

Abstract
Epigenetic dysregulation of long noncoding RNA H19 was recently found to be associated with calcific aortic valve disease (CAVD) in humans by repressing NOTCH1 transcription. This finding offers a possible epigenetic explanation for the abundance of cases of CAVD that are not explained by any clear genetic mutation. In this study, we examined the effect of age and sex on epigenetic dysregulation of H19 and subsequent aortic stenosis. Cohorts of littermate, wild‐type C57BL/6 mice were studied at developmental ages analogous to human middle age through advanced age. Cardiac and aortic valve function were assessed with M‐mode echocardiography and pulsed wave Doppler ultrasound, respectively. Bisulfite sequencing was used to determine methylation‐based epigenetic regulation of H19, and RT‐PCR was used to determine changes in gene expression profiles. Male mice were found to have higher peak systolic velocities than females, with several of the oldest mice showing signs of early aortic stenosis. The imprinting control region of H19 was not hypomethylated with age, and H19 expression was lower in the aortic valves of older mice than in the youngest group. These results suggest that age‐related upregulation of H19 is not observed in murine aortic valves and that other factors may initiate H19‐related CAVD in humans.
Keywords: H19, calcific aortic valve disease, age, epigenetics
Introduction
Calcific aortic valve disease (CAVD) is an increasingly prevalent source of cardiovascular morbidity in the elderly, but identifiable genetic causes only explain a small fraction of disease cases (Lindman et al. 2016; Stewart et al. 1997; Osnabrugge et al. 2013). NOTCH1 loss of function mutation is one of the most widely studied genetic causes of CAVD, although such a mutation is not found in the majority of disease cases (Garg et al. 2005; Mohamed et al. 2006). Despite this discrepancy, alterations in the NOTCH1 signaling pathway offer a proven mechanism for CAVD, and downstream mechanisms for valve calcification are similar for Notch1‐driven CAVD in mice and idiopathic CAVD in humans (Hutcheson et al. 2014; Chen et al. 2015; Clark et al. 2017). As a result, upstream signaling or alternative mechanisms to mimic NOTCH1 loss of function have been sought as an explanation for idiopathic CAVD.
Recently, long noncoding RNA H19 has been found to be highly upregulated in stenotic and sclerotic human aortic valves (Hadji et al. 2016; Merryman and Clark 2016). Furthermore, H19 was shown to competitively bind to the promoter region of NOTCH1 in aortic valve interstitial cells, preventing P53 recruitment and subsequent NOTCH1 transcription (Fig. 1A). This effectively suppresses NOTCH1, leading to calcification even in the absence of a NOTCH1 mutation. However, this makes the means by which H19 expression becomes so dramatically upregulated a critical question to understand disease initiation.
Figure 1.
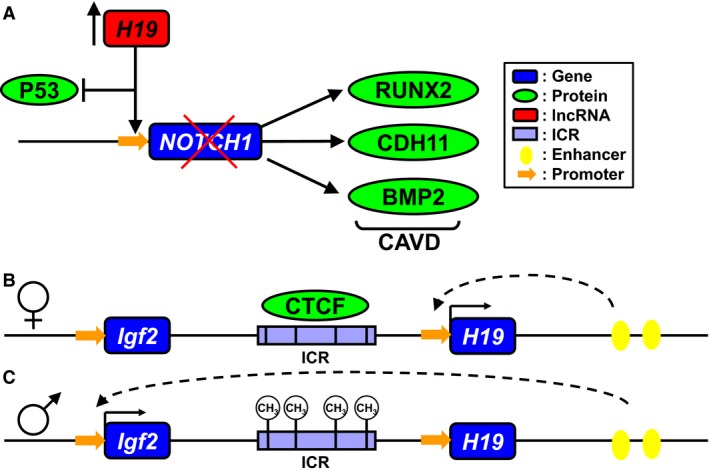
H19 imprinting and effect on CAVD. (A) High levels of H19 compete with P53 to bind the NOTCH1 promoter, decreasing expression of NOTCH1 and mimicking a loss‐of‐function mutation known to lead to CAVD. The H19/IGF2 locus contains a differentially methylated domain in the intergenic space. On the maternally inherited allele (B) CTCF binds to a series of four 21bp repeats, resulting in interaction of downstream enhancers with the H19 promoter and expression of H19. CTCF also acts as an insulator, keeping the downstream enhancers from promoting Igf2 expression. On the paternal allele (C) methylation of the differentially methylated domain prevents CTCF from binding, enabling enhancer interaction with the IGF2 promoter. IGF2 expression is increased, while H19 expression is almost entirely stopped. Hypomethylation in the ICR can lead to increased H19 expression
H19 is highly conserved among mammals and is found in an imprinted locus near insulin‐like growth factor 2 (Igf2) (Bartolomei et al. 1991). This locus is epigenetically regulated by an imprinting control region (ICR), located between the two genes (Fig. 1)B and C) (Bartolomei et al. 1991). On the maternally inherited allele, the ICR binds CCCTC‐binding factor (CTCF), which serves as an insulator between Igf2 and enhancer elements downstream of H19. As a result, shared enhancers promote expression of H19 while Igf2 is silenced (Fig. 1B). On the paternally inherited allele, methylation of the ICR prevents CTCF binding, allowing the downstream enhancers to activate Igf2 expression while H19 is silenced (Fig. 1C) (Engel et al. 2008). This imprint is established during embryonic development and persists through the life of the organism, though disruptions in this epigenetic signature could lead to rapid changes in H19 expression (Gabory et al. 2006).
Hadji et al. showed that hypomethylation in the promoter region of H19 was associated with increased expression in calcified human aortic valves, even though the imprint was maintained (Hadji et al. 2016). More recently, Agba et al. showed evidence for age‐associated hypomethylation in the H19/Igf2 ICR of rats, suggesting a loss of imprint which correlated with increased H19 expression (Agba et al. 2017). Together, these findings suggest an epigenetic mechanism by which H19 may become upregulated with advanced age and lead to CAVD via the NOTCH1 pathway, even in the absence of a genetic mutation.
The goal of this study was to determine if these findings were replicated in mouse aortic valves, and if age‐related H19 expression is a mechanism for CAVD in a mouse model. Because H19 is known to be involved in other cardiovascular diseases, expression levels were also assessed in ventricular tissue and the aortic arch, as well as the liver, which is known to express higher levels of H19 and was previously shown to exhibit age‐related loss of imprint. While many existing mouse models of CAVD consider only male mice, CAVD is also highly prevalent in women, thus motivating our investigation into the effect of sex on H19‐driven calcification (Owens et al. 2010; Simard et al. 2017). Because our primary interest was in the effects of age and sex (rather than genetic mutation or chronic injury through diet) on H19 expression in the aortic valve, these experiments were conducted in healthy C57BL/6 mice maintained on a normal diet.
Methods
Mice
Groups of five male and female C57BL/6 mice were purchased from Jackson Laboratory at 26, 52, and 78 weeks of age. Mice were assessed via echocardiography and euthanized for sample collection within two weeks of receipt. All procedures were performed in accordance with protocols approved by Institutional Animal Care and Use Committee (IACUC) at Vanderbilt University.
Echocardiography
Mice were anesthetized with isofluorane, and a Vevo 2100 imaging system was used to acquire parasternal, short axis M‐mode images of the heart and pulsed‐wave (PW) Doppler images of the aorta immediately distal to the aortic valve. An exemplary PW Doppler scan and corresponding flow profile traces are shown in Figure 2A and B. VevoLAB software was used to analyze M‐mode cardiac cycles (~9 per image) and extract PW Doppler images. A custom MATLAB script was used to isolate and average PW Doppler cardiac cycles (~10–30 per image) in order to compute the systolic transvalvular pressure gradient and peak systolic velocity (PSV) (Ferruzzi et al. 2018). Ejection fraction to velocity ratio (EFVR), a metric used as an indicator of valve disease, was calculated as EFVR = (ejection fraction)/(4*(PSV)2) (Cattaneo et al. 2009).
Figure 2.
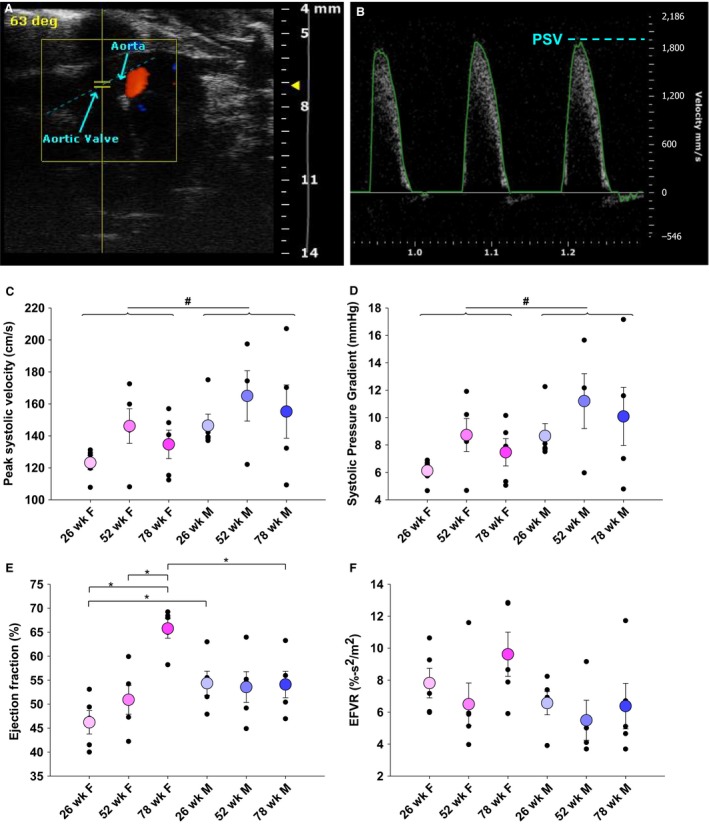
Valve health in male mice diverges with age. Pulsed wave Doppler ultrasound of aortic flow immediately distal to the aortic valve (A) with representative flow profile (B). Echocardiography found elevated PSV and transvalvular pressure gradient in male mice (P = 0.032, P = 0.032) and trends of higher and more divergent velocities and pressure gradient with increasing age in male mice (C and D). Ejection fraction was consistent for males, but increased with age in females (E). EFVR showed similar trends as PSV and pressure gradient (F). *P < 0.05 between individual groups by two‐way ANOVA; # P < 0.05 between sex by two‐way ANOVA; PSV: peak systolic velocity; EFVR: ejection fraction to velocity ratio
Microdissection and sample collection
Mice were euthanized by carbon dioxide inhalation and promptly dissected. The systemic and pulmonary circulatory systems were flushed with sterile PBS, and the aortic valve leaflets – connected to a minimal annulus of aorta – were dissected away from the ventricles. Samples of liver, left ventricle, and ascending aorta were also harvested and cleaned of external connective tissue and fat. Samples were flash frozen in liquid nitrogen and stored at −80°C until RT‐PCR analysis.
Nucleic acid purification
Tissue samples were thawed at room temperature and bead homogenized in 400 μL of RLT‐plus buffer with Reagent DX to reduce foaming (Qiagen, Hilden, Germany) in Lysing Matrix D tubes (MP Biomedicals, Santa Ana, CA) until no visible tissue remained. RNA and gDNA were purified using the Qiagen AllPrep Micro kit following manufacturer’s instructions and were stored at −80°C until further analysis.
Real‐time PCR
Reverse transcription was performed using the SuperScript IV Reverse Transcriptase kit with oligo(dT) primer (ThermoFisher Scientific, Waltham, MA). RT‐PCR was performed with equal amounts of cDNA using iQ SYBR Green Supermix (Bio‐Rad, Hercules, CA) and gene specific primer sequences (Table 1). Gene expression was normalized to the geometric mean expression of Gapdh, Tuba1b, and Actb, which was found to be more stable than any housekeeping gene in isolation. For visual clarity, gene expression was normalized to the highest expressing sample of each gene.
Table 1.
RT‐PCR primer sequences
| Gene name | Forward primer1 | Reverse primer1 |
|---|---|---|
| Gapdh | ATGACAATGAATACGGCTACAG | TCTCTTGCTCAGTGTCCTTG |
| Tuba1b | CCGGTGTCTGCTTCTATCTC | CCATGTTCCAGGCAGTAGAG |
| Actb | CAAGCAGGAGTACGATGAGTC | AACGCAGCTCAGTAACAGTC |
| H19 | GGAATGTTGAAGGACTGAGGG | GTAACCGGGATGAATGTCTGG |
| Igf2 | CGCTTCAGTTTGTCTGTTCG | GCAGCACTCTTCCACGATG |
| Notch1 | ATGTCAATGTTCGAGGACCAG | TCACTGTTGCCTGTCTCAAG |
| Bmp2 | TTATCAGGACATGGTTGTGGAG | GGGAAATATTAAAGTGTCAGCTGG |
Primer sequences are from 5′‐3′.
Methylation analysis
Pyrosequencing was performed as previously described with the following modifications. 40 ng of bisulfite treated DNA was used as input, and 5 μL of the biotinylated PCR product was used for each sequencing assay (Susiarjo et al. 2013).
Statistics
All values are presented as mean ± standard error. Two‐way ANOVA with post hoc Holm‐Sidak test for multiple comparisons was used to detect differences between age and sex. In the event that conditions of normality or equal variance were not met, one‐way ANOVA on ranks was used to detect differences due to age within each sex, and the Mann‐Whitney rank sum test was used to detect difference due to sex at a specific age. Potential correlations between measured variables were assessed by the Pearson product‐moment correlation r. For all statistical tests, a value of P < 0.05 was considered significant.
Results
Aortic valve health diverges, but does not worsen with age
Analysis of PW Doppler images revealed no changes with age in PSV or peak transvalvular pressure gradient, although as whole cohorts by sex, male mice had higher PSV than females (P = 0.032) (Fig. 2C and D). While typical values for aortic PSV in healthy BL6 mice fall below 150 cm/sec, we identified one 78‐week‐old male mouse had a PSV over 200 cm/sec, indicative of aortic stenosis, and several older male and female mice had velocities between 150 and 200 cm/sec, indicative of early stenosis (Hinton et al. 2008). Male mice also had higher systolic gradients (P = 0.032). None of the mice showed signs of left ventricular hypertrophy, indicating that observed hemodynamic changes were early stage, prior to extensive cardiac remodeling. Curiously, the 52‐ and 78‐week‐old male mice had two and three times larger variance in PSV than younger males, indicating a wide divergence of overall valve health with increased age.
Cardiac function is preserved with age
Ejection fraction was extremely consistent with age in male mice (Fig. 2E). Female mice exhibited increasing ejection fraction with age, such that 26‐week‐old females had lower ejection fraction than 26‐week‐old male mice (P = 0.042) and 78‐week‐old female mice had higher ejection fractions than 78‐week‐old males (P = 0.005). Seventy‐eight‐week‐old females also had higher ejection fractions than 26‐ and 52‐week‐old females (P = 0.017, P = 0.025). No differences were detected in EFVR, although males again showed trends toward divergent valve health with age (Fig. 2F).
H19 imprint and expression are not altered by age or sex in the aortic valve
Pyrosequencing of the H19 ICR in mouse aortic valve gDNA revealed no change in average methylation fraction due to age or sex (Fig. 3A). Likewise, there was no observed trend toward increased H19 expression in the valve with increasing age nor correlation between methylation fraction and H19 expression (Fig. 3B and C). Rather, 26‐week‐old mice had higher average H19 expression than either the 52‐ or 78‐week‐old mice (P = 0.025; P = 0.017). Expression of Igf2, which is regulated by the H19/Igf2 ICR, and Notch1, which was previously shown to be repressed by H19, also showed no clear age or gender effect, though individual comparisons reached statistical significance (Fig. 3C). Bmp2, a driver of osteogenic calcification that has been found to be increased in calcified valves, was higher in male mice than in females overall (P = 0.002) and specifically at 26 and 78 weeks of age (P = 0.015; P = 0.017). To examine if H19 may still be suppressing Notch1 on an individual level that is not revealed in group‐averaged data, we also correlated expression of H19 and Notch1 in individual mice, but no significant effect of H19 expression on Notch1 expression was detected (Fig. 3D).
Figure 3.
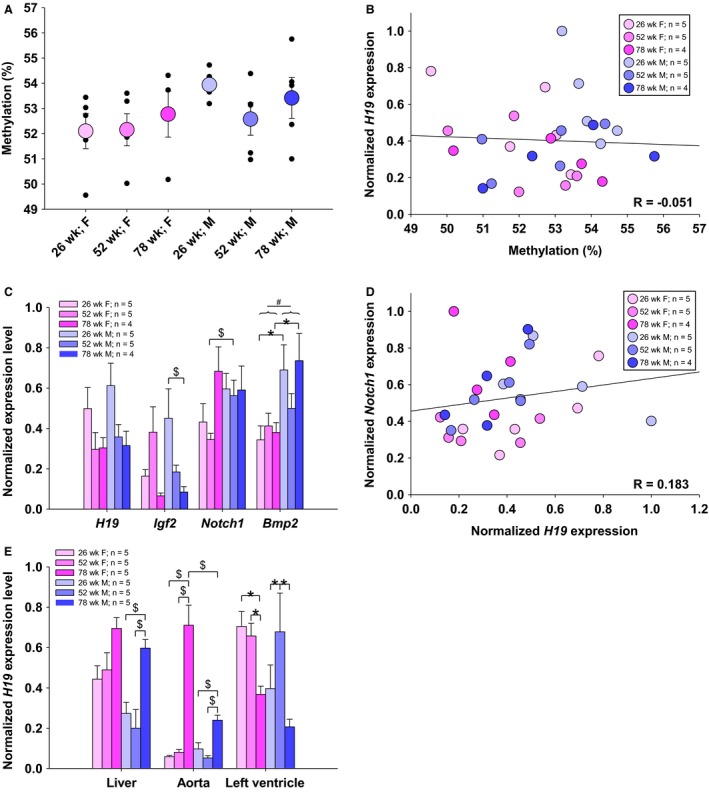
Methylation and expression of H19 and downstream genes is largely unchanged with age and sex. H19 ICR methylation was unchanged in all samples studied (A) and did not correlate with H19 expression (B). Expression of H19 was significantly higher in 26‐week‐old mice than in 52‐week‐old mice (P = 0.011) and 78‐week‐old mice (P = 0.012), but did not correlate with Notch1 expression (C, D). Igf2 and Notch1 were found to be differ in individual comparisons, and Bmp2 was higher in males than in females (P = 0.002). H19 expression was higher at 78 weeks in the liver and aorta of male mice and in the aortas of female mice. No clear trend in H19 expression was found in the left ventricle, although 78‐week‐old female mice had lower expression (P = 0.005, 78 week to 26 week; P = 0.014, 78 week to 52 week) and 52‐week‐old males had higher expression (P < 0.001, 52 week to 26 week; P < 0.001, 52 week to 78 week) than other aged mice of the same sex. *P < 0.05 between individual groups by two‐way ANOVA; $ P < 0.05 between individual groups by ANOVA on ranks or Mann‐Whitney; # P < 0.05 between sex by two‐way ANOVA
Liver and aortic tissue show increased H19 expression in oldest mice
Based on other studies showing H19 upregulation in aortic aneurysm and cardiac ischemia, as well as a study that found age‐related loss of imprint in rat liver, we also probed H19 expression changes in the liver, left ventricle, and ascending aorta (Greco et al. 2016; Agba et al. 2017; Li et al. 2018) (Fig. 3E). The 78‐week‐old male mice had higher H19 expression than other age males in both the liver and ascending aorta. H19 expression trended upwards with age in female liver, while the ascending aorta of 78‐week‐old female mice showed higher expression than the other female age groups and the 78‐week‐old male mice. In left ventricular tissue, no clear pattern of age‐related H19 upregulation was observed, though the 78‐week‐old female mice had significantly lower H19 expression than other female age groups, and 52‐week‐old males had higher expression than other male age groups.
Discussion
This study investigated the effects of age and sex on hemodynamic function and H19 expression in the aortic valve. To the best of our knowledge, this is the first cross‐sectional study of a potential CAVD‐initiating mechanism in both male and female mice that did not have a CAVD‐associated mutation or receive a chronic hypercholesteremic treatment. Furthermore, this study isolated nucleic acids from individual aortic valves, enabling expression profiling and methylation analysis without requiring sample pooling. This approach represents a novel approach to probe subtle signaling which may precede or initiate valve disease in mice without introducing the biases of specific models, whereas most studies (including our own (Clark et al. 2017)) typically utilize specific mutations, diet, or cardiovascular injury to induce symptoms of CAVD, which may bias or obscure subtle signaling changes early in disease progression.
We found that the H19/Igf2 ICR does not undergo age‐related hypomethylation and H19 is not upregulated in mouse aortic valves with age alone. This contrasts with expression data from other tissues such as liver and ascending aorta, as well as with prior studies in rats that showed loss of ICR methylation and H19 upregulation in a tissues such as brain and skin (Agba et al. 2017). The rats used in that study and the mice used here included animals comparable to 60‐ to 70‐year‐old humans, well within a timeframe at which early signs of CAVD could be expected. These results suggest a mechanism for preserving H19 imprinting and low expression levels that differs by tissue type or species.
In addition to the lack of a robust increase in H19 expression, we did not observe substantial correlation between expression of Notch1 and H19 in the aortic valve. Previous studies have shown that elevated H19 can repress NOTCH1 in human aortic valve interstitial cells and mouse brain tissue (Hadji et al. 2016). A potential explanation for this discrepancy is a threshold effect, in which H19 expression must reach a certain critical level before measurably repressing Notch1. Further work with a titratable H19 overexpression system may be a useful tool to answer this question.
Despite the lack of strong H19 upregulation, a few of the oldest mice showed signs of early aortic stenosis as indicated by elevated PSV and transvalvular pressure gradient. These functional indicators of stenosis even in the absence of H19 upregulation underscore the heterogeneous nature of CAVD and suggest that H19 is not the sole initiator of disease. Nearly 30 different mouse models of CAVD exist, and while many of these models are convergent, it is clear that multiple distinct mechanism can initiate and drive CAVD (Sider et al. 2011; Hutcheson et al. 2014; Lerman et al. 2015).
Certain sex‐related differences emerged in this study which corroborate known statistics of human disease. For example, male mice had higher overall PSV values that trended upwards with age and higher Bmp2 expression, indicative of onset of stenosis with activation of calcific pathways. This matches with clinical data showing that men are more likely to develop CAVD than women and that features of the disease (fibrosis and calcification) differ between sexes (Owens et al. 2010; Simard et al. 2017). Still, CAVD is relatively prevalent in women, and the differences in disease progression between sexes may direct more personalized treatment strategies. Despite this, many studies either do not distinguish between male and female mice or use only males. The work presented here included both male and female mice in order to better capture any differences in H19 regulation and valve stenosis.
Although age‐related upregulation of H19 was not found in the aortic valve, it was confirmed in the liver and reported for the first time in the ascending aorta. It was also not found to increase in the left ventricle, showing a high degree of tissue specificity in overall expression regulation. Many studies have identified H19 as a biomarker or driver of other cardiovascular diseases such as aortic aneurysm, smooth muscle cell apoptosis, endothelial cell aging, and ischemic heart failure (Greco et al. 2016; Li et al. 2018; Hofmann et al. 2019). Our findings of tissue‐specific differences in age‐related upregulation of H19 may inform future work looking at disease initiating events in these other pathologies.
This study is not without its limitations. One of the biggest challenges was the sample size – both of individual aortic valves and the overall size of the cohort. To obtain testable quantities of RNA and DNA, valves were used entirely for nucleic acid extraction. Alternate methodologies such as histology, immunohistochemistry, or in situ hybridization may have revealed more information about the extent of valve remodeling and stenosis. Furthermore, the high cost of raising or purchasing aged mice prevented higher numbers in each cohort, which may have increased the statistical power and significance of these results. Finally, the methylation sites tested in this study were within the ICR, which is typically considered to be the site of most epigenetic regulation of H19 expression and was shown to be hypomethylated with age in work by Agba et al. (2017). However, Hadji et al. report a stronger association of H19 expression with a particular CpG site in the promoter region of H19 (Hadji et al. 2016). Despite this difference, we did not observe the increase in H19 expression that was clearly reported in their work.
This work shows a lack of age‐related epigenetic dysregulation of H19 in mouse aortic valves, even while other tissue types demonstrated consistent upregulation of H19 expression. Nevertheless, echocardiographic metrics and changes in gene expression in individual mice showed signs of valve remodeling and early stenosis. Together, these results show that H19 loss of imprint and subsequent upregulation are not common in the aortic valve of old mice, and that increased H19 levels do not appear to be a prerequisite for early stage valve disease.
Conflict of Interest
No conflicts of interest, financial or otherwise, are declared by the authors.
Acknowledgments
We thank the Vanderbilt University Mouse Cardiovascular Pathophysiology Core which performed echocardiography. We also thank Matthew Bersi and Alison Schroer for their editorial help in manuscript preparation.
Vander Roest Mark, Krapp Christopher, Thorvaldsen Joanne L., Bartolomei Marisa S., Merryman W. David. Physiol Rep, 7 (19), 2019, e14244, 10.14814/phy2.14244
Funding Information
This work was supported by the National Institute of General Medical Sciences (GM051279 and HL007411); Fondation Leducq; and National Heart, Lung, and Blood Institute (HL135790).
References
- Agba, O. B. , Lausser L., Huse K., Bergmeier C., Jahn N., Groth M., et al. 2017. Tissue‐, sex‐, and age‐specific DNA methylation of rat glucocorticoid receptor gene promoter and insulin‐like growth factor 2 imprinting control region. Physiol. Genomics 49:690–702. 10.1152/physiolgenomics.00009.2017. [DOI] [PubMed] [Google Scholar]
- Bartolomei, M. S. , Zemel S., and Tilghman S. M.. 1991. Parental imprinting of the mouse H19 gene. Nature 351:153–155. 10.1038/351153a0 [DOI] [PubMed] [Google Scholar]
- Cattaneo, P. , Marchetti P., Baravelli M., Rossi A., Mariscalco G., Ghiringhelli S., et al. 2009. Ejection fraction‐velocity ratio for the assessment of aortic bioprosthetic valves in patients with systolic dysfunction. Can. J. Cardiol. 25:e78–e81. 10.1016/s0828-282x(09)70046-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Ryzhova L. M., Sewell‐Loftin M. K., Brown C. B., Huppert S. S., Baldwin H. S.. 2015. Notch1 mutation leads to valvular calcification through enhanced myofibroblast mechanotransduction. Arterioscler. Thromb. Vasc. Biol. 35:1597–1605. 10.1161/atvbaha.114.305095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, C. R. , Bowler M. A., Snider J. C., and Merryman W. D.. 2017. Targeting cadherin‐11 Prevents Notch1‐mediated calcific aortic valve disease. Circulation 135:2448–2450. 10.1161/circulationaha.117.027771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel, N. , Raval A. K., Thorvaldsen J. L., and Bartolomei S. M.. 2008. Three‐dimensional conformation at the H19/Igf2 locus supports a model of enhancer tracking. Hum. Mol. Genet. 17:3021–3029. 10.1093/hmg/ddn200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferruzzi, J. , Di Achille P., Tellides G., and Humphrey J. D.. 2018. Combining in vivo and in vitro biomechanical data reveals key roles of perivascular tethering in central artery function. PLoS ONE 13:e0201379 10.1371/journal.pone.0201379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabory, A. , Ripoche M.‐A., Yoshimizu T., and Dandolo L.. 2006. The H19 gene: regulation and function of a non‐coding RNA. Cytogenet. Genome Res. 113:188–193. 10.1159/000090831 [DOI] [PubMed] [Google Scholar]
- Garg, V. , Muth A. N., Ransom J. F., Schluterman M. K., Barnes R., King I. N., et al. 2005. Mutations in NOTCH1 cause aortic valve disease. Nature 437:270–274. 10.1038/nature03940 [DOI] [PubMed] [Google Scholar]
- Greco, S. , Zaccagnini G., Perfetti A., Fuschi P., Valaperta R., Voellenkle C., et al. 2016. Long noncoding RNA dysregulation in ischemic heart failure. J. Transl. Med. 14:183 10.1186/s12967-016-0926-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadji, F. , Boulanger M. C., Guay S. P., Gaudreault N., Amellah S., Mkannez G., et al. 2016. Altered DNA methylation of long noncoding RNA H19 in calcific aortic valve disease promotes mineralization by silencing NOTCH1 . Circulation 134:1848–1862. 10.1161/circulationaha.116.023116 [DOI] [PubMed] [Google Scholar]
- Hinton, R. B. , Alfieri C. M., Witt S. A., Glascock B. J., Khoury P. R., Benson D. W., et al. 2008. Mouse heart valve structure and function: echocardiographic and morphometric analyses from the fetus through the aged adult. Am. J. Physiol. Circ. Physiol. 294:H2480–H2488. 10.1152/ajpheart.91431.2007 [DOI] [PubMed] [Google Scholar]
- Hofmann, P. , Sommer J., Theodorou K., Kirchhof L., Fischer A., Li Y., et al. 2019. Long non‐coding RNA H19 regulates endothelial cell aging via inhibition of STAT3 signalling. Cardiovasc. Res. 115:230–242. 10.1093/cvr/cvy206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutcheson, J. D. , Aikawa E., and Merryman W. D.. 2014. Potential drug targets for calcific aortic valve disease. Nat. Rev. Cardiol. 11:218–231. 10.1038/nrcardio.2014.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerman, D. A. , Prasad S., and Alotti N.. 2015. Calcific aortic valve disease: molecular mechanisms and therapeutic approaches. Eur. Cardiol. 10:108–112. 10.15420/ecr.2015.10.2.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D. Y. , Busch A., Jin H., Chernogubova E., Pelisek J., Karlsson J., et al. 2018. H19 induces abdominal aortic aneurysm development and progression. Circulation 138:1551–1568. 10.1161/circulationaha.117.032184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindman, B. R. , Clavel M.‐A., Mathieu P., Iung B., Lancellotti P., Otto C. M., et al. 2016. Calcific aortic stenosis. Nat. Rev. Dis. Prim. 2:16006 10.1038/nrdp.2016.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merryman, W. D. , and Clark C. R.. 2016. Lnc‐ing NOTCH1 to idiopathic calcific aortic valve disease. Circulation 134:1863–1865. 10.1161/circulationaha.116.025601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed, S. A. , Aherrahrou Z., Liptau H., Erasmi A. W., Hagemann C., Wrobel S., et al. 2006. Novel missense mutations (p.T596M and p.P1797H) in NOTCH1 in patients with bicuspid aortic valve. Biochem. Biophys. Res. Commun. 345:1460–1465. 10.1016/j.bbrc.2006.05.046 [DOI] [PubMed] [Google Scholar]
- Osnabrugge, R. L. J. , Mylotte D., Head S. J., Van Mieghem N. M., Nkomo V. T., LeReun C. M., et al. 2013. Aortic stenosis in the elderly. J. Am. Coll. Cardiol. 62:1002–1012. 10.1016/j.jacc.2013.05.015 [DOI] [PubMed] [Google Scholar]
- Owens, D. S. , Katz R., Takasu J., Kronmal R., Budoff M. J., and O’Brien K. D.. 2010. Incidence and progression of aortic valve calcium in the multi‐ethnic study of atherosclerosis (MESA). Am. J. Cardiol. 105:701–708. 10.1016/j.amjcard.2009.10.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sider, K. L. , Blaser M. C., and Simmons C. A.. 2011. Animal models of calcific aortic valve disease. Int. J. Inflamm. 2011:364310 10.4061/2011/364310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard, L. , Côté N., Dagenais F., Mathieu P., Couture C., Trahan S., et al. 2017. Sex‐related discordance between aortic valve calcification and hemodynamic severity of aortic stenosis. Circ. Res. 120:681–691. 10.1161/circresaha.116.309306 [DOI] [PubMed] [Google Scholar]
- Stewart, B. F. F. , Siscovick D., Lind B. K., Gardin J. M., Gottdiener J. S., and Smith V. E., et al. 1997. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J. Am. Coll. Cardiol. 29:630–634. 10.1016/s0735-1097(96)00563-3 [DOI] [PubMed] [Google Scholar]
- Susiarjo, M. , Sasson I., Mesaros C., and Bartolomei M. S.. 2013. Bisphenol A exposure disrupts genomic imprinting in the mouse. PLoS Genet. 9:e1003401 10.1371/journal.pgen.1003401 [DOI] [PMC free article] [PubMed] [Google Scholar]