

Abstract
We recently developed an orthogonal replication system (OrthoRep) in yeast that allows for the rapid continuous mutagenesis of a special plasmid without mutating the genome. Although OrthoRep has been successfully applied to evolve several proteins and enzymes, the generality of OrthoRep has not yet been systematically studied. Here, we show that OrthoRep is fully compatible with all Saccharomyces cerevisiae strains tested, demonstrate that the orthogonal plasmid can encode genetic material of at least 22 kb, and report a CRISPR/Cas9-based method for expedient genetic manipulations of OrthoRep. It was previously reported that the replication system upon which OrthoRep is based is only stable in respiration-deficient S. cerevisiae strains that have lost their mitochondrial genome (ρ0 strains). However, here we trace this biological incompatibility to the activity of the dispensable toxin/antitoxin system encoded on the wild-type orthogonal plasmid. Since the toxin/antitoxin system is replaced by genes of interest in any OrthoRep application, OrthoRep is a generally compatible platform for continuous in vivo evolution in S. cerevisiae.
Keywords: orthogonal replication, OrthoRep, in vivo mutagenesis, CRISPR/Cas9, protein-primed replication, continuous evolution
Graphical Abstract
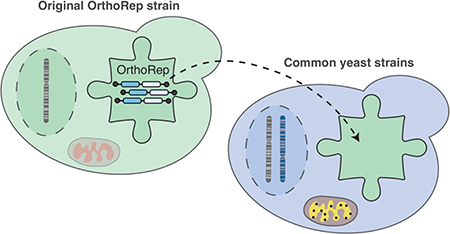
Orthogonal DNA replication (OrthoRep) is a genetic platform for the rapid, continuous, and scalable in vivo evolution of user-specified genes of interest (GOIs) in yeast.1 At its core, the OrthoRep system consists of a special DNA polymerase (DNAP) called TP-DNAP1 that stably replicates a cytoplasmic linear DNA plasmid called pGKL1 (p1) without replicating genomic DNA.2 Engineered error-prone variants of TP-DNAP1 therefore durably drive the continuous mutagenesis of p1 without elevating the mutation rate of genomic DNA, unlocking rates of extreme mutagenesis – currently up to ~10−5 substitutions per base – for p1-encoded GOIs. In addition to p1, an accessory cytoplasmic linear plasmid called pGKL2 (p2) encodes machinery responsible for the replication, transcription, and maintenance of both p1 and p2.3 Since p2 encodes its own dedicated DNAP (TP-DNAP2) and p1 replication is mechanistically insulated from p2 replication,4 rapid mutation of GOIs on p1 by engineered error-prone TP-DNAP1s occurs with complete targeting. Since the TP-DNAP1 gene, naturally found on p1, can instead by encoded on nuclear DNA to sustain p1’s replication2 and since the only other item naturally encoded on p1 is a dispensable toxin/antitoxin (TA) pair, p1 is free to contain only arbitrary genetic payloads for rapid mutation and evolution. Therefore, OrthoRep, specifically the orthogonal TP-DNAP1/p1 plasmid pair, forms an ideal genetic architecture for continuous in vivo evolution of GOIs.
We have used OrthoRep to drive the rapid in vivo evolution of several proteins, including a drug target to study mutational pathways leading to resistance in 90 replicates,1 enzymes to expand their reaction scope for both basic protein science and applied biotechnology goals, and antibodies (manuscripts in preparation). In using OrthoRep, we have found that the system is functional for all GOIs we have tested on p1, operational in all Saccharomyces cerevisiae strains we have used, and highly durable. However, the generality and behavior of OrthoRep is so far only anecdotal.
Here, we systematically study and extend aspects of OrthoRep’s generality that will impact its broad application in directed evolution and genetics. We find that OrthoRep is compatible with a wide range of S. cerevisiae strains, including standard lab strains (e.g. BY4741 and W303–1A), an industrial yeast strain (CEN.PK2–1C), and diploid yeasts (e.g. BY4743), despite previous reports indicating instability of p1 and p2 in diploids.5 We also reveal that previous reports of replicative instability of p1 and p2 in ρ+ yeast strains are due to the dispensable TA system on p1.6 In particular, we show that the p1 orthogonal plasmid is stably replicated at expected mutation rates dictated by the wild-type (wt) or an error-prone (ep) TP-DNAP1 variant, and orthogonality to genomic replication is maintained in all strains tested. Finally, we report streamlined techniques for p1 manipulation that utilize CRISPR/Cas9 and show that genetic payloads of at least 22 kb can be readily encoded on p1, demonstrating that OrthoRep is a highly general yeast system for continuous in vivo evolution of GOIs that is ready for broad application.
Results and Discussion
Generality of OrthoRep across strains
OrthoRep is based on an autonomous replication system, so its replicative properties should be consistent across different yeast hosts. Specifically, 1) the orthogonal plasmid should stably propagate in any strain, 2) the per-base mutation rate of the orthogonal plasmid’s replication by wt and ep TP-DNAP1s should be consistent across strains, matching the rates from the strain in which we have historically engineered new TP-DNAP1s1, and 3) orthogonality against genomic replication should be complete such that no increase in the host genomic error-rate is observed when GOIs on OrthoRep are undergoing rapid mutation. We therefore characterized these three features of OrthoRep in four S. cerevisiae base strains beyond the F102–2 strain that our previous reported works used. Specifically, we chose to test OrthoRep in BY4741 and W303–1A, two commonly used haploid yeast strains, BY4743, a commonly used yeast diploid strain, and CEN.PK2–1C, an industrial haploid yeast strain. Unlike F102–2, all four of these new strains are ρ+ (i.e., respiration proficient).
To be viable for continuous evolution experiments, the orthogonal p1 plasmid must remain stable when replicated not only by the wt TP-DNAP1 but also with ep variants of TP-DNAP1s. Thus, each strain was constructed with either the wt TP-DNAP1 or TP-DNAP1–4-3 (L474W, L640Y, I777K, W814N), expressed in trans from a nuclear CEN6/ARS4 plasmid. The endogenous copy of wt TP-DNAP1 on p1 was removed. Each strain was passaged under selection for a p1-encoded marker, URA3, or passaged without selection for ~100 generations (~10 generations per passage) at a dilution of 1:1000 (Figure 1A). DNA was extracted after the first and tenth passage for all 20 cultures. Agarose gel electrophoresis (Figure 1B, Figure S1) of the extracted DNA showed that OrthoRep is stably maintained with or without direct selection over ~100 generations in all cases. Notably, the p1/p2 plasmids are stable in the diploid strain (BY4743), contrary to previous experiments.5 This allows the possibility to use yeast mating with OrthoRep to introduce sexual recombination during evolution experiments, which we are actively pursuing.
Figure 1—

(A) Testing the stability of OrthoRep in common yeast strains. OrthoRep was transferred from F102–2 into BY4741, CEN.PK2–1C, W303–1A and BY4743. The ploidy and presence or absence of mitochondrial DNA (mtDNA) are indicated. After transfer, strains were passaged in media selective or non-selective (n.s.) for a p1-encoded marker (URA3) and a nuclear plasmid expressing a TP-DNAP1 variant (HIS3). (B) Stability of OrthoRep in BY4741 (see Figure S1 for similar stability data on all other strains). Agarose gel electrophoresis on DNA extracted from BY4741 strains show the stability of OrthoRep (presence of p1-mUL*) over multiple cycles of passaging even without selection. Presence of p1-mUL* is observed after the first (1) and tenth (10) passage in both selective and non-selective (n.s.) media conditions in the presence of either a wt TP-DNAP1 or error-prone TP-DNAP1–4-3 expressed from a nuclear CEN6/ARS4 plasmid. Parental BY4741 without p1/p2 is shown as a control. The 2μ plasmid is native to the base strain, BY4741. –pK conditions are controls referring to the lack of proteinase K treatment during DNA preparation; the lack of proteinase K treatment results in the lack of all p1/p2-derived bands because the terminal proteins on p1/p2 prevent migration into agarose gels as previously described.2
For OrthoRep to be useful in these S. cerevisiae strains, we needed to confirm that p1 is not only stable, but that its per-base substitution rate can be predictably elevated for rapid mutagenesis in the presence of ep TP-DNAP1s. Mutation rates for p1 were determined via fluctuation analysis for all four base strains, which were constructed to contain a p1 plasmid (p1-mUL*) encoding a fluorescent reporter (mKate), a selectable marker (URA3), and a mutation rate reporter (leu2*), and either the wt TP-DNAP1 or ep TP-DNAP1–4-3 encoded nuclearly. Fluctuation analyses were performed via leu2* (TAA stop codon at Q180) reversion to LEU2 to calculate the per-base substitution rate of p1 replication. For a given strain to be measured, replicate cultures were grown in liquid media containing leucine and then spot plated on solid media without leucine. The number of colonies grown were counted for each spot and used to determine the mutation rate via the Ma-Sandri-Sarkar method (see Methods).1,2 Strains encoding wt TP-DNAP1 had mutation rates comparable to our previously measured rate in F102–2 (Table 1). As expected, the mutation rate of the ep TP-DNAP1–4-3 was also comparable to that measured in F102–2 such that across all strains, the per-base mutation rate of ep TP-DNAP1–4-3 was at least 1000-fold higher than the wt TP-DNAP1 rate. Given this consistency, additional engineered TP-DNAP1s that have lower or higher mutation rates are expected to behave predictably in all these strains.1
Table1— p1 per-base substitution rates.
Mutation rates for TP-DNAP1 in different strains were measured via leu2* fluctuation analysis. Per-base substitution rates are reported with 95% confidence intervals indicated in parenthesis, as determined by the MSS method.17,18
| Base Strain | TP-DNAP1 (nuclearly encoded) | p1 per-base substitution rate |
|---|---|---|
| BY4741 | wt | 1.23×10−9 (0.96–2.10) |
| TP-DNAP1–4-3 | 4.48×10−6 (3.07–5.75) | |
| BY4743 | wt | 9.08×10−10 (7.59–20.1) |
| TP-DNAP1–4-3 | 1.11×10−6 (0.65–2.08) | |
| CEN.PK2–1C | wt | 2.01×10−9 (1.43–3.15) |
| TP-DNAP1–4-3 | 3.36×10−6 (2.18–4.24) | |
| W303–1A | wt | 1.73×10−9 (1.10–2.52) |
| TP-DNAP1–4-3 | 2.71×10−6 (1.99–3.41) | |
| F102–2 | wt | 1.95×10−9 (1.00–3.09) |
| TP-DNAP1–4-3 | 2.54×10−6 (1.79–3.35) |
To ensure orthogonality against host genome replication in these new strains, we measured genomic mutation rates at the URA3 locus via fluctuation analysis in the presence of replicating p1/p2 plasmids and compared the rates to those of the parental strains. BY4741, BY4743, CEN.PK2–1C, and W303–1A strains were constructed with a recombinant p1 plasmid containing a TRP1 selectable marker, mKate2, and no TP-DNAP1 (p1-mW). A wt TP-DNAP1 or ep TP-DNAP1–4-3 was expressed from a nuclear plasmid in these strains, while parental controls did not contain any component of OrthoRep. Genomic per-base mutation rates are reported in Table 2. For all strains tested, the presence of OrthoRep did not increase the genomic mutation rate. This suggests that OrthoRep operates orthogonally to host replication, thereby allowing exquisitely targeted mutagenesis of p1-encoded genes.
Table 2— Orthogonality of Orthorep.
Genomic mutation rates were measured in the presence or absence of replicating p1/p2 in different base strains. Per-base substitution rates are reported with 95% confidence intervals indicated in parenthesis, as determined by the MSS method.17,18 No statistically significant change in the genomic mutation rate is observed when OrthoRep is present.
| Base strain | TP-DNAP1/p1 | Per-base substitution rate (x10−10) |
|---|---|---|
| BY4741 | n/a (parental) | 1.06 (0.22–2.59) |
| wt | 1.43 (0.53–3.71) | |
| TP-DNAP1–4-3 | 1.81 (0.64–2.89) | |
| BY4743 | n/a (parental) | 4.28 (1.50–11.1) |
| wt | 5.66 (3.92–12.5) | |
| TP-DNAP1–4-3 | 3.30 (1.06–8.96) | |
| CEN.PK2–1C | n/a (parental) | 1.39 (0.55–2.72) |
| wt | 2.57 (1.46–4.43) | |
| TP-DNAP1–4-3 | 1.09 (0.77–3.01) | |
| W303–1A | n/a (parental) | 2.35 (1.06–2.96) |
| wt | 1.21 (0.74–2.61) | |
| TP-DNAP1–4-3 | 3.13 (2.41–5.55) | |
| AH22 | n/a (parental for F102–2) | 1.09 (0.94–2.82) |
| F102–2 | wt | 2.16 (1.01–4.90) |
| TP-DNAP1–4-3 | 1.71 (1.30–3.67) |
Compatibility of OrthoRep with ρ+ strains
Our finding that p1/p2 replication is stable in the four new base strains, all of which are respiration proficient, contradicts a previous report showing that the wt p1/p2 plasmids could not be maintained in ρ+ strains.6 Specifically, it was shown that ρ+ S. cerevisiae strains containing wt p1/p2 were lost after 25 generations of growth. While early works on creating recombinant p1 plasmids in S. cerevisiae were assembled by removing part of the TA system, the strains used were ρ0.7,8 Based on our group’s accumulated experience with OrthoRep, we suspected that the p1-encoded TA system’s activity was the source of incompatibility and that p1/p2 replication per se was in fact compatible with ρ+ strains. We therefore sought to test this hypothesis by assaying p1 plasmid stability in the presence and absence of TA activity in both ρ+ and respiration-deficient ρ0 strains. To do so, we constructed a p1 plasmid encoding the full TA system and the URA3 selection marker (p1-DNAP1-TA-U) in the S. cerevisiae strains BY4741 (ρ+) and F102–2 (ρ0). Alongside, we constructed a p1 plasmid encoding no TA system and the URA3 selection marker (p1-DNAP1-mUL*), also in both BY4741 and F102–2. Each of the four strains was passaged in triplicate without selection for the URA3 gene for ~90 generations (Figure 2A). Afterwards, DNA was extracted from all strains and agarose gel electrophoresis was used to detect the plasmids, where presence of p1 and p2 would indicate their stability. We found that p1-DNAP1-TA-U was stable only in F102–2 but not in BY4741, whereas p1-DNAP1-mUL* was stable in both (Figure 2B). This suggests that the TA system is indeed the source of incompatibility for p1 replication in ρ+ strains.
Figure 2—

(A) Design of experiments tracing source of incompatibility between p1 plasmid replication and r+ strains containing mtDNA and full mitochondrial function. A p1 plasmid with or without TA genes encoded were transferred to r0 (F102–2) or r+ (BY4741) strains and passaged in media that either suppressed (blue) or activated (orange) the toxin phenotype. (B) Stability of p1/p2 in experimental conditions described in (A) after passaging. Agarose gel electrophoresis on DNA extracted from technical triplicates of experiments outlined in (A) show the presence of the expected p1 plasmid in all conditions except for the condition where TA is encoded and activated in a r+ strain. The 2μ plasmid is native to the BY4741-derived strains.
To further test our hypothesis, we carried out a second experiment that exploits the inactivation of the p1-encoded TA system at low pH. Previous studies on the TA system revealed that its encoded killer phenotype is active in S. cerevisiae at pH ~4.5–5, but not outside this range.6,9 Therefore, we also passaged our four strains (BY4741 (ρ+) or F102–2 (ρ0) with or without a p1-encoded TA system) at a low pH condition (~4), which should suppress toxin activity. We found that when passaged at low pH, p1-DNAP1-TA-U was indeed stable in BY4741 whereas at the pH that activated the toxin phenotype, it was unstable (Figure 2B, g versus h). Assays on synthetic solid media plates indicated that all BY4741 strains from these experiments retained respiratory competence (growth on glycerol plates) (Figure S2). Taken together, this demonstrates that p1-DNAP1-TA-U is only stable in ρ+ when TA activity is inactive. As the TA system is dispensable and replaced with GOIs in applications of OrthoRep, we conclude that OrthoRep’s generality extends across all common S. cerevisiae strains.
Expedient CRISPR/Cas9 manipulation of p1
Recombinant p1 or p2 plasmids are currently created via transformation of an integration cassette with 5’ and 3’ homology arms targeting the parental p1 or p2 plasmids in a recipient cell. Typically, DNA analysis of cells shortly after transformation reveals a mixed population of parental and recombinant plasmid. This is because integration is not quantitative and the wt copy number of p1 and p2 is high. To generate a pure population of cells that only encode the recombinant plasmid, the user must passage the initial transformants for 1–2 weeks under selection for the recombinant plasmid. While straightforward, the added time is undesirable, and in the case where the recombinant plasmid is larger than the parental plasmid, the parental plasmid has a replicative advantage that slows down this process even further. We reasoned that co-transformation of our integration cassette in conjunction with a CRISPR/Cas9 system that targets the parental p1 plasmid would result in a pure population of recombinant p1 immediately after transformation, as unintegrated parental p1 plasmid would be destroyed (Figure 3A).
Figure 3—

(A) Accelerating genetic manipulation of p1 with CRISPR/Cas9. After transformation of an integration cassette containing a GOI and a selectable marker, transformants are plated on media selecting for the cassette and inducing CRISPR/Cas9. Parental p1 plasmids that did not receive the GOI are cut by CRISPR/Cas9, resulting in a cell containing just the desired p1 plasmid. (B) Integration of a model cassette (DNAP1-mUL*) onto p1 in strains with p1-DNAP1-MET15 as the parental plasmid. Agarose gel electrophoresis on DNA extracted from cells after transformation and plating is shown (lanes 2–7). Lane 1 is the parental strain. Biological replicates from transformation without Cas9 induction (lanes 2 and 3) show both parental and desired p1 bands whereas biological replicates from a transformation with the integration cassette and concomitant induction of Cas9 (lanes 4–7) shows only the desired p1 band. (C) Exploration of cassette sizes that can be integrated on p1. Agarose gel electrophoresis on DNA extracted from cells (BY4741 (AJ-Y92)) after transformation of three integration cassettes generating three sizes of p1 – small (14 kb), medium (18 kb), or large (22 kb). Three biological replicates are shown for each size. – pK conditions are controls referring to the lack of proteinase K treatment during DNA preparation; the lack of proteinase K treatment results in the lack of all p1/p2-derived bands because the terminal proteins on p1/p2 prevent migration into agarose gels as previously described.2
To test this approach, we modified a yeast CRISPR/Cas9 vector to encode an sgRNA targeting MET15 or mKate2 and a galactose-inducible Cas9, localized to the cytoplasm.10 These constructs were co-transformed with various p1 integration cassettes of different sizes containing a unique selectable marker into strains of BY4741 containing OrthoRep components and a parental p1 plasmid encoding MET15 or mKate2. Induction of Cas9 and selection for the desired p1 plasmid resulting from integration onto the parental p1 plasmid were directly imposed on solid media after transformants were plated. Control transformations were performed in parallel but without induction of Cas9. Transformed colonies were then grown to saturation in liquid culture and DNA was extracted and analyzed by gel electrophoresis to determine if a pure population of the desired p1 plasmid was obtained. Indeed, transformants that were induced for Cas9 expression displayed only desired p1 plasmid bands, whereas transformants that were not induced for Cas9 contained a mixed pool of parental and desired p1 plasmids (Figure 3B, Figure S3). As the CRISPR/Cas9 vector used is readily lost when not selecting for the plasmid,11 this process allows us to quickly generate a strain that only contains the desired recombinant p1 (and p2) plasmids.
Exploring the possible size of recombinant orthogonal p1 plasmids
In OrthoRep, GOIs that are subjected to rapid evolution are encoded on p1. Thus far, we have encoded up to 4 genes simultaneously on p1, but it may be desirable in future applications to encode larger genetic payloads, such as entire metabolic pathways or gene clusters. While the minimal size of p1 is known – only ~400 bp of DNA are responsible for initiation of p1 replication, as most of p1’s replication origin are the terminal protein components – p1 plasmids beyond its wt size (8.9 kb) have not been constructed.12 We sought to determine if we could create pure populations of recombinant p1s that far exceed the size of wt p1 while simultaneously removing the parental p1 plasmid. To explore this possibility, we created three integration cassettes containing URA3, mKate2, and varying lengths of stuffer DNA, taken from Kluveromyces lactis genomic DNA. These cassettes would generate p1 plasmid sizes of 14, 18, and 22 kb. Using the aforementioned CRISPR/Cas9 technique, transformation of these integration cassettes into a BY4741 strain containing p1/p2 yielded strains containing only the desired recombinant p1 for all three sizes, as determined via DNA miniprep and gel electrophoresis (Figure 3C). Therefore, it is possible to encode and maintain at least 22 kb of DNA on p1. This may allow the continuous evolution of gene clusters and pathways using OrthoRep. However, we note that in the experiment presented, what is encoded on the 22 kb p1 includes the wild-type TP-DNAP1, whereas in the ideal setup of a metabolic pathway evolution experiment, p1 should only encode metabolic GOIs and be replicated by an error-prone orthogonal TP-DNAP1. We expect that the stability of a 22 kb p1 will be lower in these cases due to the lower activity of error-prone DNAPs. In addition, the lack of a dedicated primase may limit the efficient replication of p1 beyond a maximum size.13 Still, we can conclude that this size limit of p1 exceeds 22 kb when given a sufficiently active TP-DNAP1.
Conclusion
We have demonstrated that OrthoRep is stable and functional in all standard yeast strains tested and have resolved conflicting reports of p1 replicative instability in ρ+ and diploid strains. We have shown that the mutational properties of OrthoRep hold across all strains tested and developed a streamlined technique to install GOIs on p1, including cargos of at least 22 kb. These developments solidifying the generality of OrthoRep should make it a highly applicable system for continuous in vivo evolution.
Methods
General DNA Cloning
Plasmids used in this study are listed in Table S1. E. coli strain TG1 (Lucigen) was used for all the DNA cloning steps. All primers used in this study were purchased from IDT. All enzymes for PCR and cloning were obtained from NEB. All plasmids were cloned via Gibson assembly.14 Cloning of sgRNAs was performed as described by Ryan et al.11
Yeast strains and Media
All yeast strains used in this study are listed in Table S2. Auxotrophic selection markers used on p1 were first fully deleted from the genome via CRISPR/Cas911 before integrating genetic cassettes onto p1 or p1/p2 transfer through protoplast fusion.
Yeast strains were grown in standard media including YPD (10 g/L Bacto Yeast Extract; 20 g/L Bacto Peptone; 20 g/L Dextrose) and appropriate synthetic drop-out media (6.7 g/L Yeast Nitrogen Base w/o Amino Acids (US Biological); 2 g/L Drop-out Mix Synthetic minus the appropriate nutrients w/o Yeast Nitrogen Base (US Biological); 20 g/L Dextrose). For the TA-induced instability experiments, SC media was buffered with citrate phosphate buffer (0.05 M citric acid and 0.1 M sodium dibasic heptahydrate) and pH was adjusted with 5 M NaOH. For all fluctuation test experiments, all liquid and solid media was adjusted to pH 5.8 with 5 M NaOH.
Yeast transformation and integration
All transformations were performed via the high efficiency Gietz method.15 For p1 integrations, 2–4 μg of plasmid was linearized by digestion with ScaI, which generated blunt ends containing homologous regions to p1. For CEN6/ARS4 nuclear plasmid transformations, roughly 100–500 ng of plasmid was transformed. Transformants were selected on the appropriate selective solid SC media. Plates were grown at 30°C for 2 days for nuclear transformations and 4–5 days for p1 integrations.
CRISPR/Cas9-based p1 manipulation
The pCas plasmid, containing a KanMX selection marker and expressing both sgRNA and Cas9, was used for all CRISPR/Cas9 editing.11 Spacer sequences were designed using Yeastriction v0.1.16 Genomic knockouts were performed as described previously.11 For all p1 integrations, a version of pCas with the 8×HIS and SV-40 nuclear localization tags removed was used.
For integration of genes onto p1 in F102–2, a pCas plasmid (~3–5 μg) containing an sgRNA spacer targeting the WT p1 plasmid (GCTGATTATACATATACAGA) was co-transformed with a ScaI-linearized integration cassette (2–4μg). Transformants were recovered overnight and plated on SC dropout solid media containing 1 g/L monosodium glutamate (MSG) as the nitrogen source and 400 μg/mL G418. Transformation plates were incubated at 30°C for 4–5 days and colonies were picked into liquid media and 200 μg/mL G418. To confirm integration and loss of parental p1, DNA was extracted (see linear plasmid DNA extraction) once cultures were saturated (~2 days) and analyzed by agarose gel electrophoresis.
For the integration of genes onto p1 in BY4741 seen in Figure 3, a base strain was constructed containing a galactose-inducible Cas9 and a MET15 marker integrated onto p1. To create this strain, a pCas plasmid was cloned to contain a spacer targeting mKate2 (TCTTCAAGTTGCAGATCAGG) and Cas9 driven by the GAL2 promoter. This was subsequently transformed into a BY4741 strain containing p1-DNAP1-mUL*. Afterwards, a p1 integration cassette containing MET15 was transformed into this strain, and transformants were recovered in YPD for 1 hr and then plated on SC-MC dropout containing MSG (1 g/L), G418 (400 μg/mL), and 2% raffinose and 2% galactose as the carbon sources. A clone containing p1-DNAP1-MET15 and no parental p1-DNAP1-mUL* was re-streaked on solid media (SC-MC) to lose the pCas plasmid. Afterwards, another pCas plasmid with a galactose-inducible Cas9 was cloned containing a MET15 spacer (GCTAAGAAGTATCTATCTAA) and transformed into this BY4741 strain containing p1-DNAP1-MET15. Transformants were grown in SC-MC containing MSG (1 g/L) + G418 (400 μg/mL). All subsequent p1 integrations into this strain, shown in Figure 3, were recovered for 1 hr in YPD and then plated on appropriate SC dropout media containing MSG (1g/L), G418 (400 μg/mL), and 2% raffinose and 2% galactose.
Yeast protoplast fusion
Transfer of p1/p2 from donor strains into recipient strains was performed via protoplast fusion. Recipient strains express a genomically-encoded auxotrophic marker not found in the donor strain. The donor strain contains p1/p2 with a p1-encoded auxotrophic marker not found in the recipient strain.
Protoplast fusion was performed as described before.2 Briefly, small precultures (~3–4 mL) of donor and recipient strains were grown in appropriate SC dropout media. Strains were then diluted (1:50) into YPD (80 mL) and grown overnight. A portion (35 mL) of both cultures was spun down at 3000g for 5 min and washed with ddH2O (10 mL). After spinning down again, the mass of the cell pellets was measured to roughly ~0.3 g. Pellets were then resuspended in β-mercaptoethanol (0.2%, 1.8 mL) and EDTA (0.06 M). Cells were incubated at 30°C for 30 min with occasional rotation. Cells were then washed with KCl (0.6M, 3 mL), spun (3000g, 5 min), and then resuspended in a solution (4.8 mL) of citrate phosphate buffer (0.1 M, pH 6.1), KCl (0.6 M), EDTA (0.01 M), and Zymolyase (29 units, US Biological). Cells were incubated at 30°C for 60 min with occasional rotation. Afterwards, cells were spun down at 2000 rpm (700 × g) for 10 min at 4°C, and then washed twice with 3 mL KCl (0.6M). Cells were then gently resuspended in a solution (3 mL) of citrate phosphate buffer (0.1 M, pH 6.1), KCl (0.6 M), and EDTA (0.01 M). Half of the donor strain (1.5 mL) and half of the recipient strain (1.5 mL) were mixed together and centrifuged at 2000 rpm (700 × g) for 10 min at 4°C. The cell pellet was resuspended gently in a solution (5 mL) containing PEG 3350 (33%), KCl (0.6 M), and CaCl2 (50 mM), followed by incubation at 30°C for 30 min with occasional rotation. Cells were then spun down at 2000 rpm (700 × g) for 10 min at 4°C and then resuspended gently in a solution (5 mL) containing KCl (0.6M) and CaCl2 (50 mM). Roughly 20–150 μL of cell suspension was mixed with 10 mL of warm, liquid SC dropout media (selecting for the p1 auxotrophic marker and the auxotrophic marker genomically-encoded only in the recipient strain) containing KCl (0.6M) and Bacto-Agar (3%), and the entire mixture was plated. The appearance of colonies following protoplast fusion typically took 2–3 days of incubation at 30°C. p1/p2 plasmid DNA extraction was first performed to assess for their presence in the recipient strain. Strains that contained both plasmids were tested for respiration proficiency by plating on glycerol with appropriate SC dropout media.
Typically, six colonies were inoculated from plates into liquid SC dropout media (same dropout used for plating of the protoplast fusion) for subsequent verification. Recipient strains were verified to contain p1/p2 by agarose gel electrophoresis of extracted linear plasmid DNA. Strains that had p1 and p2 plasmids at the correct band sizes were subsequently tested for respiration proficiency by plating on glycerol media containing SC dropout media selecting for the p1-auxtrophic marker and the nuclear marker on a CEN6/ARS4 plasmid also containing a TP-DNAP1 variant.
Linear plasmid DNA extraction
p1, p2, and recombinant p1 plasmids were extracted following the yeast DNA miniprep procedure as described previously.2
p1 fluctuation test
Fluctuation analyses on p1-encoded leu2* were performed to determine per-base substitution rates of p1 replication, as described previously.1 leu2* contains a C−>T mutation at base 538 LEU2 that results in an ochre nonsense mutation at a site permissive to all single point mutants that generate missense mutations.
To perform fluctuation analyses, yeast strains containing p1-mUL* and TP-DNAP1 expressed in trans from a CEN6/ARS4 plasmid were grown in SC media lacking uracil, histidine, and tryptophan (SC-UHW, pH 5.8). Once grown to saturation, the strains were diluted 1:10,000 into the same media and split into 48 100 μl replicate cultures in 96-well trays. Trays were grown with aluminum seal covers for 2–2.5 days until saturation. Cultures were washed with 0.9% NaCl and resuspended in 0.9% NaCl (35 μl). Four replicates were pooled and titered on SC-UHW (pH 5.8) solid plates, while 10 μl of the remaining 44 replicates were spot plated on SC-UHLW (pH 5.8) solid plates. Spots were allowed to dry before incubation. Plates were incubated at 30°C. Colonies were counted from SC-UHW titer plates after 2 days and from spot plates after 4–6 days.
The number of LEU2 revertant colonies was used to calculate the number of mutants (m) using the Ma-Sandri-Sarkar (MSS) maximum likelihood estimator as implemented by the FALCOR tool.17,18 After correcting for partial plating, m was normalized by the titers, copy number (see Determination of copy number via quantitative PCR), and the number of ways to revert leu2* to LEU2 (2.33) to obtain the per-base mutation rate. 95% confidence intervals were obtained from the MSS method and were similarly scaled.1,2,4
Genomic orthogonality measurements
Fluctuation analyses on the genomically-encoded URA3 gene were performed to determine genomic per-base substitution rates, as previously described.1,2 URA3 was integrated into yeast strains its native locus. For BY4743, a hemizygous strains was constructed (URA3::kanMX/ura3Δ0) with a geneticin-resistance gene (kanMX) inserted immediately downstream of a hemizygous URA3 gene, in order to select against 5-FOA-resistant mitotic recombinants.19 These URA3 strains also contained p1-mW, a CEN6/ARS4 plasmid containing TP-DNAP1 (wt or TP-DNAP1–4-3), and a HIS3 marker. Parental control strains did not contain p1/p2 or a CEN6/ARS4 plasmid. p1/p2-containing strains were inoculated into liquid SC media lacking uracil, histidine, and tryptophan (SC-UHW, pH 5.8), while parental strains were inoculated into liquid SC media lacking only uracil (SC-U, pH 5.8). Saturated cultures were diluted 1:5,000 into the same SC media with uracil added back and split into replicate 200 μl cultures in 96-well trays. Trays were grown with sealed covers for 2–2.5 days until saturation. Cultures were washed with 0.9% NaCl and resuspended in 0.9% NaCl (200 μl). Four cultures from each strain were pooled, diluted and titered on solid SC-HW or SC medium. 190 μl of all the remaining cultures were spot plated onto solid SC (SC-HW for strains containing p1/p2) medium supplemented with 5-FOA (1 g/L) and uracil (50 mg/L), and spots were allowed to dry before incubation. Plates were incubated at 30°C. Colonies were counted from titer plates after 2 days and from spot plates after 5 days. Per-base mutation rates were determined as above (see p1 fluctuation test) but normalized by the target size of URA3 for 5-FOA resistance via substitution mutation (104 bp). 95% confidence intervals were similarly obtained as before.2
Determination of copy number via quantitative PCR
The copy numbers of p1-mUL* strains were determined by quantitative PCR (qPCR), as described previously.2,4 Whole-cell DNA extracts from 40 mL cultures were prepared by the large-scale cytoplasmic plasmid extraction protocol specific for qPCR, as described previously.2,4 All of the extracts were diluted 4,000-fold for qPCR reactions. As before, qPCR was performed using primers specific for leu2* and LEU3. qPCR reactions were performed in 20 μL mixtures containing template DNA (5 μL), forward primer (2 μl, 5 μM), reverse primer (2 μl, 5 μM), ddH2O (1 μl), and 2X Maxima SYBR Green/Fluorescein qPCR master mix (10 μl, Thermo Scientific).
Standard curves for both primer pairs were prepared using six serial fivefold dilutions of DNA extracted from the respective parental strain, which contain LEU2 and LEU3, and a non-template control with only ddH2O, in triplicates. All samples were measured in triplicate with both primer pairs. qPCR was performed using a Roche LightCycler 480 System using the following protocol:
95°C for 10min
95°C for 15s, 60°C for 1min; 40x
95°C for 1min
55°C for 1min
Cycle threshold (Ct) values were determined by the LightCycler 480 software (fit-points method, threshold = 1.75). Ct values from both standard curves were fit to a semi-log regression line plot of Ct versus log([DNA]). From this, a slope and y intercept were calculated, and relative copy numbers of samples were determined by the equation: copy number = 10((sample Ct – y intercept)/slope).2 Triplicate samples were averaged, and p1-derived plasmid copy numbers were normalized to those of the genome by dividing the average leu2* copy number by the average LEU3 copy number.
Supplementary Material
Acknowledgements
We thank members of our group for helpful discussions and suggestions. S. cerevisiae strains BY4741, CEN.PK2-1C, and W303-1A were generously gifted from J.E. Dueber (University of California, Berkeley). BY4743 was generously gifted from S. Sandmeyer (University of California, Irvine). We thank DARPA (HR0011-15-2-0031), the National Institutes of Health (1DP2GM119163-01), and the National Science Foundation (MCB1545158) for funding this work.
Footnotes
Supporting Information
Figures S1-S3: Additional analysis of p1/p2 for stability testing, testing for respiration competency in compatibility studies in r+ strains, further analysis of CRISPR/Cas9-based integration experiments
Tables S1-S2: List of plasmids and strains used in this study
References
- (1).Ravikumar A, Arzumanyan GA, Obadi MKA, Javanpour AA, and Liu CC (2018) Scalable, Continuous Evolution of Genes at Mutation Rates above Genomic Error Thresholds. Cell 175, 1946–1957.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ravikumar A, Arrieta A, and Liu CC (2014) An orthogonal DNA replication system in yeast. Nat. Chem. Biol 10, 175–177. [DOI] [PubMed] [Google Scholar]
- (3).Klassen Rolandand Meinhardt F (2007) Linear Protein-Primed Replicating Plasmids in Eukaryotic Microbes, in Microbial Linear Plasmids (Meinhardt Friedhelmand Klassen R, Ed.), pp 187–226. Springer Berlin Heidelberg, Berlin, Heidelberg. [Google Scholar]
- (4).Arzumanyan GA, Gabriel KN, Ravikumar A, Javanpour AA, and Liu CC (2018) Mutually Orthogonal DNA Replication Systems In Vivo. ACS Synth. Biol 7, 1722–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Gunge N, Murakami K, Takesako T, and Moriyama H (1990) Mating type locus-dependent stability of the Kluyveromyces linear pGKL plasmids in Saccharomyces cerevisiae. Yeast 6, 417–427. [DOI] [PubMed] [Google Scholar]
- (6).Gunge N, and Yamane C (1984) Incompatibility of linear DNA killer plasmids pGKL1 and pGKL2 from Kluyveromyces lactis with mitochondrial DNA from Saccharomyces cerevisiae. J. Bacteriol 159, 533–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kämper J, Esser K, Gunge N, and Meinhardt F (1991) Heterologous gene expression on the linear DNA killer plasmid from Kluyveromyces lactis. Curr. Genet 19, 109–118. [DOI] [PubMed] [Google Scholar]
- (8).Schickel J, Helmig C, and Meinhardt F (1996) Kluyveromyces lactis Killer System: Analysis of Cytoplasmic Promoters of the Linear Plasmids. Nucleic Acids Res. 24, 1879–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gunge N, and Sakaguchi K (1981) Intergeneric transfer of deoxyribonucleic acid killer plasmids, pGKl1 and pGKl2, from Kluyveromyces lactis into Saccharomyces cerevisiae by cell fusion. J. Bacteriol 147, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ryan OW, Skerker JM, Maurer MJ, Li X, Tsai JC, Poddar S, Lee ME, DeLoache W, Dueber JE, Arkin AP, and Cate JHD (2014) Selection of chromosomal DNA libraries using a multiplex CRISPR system. Elife 3, e03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ryan OW, and Cate JHD (2014) Chapter Twenty-Two - Multiplex Engineering of Industrial Yeast Genomes Using CRISPRm, in Methods in Enzymology (Doudna JA, and Sontheimer EJ, Eds.), pp 473–489. Academic Press. [DOI] [PubMed] [Google Scholar]
- (12).McNeel DG, and Tamanoi F (1991) Terminal region recognition factor 1, a DNA-binding protein recognizing the inverted terminal repeats of the pGKl linear DNA plasmids. Proc. Natl. Acad. Sci 88, 11398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Krupovic M, and Koonin EV (2014) Polintons: a hotbed of eukaryotic virus, transposon and plasmid evolution. Nat. Rev. Microbiol 13, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, and Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- (15).Gietz RD, and Schiestl RH (2007) High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc 2, 31. [DOI] [PubMed] [Google Scholar]
- (16).van Maris AJA, Backx A, van Rossum HM, Pronk JT, van den Broek M, Wijsman M, Kuijpers NGA, Daran-Lapujade P, Mans R, and Daran J-MG (2015) CRISPR/Cas9: a molecular Swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae. FEMS Yeast Res. 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sarkar S, Ma WT, and Sandri G v. H. (1992) On fluctuation analysis: a new, simple and efficient method for computing the expected number of mutants. Genetica 85, 173–179. [DOI] [PubMed] [Google Scholar]
- (18).Hall BM, Ma C-X, Singh KK, and Liang P (2009) Fluctuation AnaLysis CalculatOR: a web tool for the determination of mutation rate using Luria–Delbrück fluctuation analysis. Bioinformatics 25, 1564–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Herr AJ, Kennedy SR, Knowels GM, Schultz EM, and Preston BD (2014) DNA Replication Error-Induced Extinction of Diploid Yeast. Genetics 196, 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.