflammatory. Nevertheless, one may conclude that NETs are immunomodulatory. Barrera-Vargas et al. (60) reported, as the current study was underway, that stimulation with LPS-induced NETs led to an enhanced production of TNF-α and IL-10 in macrophages from patients with SLE when compared with controls, and this was dampened when macrophages were treated with chloroquine, an inhibitor of endosomal acidification and lysosomal enzyme activity. Because acidic pH of endosomes is a prerequisite of endosomal TLR activation, chloroquine is frequently used as an antagonist for endosomal TLRs, and the results thus suggest that TLR signaling is partly implicated (60). Moreover, these findings underscore that responses to NETs in healthy individuals and in patients with autoimmune diseases may differ. Papadaki et al. (61) recently reported that exposure of DCs to NETs from mice with collagen-induced arthritis induced DC maturation characterized by upregulation of costimulatory molecules, and NETs from rheumatoid arthritis patients also had the potential to induce the maturation of DCs from healthy individuals. In contrast, NETs from healthy individuals did not alter the maturation status of DCs (62). We confirmed that NETs from healthy donors did not affect maturation of MDDCs and, moreover, that the LPS-induced upregulation of CD80, CD83, and CD86 was greatly suppressed by NETs. The potential impact of residual amounts of PMA in the purified NET samples was excluded (data not shown). NETs were also able to decrease the effect of LPS on the secretion of IL-10 and IL-12 significantly. In contrast, NETs promoted the production of IFN-γ, a type II IFN, in MDDCs coexposed to LPS, whereas NETs alone did not induce significant amounts of IFN-γ. There is a large and consistent body of evidence supporting the presence of dysregulated IFN-related pathways in SLE (63), and a previous study has shown that NETs produced by neutrophils from SLE patients activated pDCs to produce IFN-α, a type I IFN (12). Type I IFN, in turn, primed neutrophils for NET release in patients with SLE, suggesting a positive feedback loop. The role of NET-induced type II IFN release in DCs exposed to LPS remains to be studied, but it is noted that not only IFN-α, but also IFN-γ, is implicated in the pathogenesis of SLE (63). We also found that NETs remarkably abolished LPS-triggered release of VEGF both in HMDMs and MDDCs. Macrophages are not only involved in “waste disposal,” but also play a key role in the regulation of tissue regeneration with secretion of various growth factors, including VEGF, that promote proliferation of neighboring cells (64). Macrophage phagocytosis of apoptotic cells was shown to lead to VEGF secretion and promotion of the proliferation of endothelial cells (65). One may speculate on the implications of the marked reduction of VEGF secretion in macrophages and DCs costimulated with LPS and NETs. Previous work has shown that so-called low-density granulocytes from SLE patients that have an increased propensity to release NETs not only stimulated IFN-α synthesis by pDCs, but also had the ability to kill endothelial cells (66). Moreover, it has been shown that neutrophils released NETs into the liver vasculature during systemic infection with methicillin-resistant Staphylococcus aureus, resulting in profound tissue damage (67). The dysregulation of angiogenic factors such as VEGF may conceivably play a role in this context. Sensitization of mice with the common indoor allergen house dust mite plus endotoxin was recently shown to result in NETosis in the lungs with formation of NETs and so-called neutrophil cytoplasts, and the cytoplasts were implicated as an underlying mechanism for the allergen-mediated TH17 responses (68). The latter study suggests that it is highly relevant to study responses of immune cells to NETs in the absence and presence of LPS. Furthermore, we believe it is critically important to study primary human immune cells to complement findings obtained using animal models (69).
The mechanisms of NET formation have been studied in considerable detail (70, 71), but less is known about the interplay between NETs and other immune-competent cells and, in particular, regarding the clearance of these extracellular structures. However, deciphering the mechanisms that control the clearance and degradation of NETs may contribute to our understanding of inflammatory and autoimmune diseases (72). Using primary human cells, we conducted a systematic analysis of the interactions between NETs and monocyte-derived macrophages and DCs, respectively, in terms of cytokine release and with regard to degradation of the DNA component of NETs. These studies have disclosed that NETs are, overall, noninflammatory, but they are, nevertheless, capable of modulating the immune response to bacterial LPS, and several commonalities were noted between HMDMs and MDDCs, including the upregulation of IL-1β secretion and suppression of VEGF secretion in cells coexposed to LPS. We also showed for the first time, to our knowledge, that NETs are degraded by the cytosolic exonuclease TREX1 in macrophages, whereas DNase1L3 released from DCs was found to mediate extracellular degradation of NETs. These findings shed light on the interactions between NETs and phagocytic cells and provide insights regarding the disposal of NETs.
Supplementary Material
Acknowledgments
We thank Dr. Katharina Klöditz and Dr. Guotao Peng, Karolinska Institutet, for assistance with Western blot and PCR analysis, respectively, and past and present members of the Fadeel laboratory for helpful discussions during the course of this work.
This work was supported by the Swedish Research Council (Grant 2016-02040).
The online version of this article contains supplemental material.
- DC
- dendritic cell
- HMDM
- human monocyte–derived macrophage
- MDDC
- monocyte-derived DC
- MPO
- myeloperoxidase
- NET
- neutrophil extracellular trap
- pDC
- plasmacytoid dendritic cell
- PFA
- paraformaldehyde
- SLE
- systemic lupus erythematosus
- VEGF
- vascular endothelial growth factor.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Papayannopoulos V. 2018. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 18: 134–147. [DOI] [PubMed] [Google Scholar]
- 2.Jorch S. K., Kubes P. 2017. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 23: 279–287. [DOI] [PubMed] [Google Scholar]
- 3.Brinkmann V., Reichard U., Goosmann C., Fauler B., Uhlemann Y., Weiss D. S., Weinrauch Y., Zychlinsky A. 2004. Neutrophil extracellular traps kill bacteria. Science 303: 1532–1535. [DOI] [PubMed] [Google Scholar]
- 4.Fuchs T. A., Brill A., Duerschmied D., Schatzberg D., Monestier M., Myers D. D., Jr., Wrobleski S. K., Wakefield T. W., Hartwig J. H., Wagner D. D. 2010. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 107: 15880–15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martinod K., Demers M., Fuchs T. A., Wong S. L., Brill A., Gallant M., Hu J., Wang Y., Wagner D. D. 2013. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc. Natl. Acad. Sci. USA 110: 8674–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cools-Lartigue J., Spicer J., McDonald B., Gowing S., Chow S., Giannias B., Bourdeau F., Kubes P., Ferri L. 2013. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Invest. 123: 3446–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Demers M., Krause D. S., Schatzberg D., Martinod K., Voorhees J. R., Fuchs T. A., Scadden D. T., Wagner D. D. 2012. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc. Natl. Acad. Sci. USA 109: 13076–13081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warnatsch A., Ioannou M., Wang Q., Papayannopoulos V. 2015. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 349: 316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schauer C., Janko C., Munoz L. E., Zhao Y., Kienhöfer D., Frey B., Lell M., Manger B., Rech J., Naschberger E., et al. 2014. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 20: 511–517. [DOI] [PubMed] [Google Scholar]
- 10.Hakkim A., Fürnrohr B. G., Amann K., Laube B., Abed U. A., Brinkmann V., Herrmann M., Voll R. E., Zychlinsky A. 2010. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 107: 9813–9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lande R., Ganguly D., Facchinetti V., Frasca L., Conrad C., Gregorio J., Meller S., Chamilos G., Sebasigari R., Riccieri V., et al. 2011. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 3: 73ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia-Romo G. S., Caielli S., Vega B., Connolly J., Allantaz F., Xu Z., Punaro M., Baisch J., Guiducci C., Coffman R. L., et al. 2011. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 3: 73ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leffler J., Martin M., Gullstrand B., Tydén H., Lood C., Truedsson L., Bengtsson A. A., Blom A. M. 2012. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J. Immunol. 188: 3522–3531. [DOI] [PubMed] [Google Scholar]
- 14.Farrera C., Fadeel B. 2013. Macrophage clearance of neutrophil extracellular traps is a silent process. J. Immunol. 191: 2647–2656. [DOI] [PubMed] [Google Scholar]
- 15.Kaplan M. J., Radic M. 2012. Neutrophil extracellular traps: double-edged swords of innate immunity. J. Immunol. 189: 2689–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neumann A., Völlger L., Berends E. T., Molhoek E. M., Stapels D. A., Midon M., Friães A., Pingoud A., Rooijakkers S. H., Gallo R. L., et al. 2014. Novel role of the antimicrobial peptide LL-37 in the protection of neutrophil extracellular traps against degradation by bacterial nucleases. J. Innate Immun. 6: 860–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neumann A., Berends E. T., Nerlich A., Molhoek E. M., Gallo R. L., Meerloo T., Nizet V., Naim H. Y., von Köckritz-Blickwede M. 2014. The antimicrobial peptide LL-37 facilitates the formation of neutrophil extracellular traps. Biochem. J. 464: 3–11. [DOI] [PubMed] [Google Scholar]
- 18.von Köckritz-Blickwede M., Chow O. A., Nizet V. 2009. Fetal calf serum contains heat-stable nucleases that degrade neutrophil extracellular traps. Blood 114: 5245–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kagan V. E., Gleiss B., Tyurina Y. Y., Tyurin V. A., Elenström-Magnusson C., Liu S. X., Serinkan F. B., Arroyo A., Chandra J., Orrenius S., Fadeel B. 2002. A role for oxidative stress in apoptosis: oxidation and externalization of phosphatidylserine is required for macrophage clearance of cells undergoing Fas-mediated apoptosis. J. Immunol. 169: 487–499. [DOI] [PubMed] [Google Scholar]
- 20.Mukherjee S. P., Kostarelos K., Fadeel B. 2018. Cytokine profiling of primary human macrophages exposed to endotoxin-free graphene oxide: size-independent NLRP3 inflammasome activation. Adv. Healthc. Mater. 7: 1700815. [DOI] [PubMed] [Google Scholar]
- 21.Napirei M., Wulf S., Eulitz D., Mannherz H. G., Kloeckl T. 2005. Comparative characterization of rat deoxyribonuclease 1 (Dnase1) and murine deoxyribonuclease 1-like 3 (Dnase1l3). Biochem. J. 389: 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kilpinen S., Autio R., Ojala K., Iljin K., Bucher E., Sara H., Pisto T., Saarela M., Skotheim R. I., Björkman M., et al. 2008. Systematic bioinformatic analysis of expression levels of 17,330 human genes across 9,783 samples from 175 types of healthy and pathological tissues. Genome Biol. 9: R139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kenny E. F., Herzig A., Krüger R., Muth A., Mondal S., Thompson P. R., Brinkmann V., Bernuth H. V., Zychlinsky A. 2017. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife 6: e24437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van der Linden M., Westerlaken G. H. A., van der Vlist M., van Montfrans J., Meyaard L. 2017. Differential signalling and kinetics of neutrophil extracellular trap release revealed by quantitative live imaging. Sci. Rep. 7: 6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Donis-Maturano L., Sánchez-Torres L. E., Cerbulo-Vázquez A., Chacón-Salinas R., García-Romo G. S., Orozco-Uribe M. C., Yam-Puc J. C., González-Jiménez M. A., Paredes-Vivas Y. L., Calderón-Amador J., et al. 2015. Prolonged exposure to neutrophil extracellular traps can induce mitochondrial damage in macrophages and dendritic cells. Springerplus 4: 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kahlenberg J. M., Carmona-Rivera C., Smith C. K., Kaplan M. J. 2013. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J. Immunol. 190: 1217–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atianand M. K., Fitzgerald K. A. 2013. Molecular basis of DNA recognition in the immune system. J. Immunol. 190: 1911–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sisirak V., Sally B., D’Agati V., Martinez-Ortiz W., Özçakar Z. B., David J., Rashidfarrokhi A., Yeste A., Panea C., Chida A. S., et al. 2016. Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell 166: 88–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan M. P., Onji M., Fukui R., Kawane K., Shibata T., Saitoh S., Ohto U., Shimizu T., Barber G. N., Miyake K. 2015. DNase II-dependent DNA digestion is required for DNA sensing by TLR9. Nat. Commun. 6: 5853. [DOI] [PubMed] [Google Scholar]
- 30.Shiokawa D., Shika Y., Tanuma S. 2003. Identification of two functional nuclear localization signals in DNase γ and their roles in its apoptotic DNase activity. Biochem. J. 376: 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yasutomo K., Horiuchi T., Kagami S., Tsukamoto H., Hashimura C., Urushihara M., Kuroda Y. 2001. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat. Genet. 28: 313–314. [DOI] [PubMed] [Google Scholar]
- 32.Lee-Kirsch M. A., Gong M., Chowdhury D., Senenko L., Engel K., Lee Y. A., de Silva U., Bailey S. L., Witte T., Vyse T. J., et al. 2007. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet. 39: 1065–1067. [DOI] [PubMed] [Google Scholar]
- 33.Al-Mayouf S. M., Sunker A., Abdwani R., Abrawi S. A., Almurshedi F., Alhashmi N., Al Sonbul A., Sewairi W., Qari A., Abdallah E., et al. 2011. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat. Genet. 43: 1186–1188. [DOI] [PubMed] [Google Scholar]
- 34.Napirei M., Karsunky H., Zevnik B., Stephan H., Mannherz H. G., Möröy T. 2000. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat. Genet. 25: 177–181. [DOI] [PubMed] [Google Scholar]
- 35.Grieves J. L., Fye J. M., Harvey S., Grayson J. M., Hollis T., Perrino F. W. 2015. Exonuclease TREX1 degrades double-stranded DNA to prevent spontaneous lupus-like inflammatory disease. Proc. Natl. Acad. Sci. USA 112: 5117–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawane K., Fukuyama H., Kondoh G., Takeda J., Ohsawa Y., Uchiyama Y., Nagata S. 2001. Requirement of DNase II for definitive erythropoiesis in the mouse fetal liver. Science 292: 1546–1549. [DOI] [PubMed] [Google Scholar]
- 37.Thammavongsa V., Missiakas D. M., Schneewind O. 2013. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science 342: 863–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahn J., Ruiz P., Barber G. N. 2014. Intrinsic self-DNA triggers inflammatory disease dependent on STING. J. Immunol. 193: 4634–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stetson D. B., Ko J. S., Heidmann T., Medzhitov R. 2008. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134: 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiménez-Alcázar M., Rangaswamy C., Panda R., Bitterling J., Simsek Y. J., Long A. T., Bilyy R., Krenn V., Renné C., Renné T., et al. 2017. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 358: 1202–1206. [DOI] [PubMed] [Google Scholar]
- 41.Errami Y., Naura A. S., Kim H., Ju J., Suzuki Y., El-Bahrawy A. H., Ghonim M. A., Hemeida R. A., Mansy M. S., Zhang J., et al. 2013. Apoptotic DNA fragmentation may be a cooperative activity between caspase-activated deoxyribonuclease and the poly(ADP-ribose) polymerase-regulated DNAS1L3, an endoplasmic reticulum-localized endonuclease that translocates to the nucleus during apoptosis. J. Biol. Chem. 288: 3460–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Napirei M., Wulf S., Mannherz H. G. 2004. Chromatin breakdown during necrosis by serum Dnase1 and the plasminogen system. Arthritis Rheum. 50: 1873–1883. [DOI] [PubMed] [Google Scholar]
- 43.Shiokawa D., Tanuma S. 1998. Molecular cloning and expression of a cDNA encoding an apoptotic endonuclease DNase γ. Biochem. J. 332: 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Z., Meng P., Han Y., Shen C., Li B., Hakim M. A., Zhang X., Lu Q., Rong M., Lai R. 2015. Mitochondrial DNA-LL-37 complex promotes atherosclerosis by escaping from autophagic recognition. Immunity 43: 1137–1147. [DOI] [PubMed] [Google Scholar]
- 45.Chamilos G., Gregorio J., Meller S., Lande R., Kontoyiannis D. P., Modlin R. L., Gilliet M. 2012. Cytosolic sensing of extracellular self-DNA transported into monocytes by the antimicrobial peptide LL37. Blood 120: 3699–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sandgren S., Wittrup A., Cheng F., Jönsson M., Eklund E., Busch S., Belting M. 2004. The human antimicrobial peptide LL-37 transfers extracellular DNA plasmid to the nuclear compartment of mammalian cells via lipid rafts and proteoglycan-dependent endocytosis. J. Biol. Chem. 279: 17951–17956. [DOI] [PubMed] [Google Scholar]
- 47.Wittrup A., Sandgren S., Lilja J., Bratt C., Gustavsson N., Mörgelin M., Belting M. 2007. Identification of proteins released by mammalian cells that mediate DNA internalization through proteoglycan-dependent macropinocytosis. J. Biol. Chem. 282: 27897–27904. [DOI] [PubMed] [Google Scholar]
- 48.Wong A., Bryzek D., Dobosz E., Scavenius C., Svoboda P., Rapala-Kozik M., Lesner A., Frydrych I., Enghild J., Mydel P., et al. 2018. A novel biological role for peptidyl-arginine deiminases: citrullination of cathelicidin LL-37 controls the immunostimulatory potential of cell-free DNA. J. Immunol. 200: 2327–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bergsmedh A., Szeles A., Henriksson M., Bratt A., Folkman M. J., Spetz A. L., Holmgren L. 2001. Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc. Natl. Acad. Sci. USA 98: 6407–6411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bergsmedh A., Ehnfors J., Kawane K., Motoyama N., Nagata S., Holmgren L. 2006. DNase II and the Chk2 DNA damage pathway form a genetic barrier blocking replication of horizontally transferred DNA. Mol. Cancer Res. 4: 187–195. [DOI] [PubMed] [Google Scholar]
- 51.Carmona-Rivera C., Carlucci P. M., Moore E., Lingampalli N., Uchtenhagen H., James E., Liu Y., Bicker K. L., Wahamaa H., Hoffmann V., et al. 2017. Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci. Immunol. 2: eaag3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Richards A., van den Maagdenberg A. M., Jen J. C., Kavanagh D., Bertram P., Spitzer D., Liszewski M. K., Barilla-Labarca M. L., Terwindt G. M., Kasai Y., et al. 2007. C-terminal truncations in human 3′-5′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat. Genet. 39: 1068–1070. [DOI] [PubMed] [Google Scholar]
- 53.Gehrke N., Mertens C., Zillinger T., Wenzel J., Bald T., Zahn S., Tüting T., Hartmann G., Barchet W. 2013. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity 39: 482–495. [DOI] [PubMed] [Google Scholar]
- 54.Costello M., Pugh T. J., Fennell T. J., Stewart C., Lichtenstein L., Meldrim J. C., Fostel J. L., Friedrich D. C., Perrin D., Dionne D., et al. 2013. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 41: e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lood C., Blanco L. P., Purmalek M. M., Carmona-Rivera C., De Ravin S. S., Smith C. K., Malech H. L., Ledbetter J. A., Elkon K. B., Kaplan M. J. 2016. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 22: 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brinkmann V., Zychlinsky A. 2007. Beneficial suicide: why neutrophils die to make NETs. Nat. Rev. Microbiol. 5: 577–582. [DOI] [PubMed] [Google Scholar]
- 57.Guimarães-Costa A. B., Rochael N. C., Oliveira F., Echevarria-Lima J., Saraiva E. M. 2017. Neutrophil extracellular traps reprogram IL-4/GM-CSF-induced monocyte differentiation to anti-inflammatory macrophages. Front. Immunol. 8: 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu Z., Murakami T., Tamura H., Reich J., Kuwahara-Arai K., Iba T., Tabe Y., Nagaoka I. 2017. Neutrophil extracellular traps induce IL-1β production by macrophages in combination with lipopolysaccharide. Int. J. Mol. Med. 39: 549–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakazawa D., Shida H., Kusunoki Y., Miyoshi A., Nishio S., Tomaru U., Atsumi T., Ishizu A. 2016. The responses of macrophages in interaction with neutrophils that undergo NETosis. J. Autoimmun. 67: 19–28. [DOI] [PubMed] [Google Scholar]
- 60.Barrera-Vargas A., Gómez-Martín D., Carmona-Rivera C., Merayo-Chalico J., Torres-Ruiz J., Manna Z., Hasni S., Alcocer-Varela J., Kaplan M. J. 2018. Differential ubiquitination in NETs regulates macrophage responses in systemic lupus erythematosus. Ann. Rheum. Dis. 77: 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Papadaki G., Kambas K., Choulaki C., Vlachou K., Drakos E., Bertsias G., Ritis K., Boumpas D. T., Thompson P. R., Verginis P., Sidiropoulos P. 2016. Neutrophil extracellular traps exacerbate Th1-mediated autoimmune responses in rheumatoid arthritis by promoting DC maturation. Eur. J. Immunol. 46: 2542–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barrientos L., Bignon A., Gueguen C., de Chaisemartin L., Gorges R., Sandré C., Mascarell L., Balabanian K., Kerdine-Römer S., Pallardy M., et al. 2014. Neutrophil extracellular traps downregulate lipopolysaccharide-induced activation of monocyte-derived dendritic cells. J. Immunol. 193: 5689–5698. [DOI] [PubMed] [Google Scholar]
- 63.Tsokos G. C., Lo M. S., Costa Reis P., Sullivan K. E. 2016. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 12: 716–730. [DOI] [PubMed] [Google Scholar]
- 64.Vannella K. M., Wynn T. A. 2017. Mechanisms of organ injury and repair by macrophages. Annu. Rev. Physiol. 79: 593–617. [DOI] [PubMed] [Google Scholar]
- 65.Golpon H. A., Fadok V. A., Taraseviciene-Stewart L., Scerbavicius R., Sauer C., Welte T., Henson P. M., Voelkel N. F. 2004. Life after corpse engulfment: phagocytosis of apoptotic cells leads to VEGF secretion and cell growth. FASEB J. 18: 1716–1718. [DOI] [PubMed] [Google Scholar]
- 66.Villanueva E., Yalavarthi S., Berthier C. C., Hodgin J. B., Khandpur R., Lin A. M., Rubin C. J., Zhao W., Olsen S. H., Klinker M., et al. 2011. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 187: 538–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kolaczkowska E., Jenne C. N., Surewaard B. G., Thanabalasuriar A., Lee W. Y., Sanz M. J., Mowen K., Opdenakker G., Kubes P. 2015. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat. Commun. 6: 6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krishnamoorthy N., Douda D. N., Brüggemann T. R., Ricklefs I., Duvall M. G., Abdulnour R. E., Martinod K., Tavares L., Wang X., Cernadas M., et al. National Heart, Lung, and Blood Institute Severe Asthma Research Program-3 Investigators 2018. Neutrophil cytoplasts induce TH17 differentiation and skew inflammation toward neutrophilia in severe asthma. Sci. Immunol. 3: eaao4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khandagale A., Lazzaretto B., Carlsson G., Sundin M., Shafeeq S., Römling U., Fadeel B. 2018. JAGN1 is required for fungal killing in neutrophil extracellular traps: implications for severe congenital neutropenia. J. Leukoc. Biol. 104: 1199–1213. [DOI] [PubMed] [Google Scholar]
- 70.Metzler K. D., Goosmann C., Lubojemska A., Zychlinsky A., Papayannopoulos V. 2014. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 8: 883–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Amulic B., Knackstedt S. L., Abu Abed U., Deigendesch N., Harbort C. J., Caffrey B. E., Brinkmann V., Heppner F. L., Hinds P. W., Zychlinsky A. 2017. Cell-cycle proteins control production of neutrophil extracellular traps. Dev. Cell 43: 449–462.e5. [DOI] [PubMed] [Google Scholar]
- 72.Boe D. M., Curtis B. J., Chen M. M., Ippolito J. A., Kovacs E. J. 2015. Extracellular traps and macrophages: new roles for the versatile phagocyte. J. Leukoc. Biol. 97: 1023–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.